
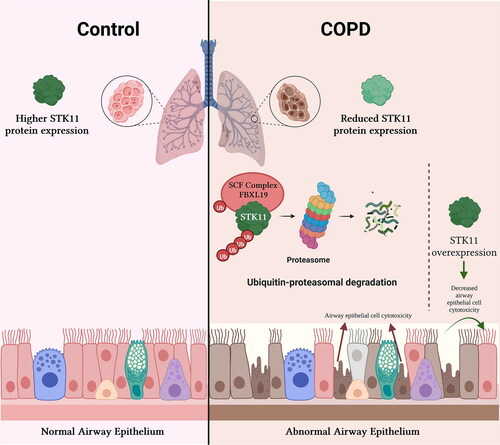
Abstract
Objective: To investigate the effects of cigarette smoke (CS) on Serine/Threonine Kinase 11 (STK11) and to determine STK11’s role in CS-induced airway epithelial cell cytotoxicity.Methods: STK11 expression levels in the lung tissues of smokers with or without COPD and mice exposed to CS or room air (RA) were determined by immunoblotting and RT-PCR. BEAS-2Bs–human bronchial airway epithelial cells were exposed to CS extract (CSE), and the changes in STK11 expression levels were determined by immunoblotting and RT-PCR. BEAS-2B cells were transfected with STK11-specific siRNA or STK11 expression plasmid, and the effects of CSE on airway epithelial cell cytotoxicity were measured. To determine the specific STK11 degradation-proteolytic pathway, BEAS-2Bs were treated with cycloheximide alone or combined with MG132 or leupeptin. Finally, to identify the F-box protein mediating the STK11 degradation, a screening assay was performed using transfection with a panel of FBXL E3 ligase subunits.Results: STK11 protein levels were significantly decreased in the lung tissues of smokers with COPD relative to smokers without COPD. STK11 protein levels were also significantly decreased in mouse lung tissues exposed to CS compared to RA. Exposure to CSE shortened the STK11 mRNA and protein half-life to 4 h in BEAS-2B cells. STK11 protein overexpression attenuated the CSE-induced cytotoxicity; in contrast, its knockdown augmented CSE-induced cytotoxicity. FBXL19 mediates CSE-induced STK11 protein degradation via the ubiquitin-proteasome pathway in cultured BEAS-2B cells. FBXL19 overexpression led to accelerated STK11 ubiquitination and degradation in a dose-dependent manner.Conclusions: Our results suggest that CSE enhances the degradation of STK11 protein in airway epithelial cells via the FBXL19-mediated ubiquitin-proteasomal pathway, leading to augmented cell death.
Lung tissues of COPD-smokers exhibited a decreased STK11 RNA and protein expression.
STK11 overexpression attenuates CS-induced airway epithelial cell cytotoxicity.
STK11 depletion augments CS-induced airway epithelial cell cytotoxicity.
CS diminishes STK11 via FBXL19-mediated ubiquitin-proteasome degradation.
HIGHLIGHTS
Graphical Abstract
Introduction
Serine-Threonine Kinase 11 (STK11), also called liver kinase B1 (LKB1), is a highly preserved intracellular serine-threonine kinase that acts as a cellular energy sensor and plays a vital part in cellular metabolism, cell-polarization, DNA repair, and regulation of cytotoxicity. STK11 is located on chromosome 19 and comprises ten exons, nine of which code STK11 protein. STK11 consists of 433 amino acid residues with a 50 kDa protein weight, and its catalytic domain relates to other protein kinases [Citation1]. STK11 functions as a primary upstream serine/threonine kinase that stimulates the adenosine monophosphate-activated protein kinase (AMPK), a crucial metabolic sensor that controls intracellular energy alterations via diverse cellular processes [Citation2], by phosphorylating the distinctive T-loop activation domains [Citation3] in response to intracellular energy alterations and other stressors [Citation4–6].
STK11 is classically regarded as a tumor suppressor gene. STK11’s loss of function has been associated with several cancer types and the cold immunosuppressive tumor microenvironment in murine models of NSCLC [Citation7–13]. STK11 also regulates embryogenesis, and the genetic deletion causes death at the midgestational period, associated with vascular and neural tube abnormalities [Citation14, Citation15]. Some germline STK11 gene mutations cause Peutz-Jeghers syndrome, a hereditary polyposis syndrome characterized by gastrointestinal hamartomas and mucocutaneous pigmentations [Citation16]. Further, STK11 is frequently downregulated by the loss-of-function somatic mutations (e.g. non-sense, missense mutations, and deletions) in various human neoplastic diseases, including non-small cell lung cancer, reemphasizing the role of STK11 in tumor suppression [Citation17].
Chronic obstructive pulmonary disease (COPD) is the third leading cause of death worldwide, characterized by progressive and irreversible airflow limitation associated with an abnormal inflammatory response in the lungs and associated with comorbidities such as lung cancer. Cigarette smoke (CS) is a complex mixture of various noxious gases containing more than 7,000 chemicals, including reactive oxygen species (ROS), reactive nitrogen species (RNS), and carcinogens [Citation18]. The hazardous effects of CS on the respiratory system have been well documented. The primary risk factor for COPD is CS which elicits pulmonary oxidative stress by constantly generating ROS and various inflammatory mediators by activating a wide variety of redox-sensitive transcription factors, signal transduction proteins, and serine-threonine kinases [Citation19, Citation20]. CS inhalation introduces exogenous oxidants, while inflammatory and structural cells contribute to endogenous oxidants in the lower airways of COPD patients, underscoring the significant role of oxidative stress in the COPD’s pathogenesis [Citation20].
However, evidence regarding the effects of CS on STK11 is limited. Therefore, we investigated the effects of CS on STK11 protein and the mechanisms by which CS regulates STK11 protein levels and cytotoxicity. In this study, we found that lung tissues from smokers with COPD displayed a reduced STK11 expression relative to smokers without COPD. Similarly, CS exposure diminished the STK11 protein half-life to 4 h via the F-Box and Leucine-Rich Repeat Protein 19 (FBXL19)-mediated ubiquitin-proteasome degradation pathway in airway epithelial cells. We also found that STK11 protein depletion augmented CSE-induced cytotoxicity. Furthermore, we discovered that STK11 overexpression attenuated the CS-induced cytotoxicity. Our findings suggest that CS enhances STK11 degradation by the E3 ligase subunit FBXL19, which augments CS-induced cytotoxicity in airway epithelial cells.
Materials and methods
Cell lines and reagents
BEAS-2B, human bronchial airway epithelial cells were maintained in Dulbecco’s-modified Eagle’s medium (DMEM)/F12 containing hydrocortisone, insulin, transferrin, estrogen, and selenium, 10% fetal bovine serum. Cells were cultured in a 5% CO2 incubator at 37 °C. V5 antibody (#PA1-993) was purchased from Invitrogen (St. Louis, MO). STK11 (#3050) antibody was purchased from Cell Signaling (Danvers, MA). Anti-ubiquitin (#sc-166553) antibody was acquired from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-HPRT1 (#ab109021) antibody was purchased from Abcam (Waltham, MA). FBXL–V5-tagged family plasmids were obtained from Addgene (Watertown, MA). Protein synthesis inhibitor cycloheximide (#ALX-380-269-G001) and ubiquitin aldehyde (#BML-UW8450-0050), an inhibitor of deubiquitinylating enzymes, were from Enzo Life Sciences (Farmingdale, NY). Thiazolyl Blue Tetrazolium Bromide (#M5655), leupeptin (CAS# 103476-89-7), MG132 (CAS# 133407-82-6), and anti-β-Actin antibody (#A3853) were obtained from Sigma (Carlsbad, CA). Anti-Rabbit IgG (#1706515) and Anti-Mouse IgG (#1706516) raised in goat were purchased from Bio-Rad Laboratories (Hercules, CA). STK11 siRNA were from Integrated DNA Technologies (Coralville, IA). RNeasy mini kit (#74106) and QIAprep Spin Miniprep Kit (#27106) were purchased from Qiagen (Valencia, CA). iTaq Universal Probes One-Step Kit was purchased from Bio-Rad (Hercules, CA).
Human lung tissue samples
The University of Pittsburgh and VA Pittsburgh Healthcare System Institutional Review Board (IRB) approved using human subject samples in this study. The Lung Tissue Research Consortium (LTRC) and the Lovelace Respiratory Research Institute provided the frozen lung tissues from nine smokers with very severe COPD and six control smokers without COPD, respectively. The individuals’ spirometry data was measured after bronchodilation. Additionally, a chest CT scan was conducted to rule out any concomitant bronchiectasis or interstitial lung disease. For the subjects with COPD, lung resection was performed for lung volume reduction surgery. Meanwhile, the controlled subjects underwent surgery for primary lung cancer. The demographic data are presented in for STK11 protein expression analysis, in for STK11 mRNA expression analysis, respectively. Due to a limited availability of some individual samples, STK11 protein expression analysis was performed using the lung tissue samples obtained from control (n = 4) and COPD (n = 9); STK11 mRNA expression analysis from control (n = 6) and COPD (n = 6).
Table 1. Demographic data used to assess STK11 protein levels.
Table 2. Demographic data used to assess STK11 mRNA levels.
Animals
The Institutional Animal Care and Use Committee (IACUC) at the University of Pittsburgh approved the use of animals and animal models in this study. Mice aged 8-10 weeks on a C57BL/6 background (n = 6/group) were exposed to either CS or RA. CS was produced using ten filter-cut and ten filtered Kentucky 1R6F reference cigarettes from the Cigarette Laboratory at the Tobacco and Health Research Institute, University of Kentucky, Lexington, KY. The SCIREQ inExpose inhalation exposure system was used to generate CS. The CS was diluted with air to reduce its toxicity, and the concentration of total particulate matter was maintained at an average level of 10.05 g/m3. Mice were exposed to CS for four consecutive days (for 1 h per day, non-filtered 20 cigarettes) and were euthanized 24 h after their last exposure.
CSE preparation and treatment
CSE was produced as we described [Citation21, Citation22] using 3R4F research-grade cigarettes (with filter) obtained from the Kentucky Tobacco Research and Development Center (Lexington, KY). BEAS-2B cells were cultured in the normal or media containing different CSE concentrations for different hour intervals, as indicated.
Plasmid and siRNA transfection
BEAS-2B cells are transfected with about 2 μg of plasmid or 40 pm of siRNA by electroporation using Amaxa Biosystems Nucleofector (Gaithersburg, MD). Immediately after electroporation, cells were transferred to a six-well plate containing a 2 ml warm BEAS-2B growth medium in each well. The monolayers were then cultured in a 5% CO2 humidified incubator at 37 °C for 48 h of transfection with plasmid and 72 h for transfection with siRNA for additional assessments.
MTT assay
The viability of BEAS-2B cells was assessed using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Cells were cultured at a density of 5,000 cells per well in a 96-well plate and cultured and treated as described above. MTT solution (10 μL at a concentration of 5 mg/mL; Life technologies) was added to each well and incubated for 4h at 37° C. Following the incubation, 200 μL of DMSO was added to dissolve the crystals and the absorbance at 560 nm was read using SpectraMax M2 microplate reader (Molecular Devices, CA).
Immunoblotting
Human or mice lung tissue was sliced into small pieces, resuspended in RIPA buffer containing protease and phosphatase inhibitors, and minced with a homogenizer at a speed and time settings of 10 and 4, respectively. The lysates were centrifuged at 13,000 × g for 10 min and analyzed by gel electrophoresis. BEAS-2B cells at approximately 80% confluence were treated with CSE; as indicated, whole-cell proteins were prepared in RIPA buffer containing a protein–phosphatase inhibitor mix. Around 20 μg of protein from tissue or cell lysates were electrophoresed using SDS-PAGE gels and electro-blotted onto a nitrocellulose membrane. The electro-blotted-membranes were blocked with nonfat milk and incubated with specified primary and HRP-conjugated-anti-secondary antibodies to determine the expression levels of specific proteins (). The signal from the membrane was developed and enhanced using a chemiluminescence (ECL) substrate and quantified using a ChemiDoc XRS + System (Bio-Rad).
Table 3. List of antibodies used.
Quantitative polymerase chain reaction (qPCR)
Total RNA from human or mouse samples was isolated using a Qiagen RNeasy kit, following the manufacturer’s instructions. RNA was eluted in RNase-free water, and the quantity and quality were determined using a Nanodrop 1000 (Thermo Fisher Scientific, Wilmington, DE). One hundred ug of Total RNA was used as the template for the qPCR reaction with iTaq Universal Probes One-Step Kit (Bio-Rad, USA) containing specific primers purchased from Thermo Fisher Scientific (). The RT reaction was performed using CFX96TM Real-Time System (Bio-Rad, USA) with the following cycling conditions, 95 °C for 2 min; 95 °C for 15 s for 40 cycles; 58 °C for 15 s; and 72 °C for 1 min. Melt curve analysis was performed to determine any nonspecific amplification. The fluorescence detection threshold was set beyond the control background. The target-gene cycle threshold (Ct) for each sample ran in triplicates was determined and normalized against the reference gene’s expression.
Table 4. List of primers used.
Statistical analysis
GraphPad Prism (La Jolla, CA) was used to analyze the experimental data statistically. Results are represented as the mean ± SEM. A statistical variation between the two groups or four groups was calculated by a student’s t-test and one-way ANOVA followed by a Bonferroni’s multiple-comparison correction test, respectively. A P value < 0.05 was accepted as statistically significant.
Results
STK11 protein expression is reduced in the lung tissues of smokers with COPD
STK11 expression is decreased in various tumors, and loss of function has been observed in several cancers [Citation7–11]. However, its role in COPD pathogenesis needs to be investigated. Thus, we tested whether STK11 expression is changed in the lungs of smokers with COPD compared to smokers with normal lungs. The protein expression levels of STK11 were significantly reduced in the lung tissues of COPD patients relative to the lung tissues of control smokers (). In contrast, the STK11 mRNA expression levels remained unchanged in COPD and control smoker lung tissues (). These results indicate that post-transcriptional modifications may accompany the downregulation of STK11 protein levels.
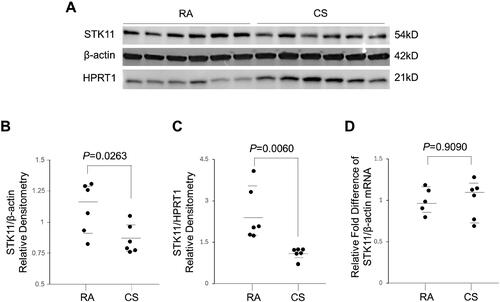
Figure 1. Smokers with COPD display diminished pulmonary STK11 protein expression. (A) Immunoblot analysis showing the STK11 protein levels in the lung tissues of smokers with normal lung function (control) and COPD. (B) The STK11 densitometry data (normalized to Hypoxanthine Phosphoribosyltransferase 1 [HPRT1]) derived from (A). (C) Expression of STK11 mRNA from the lung tissues of smokers with normal lung function (control) and COPD. The expression was determined by RT-PCR, and the change in expression relative to GAPDH (control gene) was presented. Data are represented as median with IQR and analyzed using by student’s t-test.
![Figure 1. Smokers with COPD display diminished pulmonary STK11 protein expression. (A) Immunoblot analysis showing the STK11 protein levels in the lung tissues of smokers with normal lung function (control) and COPD. (B) The STK11 densitometry data (normalized to Hypoxanthine Phosphoribosyltransferase 1 [HPRT1]) derived from (A). (C) Expression of STK11 mRNA from the lung tissues of smokers with normal lung function (control) and COPD. The expression was determined by RT-PCR, and the change in expression relative to GAPDH (control gene) was presented. Data are represented as median with IQR and analyzed using by student’s t-test.](/cms/asset/87b543c3-184b-41c0-95dd-d677e9aed349/icop_a_2342797_f0001_b.jpg)
STK11 protein expression is decreased in mouse lungs exposed to cigarette smoke
To verify the effect of CS exposure on STK11 protein levels in a murine-CS exposed model, mice were exposed to CS or RA for four days, and the alteration in STK11 expression was evaluated. As shown in , a significant downregulation in STK11 protein expression was detected in the mice lungs exposed to CS compared to those exposed to RA. Consistent with human sample data, there was no significant variation in STK11 mRNA expression levels in mouse lung tissues exposed to CS or RA (). These results further confirm that CS exposure decreases STK11 expression via a post-transcription mechanism.
Figure 2. STK11 protein expression is decreased in CS-exposed mice. Mice were exposed to CS or RA for four days. (A) Whole-lung tissue protein lysates were prepared, and the STK11 protein expression was determined by immunoblotting. (B) The STK11 densitometry data normalized to β-Actin. (C) The STK11 densitometry data normalized to HPRT1. (D) Total RNA from whole lung tissue was prepared, and the expression levels of STK11 mRNA were determined by RT-PCR. The change in expression relative to β-Actin (control gene) was presented. Data are represented as median with IQR and analyzed using by student’s t-test.
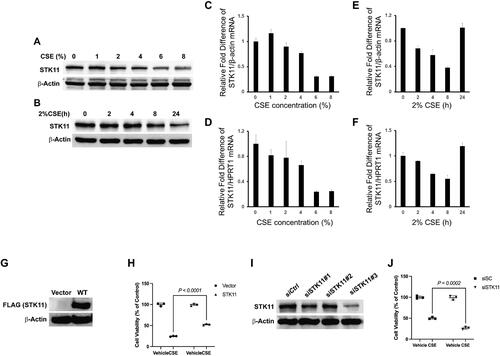
STK11 attenuates cigarette smoke-induced BEAS-2B cell cytotoxicity
The airway epithelium acts as the primary defense against various inhaled challenges and is crucial in host defense [Citation23]. Therefore, we determined the effects of CS on STK11 expression and cell-cytotoxicity in cultured human airway epithelial cells (BEAS-2Bs) incubated with or without CSE, as indicated. CSE exposure dose-dependently decreased the STK11 protein and mRNA expression (). CSE also progressively reduced STK11 protein expression for up to 24 h at a 2% concentration (), however, STK11 mRNA expression levels were recovered after 24 h at a 2% concentration (). Cells were transfected with STK11-Flag plasmid (), and the effects of STK11 overexpression on CSE-altered cell viability were determined. STK11 overexpression significantly attenuated the CSE-induced cytotoxicity (). To determine whether STK11 protein depletion augments CSE-induced cell cytotoxicity, we transfected BEAS-2B cells with STK11 siRNA to knockdown its expression. As shown in , > 70% knockdown of STK11 was achieved with siSTK11#3. STK11 knockdown using siSTK11#3 further augmented the BEAS-2Bs cytotoxicity induced by CSE (). Together, these results indicate that CSE augments the degradation of STK11 expression in airway epithelial cells in a time- and dose-dependent way and STK11 overexpression protects against CSE-induced airway epithelial cell-cytotoxicity. In contrast, its knockdown enhances the CSE-induced airway epithelial cell cytotoxicity. Thus, concluding that STK11 is a protective factor against CSE-induced cell toxicity.
Figure 3. STK11 attenuates CS-induced BEAS-2B cell cytotoxicity. BEAS-2B cells were cultured in the presence and absence of CSE, as indicated above. Whole-cell proteins and Total RNA were isolated, and STK11 expression was determined by immunoblotting (A and B) and RT-PCR (C–F). The mRNA expression levels were measured in triplicate using the same BEAS-2B cells. The bar graph indicates mean ± SE. STK11 was overexpressed in cells by transfecting with STK11-FLAG tag plasmid for 48 h. (G) Whole-cell proteins were isolated and immunoblotted with anti-FLAG (STK11) or anti-β-actin antibodies to detect the STK11 overexpression. (H) MTT assay was performed to measure the cell viability following transfection and culture in the presence or absence of CSE (2%) for 24 h. BEAS-2B cells were transfected with scrambled siRNA (siCtrl) or STK11-targeted siRNAs for 72 h to knockdown STK11. (I) Whole-cell proteins were isolated and immunoblotted with STK11 or β-actin antibodies to detect the STK11 knockdown. (J) MTT assay was performed to measure the cell viability, following siSTK11#3 transfection and culture in the presence or absence of 2% of CSE (2%) for 24 h. All experiments were repeated three times. Data are expressed as mean ± SEM and analyzed using one-way ANOVA followed by a Bonferroni’s multiple-comparison correction test.
FBXL19 targets STK11 degradation via the ubiquitin-proteasomal system
In COPD, the ubiquitin-proteasome system (UPS) is often dysregulated, with elevated ubiquitinated proteins in the lung tissues of severe COPD patients. Several proteins are targeted by UPS-mediated degradation in COPD [Citation24, Citation25]. We performed cycloheximide chase assays to determine the STK11 stability in BEAS-2B cells. The results demonstrate that in BEAS-2B cells, the half-life of STK11 was about 8 h. To further elucidate the degradation pathway participating in STK11 degradation, cells were treated with MG132 (proteasome inhibitor) or leupeptin (lysosomal inhibitor). The accumulation of STK11 protein, specifically in the presence of MG132, indicates that the UPS pathway facilitates STK11 degradation in BEAS-2B cells (). In addition, we also discovered that STK11 protein dose-dependently ubiquitinated () when cells were transfected with ubiquitin-HA plasmid, suggesting that STK11 degradation was directed by ubiquitin.
Figure 4. FBXL19 targets STK11 for proteasomal degradation. (A) To determine STK11 stability in BEAS-2B cells, cells were treated with 20 μg/mL of cycloheximide in the presence of 40 μM of MG132 or 20 μM of leupeptin indicated in the figure. Whole-cell proteins were isolated and immunoblotted with STK11 or β-actin antibodies. (B): Cells were transfected with ubiquitin-HA plasmids as indicated, whole cell proteins were isolated, and the expression of STK11, HA, or β-actin was determined by immunoblotting. To find the E3 ligase that explicitly recognizes and facilitates STK11 degradation, (C) FBXL–V5-tagged family plasmids were transfected in cells, and whole-cell proteins were immunoblotted with anti-STK11, anti-V5 or anti-β-actin antibodies. (D) BEAS-2B cells were transfected as indicated with FBXL19 or FBXL14 –V5 tagged plasmids, and whole-cell proteins were immunoblotted with anti-STK11, anti-V5, or anti-β-actin antibodies. All experiments were repeated three times.
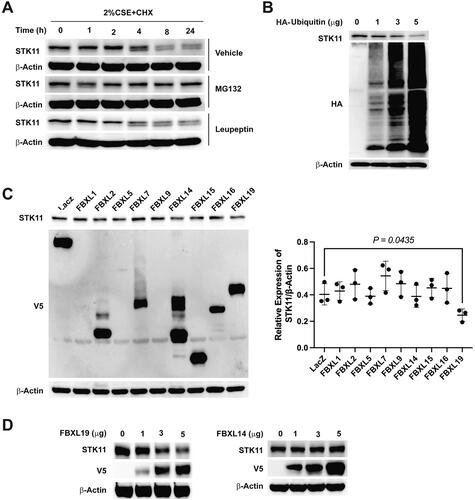
We examined the STK11 protein stability in the presence of various F-box protein–V5-tagged overexpression plasmids to detect the definite E3-ligase that identifies and facilitates ubiquitin-mediated STK11 degradation. shows that FBXL19 was linked with decreased STK11 expression levels compared with all other FBXL subfamily members. To further validate the FBXL19 participation in STK11 degradation, we overexpressed FBXL19 in cells and determined its effects. The results show that FBXL19 overexpression dose-dependently decreased STK11 protein levels compared to FBXL14 control, which did not affect the STK11 protein levels ().
Discussion
In the present study, we have shown various distinctive discoveries that (1) steady-state levels of STK11 protein expression, but not the mRNA expression, were significantly decreased in the lungs of COPD-smokers relative to smokers with normal lung function (control); (2) we demonstrated that the STK11 expression was decreased significantly in the mouse lung tissue exposed to CS. Reliable with human sample data, the STK11 mRNA expression was unaltered in the mouse model; (3) STK11 depletion augmented CSE-induced cytotoxicity in cultured immortalized HBECs, whereas STK11 overexpression mitigates CSE effects on cytotoxicity; (4) mechanistically, CSE depletes STK11 protein expression levels by posttranslational degradation pathway that augments STK11 degradation via FBXL19-E3 ligase activity. Our findings suggest the likelihood of alteration of STK11 to increase its cellular expression levels could provide a unique approach to diminish cell death in the airway epithelium, thereby alleviating the emphysema severity (Graphical Abstract).
Numerous in vitro and animal-model studies have demonstrated the cytotoxic effects of CS [Citation26] and the cytoprotective effects of STK11 [Citation27]. Increased oxidative stress and cellular ROS levels activate the STK11-AMPK pathway, further inhibiting the mammalian target of rapamycin (mTOR) and promoting autophagy via UNC 51 like kinase 1 (ULK1) [Citation28]. Prior investigations have identified that the AMPK, a major target of STK11, activation attenuates CS or elastase-induces lung inflammation and emphysema, abnormal enlargement of airspaces, a pathologic change often associated with COPD [Citation29]. Besides altering cellular metabolism by AMPK activation, STK11 plays a defensive role against ROS-induced oxidative stress. Increased cellular ROS levels initiate the STK11-AMPK pathway and increase NADPH production, an essential electron donor. Thus, diminishing ROS levels to inhibit ROS-stimulated cell death [Citation30, Citation31]. Moreover, STK11 loss augments cellular ROS and might cause an enhanced susceptibility to ROS-stimulated chemotherapies [Citation32]. CS exposure also induces oxidative stress in vitro and in vivo models [Citation33, Citation34]. Consistent with the previous studies showing the cytoprotective effects of STK11 against oxidative stress, we found that STK11 overexpression effectively and significantly attenuated the CSE-induced cytotoxicity, whereas STK11 knockdown enhanced the CSE-induced airway epithelial cell cytotoxicity.
F-box proteins promote ubiquitin-mediated protein degradation by explicitly providing target proteins to E1, E2, and E3 ligase superfamily. These conjugating enzymes then direct target protein degradation via the proteasomal or lysosomal pathway [Citation35] and maintain protein homeostasis. CS induces the degradation of several proteins via the USP pathway, facilitated explicitly by F-box proteins [Citation25]. To investigate the pathway involved in STK11 degradation and to identify the specific F-box facilitating the degradation of STK11, we performed various in vitro experiments. For the first time, we reported that STK11 protein is degraded via a proteasome-pathway exclusively facilitated by F-box protein FBXL19.
In summary, our present study reveals a novel molecular pathway providing experimental evidence that CS exposure depletes STK11 protein via FBXL19-mediated proteolysis, thereby enhancing epithelial cell death. Additional investigations will be needed toward verifying the causality of CS-stimulated STK11 degradation in the COPD progression.
Consent for publication
All authors in this manuscript consent to the publication in the Journal.
Ethical approval
The University of Pittsburgh Institutional Review Board (IRB) approved the use of human subject samples was approved by the University of Pittsburgh Institutional Review Board (IRB) in this study.
Acknowledgments
All frozen lung tissue samples used in this study were provided by the tissue core of the Lovelace Respiratory Research Institute and the Lung Tissue Research Consortium (LTRC).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The datasets used and analyzed in this study are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem. 200 6;75(1):1–9. doi: 10.1146/annurev.biochem.75.103004.142702.
- Hardie DG. AMP-activated protein kinase—an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25(18):1895–1908. doi: 10.1101/gad.17420111.
- Jansen M, Ten Klooster JP, Offerhaus GJ, et al. LKB1 and AMPK family signaling: the intimate link between cell polarity and energy metabolism. Physiol Rev. 2009;89(3):777–798. doi: 10.1152/physrev.00026.2008.
- Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9(8):563–575. doi: 10.1038/nrc2676.
- Shaw RJ, Kosmatka M, Bardeesy N, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101(10):3329–3335. doi: 10.1073/pnas.0308061100.
- Woods A, Johnstone SR, Dickerson K, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase Cascade. Curr Biol. 2003;13(22):2004–2008. doi: 10.1016/j.cub.2003.10.031.
- Gill RK, Yang SH, Meerzaman D, et al. Frequent homozygous deletion of the LKB1/STK11 gene in non-small cell lung cancer. Oncogene. 2011;30(35):3784–3791. doi: 10.1038/onc.2011.98.
- Hezel AF, Gurumurthy S, Granot Z, et al. Pancreatic LKB1 deletion leads to acinar polarity defects and cystic neoplasms. Mol Cell Biol. 2008;28(7):2414–2425. doi: 10.1128/MCB.01621-07.
- Koyama S, Akbay EA, Li YY, et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment immunosuppressive effects of STK11/LKB1 loss in lung. Cancer Res. 2016;76(5):999–1008. doi: 10.1158/0008-5472.CAN-15-1439.
- Li J, Liu J, Li P, et al. Loss of LKB1 disrupts breast epithelial cell polarity and promotes breast cancer metastasis and invasion. J Exp Clin Cancer Res. 2014;33(1):70. doi: 10.1186/PREACCEPT-6783991421275553.
- Sanchez-Cespedes M, Parrella P, Esteller M, et al. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Research. 2002;62(13):3659–3662.
- Tanwar PS, Mohapatra G, Chiang S, et al. Loss of LKB1 and PTEN tumor suppressor genes in the ovarian surface epithelium induces papillary serous ovarian cancer. Carcinogenesis. 2014;35(3):546–553. doi: 10.1093/carcin/bgt357.
- Wingo SN, Gallardo TD, Akbay EA, et al. Somatic LKB1 mutations promote cervical cancer progression. PLoS One. 2009;4(4):e5137. doi: 10.1371/journal.pone.0005137.
- Jishage K, Nezu J, Kawase Y, et al. Role of Lkb1, the causative gene of Peutz-Jegher’s syndrome, in embryogenesis and polyposis. Proc Natl Acad Sci U S A. 2002;99(13):8903–8908. doi: 10.1073/pnas.122254599.
- Ylikorkala A, Rossi DJ, Korsisaari N, et al. Vascular abnormalities and deregulation of VEGF in Lkb1-deficient mice. Science. 2001;293(5533):1323–1326. doi: 10.1126/science.1062074.
- Aretz S, Stienen D, Uhlhaas S, et al. High proportion of large genomic STK11 deletions in Peutz-Jeghers syndrome. Hum Mutat. 2005;26(6):513–519. doi: 10.1002/humu.20253.
- Mograbi B, Heeke S, Hofman P. The importance of STK11/LKB1 assessment in non-small cell lung carcinomas. Diagnostics (Basel). 2021;11(2):196. doi: 10.3390/diagnostics11020196.
- Health NCfCDPaHPUOoSa. The Health Consequences of Smoking-50 Years of Progress: a Report of the Surgeon General. The Health Consequences of Smoking-50 Years of Progress: A Report of the Surgeon General. Reports of the Surgeon General. Atlanta (GA); 2014.
- Chen ZH, Lam HC, Jin Y, et al. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci U S A. 2010;107(44):18880–18885. doi: 10.1073/pnas.1005574107.
- Nucera F, Mumby S, Paudel KR, et al. Role of oxidative stress in the pathogenesis of COPD. Minerva Med. 2022;113(3):370–404. doi: 10.23736/S0026-4806.22.07972-1.
- Nyunoya T, Monick MM, Klingelhutz A, et al. Cigarette smoke induces cellular senescence. Am J Respir Cell Mol Biol. 2006;35(6):681–688. doi: 10.1165/rcmb.2006-0169OC.
- Nyunoya T, Monick MM, Klingelhutz AL, et al. Cigarette smoke induces cellular senescence via werner’s syndrome protein down-regulation. Am J Respir Crit Care Med. 2009;179(4):279–287. doi: 10.1164/rccm.200802-320OC.
- Gao W, Li L, Wang Y, et al. Bronchial epithelial cells: the key effector cells in the pathogenesis of chronic obstructive pulmonary disease? Respirology. 2015;20(5):722–729. doi: 10.1111/resp.12542.
- Min T, Bodas M, Mazur S, et al. Critical role of proteostasis-imbalance in pathogenesis of COPD and severe emphysema. J Mol Med (Berl). 2011;89(6):577–593. doi: 10.1007/s00109-011-0732-8.
- Mallampalli RK, Li X, Jang JH, et al. Cigarette smoke exposure enhances transforming acidic coiled-coil-containing protein 2 turnover and thereby promotes emphysema. JCI Insight. 2020;5(2). doi: 10.1172/jci.insight.125895.
- Pouli AE, Hatzinikolaou DG, Piperi C, et al. The cytotoxic effect of volatile organic compounds of the gas phase of cigarette smoke on lung epithelial cells. Free Radic Biol Med. 2003;34(3):345–355. doi: 10.1016/s0891-5849(02)01289-3.
- Muniraj N, Siddharth S, Shriver M, et al. Induction of STK11-dependent cytoprotective autophagy in breast cancer cells upon honokiol treatment. Cell Death Discov. 2020;6(1):81. doi: 10.1038/s41420-020-00315-w.
- Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. doi: 10.1038/ncb2152.
- Lee JS, Park SJ, Cho YS, et al. Role of AMP-activated protein kinase (AMPK) in smoking-induced lung inflammation and emphysema. Tuberc Respir Dis (Seoul). 2015;78(1):8–17. doi: 10.4046/trd.2015.78.1.8.
- Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485(7400):661–665. doi: 10.1038/nature11066.
- Whang YM, Park SI, Trenary IA, et al. LKB1 deficiency enhances sensitivity to energetic stress induced by erlotinib treatment in non-small-cell lung cancer (NSCLC) cells. Oncogene. 2016;35(7):856–866. doi: 10.1038/onc.2015.140.
- Bonanno L, Zulato E, Pavan A, et al. LKB1 and tumor metabolism: the interplay of immune and angiogenic microenvironment in lung cancer. Int J Mol Sci. 2019;20(8):1874. doi: 10.3390/ijms20081874.
- Reddy AT, Lakshmi SP, Banno A, et al. Role of GPx3 in PPARgamma-induced protection against COPD-associated oxidative stress. Free Radic Biol Med. 2018;126:350–357. doi: 10.1016/j.freeradbiomed.2018.08.014.
- Talukder MA, Johnson WM, Varadharaj S, et al. Chronic cigarette smoking causes hypertension, increased oxidative stress, impaired NO bioavailability, endothelial dysfunction, and cardiac remodeling in mice. Am J Physiol Heart Circ Physiol. 2011;300(1):H388–H396. doi: 10.1152/ajpheart.00868.2010.
- Hochstrasser M. Biochemistry. All in the ubiquitin family. Science. 2000;289(5479):563–564. doi: 10.1126/science.289.5479.563.