
Abstract
A high smoking-independent chronic obstructive pulmonary disease (COPD) prevalence is observed in lung cancer patients. However, the underlying connection between these two diseases still remains unclear. Cigarette smoking and ambient air pollution are common risk factors for COPD and lung cancer. In this study, we established rat COPD model through exposure to cigarette smoke (CS) or motor vehicle exhaust (MVE). The model rats developed COPD-like phenotypes, manifested as lung functions decline, lung inflammation, emphysema-like alveolar enlargement and airway remodeling. The programmed death-ligand 1 (PD-L1), a factor contributing to immune escape of tumor cells, was overexpressed in lungs from COPD model rats, though more severe COPD phenotypes did not bring with further PD-L1 overexpression in lung. The upregulations of proinflammatory cytokines and PD-L1 were also observed in cultured human bronchial epithelial cells BEAS-2B upon treatment with cigarette smoke extract (CSE) or diesel-related particulate matter 2.5 (PM2.5, SEM1650b). The inflammatory cytokines produced in BEAS-2B cells reflected the PD-L1 levels. Furthermore, ERK1/2, a kinase mediating PD-L1 upregulation in premalignant bronchial cells or NSCLC cells, and STAT1/3, which was reportedly associated with PD-L1 expression in lung tumors, were activated in COPD rats’ lungs or in BEAS-2B cells treated with CSE or PM2.5. Therefore, we proposed that inflammation associated PD-L1 overexpression in airway epithelial cells could be the underlying factor facilitating lung cancer incidence in COPD.
Introduction
Several epidemiological studies highlighted the link between chronic obstructive pulmonary disease (COPD) and lung cancer, including the association between airflow limitation and increased lung cancer incidence and mortality [Citation1]. Further study indicated the prevalence of COPD in lung cancer cases was independent of age, sex or smoking history [Citation2]. The main risk factors for COPD include but are not limited to cigarette smoke (CS) exposure and ambient air pollution, both of which are common risk factors for lung cancer. However, the operative mechanisms between COPD prevalence and lung cancer are remained elusive [Citation3–5].
Transmembrane protein PD-L1 is overexpressed in many cancer types including lung cancer. Programmed death-ligand 1 (PD-L1) inhibited the functions of activated cytotoxic CD8+ T cells through interaction with PD-1 on cell surface of T cells [Citation6]. The overexpression of PD-L1 was supposed contribute to lung cancer initiation by facilitating immune escape in premalignant human bronchial epithelial cells [Citation7]. Clinical study showed that immunotherapy targeting on immune-checkpoint proteins PD-1/PD-L1 displayed promising clinical effects on non-small cell lung cancer [Citation8]. The associations among COPD, PD-1/PD-L1 axis activation and lung cancer therapy were revealed in clinical trial that immune checkpoint inhibitors (ICIs) displayed better therapeutic outcome in lung cancer patients underlying COPD than those without COPD [Citation9]. It was proposed that chronic lung inflammations commonly seen in COPD patients enhance the risk of tumorigenesis by weakening immune surveillance in lung. This hypothesis was partially verified by the findings that lipopolysaccharide-mediated chronic inflammation upregulated PD-L1 expression through TLR4/ERK axis in lung cancer cells, or activated PD-1/PD-L1 axis in lung cancer model mice lung in which therapy coupled with anti-PD-1 treatment elicited better anti-tumor efficacy [Citation10, Citation11].
As the main risk factors for COPD, both CS and MVE exposure induced lung inflammation in COPD rodent animal models [Citation12, Citation13]. However, the PD-L1 overexpression in COPD rodent animals has not been fully studied. Tobacco smoke was reported induce PD-L1 increases in lung of A/J mice, a mouse strain having comparatively high lung tumor rate upon carcinogen treatments [Citation14]. Furthermore, although the air pollutant particles activated some proteins, such as ERK1/2 and STAT, which are able to promote PD-L1 expression in bronchial epithelial cells or lung tumor cells, whether air pollutants affected lung PD-L1 expression in COPD patients or in COPD rodent animals are not fully explored [Citation7, Citation15–17].
In this study, we established rat COPD models through exposure to CS or MVE mimicking air pollution and reported that PD-L1 was upregulated in lung tissues from the two types of COPD model rats. PD-L1 levels in cultured human bronchial epithelial cells BEAS-2B were increased by CSE or diesel-related PM2.5. In lung tissues and cells, PD-L1 was upregulated along with production of proinflammatory cytokines and the activation of ERK and STAT. Our findings gave a clue that inflammation associated PD-L1 upregulation induced by CS or MVE in lung epithelial cells might contribute to the increased lung cancer incidence in COPD patients.
Methods and materials
Animals
Male SD rats (8–12 weeks old) were purchased from the Guangdong Medical Laboratory Animal Center (Guangzhou, China) and housed in the specific pathogen-free facilities accessible to food and water freely. The experimental methods were approved by the Ethics Committee for Animal Experiments of the General Hospital of Southern Theater Command of PLA. The rats were exposed to CS or MVE according to previous methods [Citation12, Citation13] and detailed steps described in Supplemental Materials.
Lung function measurement
The rats exposed to CS or MVE were subjected to lung function measurement by using a Forced Pulmonary Maneuver System (Buxco Research Systems, Wilmington, NC) following a protocol described previously [Citation13]. The values of parameters airway resistance, chord compliance, total lung capacity and the ratio of FEV50/FVC were determined.
Cell culture
The human bronchial epithelial cells BEAS-2B were subjected to treatments with CS extract (CSE, 0–20%) or diesel-related PM2.5 Standard Reference DEP material – SRM 1650 b (0–300 µg/ml), respectively. Cell viability was determined by using a CCK-8 test kit and presented as percentage of control.
Quantitative RT-PCR assay
The relative mRNA levels of rat or human PD-L1 were quantified, normalized to GAPDH and expressed as percentage of control.
Western blotting
The protein levels of ERK1/2, p-ERK1/2, STATs and p-STATs in total proteins extracted from cultured cells or lung tissues were determined by western blotting and semi-quantitated by using the software Image J.
Immunofluorescence microscopy
The distribution of PD-L1 in lung tissue sections was indicated by immunofluorescence staining with a PD-L1 antibody.
ELISA
The ELISA kits from eBioscience (San Diego, CA) were used to measure the levels of rat IL6 (88-50,625) and rat TNF-α (88-7,340) in BALF and the levels of human IL6 (88–7,066) and human IL8 (88–8,086) in cell culture medium according to the manufacturer’s instructions. The levels of Muc5ac in BALF were measured by using a capture antibody for Muc5ac (sc-21701, Santa Cruz Biotechnology, Santa Cruz, CA) and corresponding detection antibody obtained from KPL, Inc. (Gaithersburg, MD) according to protocol described previously [Citation18].
Statistical analysis
All in vitro experiments were carried out in triplicate and repeated three times. Comparison between two groups was determined by Students t-test, one-way ANOVA further followed by Bonferroni post-hoc test was adopted for comparison in experiments with ≥ 3 groups. SPSS version 16.0 (SPSS Inc., Chicago, IL) and Graphpad prism version 5.0 (Graphpad Inc., La Jolla, CA) were used for statistics and graphing, respectively. Data were presented as mean ± SEM. N denotes the number of repeats in cell experiments or the number of animals. p Values less than .05 or .01 were considered as statistically significant or extremely statistically significant respectively in all cases.
A detailed description of the study methods can be found in the Supplementary Information section.
Results
COPD-like symptoms developed in rats exposed to CS or MVE
The COPD-like symptoms were observed in rats exposed to CS or MVE. The lung functions were declined in rats exposed to CS or MVE indicated by the significantly increased parameters of resistance, chord compliance and total lung capacity and the decreased ratios of FEV50/FVC when compared with those rats in respective control group (). The mean linear intercept values, a histological measure of emphysema-like alveolar enlargement, were also increased in rats exposed to CS or MVE (). Furthermore, significant differences were observed in parameters resistance, total lung capacity, FEV50/FVC and mean linear intercept value between CS and MVE exposed rats, revealed more severe lung injury in CS exposed rats.
Figure 1. COPD like lung function loss was found in rats exposed to cigarette smoke or motor vehicle exhaust. Male SD rats were exposed to cigarette smoke (CS) for 60 d in a whole body exposure chamber or motor vehicle exhaust (MVE) in an exposure system for 24 weeks. (A) The values of lung function parameters, including airway resistance, chord compliance, total lung capacity and the ratio of FEV50/FVC were determined. (B) Representative H&E staining images of rat tissues sections with or without CS or MVE exposure treatment. Each calculation for mean linear intercept (Lm) in lung was based on six rats and five representing slides across different regions of the lung from each rat. Data are represented as mean ± SEM (n = 6). **p < .01 for CS or MVE group versus respective CTL group; #p < .05 and ##p < .01 for CS group versus MVE group.
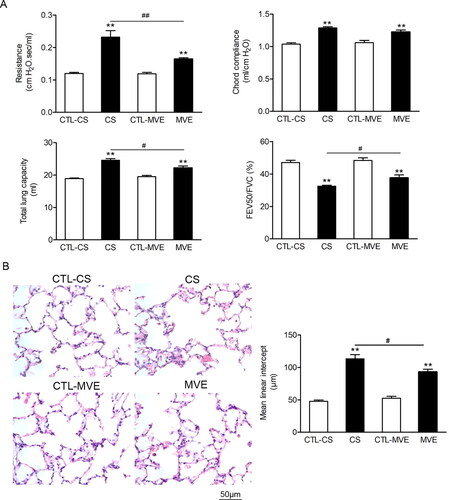
The inflammatory responses developed in lungs from rats exposed to CS or MVE, manifested by increased leukocytes and pro-inflammatory cytokine levels in BALF. When compared CS- with MVE-exposed rats, we found although CS-exposed rats had much more inflammatory cells in BAL fluid than MVE-exposed rats (p < .01), the levels of proinflammatory cytokines IL-6 (p < .05) and TNF-α (p > .1) in BAL fluid did not show similarly obvious difference between the two COPD models (). Mucus hypersecretion in model rats was characterized by goblet cell hyperplasia indicated by positive PAS staining and upregulated Muc5ac levels in BALF. In the two COPD models, CS exposure induced more obvious mucus hypersecretion than MVE exposure (). Furthermore, the increased ratios of wall area/total bronchial area were seen in both COPD models, though no difference existed between them (). It meant that the two COPD models presented the same extent in airway remodeling.
Figure 2. Cigarette smoke and motor vehicle exhaust exposure-induced inflammation and mucus hypersecretion in airway. The bronchial alveolar lavage fluid (BALF) was collected. (A) The cells in BALF were separated, subjected to H&E staining and differential counting. (B) The levels of IL-6 and TNF-α in supernatants of BALF were determined with ELISA. (C) The Muc5ac levels in BALF supernatants were determined with ELISA. (D) The PAS staining of lung sections and the PAS staining positive areas in bronchia were semi-quantitatively analyzed and presented as ratios to the total bronchial area. (E) Lung sections were subjected to H&E staining and the calculation of bronchial wall area ratio to the total bronchial area. The ratio analyses for PAS staining positive area or bronchial wall area were performed in six rats per group with five representing slides in each rat. Data are represented as mean ± SEM (n = 6). **p < .01 for CS or MVE group versus respective CTL group; #p < .05 and ##p < .01 for CS group versus MVE group.
PD-L1 upregulations in lungs exposed to CS or MVE
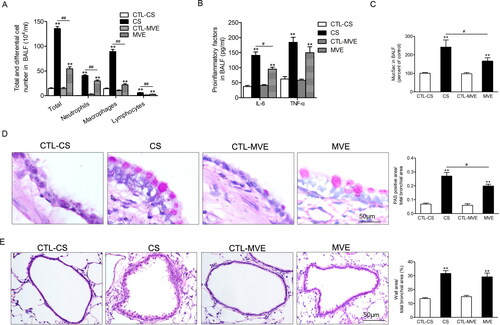
The lysates of lung tissues were subjected to quantitative RT-PCR or western blotting for measurements of mRNA or protein levels of PD-L1. As shown in , both mRNA and protein levels of PD-L1 were upregulated in rats’ lungs exposed to CS or MVE. However, concerning the changes of PD-L1 mRNA or protein levels, no difference existed between the two COPD models. Immunofluorescence staining with antibody against PD-L1 indicated PD-L1 predominantly expressed in bronchial epithelial cells. When compared with the normal rats, enhanced immunofluorescence staining was observed in sections from COPD model rats’ lungs ().
Figure 3. PD-L1 was increased in lung exposed to CS or MVE. The relative mRNA levels of PD-L1 in lung tissues were presented as percentage of the control group. (B) Protein levels of PD-L1 in lung tissues were determined by using western blotting. The blotted bands were relatively semi-quantitated to internal control (GAPDH) by densitometry and presented as relative amount to control groups. Data are presented as mean ± SEM. **p < .01 for CS or MVE group versus respective CTL group. (C) Representative images from lung tissue sections with immunofluorescence staining of PD-L1. Arrows indicated the cells with relatively brighter fluorescence staining than average.
Both CSE and PM2.5 induced inflammatory responses in lung epithelial cells
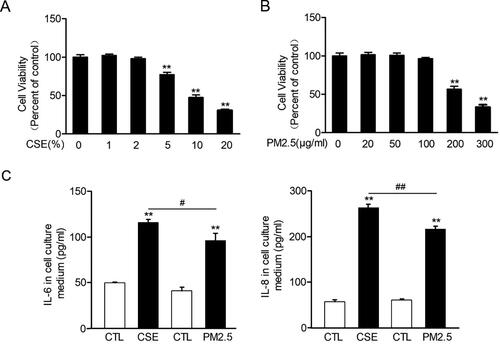
In order to investigate the effects of CS or MVE exposure on bronchial epithelial cells, we treated the cultured immortal normal human bronchial epithelial cells BEAS-2B with cigarette smoke extracts (CSEs) or diesel-related PM2.5 which mimicked the effects of MVE. Both CSE and PM2.5 in high levels, namely more than 10% for CSE and more than 200 µg/ml for PM2.5, reduced cell viability for more than 50% (). Therefore, we used 2% CSE or 100 µg/ml PM2.5 in cell treatments hereafter. In these dosages, both CSE and PM2.5 induced inflammatory responses in BEAS-2B cells manifested by upregulated cytokine levels of IL-6 and IL-8 in cell culture medium. Moreover, CSE induced more robust productions of IL-6 and IL-8 than PM2.5 ().
Figure 4. Cigarette smoke extract and PM2.5 reduced cell viability and induced proinflammatory cytokines production in airway epithelial cells. (A,B) Human normal bronchial cells BEAS-2B were incubated with 0–20% CSE or 0–300 µg/ml PM2.5 for 48 h. Cell viability was measured with CCK-8 kit and presented as percentage of control group. (C) BEAS-2B cells were incubated with 2% CSE or 100 µg/ml PM2.5 for 48 h. The levels of IL6 and IL8 in cell culture medium were determined with ELISA. Data are presented as mean ± SEM (n = 5). **p < .01 for CS or PM2.5 group versus respective CTL group; #p < .05 and ##p < .01 for CSE group versus PM2.5 group.
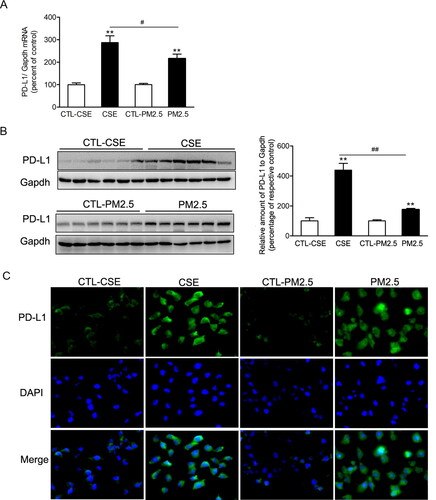
PD-L1 upregulations in bronchial epithelial cells treated with CSE or PM2.5
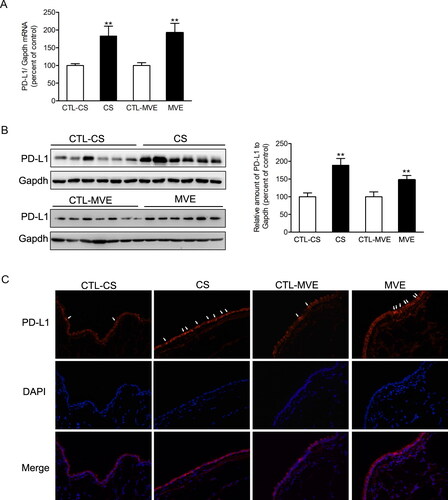
Both CSE and PM2.5 enhanced PD-L1 expression in mRNA and protein levels in BEAS-2B cells. The mRNA and protein levels of PD-L1 were higher in cells exposed to CSE than those cells exposed to PM2.5 (). The upregulations of PD-L1 induced by CSE or PM2.5 were confirmed by the immunofluorescence staining ().
Figure 5. Cigarette smoke extract and PM2.5 induced PD-L1 increases in airway epithelial cells. BEAS-2B cells were incubated with 2% CSE or 100 µg/ml PM2.5 for 48 h. (A) The relative mRNA levels of PD-L1 were presented as percentage of the control cells. (B) Protein levels of PD-L1 were determined by western blotting. The blotted bands were relatively semi-quantitated to internal control (GAPDH) by densitometry and presented as percentage amount to control cells. Data are presented as mean ± SEM (n = 6). **p < .01 for CSE or PM2.5 group versus respective CTL group; #p < .05 and ##p < .01 for CSE group versus PM2.5 group. (C) Representative images from immunofluorescence staining of PD-L1 in cultured BEAS-2B cells.
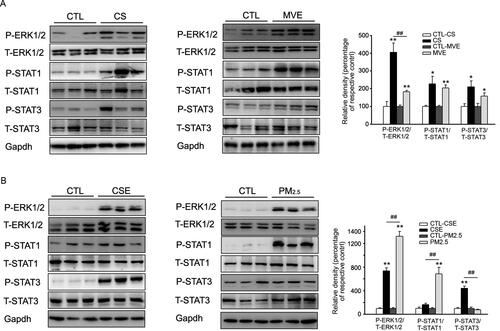
In vitro and in vivo activation of ERK and STAT upon CS/CSE or MVE/PM2.5 exposure
The signaling mediated by ERK or STAT was activated in lung from COPD model rats. Here we found that both CS and MVE induced phosphorylations of ERK1/2, STAT1 and STAT3 in lung tissues from the two rat COPD models in which more obvious ERK1/2 phosphorylation found in CS-induced model (). In cultured cells, both CSE and PM2.5 induced very obvious ERK1/2 phosphorylation. However, CSE preferred to induce STAT3 but not STAT1 phosphorylation, whereas PM2.5 inclined to induce STAT1 phosphorylation in BEAS-2B cells ().
Figure 6. Phosphorylation of ERK and STAT in lung or BEAS-2B cells in respond to CS/CSE or MVE/PM2.5 treatments. Western blotting was used to determine the levels of total and phosphorylated ERK, STAT1 and STAT3 proteins in (A) lung tissues exposed to CS or MVE, or in (B) BEAS-2B cells treated with 2% CSE or 100 µg/ml PM2.5. For lung tissues, samples were randomly selected from six rats per group. The results for cell lysates were from one representative experiment performed in triplicates and repeated for three times. Relative semi-quantitation of the bands to internal control (GAPDH) was performed by densitometry. The ratios of phosphorylated protein to total protein were calculated and presented as percentage of respective control. Data are presented as mean ± SEM. *p < .05 or **p < .01 for CS/CSE or MVE/PM2.5 group versus respective CTL group; ##p < .01 for CS/CSE group versus MVE/PM2.5 group.
Discussion
The prevalence of COPD in lung cancer patients is greater than that in both smokers and randomly recruited control group [Citation2]. In order to elucidate the association between COPD independent of smoking and lung carcinogenesis, we established COPD model in rats by CS or MVE exposure and treated airway epithelial cells with CSE or diesel-related PM2.5. PD-L1 upregulation was observed in COPD model rats’ lung, accompanied with lung function decline and lung inflammation. ERK1/2 and STAT1/3, both of which reportedly modulated PD-L1 expression in lung inflammation and lung cancer cells, were activated in COPD rats’ lung. Inflammatory responses, PD-L1 upregulation and activations of ERK1/2 and STAT1/3 were reproduced in airway epithelial cells upon CSE or PM2.5 treatment. Our results suggested a possible interpretation for the comorbid of COPD and lung cancer that overexpression of PD-L1 under inflammatory condition might contribute to the lung cancer initiation in COPD patients.
The increase of PD-1/PD-L1 axis in lung cancer generally exhibited the upregulated expressions of PD-1 in T cells and of PD-L1 in tumor cells which lead to immune escape of tumor cells [Citation19, Citation20]. The main immunological dysfunction in COPD patients’ lung is T-cell exhaustion due to the binding of immune checkpoints on T-cells. The interaction of PD-1/PD-L1 established an immunosuppressive microenvironment facilitating the tumor initiation, development and progression [Citation21]. However, despite the increased CD8+ PD1 levels in COPD patients’ lung [Citation22], at least one study mentioned no difference found in CD4/8+ T cells PD-1 levels in resected tumor tissues between COPD and non-COPD NSCLC patients [Citation9]. PD-L1 overexpression alone is unable to induce cancer genesis, unless with the presence of carcinogen [Citation14]. Therefore, PD-Ll overexpression in lung epithelial cells is critical for establishment of immune escape contributing to increased lung cancer incidence in COPD patients.
By comparing the status of lung functions decline and lung PD-L1 upregulations between the two COPD models, we noticed that more severe lung functions decline in CS model did not bring higher PD-L1 expressions in lung ( and ). Although the PD-L1 upregulation occurred in COPD, the extent of lung function decline was not correlated with PD-L1 level. Concerning the PD-L1 overexpression in lung cancer, one point of view supposed the PD-L1 overexpression was modulated by crosstalk between NSCLC and activated macrophages [Citation17]. In a study focusing on resected lung cancer tissues, tumor PD-L1 expression was associated with the tumor infiltrated macrophages which accumulated around PD-L1-positive carcinoma cell areas in early lung adenocarcinoma patients. The cytokines secreted by tumor-associated macrophages, including TGF-β1, TNF-α, IL-10, IL-1α, IL-1β and IL-27, are supposed to be extrinsic regulators of tumor PD-L1 expression and contribute to tumor development in early stage [Citation23]. However, in our study, the PD-L1 levels showed no difference between the two COPD model, though CS exposed rats had much more airway macrophages. These results reflected that macrophages infiltration in lung might correlate with PD-L1 upregulation but more macrophages infiltration did not lead to higher lung PD-L1 expression in COPD ().
A prospective study supposed the elevated serum IL-6 level mean poor response to immune checkpoint therapy [Citation24]. In epithelial inflammation, PD-L1 upregulated the production of proinflammatory cytokines [Citation25]. We would like to evaluate the association between BAL fluid cytokine increases and PD-L1 upregulation. Both Inflammatory cytokines increase and PD-L1 upregulation were found in COPD lungs. When compared the two COPD models, higher IL-6 levels or relative higher TNF-α and PD-L1 levels presented in CS model lung, though no statistically significant difference found in TNF-α and PD-L1 between the two models ( and 3). More consistent changes between proinflammatory cytokines increases and PD-L1 upregulation were found in culture cells, in which more cytokines production meant higher PD-L1 expression ( and 5). Therefore, inflammatory cytokines production in COPD lung meant PD-L1 upregultion. But the levels of proinflammatory cytokines in BAL fluid were not correlated with the PD-L1 expression level. The proinflammatory cytokine levels in airway epithelial cells might be more suitable to predict the lung PD-L1 level than other parameters in COPD.
The phosphorylation of ERK was triggered by CS/CSE or diesel-related PM2.5 in in vivo and in vitro study [Citation26–28]. A speculate on investigating the relationship between inflammation and tumor development suggested that the activation of TLR4/ERK/PD-L1 axis contribute to the NSCLC initiation [Citation10]. The ratio of p-ERK/ERK, a hallmark of lung inflammation, was increased in COPD rats’ lungs and in cultured bronchial epithelial cells upon CSE or diesel-related PM2.5 treatments (). Unlike previously report in which diesel related PM2.5 induced phosphorylation of ERK in bronchial epithelial cells in dosage bringing cell cycle arrest [Citation29], our results showed that both CSE and diesel related PM2.5 activated ERK and upregulated PD-L1 in a dosage without inducing obvious cell viability loss but enough to induce inflammatory responses ( and 5). It was supposed that the carcinogens like polycyclic aromatic hydrocarbon (PAHs) in CS or PM2.5-bound PAHs induced ERK activation and cell transformation in bronchial epithelial cells [Citation15, Citation30]. However, it should be noted that higher p-ERK/ERK ratio in lung did not bring further PD-L1 overexpression when compared the two COPD models (). In culture cells received CSE or PM2.5 treatment, more robust PD-L1 increase even emerged in CSE-treated cells in spite of less p-ERK augment. Thus, we proposed p-ERK is correlated with PD-L1 upregulation in COPD lung epithelial cells. But PD-L1 expression is probably modulated by elements unlimited to p-ERK.
We found both CS and MVE induced similar phosphorylation of STAT1 and 3 in lungs. However, in cultured bronchial epithelial cells BEAS-2B, CSE or diesel-related PM2.5 activated STAT family members preferentially, namely STAT3 for CSE and STAT1 for diesel-related PM2.5. The bias between in vivo and in vitro effects could be attributed to the inability of CSE or diesel-related PM2.5 to contain all components in CS or MVE. Concerning our results, it is not known whether the difference in PD-L1 levels between CSE and diesel-related PM2.5 treatment was due to alternative activation of STAT family members. Conflicting results have been obtained regarding the involvement of p-STAT in modulating PD-L1 expression in lung epithelial cells. Both IFNγ/STAT1/STAT3 and JAK/STAT1/STAT3 signaling axis potentiated PD-L1 overexpression upon different stimuli [Citation17, Citation31, Citation32]. STAT3 activation also participated in eliminating PD-L1 expression [Citation33]. That means the regulation of STATs on PD-L1 could be complex. Further research is needed to explore the effects of STAT phosphorylation on PD-L1 upregulation in COPD developed from various risk factors.
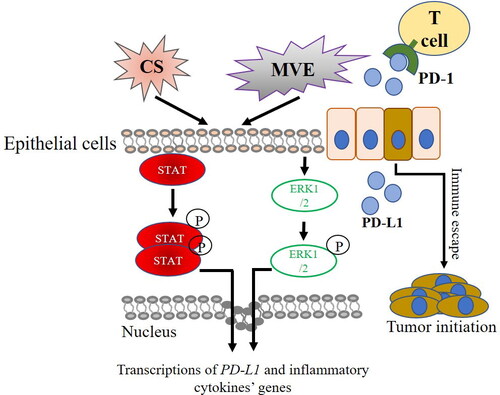
In conclusion, our study presented the effects of CS and air pollutant on establishing COPD animal models with simultaneous PD-L1 upregulation in lung. However, more severe COPD phenotypes did not bring higher PD-L1 expression in lung. The inflammatory cytokine levels in airway epithelial cells were likely to reflect the PD-L1 level. Our hypothesis illustrated in suggested that PD-L1 upregulation was accompanied by activation of ERK1/2 and STAT family members and might potentially facilitate the high prevalence of lung cancer in COPD patients.
Figure 7. PD-L1 overexpression in COPD and lung cancer initiation. Compounds in CS or MVE activate STATs or EEK1/2-mediated pathways which trigger the overexpression of pro-inflammatory cytokines and PD-L1 in COPD. PD-L1 potentiates the initiation of lung cancer by promoting immune escape of cells bearing oncogenic mutation from T cells.
Ethics approval
The experimental methods were approved by the Ethics Committee for Animal Experiments of the General Hospital of Southern Theater Command of PLA.
Consent for publication
All authors have read the manuscript and approved the submission.
What is the Central question of this study?
Our study presented the effects of cigarette smoke and air pollutant on establishing COPD animal models with simultaneous PD-L1 upregulation in lung and suggested the premalignant PD-L1 upregulation in lung epithelial cells could be the link between COPD prevalence and lung cancer.
What is the main finding and its importance?
PD-L1 upregulation in airway epithelial cells exposed to various COPD risk factors might be associated with activation of ERK1/2 and alternative STAT family members.
Availability of data and material
This manuscript or the results included are original and have not been submitted for publication elsewhere.
| Abbreviations: | ||
| BALF | = | bronchoalveolar lavage fluid |
| COPD | = | Chronic obstructive pulmonary disease |
| CS | = | cigarette smoke |
| CSE | = | cigarette smoke extract |
| ERK | = | extracellular regulated protein kinases |
| MVE | = | motor vehicle exhaust |
| PD-L1 | = | programmed death-ligand 1 |
| PM2.5 | = | particulate matter 2.5 |
| STAT | = | signal transducer and activator of transcription |
Acknowledgments
We would like to thank Professor Wenju Lu for technical assistance in the establishment of MVE-induced COPD animal models.
Disclosure statement
The authors have no conflict of interest to declare.
Additional information
Funding
Notes on contributors
Huang Wenjie
Wenjie Huang conceived the idea for the project. Zhenhui Guo contributed to the design of the protocol. Rui Chen and Li Defu built the animal models, collected samples and drafted the manuscript. Lingling Wang and Yuanyuan Ye analyzed the data. Jiahui Dong and Richeng Xiong took part in the collection of samples. Wenjie Huang revised the manuscript critically. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
References
- Tockman MS, Anthonisen NR, Wright EC, et al. Airways obstruction and the risk for lung cancer. Ann Intern Med. 1987;106(4):512–518. DOI:10.7326/0003-4819-106-4-512
- Young RP, Hopkins RJ, Christmas T, et al. COPD prevalence is increased in lung cancer, independent of age, sex and smoking history. Eur Respir J. 2009;34(2):380–386. DOI:10.1183/09031936.00144208
- Fajersztajn L, Veras M, Barrozo LV, et al. Air pollution: a potentially modifiable risk factor for lung cancer. Nat Rev Cancer. 2013;13(9):674–678.
- Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91(14):1194–1210. DOI:10.1093/jnci/91.14.1194
- Wang C, Xu J, Yang L, et al. Prevalence and risk factors of chronic obstructive pulmonary disease in China (the China pulmonary health [CPH] study): a national cross-sectional study. Lancet. 2018;391(10131):1706–1717. DOI:10.1016/S0140-6736(18)30841-9
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264.
- Lee MH, Yanagawa J, Tran L, et al. FRA1 contributes to MEK-ERK pathway-dependent PD-L1 upregulation by KRAS mutation in premalignant human bronchial epithelial cells. Am J Transl Res. 2020;12(2):409–427.
- Jia M, Feng W, Kang S, et al. Evaluation of the efficacy and safety of anti-PD-1 and anti-PD-L1 antibody in the treatment of non-small cell lung cancer (NSCLC): a Meta-analysis. J Thorac Dis. 2015;7(3):455–461.
- Mark NM, Kargl J, Busch SE, et al. Chronic obstructive pulmonary disease alters immune cell composition and immune checkpoint inhibitor efficacy in Non-Small cell lung cancer. Am J Respir Crit Care Med. 2018;197(3):325–336. DOI:10.1164/rccm.201704-0795OC
- Kang X, Li P, Zhang C, et al. The TLR4/ERK/PDL1 axis may contribute to NSCLC initiation. Int J Oncol. 2020;57(2):456–465.
- Liu CH, Chen Z, Chen K, et al. Lipopolysaccharide-mediated chronic inflammation promotes tobacco carcinogen-induced lung cancer and determines the efficacy of immunotherapy. Cancer Res. 2020;81:144–157. DOI:10.1158/0008-5472.CAN-20-1994
- Shu J, Lu W, Yang K, et al. Establishment and evaluation of chronic obstructive pulmonary disease model by chronic exposure to motor vehicle exhaust combined with lipopolysaccharide instillation. Exp Physiol. 2018;103(11):1532–1542.
- Li D, Wang J, Sun D, et al. Tanshinone IIA sulfonate protects against cigarette smoke-induced COPD and down-regulation of CFTR in mice. Sci Rep. 2018;8(1):376.
- Wang GZ, Zhang L, Zhao XC, et al. The aryl hydrocarbon receptor mediates tobacco-induced PD-L1 expression and is associated with response to immunotherapy. Nat Commun. 2019;10(1):1125.
- Chen S, Li D, Zhang H, et al. The development of a cell-based model for the assessment of carcinogenic potential upon long-term PM2.5 exposure. Environ Int. 2019;131:104943. DOI:10.1016/j.envint.2019.104943
- Xu Z, Wu H, Zhang H, et al. Interleukins 6/8 and cyclooxygenase-2 release and expressions are regulated by oxidative stress-JAK2/STAT3 signaling pathway in human bronchial epithelial cells exposed to particulate matter < = 2.5 mum. J Appl Toxicol. 2020;40(9):1210–1218.
- Narayanapillai SC, Han YH, Song JM, et al. Modulation of the PD-1/PD-L1 immune checkpoint axis during inflammation-associated lung tumorigenesis. Carcinogenesis. 2020;41(11):1518–1528.
- Shao MX, Ueki IF, Nadel JA. Tumor necrosis factor alpha-converting enzyme mediates MUC5AC mucin expression in cultured human airway epithelial cells. Proc Natl Acad Sci USA. 2003;100(20):11618–11623. DOI:10.1073/pnas.1534804100
- Aldarouish M, Su X, Qiao J, et al. Immunomodulatory effects of chemotherapy on blood lymphocytes and survival of patients with advanced non-small cell lung cancer. Int J Immunopathol Pharmacol. 2019;33:2058738419839592.
- Kim GJ, Lee JH, Park WJ, et al. The prognostic value of measuring PD-L1 mRNA expression levels in surgically resected Non-Small cell lung cancer. Ann Clin Lab Sci. 2019;49(3):317–323.
- Chen N, Fang W, Lin Z, et al. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol Immunother. 2017;66(9):1175–1187.
- McKendry RT, Spalluto CM, Burke H, et al. Dysregulation of antiviral function of CD8(+) T cells in the chronic obstructive pulmonary disease lung. Role of the PD-1-PD-L1 axis. Am J Respir Crit Care Med. 2016;193(6):642–651.
- Shima T, Shimoda M, Shigenobu T, et al. Infiltration of tumor-associated macrophages is involved in tumor programmed death-ligand 1 expression in early lung adenocarcinoma. Cancer Sci. 2020;111(2):727–738.
- Kauffmann-Guerrero D, Kahnert K, Kiefl R, et al. Systemic inflammation and pro-inflammatory cytokine profile predict response to checkpoint inhibitor treatment in NSCLC: a prospective study. Sci Rep. 2021;11(1):10919.
- Zhao S, Gao J, Li J, et al. PD-L1 regulates inflammation in LPS-Induced lung epithelial cells and vascular endothelial cells by interacting with the HIF-1α Signaling Pathway. Inflammation. 2021;44(5):1969–1981.
- Guan R, Wang J, Li D, et al. Hydrogen sulfide inhibits cigarette smoke-induced inflammation and injury in alveolar epithelial cells by suppressing PHD2/HIF-1α/MAPK signaling pathway. Int Immunopharmacol. 2020;81:105979. DOI:10.1016/j.intimp.2019.105979
- Park EJ, Park YJ, Lee SJ, et al. Whole cigarette smoke condensates induce ferroptosis in human bronchial epithelial cells. Toxicol Lett. 2019;303:55–66. DOI:10.1016/j.toxlet.2018.12.007
- Blanchet S, Ramgolam K, Baulig A, et al. Fine particulate matter induces amphiregulin secretion by bronchial epithelial cells. Am J Respir Cell Mol Biol. 2004;30(4):421–427. DOI:10.1165/rcmb.2003-0281RC
- Wu J, Shi Y, Asweto CO, et al. Fine particle matters induce DNA damage and G2/M cell cycle arrest in human bronchial epithelial BEAS-2B cells. Environ Sci Pollut Res Int. 2017;24(32):25071–25081.
- Miller A, Brooks GD, McLeod L, et al. Differential involvement of gp130 signalling pathways in modulating tobacco carcinogen-induced lung tumourigenesis. Oncogene. 2015;34(12):1510–1519.
- Zhang X, Zeng Y, Qu Q, et al. PD-L1 induced by IFN-gamma from tumor-associated macrophages via the JAK/STAT3 and PI3K/AKT signaling pathways promoted progression of lung cancer. Int J Clin Oncol. 2017;22(6):1026–1033.
- Li YM, Yu JM, Liu ZY, et al. Programmed death ligand 1 indicates pre-existing adaptive immune response by tumor-infiltrating CD8(+) T cells in non-small cell lung cancer. Int J Mol Sci. 2019:20:20.
- Seki N, Kan OK, Matsumoto K, et al. Interleukin-22 attenuates double-stranded RNA-induced upregulation of PD-L1 in airway epithelial cells via a STAT3-dependent mechanism. Biochem Biophys Res Commun. 2017;494(1–2):242–248. DOI:10.1016/j.bbrc.2017.10.045