
Abstract
Cigarette smoking-related inflammation, cellular stresses, and tissue destruction play a key role in lung disease, such as chronic obstructive pulmonary disease (COPD). Notably, augmented apoptosis and impaired clearance of apoptotic cells, efferocytosis, contribute to the chronic inflammatory response and tissue destruction in patients with COPD. Of note, exposure to cigarette smoke can impair alveolar macrophages efferocytosis activity, which leads to secondary necrosis formation and tissue inflammation. A better understanding of the processes behind the effect of cigarette smoke on efferocytosis concerning lung disorders can help to design more efficient treatment approaches and also delay the development of lung disease, such as COPD. To this end, we aimed to seek mechanisms underlying the impairing effect of cigarette smoke on macrophages-mediated efferocytosis in COPD. Further, available therapeutic opportunities for restoring efferocytosis activity and ameliorating respiratory tract inflammation in smokers with COPD were also discussed.
Abbreviations
| A1AT | = | Acute phase-reactant alpha-1 antitrypsin |
| ACs | = | Apoptotic cells |
| AMs | = | Alveolar macrophages |
| aSMase | = | Acid sphingomyelinase |
| COPD | = | Chronic obstructive pulmonary disease |
| CS | = | Cigarette smoke |
| CSE | = | CS extract |
| CXCL8 | = | C-X-C Motif Chemokine Ligand 8 |
| DAMPs | = | Damage-associated molecular patterns |
| FDA | = | Food and Drug Administration |
| EGFR | = | Epidermal growth factor receptor |
| EMPs | = | Endothelial microparticles |
| ERK1/2 | = | Extracellular signal-regulated kinase 1/2 |
| EVs | = | Extracellular vesicles |
| GM-CSF | = | Granulocyte–macrophage colony-stimulating factor |
| GSH | = | Glutathione |
| HDAC | = | Histone deacetylase |
| HIF-1 | = | Hypoxia inducible factor-1 |
| HMGB1 | = | High mobility group box 1 |
| Hsp27 | = | Heat shock protein 27 |
| IL | = | Interleukin |
| MBL | = | Mannose binding lectin |
| MerTK | = | Mer tyrosine kinase |
| MGF-E8 | = | Milk fat globule‑EGF factor 8 |
| MAPKAPK | = | Mitogen-activated protein kinase-activated protein kinase |
| MMP | = | Matrix metalloproteinases |
| NK | = | Natural killer |
| pDCs | = | plasmacytoid dendritic cells |
| PGE2 | = | Prostaglandin E2 |
| PPAR-γ | = | Peroxisome proliferator-activated receptor gammaPtdSerPhosphatidylserine |
| rmMFG-E8 | = | Recombinant mouse MFG-E8 |
| RNS | = | Reactive nitrogen species |
| ROS | = | Reactive oxygen species |
| RvD1 | = | Resolvin D1 |
| SHS | = | Secondhand smoke |
| SK | = | Sphingosine kinase |
| SMase | = | Sphingomyelinase |
| S1P | = | Sphingosine 1-phosphate |
| S1PR5 | = | Sphingosine-1 phosphate receptor 5 |
| S1PRs | = | S1P receptors |
| TSA | = | Trichostatin A |
| Tc | = | Cytotoxic T cell |
| TGF-β | = | Transforming growth factor-beta |
| TNF-α | = | Tumour necrosis factor-alpha |
Introduction
The pro-inflammatory immune responses are controlled through the inflammation resolution process. This involves the clearance of damaged cells and pro-inflammatory immune cells through coordinated processes of apoptosis and efferocytosis. Impaired efferocytosis [apoptotic cell (AC) clearance], augmented apoptosis, and abnormal tissue remodeling result in the chronic inflammatory response and tissue destruction in lung disease, particularly chronic obstructive pulmonary disease (COPD) () [Citation1–5]. COPD is a chronic lung disease that includes emphysema and chronic bronchitis as two main forms of COPD [Citation6]. Immunity, genetic, and environmental factors are important elements for the development of COPD. The association between exposure to respiratory contaminants and disease progression is clear for COPD. Cigarette smoke (CS), as an environmental factor initiating inflammatory cascades, is a major risk factor of COPD [Citation7–9], in which a long-time chronic causes the expansion of COPD [Citation7]. The “danger signals” elicited by the injured cells of nonspecific immunity stimulate the activation of the downstream pro-inflammatory cascades and antigen-specific adaptive immune responses [Citation1]. Moreover, CS activates c-Src and thereby augments airway inflammation and the destruction of lung tissue. c-Src can modulate the tyrosine kinase receptor epidermal growth factor receptor (EGFR) and also induce interleukin (IL)-17, matrix metalloproteinases (MMP)-9, and MMP-12 signals in the lung [Citation10, Citation11]. MMPs involve in promoting inflammation, inducing proteolytic attack on the alveolar wall matrix, and modifying the release of fibrogenic growth factors in COPD [Citation12]. MMP-1, MMP-8, MMP-9, and MMP-12 are associated with the pathophysiological processes underlying COPD, particularly in emphysema [Citation12–14].
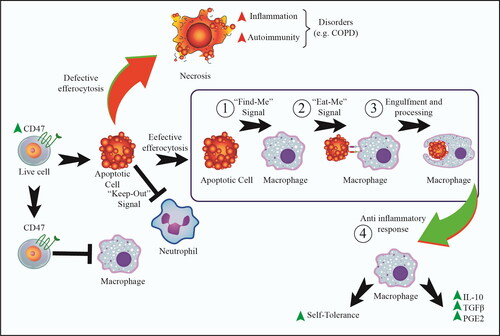
Figure 1. The main efferocytosis signaling and steps. These signals are related to the apoptotic cells (ACs), characterizing by ‘Find-Me’ signals and also the absence of ‘Don’t Eat-Me’ signals. ‘Don’t Eat-Me’ signals, such as CD47 and CD31 commonly found on living cells. However, there are major steps in efferocytosis including: (1): ‘Find-Me’ signals (to attract phagocytes to sites of apoptotic cell death), (2): ‘Eat-Me’ signal (to catch ACs), and (3): the processes of engulfment and digestion. Ultimately, effective efferocytosis results in 4): the production of anti-inflammatory cytokines, such as IL-10 and TGF-β. Moreover, an additional steps include ‘Keep-Out’ signal helps to efferocytosis to reduce number of unwanted neutrophils. On the other hand, defective efferocytosis can lead to necrosis, which in turn increases inflammation and inflammatory diseases, such as COPD. In this line, cigarette smoke can impair efferocytosis activity, which leads to necrosis and tissue inflammation.
Further, Notch signaling is diminished in the airway epithelium of COPD through the reduced expression of Notch-related genes, such as NOTCH3, DLL1, HES5, HEY1, and HEY2 in smokers with COPD [Citation15]. The Notch signaling pathway serves multiple functions, such as determining the fate of cells in different organ systems and as a gatekeeper for epithelial differentiation [Citation16–18]. It is suggested that, by continuing epithelial injury mediated by CS, Notch signaling is down-regulated through COPD to facilitate differentiation and restoration of the epithelium of the airways [Citation15].
CS also induces hypoxia-inducible factor-1 (HIF-1)-mediated signaling, which leads to the promotion of airway inflammation and activation of fibroblasts and myofibroblasts, resulting in fibrous remodeling of the subepithelium [Citation19, Citation20]. Moreover, remodeling of subepithelial connective tissue and the decreased microvasculature affect the bronchial epithelium microenvironment and provide conditions for the activation of hypoxia-inducible signaling in epithelial cells [Citation21], thus promoting hyperplasia of mucin-producing goblet cells and progression of epithelial structural abnormalities in COPD [Citation22, Citation23].
Of note, CS can also impair efferocytosis; exposure to CS alters the phenotype and function of alveolar macrophages (AMs) and inhibits their efferocytosis activity [Citation24–29]. Defective efferocytosis can lead to secondary necrosis formation and release damage-associated molecular patterns (DAMPs), such as high mobility group box 1 (HMGB1) [Citation30, Citation31]. Several efferocytosis-related factors/pathways are associated with CS and lung disorders, such as lectins, RhoA, Rac, Rab5 and Rab7, CD36, CD44, peroxisome proliferator-activated receptor gamma (PPAR-γ), macrophage polarization, oxidative stress, the resolution of inflammation, sphingosine 1-phosphate (S1P), phosphatidylserine (PtdSer), Mer tyrosine kinase (MerTK), galectin-3, milk fat globule‑EGF factor 8 (MGF-E8), formyl peptide receptor 2/3 (FPR2/3), annexin A1 and endothelial function in efferocytosis [Citation25, Citation26, Citation32–39] (). Here, we thus attempted to seek factors/pathways and mechanisms that govern the effect of CS on efferocytosis in COPD.
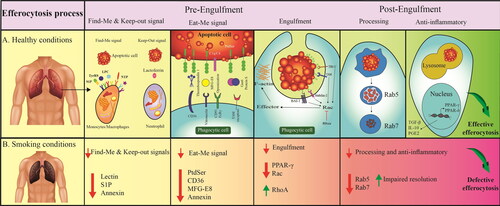
Figure 2. Effective and defective efferocytosis signaling. A: Effective efferocytosis in the physiologic condition that is regulated by certain pathways/factors; B: Defective efferocytosis in pathophysiological conditions caused by cigarette smoking. Abbreviations: BAI-1: Brain-specific angiogenesis factor 1; C1q: Complement component 1q; CD36: Cluster of differentiation 36; FCRγ: Fc receptor γ-chain; Gas6: Growth Arrest Specific 6; IL-10: Interleukin 10; LPC: Lysophosphatidylcholine; MerTK: Mer tyrosine kinase; MGF-E8: Milk fat globule‑EGF factor 8; NTP: Nucleoside triphosphate; PGE2: Prostaglandin E2; PPAR: Peroxisome proliferator activated receptor; PtdSer: Phosphatidylserine; S1P: Sphingosine 1-phosphate; TGF-β: Transforming growth factor beta; TIM-1: T-cell immunoglobulin and mucin domain 1; TyrRS: Tyrosyl-tRNA synthetase.
Efferocytosis regulation and its mechanisms
Efferocytosis is the homeostatic phagocytosis of ACs, preventing the release of dangerous intracellular contents and subsequent tissue damage. Efferocytosis includes two main steps; pre-engulfment (“Find-Me” signals, “Eat-Me” signals, and engulfment) and post-engulfment (anti-inflammatory responses and self-tolerance) ( and ). All stages are important and, hence, a defect in one of these steps triggers the failure in the whole process. Efferocytosis as an anti-inflammatory process by releasing IL-10, transforming growth factor-beta (TGF-β) as well as prostaglandin E2 (PGE2) eventually results in decreased pro-inflammatory and increased anti-inflammatory responses as well as increased self-tolerance. Thus, defective efferocytosis will result in chronic inflammation that in turn causes autoimmune and inflammatory disorders, such as lung disorder COPD ( and ) [Citation30, Citation40–44].
Table 1. The main efferocytosis related-molecules.
COPD and defective efferocytosis
The impaired phagocytic function of AMs specific for uptaking and the increased numbers of ACs in COPD patients are the signs of defective efferocytosis [Citation45, Citation46]. COPD smoker patients have greater efferocytosis deficiency [Citation27]. CS decreases efferocytosis of ACs by AM. This deficit of AM engulfment exacerbates outcomes of COPD pathology in active smokers or AM exposed to CS ex-vivo [Citation47]. The effect of CS in the regulation of phagocytosis has been shown by the ability of AM to engulf apoptotic bronchial epithelial cells. This engulfment is reduced in current smokers with COPD and in healthy smokers compared with never-smoker control individuals [Citation24]. CS also alters the functional activity of macrophages. In this case, CS has been shown to reduce the expression of CD31 (LDL receptor–related protein) and CD91 (PECAM) in AMs, leading to a decrease in the phagocytic capacity of AMs [Citation24].
Defective efferocytosis of ACs leads to sustained lung inflammation in COPD [Citation48]. The involvement of an autoimmune response, destruction of the extracellular matrix, defective efferocytosis, and tissue hypoxia have been proposed to be factors contributing to the persistence of inflammation after the cessation of smoking [Citation49]. However, defective efferocytosis can be improved upon give up smoking. These findings suggest a direct role of CS in the pathogenesis of defective efferocytosis [Citation24]. In this respect, efferocytosis has been higher in COPD ex-smokers compared with COPD current smokers [Citation24, Citation50]. Compared with COPD ex-smokers, current COPD smokers have impaired AM phagocytosis and have lower levels of CD163 and CD36 in AMs [Citation50]. Moreover, the expression level of CD91 was indicated to be reduced in AMs from non-COPD smokers and COPD smokers compared with nonsmokers [Citation24]. It was also indicated that CD169 levels decreased in COPD ex-smokers compared with healthy ex-smokers or never-smokers [Citation51]. However, in this study, current and recent ex-smokers were not evaluated [Citation51]. It is worthy to note that CD36, CD163, and CD169 play a key role in efferocytosis [Citation52, Citation53].
Inhibitory effects of CS on efferocytosis
Based on the previous studies, there are several efferocytosis-related factors/pathways that are affected by CS in COPD (). Such mediators will be discussed as follows.
Table 2. The main defective efferocytosis-related factors/pathways that are affected by CS in COPD.
Sphingolipid-mediated inhibitory effect of CS on efferocytosis
Sphingolipids including ceramide, sphingosine, and sphingosine-1-phosphate (S1P) are signaling mediators involved in various cellular processes, including growth arrest, differentiation, and apoptosis [Citation57]. Once sphingosine is phosphorylated by the sphingosine kinases, S1P is produced, while its acetylation by ceramide synthase generates ceramide and then sphingomyelin after sphingomyelin synthase catalyzed coupling to phosphocholine. Sphingolipid mediators are linked to inflammatory lung disorders and are used as potential therapeutic targets in various lung diseases, such as acute lung injury [Citation62], cystic fibrosis [Citation63], as well as COPD [Citation64].
S1P has been known to involve in defective efferocytosis in AMs of COPD patients [Citation54]. S1P plays a role in actin cytoskeletal dynamics required for macrophage phagocytic function [Citation65]. S1P binding to S1P receptors (S1PRs) triggers the rapid and complex signaling and externalization events that precede and control actin assembly in macrophages [Citation65, Citation66]. After CS exposure, a reduction of sphingosine kinase (SK)-1 and SK2 was detected in macrophages. Macrophage SK1 protein is localized to the plasma membrane and cytoplasm, while SK2 is localized to the nucleus and cytoplasm of the cell. CSE was exhibited to promote a retraction of SK1 from the plasma membrane and SK1/2 clumping in the Golgi domain and, thereby, suppress efferocytosis. Supporting this, CS‐inhibited efferocytosis was reversed to near‐control levels in the presence of exogenous S1P or FTY720 (fingolimod, a modulator of S1P signaling, which was found to reduce CS‐induced clumping of SK1/2) [Citation37]. Moreover, CS was also found to decrease the gene expression of S1PR5, through epigenetic modification by inducing differential methylation of the S1PR5 promoter CpG island containing transcription factor binding motifs. Of note, DNA de-methylation with 5-azacytidine could enhance efferocytosis in macrophages and rescue the cells from the CS-induced defect in efferocytosis [Citation54]. Supporting this, thymoquinone, an antioxidant/anti-inflammatory agent, was shown to antagonize the impairing effect of CS on efferocytosis and protect bronchial epithelial cells from CS-induced apoptosis through modulating the S1P system, particularly increasing S1PR-5 expression [Citation67].
Furthermore, ceramide, the precursor of S1P, has an opposing role to S1P and has been found to decrease macrophage efferocytosis in COPD [Citation55]. Ceramide levels are regulated by sphingomyelinase (SMase)-mediated sphingomyelin hydrolysis [Citation68].
SMase is an enzyme which involves in stress-induced apoptosis in various cell types and shows elevated levels and higher activity in the plasma of COPD patients and CS-exposed mice [Citation69]. CS was shown to induce acid sphingomyelinase (aSMase) and, thereby, increase the generation of ceramides [Citation69]. Increased levels of ceramides in the lung in response to CS has been found to be associated with accumulation of AMs with impaired phagocytosis of ACs as well as increased detection of apoptotic alveolar epithelial and endothelial cells in the lung parenchyma [Citation55, Citation70]. The mechanism of ceramide-induced efferocytosis impairment depends on the generation of sphingosine by ceramidase. Sphingosine can hinder efferocytosis, and CS was shown to inhibit AM efferocytosis by ceramide-dependent sphingosine production [Citation55]. The effect of ceramide on efferocytosis is associated with decreased membrane ruffle formation and attenuated Rac1 plasma membrane recruitment. CS exposure increases AM ceramides and recapitulates the effect of ceramides on Rac1 membrane recruitment in a sphingosine-dependent manner. In this line, active Rac1 overexpression was shown to rescue AM efferocytosis against the effects of CS-induced ceramide [Citation55]. Thus, CS-related excessive lung ceramides can increase lung injury by causing apoptosis of structural cells and inhibiting their efferocytosis via AM.
Zinc deficiency-mediated inhibitory effect of CS on efferocytosis
Nutritional zinc restriction exacerbates the inflammation of the airway followed by increased activation of caspase-3 and aggregation of apoptotic epithelial cells in bronchioles [Citation56]. Zinc homeostasis is regulated by Zrt-/Irt-like protein proteins (ZIPs) located in the plasma membrane of alveolar lung macrophages [Citation56].
Zinc homeostasis in macrophages requires the coordinated action of ZIP1 and ZIP2 transporters responding to zinc deficiency signals that play key roles in macrophage efferocytosis. Interestingly, abnormal expression of ZIP1 protein was found in airway epithelial or AMs, without systemic zinc deficiency, in animal models of airway inflammation or acute lung injury [Citation71, Citation72]. Zinc deficiency is associated with the defective efferocytosis by macrophages in COPD [Citation56]. Inflammation and lung injury results in a down-regulation of the zinc transporter expression in the lung fluids, airways, and AM and causes a defective efferocytosis [Citation56]. Of note, lavage zinc is positively correlated with AM efferocytosis and zinc-depleted macrophages show a decreased efferocytosis. Notably, the lower bronchoalveolar fluid zinc was observed in COPD patients who were active smokers relative to healthy volunteers and COPD patients with no recent history of smoking [Citation56]. Exposure to CS can sustain low bronchoalveolar fluid zinc levels. These events might contribute to the loss of AMs zinc via the replacement of zinc with cadmium, a major component of CS, leading to reduced efferocytosis in the AM [Citation73]. Cadmium reduces efferocytosis and stimulates emphysema by the upregulation of peptidyl arginine deiminase 4 (PAD4). In this line, COPD patients show the increased expression of PAD4 in Bronchoalveolar lavages as well as lung tissue. It is further supported by the finding that indicates GSK484, as a PAD4 inhibitor, can restore efferocytosis ability of cadmium-treated macrophages [Citation74].
Endothelial microparticle-mediated inhibitory effect of CS on efferocytosis
Acute exposure to CS has been shown to stimulate the release of endothelial microparticles (EMPs) consisting of a mixed population of exosomes and membrane particles of much smaller size than apoptotic bodies [Citation69]. Circulating EMPs are emerging as biomarkers of COPD in CS-exposed subjects. Of note, EMPs has been found to inhibit efferocytosis by multiple mechanisms. It can be linked to high ceramide content, posttranscriptional modifications via miRNA cargo, or competition for PtdSer -binding receptors.
CS-released EMPs are ceramide-rich and require the ceramide-synthesis enzyme aSMase for their release [Citation57]. Indeed, CS exposure, by activating aSMase enzyme, raises the levels of circulating EMPs [Citation57]. Mechanistically, aSMase degrades sphingomyelin and enriches the outer membrane leaflet with plasma membrane cholesterol, which causes increased membrane fluidity and destabilization as well as membrane blebbing, resulting in the release of EMPs [Citation75, Citation76]. Ceramide-enriched EMPs can suppress macrophages efferocytosis through increasing membrane stiffness and impairing intercellular membrane fusion [Citation69, Citation77]. The mechanism of ceramide-induced efferocytosis impairment depends on generation of sphingosine by ceramidase. Sphingosine can dose-dependently hinder efferocytosis, and CS was shown to inhibit AM efferocytosis by ceramide-dependent sphingosine production [Citation55].
Besides, CS can also hinder efferocytosis of ACs through profiting off the competitive effect of EMPs with ACs for phagocyte engulfment by AMs. PtdSer, an anionic phospholipid located in the inner leaflet of the cell membrane, is an immunosuppressive signal in efferocytosis. In ACs, it is exposed on the outer leaflet of the plasma membrane [Citation78]. PtdSer is known to be externalized on the surface of apoptotic bodies and extracellular vesicles (EVs) like EMPs [Citation69]. Indeed, PtdSer exposure is an “Eat-Me” signal that facilitates efferocytosis [Citation79–81]. CSE was identified to promote the release of PtdSer-positive EVs from different cell types in-vitro and increase the circulating levels of PtdSer+ EVs in-vivo [Citation69, Citation82]. CSE-induced secretion of PtdSer+ EMPs were shown to inhibit efferocytosis of ACs via PtdSer-dependent blocking of the phagocyte receptors, such as MerTK, that compete for engulfment [Citation69]. MerTK is a cell surface receptor belonging to AC clearance receptors on AMs, which recognizes ACs. It is a transmembrane tyrosine kinase receptor, acts as an opsonin and bridge molecule, binds the “Eat-Me” signal PtdSer on ACs, and facilitate efferocytosis [Citation81, Citation83]. MerTK is up-regulated in AMs obtained from bronchoalveolar lavage of cigarette smokers [Citation32, Citation81, Citation83], which might increase demand for efferocytosis in smokers in the development of COPD. Indeed, up-regulation of the MerTK reflects a lung reaction to smoking stress with an increase in parenchymal cell turnover and an increase in the number of ACs [Citation7, Citation32, Citation84]. However, since PtdSer+ EMPs can engage specific receptors, like MerTK, for AC engulfment, EMPs can compete with other PtdSer-expressing targets, such as ACs, for efferocytosis [Citation69]. Moreover, CS, by promoting aSMase, induces release of circulating EMPs with distinct microRNA (miR) cargo (let-7d, miR- 22, miR- 125-5p, miR-126, and miR-191) by which EMPs affect the clearance of ACs via specialized macrophages [Citation69]. Thus, EMPs may be a valuable indication of endothelial function in smokers and may play important roles in the inflammation and vascular remodeling alterations in response to CS.
Oxidative stress-mediated inhibitory effect of CS on efferocytosis
Excessive oxidative stress is deleterious to AM function and results in defective efferocytosis [Citation24]. Distractive oxidative stress, together with inflammation, increased bronchial epithelial cell apoptosis, and deficient efferocytosis by AMs, are found in smoker COPD patients. Moreover, in-vivo studies on animal models show that administration with anti-oxidants, such as procysteine, can improve efferocytic function of AMs in COPD [Citation58]. In a mechanism point of view, CS is known to, through oxidant-dependent activation of RhoA, a negative regulator of phagocytosis, and inhibition of Rac1, impair actin polymerization required for efficient efferocytosis [Citation25, Citation85]. Moreover, RhoA has been found to be increased by the generation of reactive oxygen species (ROS) and subsequently reduce efferocytosis in-vivo [Citation86]. CS produces ROS that activates nuclear factor-κB (NF-κB). In this respect, NF-κB triggers the pro-inflammatory cytokines production, such as TNF-α, TGF-β1, IL-6, IL-8, and granulocyte–macrophage colony-stimulating factor (GM-CSF), monocyte chemoattractant protein (MCP) −1, and also proteases [Citation87].
The other possible mechanism underlying oxidative-dependent deleterious effect of CS in COPD is mediated by glutathione (GSH) depletion. GSH as an important protective antioxidant is critical for effective efferocytosis [Citation88, Citation89] and plays a key role in the regulation of pro-inflammatory processes in the lung [Citation90]. Of note, CSE has been found to deplete intracellular GSH stores [Citation8], which accelerates CS-induced inflammation [Citation91]. Decreased GSH leads to the reduction of cellular functions including efferocytosis and respiratory burst [Citation88].
Notably, efferocytosis can be facilitated by carbohydrate-binding proteins known as lectins, such as mannose binding lectin (MBL), galectins, and pentraxins, which contribute to intracellular production of GSH [Citation60, Citation88, Citation89]. Galectin-3 and MBL are found to bind CD98 and consequently induce the M2 phenotype and also improve efferocytosis [Citation88, Citation92]. In COPD, it has been indicated that oxidative stress decreases MBL, resulting in pulmonary macrophage dysfunction [Citation60]. Galectin-3 is secreted by macrophages and derives MerTK-specific “Eat-Me” signal, involving phagocytosis and efferocytosis [Citation93]. In COPD, galectin-3 affects the impaired efferocytosis through influencing cytoskeletal remodeling, macrophage phenotype, and GSH availability [Citation94]. Of note, galectin-3 and CD98 have been detected to be decreased in the airways of both current and ex-smoker COPD. Galectin-3 have been also found to be reduced in bronchoalveolar lavage samples from healthy smokers, which can support at least a partial potential effect of CS [Citation94]. Galectin-3 has been shown to regulate CSE-induced endothelial progenitor cells autophagy and dysfunction by the mammalian target of rapamycin (mTOR) and AMP-activated protein kinase (AMPK) signaling pathways [Citation95]. Galectin-3 inhibition may be a promising strategy for preventing CS-induced endothelial progenitor cell failure and dysregulated autophagy. Furthermore, endoplasmic reticulum stress and oxidative stress stimulated via smoking activate the protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK)-eukaryotic initiation factor-2α (eIF2α) pathway, whereby inhibiting efferocytosis in AMs in-vivo, and GSK2606414 could be able to restore this process [Citation59].
Effect of CS-mediated efferocytosis impairment on neutrophilic inflammation in COPD
The chronic and pathogenic inflammation in the lungs of patients with COPD is predominantly characterized by neutrophilic infiltration, and neutrophil numbers show a positive correlation with disease progression [Citation96]. CS exposure is known to cause neutrophilic airway inflammation in human and animal models [Citation97]. Neutrophilic inflammation is a defense mechanism to eliminate pathogens and to promote the repair of damaged tissue. However, when this host defense mechanism is exaggerated or inefficiently cleared, inflammation can become chronic, resulting in lung tissue injury and remodeling [Citation98, Citation99]. During physiological conditions, neutrophils, immediately after recruitment, are subjected to apoptosis, resulting in the inflammatory response resolution [Citation100] and inhibiting the progress of chronic inflammatory disorders [Citation101, Citation102]. Once neutrophils undergo apoptosis, these cells initially keep the plasma membrane integrity, and their intracellular contents will not be released, unless cells are not rapidly cleared by macrophages. By contrast, necrotic cells unleash the integrity of the membrane, causing the release of specific intracellular danger signals known as DAMPs that are normally hidden in the interior of cells [Citation103]. Thus, the inability to efficiently remove apoptotic neutrophils has damaging implications in COPD.
Notably, CS-mediated impaired efferocytosis of apoptotic neutrophils by macrophages was shown to result in the accumulation of necrotic neutrophils in the lung and subsequent increased release of DAMPs [Citation25, Citation26, Citation85]. Besides imparting macrophages-mediated efferocytosis, CS exposure has also been found to induce apoptotic, as well as necrotic, cell death in neutrophils [Citation104]. It is further supported by observations that show cigarette smoking leads to a marked decrease in sputum neutrophil numbers [Citation97]. Of note, it was found that CS exposure disturbs mitochondrial function of lung neutrophils, causing a switch from apoptotic to necrotic cell death with concomitant release of DAMPs, particularly HMGB1, which leads to elevated release of proinflammatory cytokines, including C-X-C Motif Chemokine Ligand 8 (CXCL8), in normal human bronchial epithelial cells, promoting airway inflammation [Citation97]. Upon cell death, mitochondria show a rich source of DAMPs [Citation105, Citation106]. During necrosis, membranes of mitochondria became disrupted and mitochondrial DAMPs are released [Citation107]. Upon release into the extracellular space, DAMPs induce inflammation by activating pattern recognition receptors that are present on airway epithelial cells and activate gene expression and synthesis of a wide spectrum of proinflammatory cytokines, particularly CXCL8, in the lungs [Citation97, Citation108–110]. The role of DAMPs in the pathogenesis of COPD has already been shown, as multiple DAMPs, particularly HMGB1, were found to be increased in lung fluid of COPD patients compared with control smokers [Citation105]. Besides its role as an inflammatory mediator, HMGB1 has also been known as a negative regulator of efferocytosis that suppresses the uptake of apoptotic neutrophils by macrophages [Citation111, Citation112] through binding to PtdSer on the surface of apoptotic neutrophils and inhibiting the binding of MFG-E8 to αvβ3-integrin on the phagocytic cell surface [Citation113]. Furthermore, the accumulation of necrotic neutrophils can also contribute to the release of granule protease, such as neutrophil elastase that localizes to lung elastic fibers in emphysematic patients. Neutrophil elastase degrades extracellular matrix components and promotes the excessive release of mucins by EGFR-dependent mechanisms [Citation114, Citation115], leading to COPD and chronic bronchitis phenotype [Citation61].
PPAR-γ is a member of the nuclear hormone receptor family, which regulates the macrophage efferocytosis and clearance of ACs. PPAR-γ controls the macrophage program of alternative activation with effective efferocytic surface receptors, such as CD36, macrophage mannose receptor, and arginase-I [Citation52, Citation116–118]. PPAR-γ agonist activity is reduced in the plasma of patients with emphysema [Citation119]. Activated PPAR-γ increases the efferocytic ability of AMs while reduces the production of pulmonary inflammatory cytokines and neutrophil recruitment [Citation120, Citation121]. Notably, CS has been found to down-regulate PPAR-γ activity [Citation122, Citation123], while PPAR-γ activation could decrease lung inflammation and fibrosis by the regulation of efferocytosis [Citation120].
To sum up, CS promotes neutrophilic inflammation in COPD through inhibiting macrophage-mediated efferocytosis and causing the accumulation of necrotic neutrophils, which results in the release of DAMPs and granule protease causing chronic airway inflammation.
The effect of CS on formyl peptide receptor 2/3 and annexin A1
The polarization of M1/M2 macrophages is found to involve in COPD [Citation39, Citation94, Citation124, Citation125]. It was indicated that a defective expression of FPR2/3 and annexin A1 is associated with decreased polarization of M2a macrophage (as an anti-inflammatory phenotype) and efferoytosis [Citation39, Citation126, Citation127]. Annexin A1, as a bridging molecule in efferocytosis, stimulates macrophage reprogramming toward a M2 phenotype, which results in reduced production of pro-inflammatory mediators as well as secondary necrosis [Citation128, Citation129]. FPR2 expression is a marker of efferocytosis and a mediator of M1/M2 macrophage polarization. ACs release annexin A1, an endogenous FPR2 ligand, that acts together with FPRs to modulate the activation of inflammatory monocytes [Citation130]. Annexin A1 activates FPR2/FPR2 homodimerization that triggers the p38/mitogen-activated protein kinase-activated protein kinase (MAPKAPK)/heat shock protein 27 (Hsp27) pathway mediating anti-inflammatory responses, whereas the activation of FPR2/FPR1 heterodimerization stimulates JNK pathway that is associated with the delay of neutrophil apoptosis and the induction of pro-inflammatory responses [Citation131]. The smokers with COPD have a reduced number of M2a phenotype and an increased number of natural killer (NK) cells, NK T (NKT) cells, T helper (Th) cells, and cytotoxic T (CT) cells. FPR2 expression in Th/CT cells, FPR3 expression in M1, M2a, NK, NKT, Th, and CT cells, and serum level of annexin A1 have been detected to be decreased in smoking-related COPD patients [Citation39]. Thus, altered expressions of FPR2/3 and annexin A1 is associated with decreased M2a polarization and defective efferocytosis in the development of COPD.
Treatment opportunities for restoring CS-induced efferocytosis impairment
Well-understanding of disorders related-pathways and factors can help the researcher to find more effective treatments. Several pharmacological regents have been found to have potential therapeutic effects for CS-related lung disorders ().
Table 3. The regents that have the potential therapeutic effect for treating CS-related lung injury.
The drug-development process is complex, and as a result, expensive, lengthy, and risky, while new-indication for already approved compounds can overcome such limitations [Citation132]. Indeed, the drug re-purposing strategy, a process of finding new indications for existing Food and Drug Administration (FDA)-approved drugs, can reduce concerns about safety/toxicity, because these agents have already passed toxicity and safety tests in humans. Moreover, development costs are lower for new indications in general than for what is necessary to get an original indication approved. This seems likely because discovery, preclinical development, and clinical safety issues may have been largely resolved by the initial development. Of note, there are some FDA-approved drugs that have been reported to modulate molecular targets involving in efferocytosis impairment and thereby enhance this process, as discussed in the following sections.
Restoring CS-induced efferocytosis impairment by statins
Statins have been found to decrease the incidence of COPD exacerbations [Citation133, Citation134] and prevent the development of CS-mediated emphysema [Citation135]. Intervention with simvastatin was found to reverse pulmonary hypertension and prevent emphysema [Citation136]. Of note, as the RhoA-Rho kinase pathway implicates in CS-induced efferocytosis impairment, statins, as Rho-kinase inhibitors, have the potential to increase efferocytosis [Citation137, Citation138]. Notably, lovastatin was shown to increases efferocytosis by AMs from COPD patients in a 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase dependent manner [Citation137].
Restoring CS-induced efferocytosis impairment by azithromycin
It has been shown that macrolide antibiotics are associated with enhanced efferocytosis in COPD [Citation139–142]. Azithromycin could improve AM efferocytosis ability, reduce bronchial epithelial cell apoptosis, as well as increase the surface expression of mannose receptors (CD206), as the M2 marker, and reduce inflammatory markers in the peripheral blood of COPD smokers patients [Citation139]. The mannose receptors, one of the genes stimulated by IL-4, involved in the defective efferocytosis function of AMs are decreased in AMs from current- and ex-smoker patients with COPD and has been found to be targets for the azithromycin-mediated improvement in efferocytosis ability [Citation24, Citation139, Citation143]. In this line, azithromycin leads to the polarization of macrophages toward the alternatively activated phenotype, modulating inflammatory process [Citation144]. Alternatively activated phenotype of macrophages show the high expression of surface mannose receptor [Citation145]. In-vivo interventional low-dose azithromycin therapy has shown to reduce CS-induced emphysema and lung inflammation [Citation146]. Thus, azithromycin can improve efferocytosis and reduced apoptosis by the polarization of macrophages toward alternatively activated phenotype.
Restoring CS-induced impairment of efferocytosis by GM-CSF
GM-CSF treatment has been found to increase uptake and efferocytosis of ACs by AMs [Citation33], through activating GTPases Rab5 and Rab7 that involve in phagocytic trafficking via mediating vesicle formation, maturation, as well as transport [Citation147–149]. To the best of our knowledge, there is no finding about direct effect of CS on the expression or activity of Rab5 and Rab7. Notably, treatment with GM-CSF enhanced the expression of Rab5 and Rab7 in AM from secondhand smoke (SHS)-exposed mice. However, no significant changes were shown in the Rab5 and Rab7 expression between naïve mice exposure to SHS and unexposed controls. In this respect, the mechanism of such an overexpression of Rab5 and Rab7 by GM-CSF remains unclear [Citation33]. Moreover, exposure to SHS was shown to reduce expression of Fcγ III/II receptors (CD16/32) on AMs, and the treatment with GM-CSF not only restored but also increased the expression of receptors on AMs and enhanced efferocytosis activity in AMs from GM-CSF-treated, SHS-exposed mice [Citation33]. FcγRs are suggested to induce efferocytosis through stimulating ACs opsonization by specific antibodies and subsequent uptake by antigen-presenting cells (APCs) [Citation150–152]. Of note, LC3 (microtubule-associated protein 1 A/1B-light chain 3)-associated phagocytosis (LAP) is required for the effective engulfment and subsequently efferocytosis. When ACs attach to extracellular receptors during phagocytosis, LAP is stimulated [Citation153]. LAP is triggered through the engagement of the FcγR via complexes of self-antigen and autoantibodies in plasmacytoid dendritic cells (pDC) [Citation154].
Restoring CS-induced impairment of efferocytosis by thymoquinone
Thymoquinone is the major constituent of Nigella sativa and has broad nutraceutical potential, such as antioxidant, anti-inflammatory, immunomodulatory, and antihistaminic biological properties [Citation155]. Thymoquinone was reported to improve macrophage efferocytosis by modulation of the S1P system and protect bronchial epithelial cells from CS-induced apoptosis [Citation67]. CS decreases phagocytosis and increases S1PR-5 expressions in macrophages, and thymoquinone was shown to enhance efferocytosis, antagonize the effects of CS on efferocytosis and S1PR5, and protect bronchial epithelial cells from CS-induced apoptosis [Citation67].
Restoring CS-induced impairment of efferocytosis by thiazolidinediones
PPAR-γ agonists, thiazolidinediones; rosiglitazone and pioglitazone, have been found to attenuate airway neutrophilia, through increasing efferocytosis of apoptotic neutrophils, in corticosteroid-resistant disease [Citation35]. Moreover, rosiglitazone-treated AMs could improve the efferocytosis of apoptotic neutrophils [Citation35]. Thiazolidinediones were shown to prevent the effect of CS on PPAR-γ; the treatment of COPD patients with PPAR-γ agonist rosiglitazone could block CS-induced production of cytokines, chemokines, and ROS, and accompanying inhibition of histone deacetylase 2 levels [Citation122], causing a significant amelioration in COPD exacerbations [Citation122, Citation123].
Conclusion
Environmental stresses, such as CS, are involved in COPD. COPD patients who quit smoking have improved phagocytosis compared to patients who continue to smoke. However, even in patients who have quit smoking for a long time, the phagocytosis of COPD patients will not return to normal levels. This result could be because of abnormality and imbalance in endoplasmic reticulum stress, oxidative stress, as well as protease and anti-protease activity after smoking cessation. This suggests that a combination of exposure and immune deficiency, like impaired efferocytosis, is required. In this line, impaired efferocytosis of apoptotic epithelial cells by AMs is a key mechanism of inflammatory lung diseases and occurs in COPD. CS reduces the efferocytosis of AMs to engulf ACs, which is impaired in COPD. This impaired efferocytosis might result in secondary necrosis formation, which increases different danger signals, pro-inflammatory mediators, oxidative stress responses and the disruption of the epithelial barrier. Thus, the effects of CE on AM may be partially resolved when quitting smoking in COPD patients. In this case, even if you completely quit smoking, you need to use other compounds to more completely restore efferocytosis. Hence, a better understanding of the processes behind the effect of CS on efferocytosis in relation to lung disorders can help to design more efficient treatment approaches and also delay the development of lung disease, such as COPD. The critical immunological and molecular effects of CS-mediated defective efferocytosis may be mimicked, which may lead to novel important therapeutic approaches to restoring impaired epithelial repair processes in CS-induced COPD. Furthermore, restoring efferocytosis can reduce morbidity following infections. Thus, considering the essential function of efferocytosis in COPD, targeting molecules/pathways-associated with efferocytosis has a strong potential to attenuate inflammatory reactions, to promote the repair of damaged tissue in patients, and to prevent the development of advanced stages of the disease.
Declaration of interest
The authors declare that there are no conflicts of interest and financial support for the present review article.
Data availability statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Funding
The author(s) reported there is no funding associated with the work featured in this article.
References
- Tzortzaki EG, Papi A, Neofytou E, et al. Immune and genetic mechanisms in COPD: possible targets for therapeutic interventions. Curr Drug Targets. 2013;14(2):141–148. DOI:https://doi.org/10.2174/1389450111314020002
- Morimoto K, Janssen WJ, Terada M. Defective efferocytosis by alveolar macrophages in IPF patients. Respir Med. 2012;106(12):1800–1803. DOI:https://doi.org/10.1016/j.rmed.2012.08.020
- Yun JH, Henson PM, Tuder RM. Phagocytic clearance of apoptotic cells: role in lung disease. Expert Rev Respir Med. 2008;2(6):753–765. DOI:https://doi.org/10.1586/17476348.2.6.753
- Henson PM, Vandivier RW, Douglas IS. Cell death, remodeling, and repair in chronic obstructive pulmonary disease?Proc Am Thorac Soc. 2006;3(8):713–717. DOI:https://doi.org/10.1513/pats.200605-104SF
- Mahida RY, Scott A, Parekh D, et al. Acute respiratory distress syndrome is associated with impaired alveolar macrophage efferocytosis. Eur Respir J. 2021; DOI:https://doi.org/10.1183/13993003.00829-2021
- Society AT. Standards for the diagnosis and care of subjects withchronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1995;152:S77–S121.
- Demedts IK, Demoor T, Bracke KR, et al. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir Res. 2006;7:53. DOI:https://doi.org/10.1186/1465-9921-7-53
- Müller T, Gebel S. The cellular stress response induced by aqueous extracts of cigarette smoke is critically dependent on the intracellular glutathione concentration. Carcinogenesis. 1998;19:797–801.
- Salvi SS, Barnes PJ. Chronic obstructive pulmonary disease in non-smokers. The Lancet. 2009;374(9691):733–743. DOI:https://doi.org/10.1016/S0140-6736(09)61303-9
- Lakshmi SP, Reddy AT, Reddy RC. Emerging pharmaceutical therapies for COPD. Int J Chron Obstruct Pulmon Dis. 2017;12:2141–2156. DOI:https://doi.org/10.2147/COPD.S121416
- Geraghty P, Hardigan A, Foronjy RF. Cigarette smoke activates the proto-oncogene c-src to promote airway inflammation and lung tissue destruction. Am J Respir Cell Mol Biol. 2014;50(3):559–570. DOI:https://doi.org/10.1165/rcmb.2013-0258OC
- Churg A, Zhou S, Wright JL. Series “matrix metalloproteinases in lung health and disease”: matrix metalloproteinases in COPD. Eur Respir J. 2012;39(1):197–209. DOI:https://doi.org/10.1183/09031936.00121611
- Kraen M, Frantz S, Nihlen U, et al. Matrix metalloproteinases in COPD and atherosclerosis with emphasis on the effects of smoking. PLoS One. 2019;14(2):e0211987. DOI:https://doi.org/10.1371/journal.pone.0211987
- Ilumets H, Rytila P, Demedts I, et al. Matrix metalloproteinases -8, -9 and -12 in smokers and patients with stage 0 COPD. Int J Chron Obstruct Pulmon Dis. 2007;2:369–379.
- Tilley AE, Harvey BG, Heguy A, et al. Down-regulation of the notch pathway in human airway epithelium in association with smoking and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2009;179(6):457–466. DOI:https://doi.org/10.1164/rccm.200705-795OC
- Hansson EM, Lendahl U, Chapman G. Notch signaling in development and disease. Semin Cancer Biol. 2004;14(5):320–328.
- Harper J, Yuan J, Tan J, et al. Notch signaling in development and disease. Clin Genet. 2003;64(6):461–472. DOI:https://doi.org/10.1046/j.1399-0004.2003.00194.x
- Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284(5415):770–776. DOI:https://doi.org/10.1126/science.284.5415.770
- Holgate ST. Epithelial damage and response. Clin Exp Allergy. 2000;30(Suppl 1):37–41. DOI:https://doi.org/10.1046/j.1365-2222.2000.00095.x
- Knight DA, Holgate ST. The airway epithelium: structural and functional properties in health and disease. Respirology. 2003;8(4):432–446. DOI:https://doi.org/10.1046/j.1440-1843.2003.00493.x
- Polosukhin VV, Lawson WE, Milstone AP, et al. Association of progressive structural changes in the bronchial epithelium with subepithelial fibrous remodeling: a potential role for hypoxia. Virchows Arch. 2007;451(4):793–803. DOI:https://doi.org/10.1007/s00428-007-0469-5
- Yu H, Li Q, Kolosov VP, et al. Regulation of cigarette smoke-mediated mucin expression by hypoxia-inducible factor-1α via epidermal growth factor receptor-mediated signaling pathways. J Appl Toxicol. 2012;32(4):282–292. DOI:https://doi.org/10.1002/jat.1679
- Polosukhin VV, Cates JM, Lawson WE, et al. Hypoxia-inducible factor-1 signalling promotes goblet cell hyperplasia in airway epithelium. J Pathol. 2011;224(2):203–211. DOI:https://doi.org/10.1002/path.2863
- Hodge S, Hodge G, Ahern J, et al. Smoking alters alveolar macrophage recognition and phagocytic ability: implications in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2007;37(6):748–755. DOI:https://doi.org/10.1165/rcmb.2007-0025OC
- Richens TR, Linderman DJ, Horstmann SA, et al. Cigarette smoke impairs clearance of apoptotic cells through oxidant-dependent activation of RhoA. Am J Respir Crit Care Med. 2009;179(11):1011–1021. DOI:https://doi.org/10.1164/rccm.200807-1148OC
- Noda N, Matsumoto K, Fukuyama S, et al. Cigarette smoke impairs phagocytosis of apoptotic neutrophils by alveolar macrophages via inhibition of the histone deacetylase/rac/CD9 pathways. Int Immunol. 2013;25(11):643–650. DOI:https://doi.org/10.1093/intimm/dxt033
- Hodge S, Hodge G, Scicchitano R, et al. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol. 2003;81(4):289–296. DOI:https://doi.org/10.1046/j.1440-1711.2003.t01-1-01170.x
- Hodge S, Jersmann H, Reynolds PN. The effect of colonization with potentially pathogenic microorganisms on efferocytosis in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2016;194(7):912–915. DOI:https://doi.org/10.1164/rccm.201601-0019LE
- Phipps JC, Aronoff DM, Curtis JL, et al. Cigarette smoke exposure impairs pulmonary bacterial clearance and alveolar macrophage complement-mediated phagocytosis of Streptococcus pneumoniae. Infect Immun. 2010;78(3):1214–1220. DOI:https://doi.org/10.1128/IAI.00963-09
- Tajbakhsh A, Rezaee M, Barreto GE, et al. The role of nuclear factors as “Find-Me”/alarmin signals and immunostimulation in defective efferocytosis and related disorders. Int Immunopharmacol. 2020;80:106134. DOI:https://doi.org/10.1016/j.intimp.2019.106134
- Tajbakhsh A, Bianconi V, Pirro M, et al. Efferocytosis and atherosclerosis: regulation of phagocyte function by MicroRNAs. Trends Endocrinol Metab. 2019;30(9):672–683. DOI:https://doi.org/10.1016/j.tem.2019.07.006
- Kazeros A, Harvey BG, Carolan BJ, et al. Overexpression of apoptotic cell removal receptor MERTK in alveolar macrophages of cigarette smokers. Am J Respir Cell Mol Biol. 2008;39(6):747–757. DOI:https://doi.org/10.1165/rcmb.2007-0306OC
- Subramaniam R, Mukherjee S, Chen H, et al. Restoring cigarette smoke-induced impairment of efferocytosis in alveolar macrophages. Mucosal Immunol. 2016;9(4):873–883. DOI:https://doi.org/10.1038/mi.2015.120
- Ween MP, Hamon R, Macowan MG, et al. Effects of E-cigarette E-liquid components on bronchial epithelial cells: demonstration of dysfunctional efferocytosis. Respirology. 2020;25(6):620–628. DOI:https://doi.org/10.1111/resp.13696
- Lea S, Plumb J, Metcalfe H, et al. The effect of peroxisome proliferator-activated receptor-γ ligands on in vitro and in vivo models of COPD. Eur Respir J. 2014;43(2):409–420. DOI:https://doi.org/10.1183/09031936.00187812
- Zhang S, Xie JG, Su BT, et al. MFG-E8, a clearance glycoprotein of apoptotic cells, as a new marker of disease severity in chronic obstructive pulmonary disease. Braz J Med Biol Res. 2015;48(11):1032–1038. DOI:https://doi.org/10.1590/1414-431x20154730
- Tran HB, Barnawi J, Ween M, et al. Cigarette smoke inhibits efferocytosis via deregulation of sphingosine kinase signaling: reversal with exogenous S1P and the S1P analogue FTY720. J Leukoc Biol. 2016;100(1):195–202. DOI:https://doi.org/10.1189/jlb.3A1015-471R
- Pappas K, Papaioannou AI, Kostikas K, et al. The role of macrophages in obstructive airways disease: chronic obstructive pulmonary disease and asthma. Cytokine. 2013;64(3):613–625. DOI:https://doi.org/10.1016/j.cyto.2013.09.010
- Chen YC, Lin MC, Lee CH, et al. Defective formyl peptide receptor 2/3 and annexin A1 expressions associated with M2a polarization of blood immune cells in patients with chronic obstructive pulmonary disease. J Transl Med. 2018;16(1):69. DOI:https://doi.org/10.1186/s12967-018-1435-5
- Tajbakhsh A, Gheibi Hayat SM, Butler AE, et al. Effect of soluble cleavage products of important receptors/ligands on efferocytosis: their role in inflammatory, autoimmune and cardiovascular disease. Ageing Res Rev. 2019;50:43–57. DOI:https://doi.org/10.1016/j.arr.2019.01.007
- Tajbakhsh A, Farahani N, Gheibihayat SM, et al. Autoantigen-specific immune tolerance in pathological and physiological cell death: nanotechnology comes into view. Int Immunopharmacol. 2021; 90:107177. DOI:https://doi.org/10.1016/j.intimp.2020.107177
- Eltboli O, Bafadhel M, Hollins F, et al. COPD exacerbation severity and frequency is associated with impaired macrophage efferocytosis of eosinophils. BMC Pulm Med. 2014;14(1):112. DOI:https://doi.org/10.1186/1471-2466-14-112
- Tajbakhsh A, Gheibi Hayat SM, Movahedpour A, et al. The complex roles of efferocytosis in cancer development, metastasis, and treatment. Biomed Pharmacother. 2021;140:111776. DOI:https://doi.org/10.1016/j.biopha.2021.111776
- Tajbakhsh A, Read M, Barreto GE, et al. Apoptotic neurons and amyloid-beta clearance by phagocytosis in Alzheimer’s disease: pathological mechanisms and therapeutic outlooks. Eur J Pharmacol. 2021;895:173873. DOI:https://doi.org/10.1016/j.ejphar.2021.173873
- Nakamura Y, Romberger DJ, Tate L, et al. Cigarette smoke inhibits lung fibroblast proliferation and chemotaxis. Am J Respir Crit Care Med. 1995;151(5):1497–1503. DOI:https://doi.org/10.1164/ajrccm.151.5.7735606
- Rennard SI, Togo S, Holz O. Cigarette smoke inhibits alveolar repair: a mechanism for the development of emphysema. Proc Am Thorac Soc. 2006;3(8):703–708. DOI:https://doi.org/10.1513/pats.200605-121SF
- Serban KA, Petrusca DN, Mikosz A, et al. Alpha-1 antitrypsin supplementation improves alveolar macrophages efferocytosis and phagocytosis following cigarette smoke exposure. PLoS One. 2017;12(4):e0176073. DOI:https://doi.org/10.1371/journal.pone.0176073
- McCubbrey AL, Curtis JL. Efferocytosis and lung disease. Chest. 2013;143(6):1750–1757. DOI:https://doi.org/10.1378/chest.12-2413
- Aoshiba K, Tsuji T. Immune responses in chronic obstructive pulmonary disease. Japanese J Chest Dis. 2013;72:1315–1320.
- Dewhurst JA, Lea S, Hardaker E, et al. Characterisation of lung macrophage subpopulations in COPD patients and controls. Sci Rep. 2017;7(1):7143. DOI:https://doi.org/10.1038/s41598-017-07101-2
- Tanno A, Fujino N, Yamada M, et al. Decreased expression of a phagocytic receptor siglec-1 on alveolar macrophages in chronic obstructive pulmonary disease. Respir Res. 2020;21(1):30. DOI:https://doi.org/10.1186/s12931-020-1297-2
- Savill J, Hogg N, Ren Y, et al. Thrombospondin cooperates with CD36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. J Clin Invest. 1992;90(4):1513–1522. DOI:https://doi.org/10.1172/JCI116019
- Ravishankar B, Shinde R, Liu H, et al. Marginal zone CD169+ macrophages coordinate apoptotic cell-driven cellular recruitment and tolerance. Proc Natl Acad Sci USA. 2014;111(11):4215–4220. DOI:https://doi.org/10.1073/pnas.1320924111
- Barnawi J, Jersmann H, Haberberger R, et al. Reduced DNA methylation of sphingosine-1 phosphate receptor 5 in alveolar macrophages in COPD: a potential link to failed efferocytosis. Respirology. 2017;22(2):315–321. DOI:https://doi.org/10.1111/resp.12949
- Petrusca DN, Gu Y, Adamowicz JJ, et al. Sphingolipid-mediated inhibition of apoptotic cell clearance by alveolar macrophages. J Biol Chem. 2010;285(51):40322–40332. DOI:https://doi.org/10.1074/jbc.M110.137604
- Hamon R, Homan CC, Tran HB, et al. Zinc and zinc transporters in macrophages and their roles in efferocytosis in COPD. PLoS One. 2014;9(10):e110056. DOI:https://doi.org/10.1371/journal.pone.0110056
- Mathias S, Peña LA, Kolesnick RN. Signal transduction of stress via ceramide. Biochem J. 1998;335(Pt 3):465–480. DOI:https://doi.org/10.1042/bj3350465
- Hodge S, Matthews G, Mukaro V, et al. Cigarette smoke-induced changes to alveolar macrophage phenotype and function are improved by treatment with procysteine. Am J Respir Cell Mol Biol. 2011;44(5):673–681. DOI:https://doi.org/10.1165/rcmb.2009-0459OC
- Ito H, Yamashita Y, Tanaka T, et al. Cigarette smoke induces endoplasmic reticulum stress and suppresses efferocytosis through the activation of RhoA. Sci Rep. 2020;10(1):12620. DOI:https://doi.org/10.1038/s41598-020-69610-x
- Tran HB, Ahern J, Hodge G, et al. Oxidative stress decreases functional airway mannose binding lectin in COPD. PLoS One. 2014;9(6):e98571. DOI:https://doi.org/10.1371/journal.pone.0098571
- Lu W, Zheng J. The function of mucins in the COPD airway. Curr Respir Care Rep. 2013;2(3):155–166. DOI:https://doi.org/10.1007/s13665-013-0051-3
- Kuebler WM, Yang Y, Samapati R, et al. Vascular barrier regulation by PAF, ceramide, caveolae, and NO - an intricate signaling network with discrepant effects in the pulmonary and systemic vasculature. Cell Physiol Biochem. 2010;26(1):29–40. DOI:https://doi.org/10.1159/000315103
- Becker KA, Riethmüller J, Lüth A, et al. Acid sphingomyelinase inhibitors normalize pulmonary ceramide and inflammation in cystic fibrosis. Am J Respir Cell Mol Biol. 2010;42(6):716–724. DOI:https://doi.org/10.1165/rcmb.2009-0174OC
- Filosto S, Castillo S, Danielson A, et al. Neutral sphingomyelinase 2: a novel target in cigarette smoke-induced apoptosis and lung injury. Am J Respir Cell Mol Biol. 2011;44(3):350–360. DOI:https://doi.org/10.1165/rcmb.2009-0422OC
- Kusner DJ, Thompson CR, Melrose NA, et al. The localization and activity of sphingosine kinase 1 are coordinately regulated with actin cytoskeletal dynamics in macrophages. J Biol Chem. 2007;282(32):23147–23162. DOI:https://doi.org/10.1074/jbc.M700193200
- Kuehnel MP, Reiss M, Anand PK, et al. Sphingosine-1-phosphate receptors stimulate macrophage plasma-membrane actin assembly via ADP release, ATP synthesis and P2X7R activation. J Cell Sci. 2009;122(Pt 4):505–512. DOI:https://doi.org/10.1242/jcs.034207
- Barnawi J, Tran HB, Roscioli E, et al. Pro-phagocytic effects of thymoquinone on cigarette smoke-exposed macrophages occur by modulation of the sphingosine-1-phosphate signalling system. COPD. 2016;13(5):653–661. DOI:https://doi.org/10.3109/15412555.2016.1153614
- Merrill AH, Jr., Schmelz EM, Dillehay DL, et al. Sphingolipids-the enigmatic lipid class: biochemistry, physiology, and pathophysiology. Toxicol Appl Pharmacol. 1997;142(1):208–225. DOI:https://doi.org/10.1006/taap.1996.8029
- Serban KA, Rezania S, Petrusca DN, et al. Structural and functional characterization of endothelial microparticles released by cigarette smoke. Sci Rep. 2016;6:31596. DOI:https://doi.org/10.1038/srep31596
- Petrache I, Natarajan V, Zhen L, et al. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med. 2005;11(5):491–498. DOI:https://doi.org/10.1038/nm1238
- Joshi PC, Mehta A, Jabber WS, et al. Zinc deficiency mediates alcohol-induced alveolar epithelial and macrophage dysfunction in rats. Am J Respir Cell Mol Biol. 2009;41(2):207–216. DOI:https://doi.org/10.1165/rcmb.2008-0209OC
- Murgia C, Grosser D, Truong-Tran AQ, et al. Apical localization of zinc transporter ZnT4 in human airway epithelial cells and its loss in a murine model of allergic airway inflammation. Nutrients. 2011;3(11):910–928. DOI:https://doi.org/10.3390/nu3110910
- Napolitano JR, Liu MJ, Bao S, et al. Cadmium-mediated toxicity of lung epithelia is enhanced through NF-κB-mediated transcriptional activation of the human zinc transporter ZIP8. Am J Physiol Lung Cell Mol Physiol. 2012;302(9):L909–18. DOI:https://doi.org/10.1152/ajplung.00351.2011
- Surolia R, Li F, Singh P, et al. Cadmium decreases macrophage effrocytosis and induces emphysema via PAD4 upregulation. TP65. TP065 Environmental Exposures and Lung Disease: American Thoracic Society. Am J Respir Crit Care Med 2021;203:A3134. DOI:https://doi.org/10.1164/ajrccm-conference.2021.203.1_MeetingAbstracts.A3134
- Slotte JP, Hedström G, Rannström S, et al. Effects of sphingomyelin degradation on cell cholesterol oxidizability and steady-state distribution between the cell surface and the cell interior. Biochim Biophys Acta. 1989;985(1):90–96. DOI:https://doi.org/10.1016/0005-2736(89)90108-9
- Chang MP, Mallet WG, Mostov KE, et al. Adaptor self‐aggregation, adaptor‐receptor recognition and binding of alpha‐adaptin subunits to the plasma membrane contribute to recruitment of adaptor (AP2) components of clathrin‐coated pits. Embo J. 1993;12(5):2169–2180. DOI:https://doi.org/10.1002/j.1460-2075.1993.tb05865.x
- Justice MJ, Petrusca DN, Rogozea AL, et al. Effects of lipid interactions on model vesicle engulfment by alveolar macrophages. Biophys J. 2014;106(3):598–609. DOI:https://doi.org/10.1016/j.bpj.2013.12.036
- Iguchi K, Hirano K, Hamatake M, et al. Phosphatidylserine induces apoptosis in adherent cells. Apoptosis. 2001;6(4):263–268. DOI:https://doi.org/10.1023/A:1011331424311
- Birge RB, Boeltz S, Kumar S, et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016;23(6):962–978. DOI:https://doi.org/10.1038/cdd.2016.11
- Tajbakhsh A, Kovanen PT, Rezaee M, et al. Regulation of efferocytosis by caspase-dependent apoptotic cell death in atherosclerosis. Int J Biochem Cell Biol. 2020;120:105684. DOI:https://doi.org/10.1016/j.biocel.2020.105684
- Tajbakhsh A, Rezaee M, Kovanen PT, et al. Efferocytosis in atherosclerotic lesions: malfunctioning regulatory pathways and control mechanisms. Pharmacol Ther. 2018;188:12–25. DOI:https://doi.org/10.1016/j.pharmthera.2018.02.003
- Neri T, Pergoli L, Petrini S, et al. Particulate matter induces prothrombotic microparticle shedding by human mononuclear and endothelial cells. Toxicol in Vitro. 2016;32:333–338. DOI:https://doi.org/10.1016/j.tiv.2016.02.001
- Myers KV, Amend SR, Pienta KJ. Targeting Tyro3, axl and MerTK (TAM receptors): implications for macrophages in the tumor microenvironment. Mol Cancer. 2019;18(1):94. DOI:https://doi.org/10.1186/s12943-019-1022-2
- Aoshiba K, Yokohori N, Nagai A. Alveolar wall apoptosis causes lung destruction and emphysematous changes. Am J Respir Cell Mol Biol. 2003;28(5):555–562. DOI:https://doi.org/10.1165/rcmb.2002-0090OC
- Minematsu N, Blumental-Perry A, Shapiro SD. Cigarette smoke inhibits engulfment of apoptotic cells by macrophages through inhibition of actin rearrangement. Am J Respir Cell Mol Biol. 2011;44(4):474–482. DOI:https://doi.org/10.1165/rcmb.2009-0463OC
- Bianchi SM, Prince LR, McPhillips K, et al. Impairment of apoptotic cell engulfment by pyocyanin, a toxic metabolite of Pseudomonas aeruginosa. Am J Respir Crit Care Med. 2008;177(1):35–43. DOI:https://doi.org/10.1164/rccm.200612-1804OC
- Ji X, Yao H, Meister M, et al. Tocotrienols: dietary supplements for chronic obstructive pulmonary disease. Antioxidants. 2021;10:883. DOI:https://doi.org/10.3390/antiox10060883
- Brown LA, Ping XD, Harris FL, et al. Glutathione availability modulates alveolar macrophage function in the chronic ethanol-fed rat. Am J Physiol Lung Cell Mol Physiol. 2007;292(4):L824–32. DOI:https://doi.org/10.1152/ajplung.00346.2006
- Dobashi K, Aihara M, Araki T, et al. Regulation of LPS induced IL-12 production by IFN-gamma and IL-4 through intracellular glutathione status in human alveolar macrophages . Clin Exp Immunol. 2001;124(2):290–296. DOI:https://doi.org/10.1046/j.1365-2249.2001.01535.x
- Rahman I, MacNee W. Oxidative stress and regulation of glutathione in lung inflammation. Eur Respir J. 2000;16(3):534–554. DOI:https://doi.org/10.1034/j.1399-3003.2000.016003534.x
- Gould NS, Min E, Huang J, et al. Glutathione depletion accelerates cigarette smoke-induced inflammation and airspace enlargement. Toxicol Sci. 2015;147(2):466–474. DOI:https://doi.org/10.1093/toxsci/kfv143
- MacKinnon AC, Farnworth SL, Hodkinson PS, et al. Regulation of alternative macrophage activation by galectin-3. J Immunol. 2008;180(4):2650–2658. DOI:https://doi.org/10.4049/jimmunol.180.4.2650
- Caberoy NB, Alvarado G, Bigcas JL, et al. Galectin-3 is a new MerTK-specific eat-me signal. J Cell Physiol. 2012;227(2):401–407. DOI:https://doi.org/10.1002/jcp.22955
- Mukaro VR, Bylund J, Hodge G, et al. Lectins offer new perspectives in the development of macrophage-targeted therapies for COPD/emphysema. PLoS One. 2013;8(2):e56147. DOI:https://doi.org/10.1371/journal.pone.0056147
- Pei C, Wang X, Lin Y, et al. Inhibition of galectin-3 alleviates cigarette smoke Extract-Induced autophagy and dysfunction in endothelial progenitor cells. Oxid Med Cell Longev. 2019;2019:7252943. DOI:https://doi.org/10.1155/2019/7252943
- Grootendorst DC, Gauw SA, Verhoosel RM, et al. Reduction in sputum neutrophil and eosinophil numbers by the PDE4 inhibitor roflumilast in patients with COPD. Thorax. 2007;62(12):1081–1087. DOI:https://doi.org/10.1136/thx.2006.075937
- Heijink IH, Pouwels SD, Leijendekker C, et al. Cigarette smoke-induced damage-associated molecular pattern release from necrotic neutrophils triggers proinflammatory mediator release. Am J Respir Cell Mol Biol. 2015;52(5):554–562. DOI:https://doi.org/10.1165/rcmb.2013-0505OC
- Vandivier RW, Henson PM, Douglas IS. Burying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest. 2006;129(6):1673–1682. DOI:https://doi.org/10.1378/chest.129.6.1673
- van der Toorn M, Slebos DJ, de Bruin HG, et al. Critical role of aldehydes in cigarette smoke-induced acute airway inflammation. Respir Res. 2013;14(1):45. DOI:https://doi.org/10.1186/1465-9921-14-45
- Geering B, Stoeckle C, Conus S, et al. Living and dying for inflammation: neutrophils, eosinophils, basophils. Trends Immunol. 2013;34(8):398–409. DOI:https://doi.org/10.1016/j.it.2013.04.002
- Scheel-Toellner D, Wang KQ, Webb PR, et al. Early events in spontaneous neutrophil apoptosis. Biochem Soc Trans. 2004;32(Pt3):461–464. DOI:https://doi.org/10.1042/BST0320461
- Du H, Sun J, Chen Z, et al. Cigarette smoke-induced failure of apoptosis resulting in enhanced neoplastic transformation in human bronchial epithelial cells. J Toxicol Environ Health A. 2012;75(12):707–720. DOI:https://doi.org/10.1080/15287394.2012.690088
- Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8(4):279–289. DOI:https://doi.org/10.1038/nri2215
- Guzik K, Skret J, Smagur J, et al. Cigarette smoke-exposed neutrophils die unconventionally but are rapidly phagocytosed by macrophages. Cell Death Dis. 2011;2:e131. DOI:https://doi.org/10.1038/cddis.2011.13
- Pouwels SD, Heijink IH, ten Hacken NH, et al. DAMPs activating innate and adaptive immune responses in COPD. Mucosal Immunol. 2014;7(2):215–226. DOI:https://doi.org/10.1038/mi.2013.77
- Tait SW, Green DR. Mitochondria and cell signalling. J Cell Sci. 2012;125(Pt 4):807–815. DOI:https://doi.org/10.1242/jcs.099234
- Wen Z, Xu L, Chen X, et al. Autoantibody induction by DNA-containing immune complexes requires HMGB1 with the TLR2/microRNA-155 pathway. J Immunol. 2013;190(11):5411–5422. DOI:https://doi.org/10.4049/jimmunol.1203301
- Kunkel SL, Standiford T, Kasahara K, et al. Interleukin-8 (IL-8): the major neutrophil chemotactic factor in the lung. Exp Lung Res. 1991;17(1):17–23. DOI:https://doi.org/10.3109/01902149109063278
- Masubuchi T, Koyama S, Sato E, et al. Smoke extract stimulates lung epithelial cells to release neutrophil and monocyte chemotactic activity. Am J Pathol. 1998;153(6):1903–1912. DOI:https://doi.org/10.1016/S0002-9440(10)65704-5
- Zitvogel L, Kepp O, Kroemer G. Decoding cell death signals in inflammation and immunity. Cell. 2010;140(6):798–804. DOI:https://doi.org/10.1016/j.cell.2010.02.015
- Liu G, Wang J, Park Y-J, et al. High mobility group protein-1 inhibits phagocytosis of apoptotic neutrophils through binding to phosphatidylserine. J Immunol. 2008;181(6):4240–4246. DOI:https://doi.org/10.4049/jimmunol.181.6.4240
- Wang Y, Luo G, Chen J, et al. Cigarette smoke attenuates phagocytic ability of macrophages through down-regulating milk fat globule-EGF factor 8 (MFG-E8) expressions. Sci Rep. 2017;7:42642. DOI:https://doi.org/10.1038/srep42642
- Friggeri A, Yang Y, Banerjee S, et al. HMGB1 inhibits macrophage activity in efferocytosis through binding to the alphavbeta3-integrin. Am J Physiol Cell Physiol. 2010;299(6):C1267–C76. DOI:https://doi.org/10.1152/ajpcell.00152.2010
- Shao MX, Nadel JA. Neutrophil elastase induces MUC5AC mucin production in human airway epithelial cells via a Cascade involving protein kinase C, reactive oxygen species, and TNF-alpha-converting enzyme. J Immunol. 2005;175(6):4009–4016. DOI:https://doi.org/10.4049/jimmunol.175.6.4009
- Damiano V, Tsang A, Kucich U, et al. Immunolocalization of elastase in human emphysematous lungs. J Clin Invest. 1986;78(2):482–493. DOI:https://doi.org/10.1172/JCI112600
- Fadok VA, Warner ML, Bratton DL, et al. CD36 is required for phagocytosis of apoptotic cells by human macrophages that use either a phosphatidylserine receptor or the vitronectin receptor (αvβ3). J Immunol. 1998;161:6250–6257.
- Majai G, Sarang Z, Csomós K, et al. PPARgamma-dependent regulation of human macrophages in phagocytosis of apoptotic cells. Eur J Immunol. 2007;37(5):1343–1354. DOI:https://doi.org/10.1002/eji.200636398
- Asada K, Sasaki S, Suda T, et al. Antiinflammatory roles of peroxisome proliferator-activated receptor gamma in human alveolar macrophages. Am J Respir Crit Care Med. 2004;169(2):195–200. DOI:https://doi.org/10.1164/rccm.200207-740OC
- Shan M, You R, Yuan X, et al. Agonistic induction of PPARγ reverses cigarette smoke-induced emphysema. J Clin Invest. 2014;124(3):1371–1381. DOI:https://doi.org/10.1172/JCI70587
- Yoon YS, Kim SY, Kim MJ, et al. PPARγ activation following apoptotic cell instillation promotes resolution of lung inflammation and fibrosis via regulation of efferocytosis and proresolving cytokines. Mucosal Immunol. 2015;8(5):1031–1046. DOI:https://doi.org/10.1038/mi.2014.130
- Li Y, Wen X, Spataro BC, et al. Hepatocyte growth factor is a downstream effector that mediates the antifibrotic action of peroxisome proliferator-activated receptor-gamma agonists. J Am Soc Nephrol. 2006;17(1):54–65. DOI:https://doi.org/10.1681/ASN.2005030257
- Lakshmi SP, Reddy AT, Zhang Y, et al. Down-regulated peroxisome proliferator-activated receptor γ (PPARγ) in lung epithelial cells promotes a PPARγ agonist-reversible proinflammatory phenotype in chronic obstructive pulmonary disease (COPD). J Biol Chem. 2014;289(10):6383–6393. DOI:https://doi.org/10.1074/jbc.M113.536805
- Rinne ST, Liu CF, Feemster LC, et al. Thiazolidinediones are associated with a reduced risk of COPD exacerbations. Int J Chron Obstruct Pulmon Dis. 2015;10:1591–1597. DOI:https://doi.org/10.2147/COPD.S82643
- Moreira AR, Pereira de Castro TB, Kohler JB, et al. Chronic exposure to diesel particles worsened emphysema and increased M2-like phenotype macrophages in a PPE-induced model. PLoS One. 2020;15(1):e0228393. DOI:https://doi.org/10.1371/journal.pone.0228393
- Mehta M, DeekshaSharma N, et al. Interactions with the macrophages: an emerging targeted approach using novel drug delivery systems in respiratory diseases. Chem Biol Interact. 2019;304:10–19. DOI:https://doi.org/10.1016/j.cbi.2019.02.021
- Cooray SN, Gobbetti T, Montero-Melendez T, et al. Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses . Proc Natl Acad Sci USA. 2013;110(45):18232–18237. DOI:https://doi.org/10.1073/pnas.1308253110
- Maderna P, Cottell DC, Toivonen T, et al. FPR2/ALX receptor expression and internalization are critical for lipoxin A4 and annexin-derived peptide-stimulated phagocytosis. FASEB J. 2010;24(11):4240–4249. DOI:https://doi.org/10.1096/fj.10-159913
- Sugimoto MA, Vago JP, Teixeira MM, et al. Annexin A1 and the resolution of inflammation: modulation of neutrophil recruitment, apoptosis, and clearance. J Immunol Res. 2016;2016:8239258. DOI:https://doi.org/10.1155/2016/8239258
- Blume KE, Soeroes S, Waibel M, et al. Cell surface externalization of annexin A1 as a failsafe mechanism preventing inflammatory responses during secondary necrosis. J Immunol. 2009;183(12):8138–8147. DOI:https://doi.org/10.4049/jimmunol.0902250
- Pupjalis D, Goetsch J, Kottas DJ, et al. Annexin A1 released from apoptotic cells acts through formyl peptide receptors to dampen inflammatory monocyte activation via JAK/STAT/SOCS signalling. EMBO Mol Med. 2011;3(2):102–114. DOI:https://doi.org/10.1002/emmm.201000113
- Filep JG. Biasing the lipoxin A4/formyl peptide receptor 2 pushes inflammatory resolution. Proc Natl Acad Sci USA. 2013;110(45):18033–18034. DOI:https://doi.org/10.1073/pnas.1317798110
- DiMasi JA. Innovating by developing new uses of already-approved drugs: trends in the marketing approval of supplemental indications. Clin Ther. 2013;35(6):808–818. DOI:https://doi.org/10.1016/j.clinthera.2013.04.004
- Mancini GB, Etminan M, Zhang B, et al. Reduction of morbidity and mortality by statins, angiotensin-converting enzyme inhibitors, and angiotensin receptor blockers in patients with chronic obstructive pulmonary disease. J Am Coll Cardiol. 2006;47(12):2554–2560. DOI:https://doi.org/10.1016/j.jacc.2006.04.039
- Søyseth V, Brekke P, Smith P, et al. Statin use is associated with reduced mortality in COPD. Eur Respir J. 2007;29(2):279–283. DOI:https://doi.org/10.1183/09031936.00106406
- Lee J-H, Lee D-S, Kim E-K, et al. Simvastatin inhibits cigarette smoking-induced emphysema and pulmonary hypertension in rat lungs. Am J Respir Crit Care Med. 2005;172(8):987–993. DOI:https://doi.org/10.1164/rccm.200501-041OC
- Wright JL, Zhou S, Preobrazhenska O, et al. Statin reverses smoke-induced pulmonary hypertension and prevents emphysema but not airway remodeling. Am J Respir Crit Care Med. 2011;183(1):50–58. DOI:https://doi.org/10.1164/rccm.201003-0399OC
- Morimoto K, Janssen WJ, Fessler MB, et al. Lovastatin enhances clearance of apoptotic cells (efferocytosis) with implications for chronic obstructive pulmonary disease. J Immunol. 2006;176(12):7657–7665. DOI:https://doi.org/10.4049/jimmunol.176.12.7657
- Janssen WJ, McPhillips KA, Dickinson MG, et al. Surfactant proteins a and D suppress alveolar macrophage phagocytosis via interaction with SIRP alpha. Am J Respir Crit Care Med. 2008;178(2):158–167. DOI:https://doi.org/10.1164/rccm.200711-1661OC
- Hodge S, Hodge G, Jersmann H, et al. Azithromycin improves macrophage phagocytic function and expression of mannose receptor in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;178(2):139–148. DOI:https://doi.org/10.1164/rccm.200711-1666OC
- Yamaryo T, Oishi K, Yoshimine H, et al. Fourteen-Member macrolides promote the phosphatidylserine receptor-dependent phagocytosis of apoptotic neutrophils by alveolar macrophages. Antimicrob Agents Chemother. 2003;47(1):48–53. DOI:https://doi.org/10.1128/AAC.47.1.48-53.2003
- Hodge S, Hodge G, Brozyna S, et al. Azithromycin increases phagocytosis of apoptotic bronchial epithelial cells by alveolar macrophages. Eur Respir J. 2006;28(3):486–495. DOI:https://doi.org/10.1183/09031936.06.00001506
- Hodge S, Tran HB, Hamon R, et al. Nonantibiotic macrolides restore airway macrophage phagocytic function with potential anti-inflammatory effects in chronic lung diseases. Am J Physiol Lung Cell Mol Physiol. 2017;312(5):L678–L87. DOI:https://doi.org/10.1152/ajplung.00518.2016
- Stein M, Keshav S, Harris N, et al. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176(1):287–292. DOI:https://doi.org/10.1084/jem.176.1.287
- Murphy BS, Sundareshan V, Cory TJ, et al. Azithromycin alters macrophage phenotype. J Antimicrob Chemother. 2008;61(3):554–560. DOI:https://doi.org/10.1093/jac/dkn007
- Cory TJ. Effect of azithromycin on macrophage phenotype during pulmonary infections and cystic fibrosis. University of Kentucky; 2011.
- Macowan MG, Liu H, Keller MD, et al. Interventional low-dose azithromycin attenuates cigarette smoke-induced emphysema and lung inflammation in mice. Physiol Rep. 2020;8(13):e14419. DOI:https://doi.org/10.14814/phy2.14419
- Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001;2(2):107–117. DOI:https://doi.org/10.1038/35052055
- Sender V, Moulakakis C, Stamme C. Pulmonary surfactant protein a enhances endolysosomal trafficking in alveolar macrophages through regulation of Rab7. J Immunol. 2011;186(4):2397–2411. DOI:https://doi.org/10.4049/jimmunol.1002446
- Rodman JS, Wandinger-Ness A. Rab GTPases coordinate endocytosis. J Cell Sci. 2000;113 Pt 2:183–192. DOI:https://doi.org/10.1242/jcs.113.2.183
- Hart SP, Alexander KM, Dransfield I. Immune complexes bind preferentially to Fc gamma RIIA (CD32) on apoptotic neutrophils, leading to augmented phagocytosis by macrophages and release of proinflammatory cytokines. J Immunol. 2004;172(3):1882–1887. DOI:https://doi.org/10.4049/jimmunol.172.3.1882
- Brencicova E, Diebold SS. Nucleic acids and endosomal pattern recognition: how to tell friend from foe?Front Cell Infect Microbiol. 2013;3:37.
- Ivan E, Colovai AI. Human Fc receptors: critical targets in the treatment of autoimmune diseases and transplant rejections. Hum Immunol. 2006;67(7):479–491. DOI:https://doi.org/10.1016/j.humimm.2005.12.001
- Green DR, Oguin TH, Martinez J. The clearance of dying cells: table for two. Cell Death Differ. 2016;23(6):915–926. DOI:https://doi.org/10.1038/cdd.2015.172
- Henault J, Martinez J, Riggs JM, et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity. 2012;37(6):986–997. DOI:https://doi.org/10.1016/j.immuni.2012.09.014
- Woo CC, Kumar AP, Sethi G, et al. Thymoquinone: potential cure for inflammatory disorders and cancer. Biochem Pharmacol. 2012;83(4):443–451. DOI:https://doi.org/10.1016/j.bcp.2011.09.029