
Abstract
Current knowledge about the respiratory microbiome is mainly based on 16S ribosomal RNA gene sequencing. Newer sequencing approaches, such as metatranscriptomics, offer the technical ability to measure the viable microbiome response to environmental conditions such as smoking as well as to explore its functional role by investigating host-microbiome interactions. However, knowledge about its feasibility in respiratory microbiome research, especially in lung biopsies, is still very limited. RNA sequencing was performed in bronchial biopsies from clinically stable smokers (n = 5) and ex-smokers (n = 6) with COPD not using (inhaled) steroids. The Trinity assembler was used to assemble non-human reads in order to allow unbiased taxonomical and microbial transcriptional analyses. Subsequently, host-microbiome interactions were analyzed based on associations with host transcriptomic data. Ultra-low levels of microbial mass (0.009%) were identified in the RNA-seq data. Overall, no differences were identified in microbiome diversity or transcriptional profiles of microbial communities or individual microbes between COPD smokers and ex-smokers in the initial test dataset as well as a larger replication dataset. We identified an upregulated host gene set, related to the simultaneous presence of Bradyrhizobium, Roseomonas, Brevibacterium.spp., which were related to PERK-mediated unfolded protein response (UPR) and expression of the microRNA-155-5p. Our results show that metatranscriptomic profiling in bronchial biopsy samples from stable COPD patients yields ultra-low levels of microbial mass. Further, this study illustrates the potential of using transcriptional profiling of the host and microbiome to gain more insight into their interaction in the airways.
Trial registration::
Introduction
The airway microbiome is altered in patients with moderate to severe Chronic Obstructive Pulmonary Diseases (COPD), compared to healthy controls, and associations between changes in the airway microbiome and development of COPD have been proposed [Citation1–Citation4]. Ramsheh and colleagues have recently shown that with increasing severity of COPD, the airway microbiomes changes in concert with downregulation of genes involved in epithelial defense and increased expression of Il-17 and TNF inflammatory pathways [Citation5].
Current knowledge about the respiratory microbiomes as well as the influence of smoke-exposure is still mainly based on 16S ribosomal RNA (rRNA) gene sequencing, focusing on the bacterial microbiome. So far, no significant differences have been found in lower respiratory tract (LRT) samples between nonsmokers and smokers [Citation2,Citation3]. 16S rRNA gene sequencing provides limited insight into the characteristics of the microbiome. Important limitations include the inability to distinguish between live and dead microbes or to detect viruses, fungi, and other eukaryotic organisms.
Applying newer sequencing techniques, such as metatranscriptomics, offers the technical ability to overcome these limitations. It enables us to assess broad microbial transcriptional signatures, which cannot be found solely at the DNA level. They allow us to evaluate the viable microbiome and to explore shifts in microbiome activity caused by environmental factors, such as smoking. Further, simultaneous transcriptomic profiling of host and microbiota can be of help to elucidate its role played in airway health and chronic pulmonary disease pathology [Citation6,Citation7].
Knowledge about the feasibility of applying metatranscriptomic sequencing in COPD lung samples for microbiome analyses is still very limited. Ren and colleagues have shown in bronchoalveolar lavage (BAL) samples that transcriptionally active microbial profiles were associated with bacterial load and Th17 immune response [Citation8]. To our best knowledge, no study has analyzed the microbiome in bronchial biopsy specimens from COPD subjects, using metatranscriptomic sequencing.
This study aims to identify a biological signature of the viable microbiome in bronchial biopsies from smokers and ex-smokers with stable COPD, using an unbiased metatranscriptomic approach. We aimed to compare the bronchial microbiome depending on smoking status as well as to study host-microbiome interactions.
Methods
Study design and subjects
Baseline RNA-seq expression was studied in bronchial biopsies from 11 COPD subjects. Two negative samples were sequenced in the same batch to assess the influence of the reagent contamination in our samples. The COPD subjects participated in the Groningen Leiden Universities and Corticosteroids in Obstructive Lung Disease (GLUCOLD) study (registered at ClinicalTrials.gov with identifier number NCT00158847) [Citation9]. In addition, expression profiles of identified microbial contigs were reevaluated in a previous RNA-seq GLUCOLD dataset consisting of 56 COPD subjects (38 smokers; 18 ex-smokers), which was initially not sequenced together with negative control samples.
Methods and patient characteristics have been previously described [Citation9]. In short, all subjects had irreversible airflow limitation and at least one of the following symptoms: chronic cough, chronic sputum production, or dyspnea on exertion. All subjects were current or ex-smokers (≥ 1 year) with at least 10 pack-years of smoking. Exclusion criteria were asthma and the use of oral corticosteroids during the last three months and maintenance treatment with inhaled or oral steroids during the last six months. All patients were in a stable clinical condition. This study was performed in a subset of the GLUCOLD study. All study protocols were approved by the medical ethics committees of the Leiden University Medical Center and the Groningen University Medical Center, and all patients gave their written informed consent.
Sample collection and processing
The sample collection has been described previously [Citation9]. Briefly, samples were obtained by fiberoptic bronchoscopy, which was performed using a standardized protocol as described previously [Citation10,Citation11]. Subjects were asked to refrain from smoking on the day of the bronchoscopy. Bronchial biopsies were randomly taken from (sub) segmental carinae in the right or left lower lobe of the lung, whereby one part was immediately snap-frozen and stored at −80 C for later sequencing analyses. Subsequent sample processing, RNA sequencing, quality control, and normalization are fully described in the online supplementary material of this manuscript.
Sequence assembly, mapping, and contamination assessment
Sequence assembly, mapping, and contamination assessment are fully described in the supplementary online material of this manuscript. In short, the de novo assembly program Trinity was applied to assemble the unmapped reads [Citation12]. This program allows a reconstruction of a full-length transcriptome without a genome reference from RNA-seq data by generating de novo transcriptome contigs. Principal component analysis (PCA) was conducted, based on GMPR (geometric means of pairwise ratios) normalized microbial contigs, to visualize variation differences of microbial expression data between bronchial biopsy and negative control samples.
Statistics
Microbial contigs were normalized using the GMPR normalization method for zero-inflated sequencing data, such as microbiome data [Citation13]. The richness and alpha-diversity indices (Shannon, Simpson, and inverse Simpson) were calculated, based on normalized microbial contigs between COPD smokers and ex-smokers, using the Bioconductor package “vegan” [Citation14]. Richness and diversity indices, as well as microbiome communities at the phylum-level, were compared between COPD smokers and ex-smokers, by Mann-Whitney U-tests. Individual microbial contigs were analyzed for differential expression between COPD smokers and ex-smokers, using Bioconductor package DESeq2 [Citation15]. We corrected for age and maintained a false discovery rate (FDR) below 0.05 to control for multiple testing [Citation16].
To determine host-microbial-specific interactions, microbial contigs were compared to bronchial inflammatory cell counts (CD3, CD4, CD8 T cells; plasma cells, macrophages, eosinophil cells, neutrophil cells) and lung function (post-bronchodilator FEV1%; postbronchodilator FEV1/IVC), using Spearman correlation testing, while maintaining a FDR below 0.05. Further, we aimed to investigate associations between host gene expression profiles and RNA abundance from microbial contigs. Therefore, microbial contigs were compared to host gene expression profiles per subject, using the quasi-likelihood F-test in edgeR package [Citation17]. The model was adjusted for smoking status and age. We maintained a false discovery rate (FDR) below 0.05 to control for multiple testing [Citation16]. Subsequently, the top 50 significant host genes were analyzed for biological and functional profiling in the web-based toolset g:profiler [Citation18]. Besides, the top 50 associated host genes were surveyed for overlap across the different microbes to assess a uniform host-microbiome interaction. All statistical analyses were performed with the R statistical software v. 3.6.1.
Results
Participant characteristics
summarizes the baseline characteristics of 5 smokers and 6 ex-smokers with COPD in GLUCOLD, who had biopsies with RNA of sufficient quality for analysis. History of smoking, post-bronchodilator FEV1 (% predicted), FEV1/IVC, and inflammatory cell counts from bronchial biopsies did not show significant differences between groups.
Table 1. Participant characteristics.
Bronchial microbiome compositions between COPD smokers and ex-smokers
In total, 6,284,287 RNA counts per participant were generated by RNA-seq of the bronchial biopsies. About 99% of the obtained RNA-seq reads mapped to the human genome (Supplemental Figure S2). The remaining 1% unmapped reads were assembled to contigs, using the Trinity assembler. In total 242,312 contigs were generated. Our analysis identified that 92.9% of contig counts belonged to human, 0.1% to fungus, 1% to other micro-eukaryotic, 5.6% to bacteria, and 0.4% to plants genetic material. For further analysis, we removed contigs mapping to human or plant genetic data. In total, 1219 microbial contigs were identified and taxonomically classified. The principal component analysis (PCA) based on normalized microbial expression data showed a clear separation between bronchial biopsy and negative control samples (Supplemental Figure S3). About 94.30% of the microbial expression data were identified as likely contaminants (i.e. contaminant microbiota during library preparation) and removed from further analysis. 69 microbial contigs were identified after contamination assessment with an average of 541 microbial RNA counts per participant, which represented 0.009% of all generated RNA counts by RNA-seq. In total, 94.06% of these contigs counts belonged to bacteria and 5.94% to other eukaryota (supplemental Figure S4). Viral or fungal contigs were not detected in this cleaned data set. At the phylum level, the most relatively abundant phyla were Proteobacteria (24.95%), Verrucomicrobia (21.08%), and Bacteroidetes (18.78%). At the genus level, the most relatively abundant bacterial genera were Methylacidiphilum (21.95%), Flavobacterium (12.44%), and Staphylococcus (6.03%), whereas Philasterides (33.54%) was the most abundant genera belonging to other eukaryotes. Next, we compared the bronchial microbiome composition between COPD smokers and COPD ex-smokers at the phylum-level (). Predominantly expressed microbial contigs belonged to the phyla Proteobacteria (COPD smokers: 25.5%; COPD ex-smokers: 24.5%), Verrucomicrobia (COPD smokers: 24.2%; COPD ex-smokers: 19.0%), Bacteroidetes (COPD smokers: 19.5%; COPD ex-smokers: 18.3%). No significant differences were found between COPD smokers and ex-smokers concerning RNA abundance of the identified phyla (p-value >0.05) ().
Figure 1. Phyla composition of the bronchial microbiome.
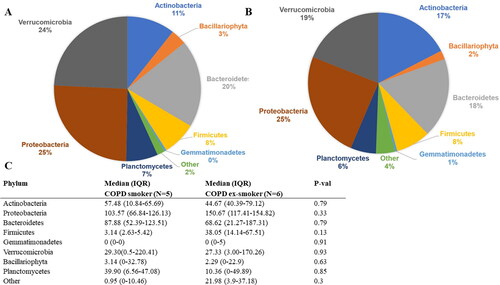
Pie chart of expression percentages of phyla in A) COPD smokers; B) COPD ex-smokers. C) Comparison of phyla RNA abundance (GMPR normalized counts) between smoker and ex-smoker with COPD, using Mann–Whitney U-tests; IQR = Interquartile range: 25th–75th percentile.
Richness and diversity
No significant differences were identified between COPD smokers and ex-smokers, regarding richness and alpha-diversity indices (Shannon, Simpson, and inverse Simpson), based on the expression data of the 69 identified microbial contigs (supplementary figure S5).
Microbial transcriptional profile analysis
Transcriptional profiles of the 69 microbial contigs were compared between COPD smokers and ex-smokers, based on differential RNA abundance. No differences between COPD smokers and ex-smokers were identified (FDR >0.05) (supplementary Table S1). In addition, expression profiles of these microbial contigs were reevaluated in a previous larger RNA-seq GLUCOLD dataset consisting of 56 COPD subjects (38 smokers; 18 ex-smokers) (patient characteristics are described in supplementary Table S3). 47 out of 69 microbial contigs were identified in this dataset. Again, no differences were found between COPD smokers and ex-smokers (FDR >0.05) (supplementary Table S4).
Host-microbiome interactions
To determine host-microbiome interactions, transcriptional profiles of 69 microbial contigs were compared to bronchial inflammatory cell counts, and lung function parameters. No significant correlations were identified while maintaining an FDR of 0.05 (supplemental Table S2). Further, we determined microbial-specific host pathways by comparing microbial contig expression to host gene expression per subject. The top 50 significant associated host genes per microbial contig (FDR < 0.05) were biologically and functionally profiled (supplemental Table S3). Upregulated host gene sets were only identified for three microbial contigs. These contigs belonged to the genera Bradyrhizobium sp. (590 associated host genes), Breivibacterium sp. (402 associated host genes), and Roseomonas sp. (1089 associated host genes) and included various gene ontology cellular components and biological pathways.
Downregulated host genes were identified for the same three microbial contigs. Various biological and functional profiles were identified for Brevibacterium sp. (119 associated host genes), including anatomical structure morphogenesis, system development, and axon guidance. No biological and functional profiles were identified for downregulated host genes, that were associated with Bradyrhizobium and Roseomonas spp.
Further, the overlap of up-and downregulated host gene sets were surveyed across the different microbes to evaluate the presence of uniform host-microbiome interactions. Therefore, overlaps between the top 50 host genes, associated with Bradyrhizobium sp, Breivibacterium sp., and Roseomonas sp., were tested and further biological and functional profiled (). Across the upregulated host gene sets, we identified eight overlapping genes, including CXCL8, TMEM33, LYPLA1, CYB5R4, NAMPT, GOLT1B, LIN7C, FAM91A1. Profiling of these genes revealed biological and functional profiles related to PERK-mediated unfolded protein response (CXCL8, TMEM33) and miR-155-5p (CXCL8, TMEM33, LYPLA1, NAMPT, GOLT1B, LIN7C, FAM91A1). Across the downregulated host gene sets, no overlap was identified.
Table 2. Uniform host-microbiome interactions related to the simultaneous presence of Bradyrhizobium sp, Breivibacterium sp., and Roseomonas sp. contigs.
Discussion
This study aimed to investigate the bronchial microbiome at RNA-level in clinically stable COPD patients and to study host-microbiome interactions. To our knowledge, this is the first study that investigated the viable microbiome in bronchial biopsy specimens from COPD subjects, using an unbiased metatranscriptomic approach. Our results indicate that RNA-sequencing of bronchial biopsy specimens in stable COPD patients yields ultra-low levels of microbial biomass, considering that only 0.009% of all generated RNA-seq data could be determined as microbiota. Furthermore, our results highlight the importance of including negative controls when assessing the lung microbiome at the RNA-level, as 94.30% of the identified microbial expression were identified as likely contaminants during library preparation. Overall, this is in line with previous studies, which showed that the lower respiratory tract (LRT) harbors low microbiome biomass, also in stable COPD patients [Citation19,Citation20]. Our findings indicate that current- compared to ex-smoking in COPD patients is associated with no differences in bronchial microbiome diversity or transcriptional profiles of microbial communities at phylum-level as well as individual microbes. Since most of the microbial expression data belonged to bacteria, these results are in line with previous studies, which also found no differences in the lung bacterial microbiome between healthy nonsmokers and smokers [Citation2,Citation3]. Importantly, we cannot draw any conclusions regarding differences of the virome or mycobiome, since no viral contigs were detected and the identified fungal contigs were removed after contamination assessment.
Surprisingly, in our study, we identified Bacillariophyta and Philasterides sp. as the most abundant non-bacterial phylum and genus, respectively, which requires careful interpretation. Bacillariophyta are unicellular eukaryotic organisms, belonging to the group of diatom and Philasterides sp. is a protozoan and marine-known organism [Citation21,Citation22]. No previous microbiome study has reported the presence of such organisms in the lungs. It could be speculated that its presence was missed due to different sequencing and mapping methods. Nevertheless, the possibility of false-positive signals must be considered as well, suggesting that the microbial background noise remains high in bronchial biopsies using transcriptomic profiling, despite rigorous contamination assessment. This raises the question of whether other lung specimens with a more balanced ratio of microbiota to host cells, such as bronchial wash, BAL, or sputum samples, might be more suitable to analyze the viable microbiome, using transcriptomic profiling [Citation23].
Further, we identified host-microbiome interactions for only three out of 69 microbial contigs. The three microbial contigs belonged to the genera Bradyrhizobium, Roseomonas, and Brevibacterium. Bradyrhizobium sp. is a Gram-negative bacterium, which has been identified in BAL samples from corticosteroid-sensitive asthma patients as well as patients with lung cancer [Citation24,Citation25]. In our study, we found various upregulated host-Bradyrhizobium-specific profiles, including integrin alpha_V-beta_6 complex, which is a glycoprotein involved in wound healing and the pathogenesis of diseases including fibrosis and cancer [Citation26]. Roseomonas sp. is a Gram-negative bacterium and its presence in the lungs has been described in one case report, based on a sputum sample, causing a secondary bacterial infection in a patient with pulmonary tuberculosis [Citation27]. In our study, we found inconclusive upregulated host Roseomonas-specific profiles, such as transport or organic substance transport. Brevibacterium sp. is a Gram-positive bacterium, which colonizes the skin [Citation28]. Although it has been identified in oropharyngeal swabs from asthma and COPD patients, this organism has not been described in any other lung microbiome study, yet [Citation29]. In our study, we found inconclusive up-and downregulated host Brevibacterium-specific profiles, including cytoplasm, system development, and axon guidance.
Interestingly, we identified upregulated uniform host-microbiome interactions, related to the simultaneous presence of Bradyrhizobium, Roseomonas, Brevibacterium.spp., including PERK-mediated unfolded protein response (UPR) and miR-155-5p. The UPR is a cellular pathway that is activated upon endoplasmic reticulum (ER) stress resulting from e.g. accumulation of misfolded proteins and is aimed at restoring homeostasis [Citation30,Citation31]. The UPR can be triggered by numerous endogenous and exogenous conditions, including pathogen or smoking exposure, and is known to be involved in regulating immune responses, also in pulmonary pathology [Citation30,Citation32]. Therefore, our results suggest that either the presence of these microbes is associated with this type of host cellular stress responses or that the UPR is a prerequisite allowing the presence of these microbial species. Importantly, no causality can be derived from the identified associations between host and microbial transcriptional profiles. miR-155-5p is a noncoding microRNA and a gene regulator in immune system function [Citation33]. It has been shown that smoke-induced pulmonary inflammation is attenuated in miR-155- deficient mice, highlighting its potential role in the pathogenesis of COPD [Citation34]. Therefore, it can be speculated that the presence of Bradyrhizobium, Roseomonas, Brevibacterium. spp in bronchial airways might be involved in this pathway. Importantly, no causal conclusions can be drawn due to the cross-sectional study design.
Although we identified plausible uniform host-microbiome interactions concerning these bacteria, future studies are needed to confirm these findings as well as their presence in the lungs. These bacteria have been described in previous reagent contamination studies and remain suspicious as being false negative signals, despite rigorous contamination assessment in our study [Citation35–37].
There are several limitations related to our study as well as future recommendations. We aimed to obtain a biological signature of the microbiome using an RNA-seq dataset, which was initially intended to investigate host gene expression profiles. Therefore, a non-microbiome-specific RNA preparation kit was used during sample processing. Since we identified only ultra-low levels of microbial biomass, future studies should consider using microbiome-enrichment RNA preparation kits to increase microbial signals. In addition, we would suggest subdividing collected samples for distinct microbiome and host RNA extraction and processing to optimize host and microbiome analyses. Further, future studies should consider to include positive controls, next to negative controls, to optimize further sample-to-sample normalization of the microbiome as well as to evaluate the limit of detection and the efficiency of library preparation [Citation37].
Furthermore, microbiome differences were assessed in bronchial biopsies based on a relatively small number of COPD smokers and ex-smokers. Here, it is important to realize that different airway sampling methods are likely to provide divergent insight into smoking effects on the microbiome, as these are consistent with the niche-specificity of the airway tract [Citation38]. Besides, our study did not include never-smokers, which does not allow concluding general smoking effects on the microbiome, but only on smoking status. Further, our study failed to evaluate RNA signals from viruses. It can be speculated that viral signals were missed due to our rigorous sequence mapping approach or due to the ultra-low levels of microbial biomass in bronchial biopsies. Nevertheless, airway DNA- and RNA-based viruses are of particular interest in airway microbiome research and require further scientific attention concerning their detection by metagenomic and -transcriptomic sequencing. Future studies need to consider that their identification by metagenomic/-transcriptomic approaches require optimized strategies since the choice of sequencing platforms dictates the quality of viral transcriptome profiling [Citation39,Citation40].
Conclusion
In conclusion, our results show that metatranscriptomic profiling in bronchial biopsy samples from stable COPD patients yields ultra-low levels of microbial mass. Further, this study illustrates the potential of using transcriptional profiling of the host and microbiome to gain more insight into their interaction in the airways.
Author contributions
B.D., J.B., V.G., MvdB., A.F. contributed to the study concept and design. P.H., W.T., MvdB., H.K., coordinated patient inclusion and data collection for GLUCOLD. MvdB., C.B., M.N., M.G., G.T., organized and performed the RNA sequencing. B.D., J.B., V.G., MvdB., A.F., N.C., J.R. analyzed and interpreted data. B.D drafted the manuscript which was critically read and revised by the other coauthors.
Ethics approval and consent to participate
The ethical committee of the University Medical Center Groningen (Medisch Ethische Toetsingscommisie) approved the study and all included subjects gave their written informed consent
Consent for publication
Not applicable.
Compete of interest
HK reports unrestricted grants, from Boehringer Ingelheim, Novartis and GlaxoSmithKline. He also reports fees advisory board participation for AstraZeneca, Boehringer Ingelheim, Novartis and GlaxoSmithKline. All above paid to his institution. GWT reports and I am a full time employee of Genentech, Inc and hold stock and options in the Roche. PH reports grants from Boerhinger Ingelheim, grants from Galapagos, outside the submitted work; MAG reports I am full time employee of Genentech, Inc and hold stock and options in the Roche Group. WT reports personal fees from Roche Diagnostics/Ventana, personal fees from Merck Sharp Dohme, personal fees from Bristol-Myers-Squibb, personal fees from AbbVie, outside the submitted work. JR reports personal fees from IDbyDNA Inc., from null, from null, outside the submitted work; and Dr. Rossen is currently an employee of IDbyDNA. IDbyDNA did not have any influence on the interpretation of reviewed data and conclusions drawn, nor on the drafting of the manuscript, and did not (financial) support the study. MN reports I am a full time employee of Genentech, Inc and hold stock and options in the Roche Group. BD, JB, NC, CAB, VG, AF and MvdB have nothing to disclose.
| Abbreviations | ||
| COPD | = | Chronic Obstructive Pulmonary Diseases |
| FDR | = | False discovery rate |
| GLUCOLD | = | Groningen and Leiden Universities study of Corticosteroids in Obstructive Lung Disease |
| LRT | = | Lower respiratory tract |
| PCA | = | Principal component analysis |
| rRNA | = | ribosomal RNA |
Acknowledgements
The author would like to thank Arnau Vich Vila (University of Groningen, Groningen, The Netherlands) for his scientific input regarding the development of the RNA-seq analysis pipeline in this study.
Funding
The submitted work is cofinanced by the Ministry of Economic Affairs and Climate Policy by means of the PPP. AF was supported by a junior longfond grant (4.2.16.132JO). The study was funded by unrestricted grants from the Stichting Astma Bestrijding, the Netherlands Asthma Foundation, Netherlands Organization for Scientific Research (ZonMw), GlaxoSmithKline, the Royal Dutch Academy of Sciences and Arts, the University Medical Center Groningen and Leiden University Medical Center and the NIH R01 HL095388 (Spira/Lenburg). RNA-sequencing of the bronchial biopsies was funded by Genentech (San Francisco, CA, USA).
Availability of data and materials
The data that support the findings of this study are available from Genentech but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request and with permission of Genentech.
References
- Erb-Downward JR, Thompson DL, Han MK, et al. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS One. 2011;6(2):e16384. DOI:10.1371/journal.pone.0016384
- Einarsson GG, Comer DM, Mcilreavey L, et al. Community dynamics and the lower airway microbiota in stable chronic obstructive pulmonary disease, smokers and healthy non-smokers. Thorax. 2016;71(9):795–803. DOI:10.1136/thoraxjnl-2015-207235
- Morris A, Lung HIV Microbiome Project, Beck JM, Schloss PD, et al. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med. 2013;187(10):1067–1075. DOI:10.1164/rccm.201210-1913OC
- Dickson RP, Erb-Downward JR, Huffnagle GB. The role of the bacterial microbiome in lung disease. Expert Rev Respir Med. 2013;7(3):245–257.
- Ramsheh MY, Haldar K, Esteve-Codina A, et al. Lung microbiome composition and bronchial epithelial gene expression in patients with COPD versus healthy individuals: a bacterial 16S rRNA gene sequencing and host transcriptomic analysis. The Lancet Microbe. 2021;2(7):e300–e310. DOI:10.1016/S2666-5247(21)00035-5
- Bashiardes S, Zilberman-Schapira G, Elinav E. Use of metatranscriptomics in microbiome research. Bioinform Biol Insights. 2016;10:19–25. DOI:10.4137/BBI.S34610
- Westermann AJ, Barquist L, Vogel J. Resolving host-pathogen interactions by dual RNA-seq. PLoS Pathog. 2017;13(2):e1006033 DOI:10.1371/journal.ppat.1006033
- Ren L, Zhang R, Rao J, et al. Transcriptionally active lung microbiome and its association with bacterial biomass and host inflammatory status. mSystems. 2018;3(5):e00199–18., Oct 30DOI:10.1128/mSystems.00199-18
- Lapperre TS, Snoeck-Stroband JB, Gosman MM. Effect of fluticasone with and without salmeterol on pulmonary outcomes in chronic obstructive pulmonary disease: a randomized trial. Ann Intern Med. 2009;151(8):517–527. DOI:10.7326/0003-4819-151-8-200910200-00004
- Jeffery P, Endobronchial Biopsy Workshop, Holgate S, Wenzel S. Methods for the assessment of endobronchial biopsies in clinical research: Application to studies of pathogenesis and the effects of treatment. Am J Respir Crit Care Med. 2003;168(6 Pt 2):S1–S17. DOI:10.1164/rccm.200202-150WS
- Lapperre TS, Postma DS, Gosman MME, et al. Relation between duration of smoking cessation and bronchial inflammation in COPD. Thorax. 2006;61(2):115–121. DOI:10.1136/thx.2005.040519
- Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–652. doi:10.1038/nbt.1883.Trinity
- Chen L, Reeve J, Zhang L, et al. GMPR: a robust normalization method for zero-inflated count data with application to microbiome sequencing data. PeerJ. 2018;6:e4600 DOI:10.7717/peerj.4600
- Oksanen AJ, Blanchet FG, Kindt R, et al. Vegan: community ecology package. R package, version. R Proj Stat Comput Vienna, Austri. 2015;2.2–1.
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550550. DOI:10.1186/s13059-014-0550-8
- Benjamini Y, Drai D, Elmer G, et al. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125(1-2):279–284. DOI:10.1016/S0166-4328(01)00297-2
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. DOI:10.1093/bioinformatics/btp616
- Reimand J, Kull M, Peterson H, et al. Profiler-a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007;35:193–200. DOI:10.1093/nar/gkm226
- Charlson ES, Bittinger K, Haas AR, et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184(8):957–963. DOI:10.1164/rccm.201104-0655OC
- Sze MA, Dimitriu PA, Hayashi S, et al. The lung tissue microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;185(10):1073–1080. DOI:10.1164/rccm.201111-2075OC
- Iglesias R, Paramá A, Alvarez MF, et al. Philasterides dicentrarchi (ciliophora, scuticociliatida) as the causative agent of scuticociliatosis in farmed turbot Scophthalmus maximus in Galicia (NW Spain). Dis Aquat Organ. 2001;46(1):47–55. DOI:10.3354/dao046047
- Parks MB, Nakov T, Ruck EC, et al. Phylogenomics reveals an extensive history of genome duplication in diatoms (bacillariophyta). Am J Bot. 2018;105(3):330–347. DOI:10.1002/ajb2.1056
- Ditz B, Christenson S, Rossen J, et al. Sputum microbiome profiling in COPD: beyond singular pathogen detection. Thorax. 2020;75(4):338–344. DOI:10.1136/thoraxjnl-2019-214168
- Jin J, Gan Y, Liu H, et al. Diminishing microbiome richness and distinction in the lower respiratory tract of lung cancer patients: A multiple comparative study design with independent validation. Lung Cancer. 2019;136:129–135. DOI:10.1016/j.lungcan.2019.08.022
- Goleva E, Jackson LP, Harris JK, et al. The effects of airway microbiome on corticosteroid responsiveness in asthma. Am J Respir Crit Care Med. 2013;188(10):1193–1201. DOI:10.1164/rccm.201304-0775OC
- Meecham A, Marshall JF. The ITGB6 gene: its role in experimental and clinical biology. Gene X. 2020;5:100023. DOI:10.1016/j.gene.2019.100023
- Kaore NM, Khan Z, Aher AR, et al. Community acquired Roseomonas infection in a pre-existing Tubercular lung lesion. Lung India. 2014;31(3):289–292. DOI:10.4103/0970-2113.135785
- Grice EA, Segre JA. The skin microbiome. Nat Rev Microbiol. 2011;9(4):244–253.
- Ogorodova LM, Fedosenko SV, Popenko AS, et al. Comparison study of oropharyngeal microbiota in case of bronchial asthma and chronic obstructive pulmonary disease in different severity levels. Ann Rams. 2015;70(6):669–678. DOI:10.15690/vramn532
- Grootjans J, Kaser A, Kaufman RJ, et al. The unfolded protein response in immunity and inflammation. Nat Rev Immunol. 2016;16(8):469–484.
- Marciniak SJ. Endoplasmic reticulum stress : a key player in human disease. FEBS J. 2019;286(2):228–231. DOI:10.1111/febs.14740
- Dickens JA, Malzer E, Chambers JE, et al. Pulmonary endoplasmic reticulum stress-scars, smoke, and suffocation. FEBS J. 2019;286(2):322–341. DOI:10.1111/febs.14381
- Conickx G, Avila Cobos F, Van Den Berge M, et al. MicroRNA profiling in lung tissue and bronchoalveolar lavage of cigarette smoke-exposed mice and in COPD patients: a translational approach. Sci Rep. 2017;7(1):1–14. DOI:10.1038/s41598-017-13265-8
- Smet EG, DeEeckhoutte HP, VanCobos FA, et al. The role of miR-155 in cigarette smoke-induced pulmonary in fl ammation and COPD. Mucosal Immunol. 2020;13(3):423–436. DOI:10.1038/s41385-019-0241-6
- de Goffau MC, Lager S, Salter SJ, et al. Recognizing the reagent microbiome. Nat Microbiol. 2018;3(8):851–853. DOI:10.1038/s41564-018-0202-y
- Salter SJ, Cox MJ, Turek EM, et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014;12:87. DOI:10.1186/s12915-014-0087-z
- Eisenhofer R, Minich JJ, Marotz C, et al. Contamination in low microbial biomass microbiome studies: Issues and recommendations. Trends Microbiol. 2019;27(2):105–117. DOI:10.1016/j.tim.2018.11.003
- Man WH, De Steenhuijsen Piters WAA, Bogaert D. The microbiota of the respiratory tract: Gatekeeper to respiratory health. Nat Rev Microbiol. 2017;15(5):259–270. DOI:10.1038/nrmicro.2017.14
- Wongsurawat T, Jenjaroenpun P, Taylor MK, et al. Rapid sequencing of multiple RNA viruses in their native form. Front Microbiol. 2019;10:260. DOI:10.3389/fmicb.2019.00260
- Depledge DP, Mohr I, Wilson AC. Going the distance : Optimizing RNA-Seq strategies for transcriptomic analysis of complex viral genomes. J Virol. 2019;93(1):18. pii: e01342. DOI:10.1128/JVI.01342-18