Abstract
Adhesion to the extracellular matrix (ECM) proteins collagen I and vitronectin is sufficient to drive human mesenchymal stem cells (hMSCs) into an osteogenic differentiation pathway, but the mechanisms underlying this stimulation are not well understood. We found that addition of β1 and α vβ3 integrin blocking antibodies inhibited ECM-induced ERK activation, while addition of the MEK inhibitor PD98059 blocked ERK activation, serine phosphorylation of the osteogenic transcription factor runx2/cbfa-1, osteogenic gene expression, and calcium deposition. These results suggest that ERK plays an important role in driving the ECM-induced osteogenic differentiation of hMSC.
INTRODUCTION
How undifferentiated stem cells commit to a particular phenotypic fate remains one of the major questions in developmental biology and tissue engineering. Ample evidence implicates extracellular matrix (ECM) proteins in regulating this process, though the mechanisms underlying ECM-induced differentiation are poorly understood. For example, one of the hallmark events regulating the differentiation of human mesenchymal stem cells (hMSC) along the osteogenic lineage is the deposition of a type I collagen (COLL-I) containing extracellular matrix (ECM) (Citation1, Citation2). Production of this collagenous matrix is essential in the induction of osteoblast-specific gene expression of alkaline phosphatase and osteocalcin mRNAs, and ultimately, mineralization of bone tissue (Citation3, Citation4). Improper synthesis of the heterotrimeric COLL-I molecule, caused by mutations in the loci encoding its COLL1α 1 and COLL1α 2 chains, leads to inhibition of preosteoblast maturation (Citation5) and the condition known as osteogenesis imperfecta (Citation6).
The interactions between cells and the ECM cause the activation of integrins on the cell surface resulting in the transduction of signals through various intracellular pathways mediating cell adhesion, proliferation, migration, and differentiation (Citation7). The hMSC express several integrin subunits including α 1, α 2, α 3, α a, α v, β 1, β 3, and β 4 (Citation8). COLL-I is known to associate with α 1β 1, α 2β 1, α 3β 1 integrins, whereas α vβ 3 is the predominant cell surface receptor for vitronectin (VN). ECM proteins and their integrin receptors contribute to the osteogenic differentiation of bone marrow stromal cells (Citation1, Citation9) and hMSC (Citation10). However, the signaling mechanisms linking integrin binding to the commitment to osteogenic differentiation of hMSC are essentially unknown.
Most of what is known about integrin signaling and osteoegenesis comes from studies of fully differentiated osteoblasts or preosteoblasts, cells that have already committed to the osteogenic phenotype. In these cells, the interaction of COLL-I with the α 2β 1 integrin mediates the development of osteoblastic phenotypes (Citation11), (Citation12), (Citation13). Disruption of the α 2-integrin interaction with COLL-I, through the administration of specific inhibitory peptides or blocking antibodies, disrupts osteogenic gene expression and bone formation (Citation12, Citation14). Also, matrix mineralization is significantly reduced in osteoblastic cells cultured with blocking antibodies raised against the α 2β 1 95%) and α vβ 3 (65%) integrin dimers (Citation15).
In these committed cells, adhesion to COLL-I and VN induces activation of the ERK 1/2 isoforms of the Mitogen Activated Protein Kinase superfamily and drives progression of osteoblasts into mature osteocytes via a Ras-dependent pathway (Citation16). Disruption of ERK 1/2 signaling by inhibitory drugs or retroviral transfection encoding a mutated form of the enzyme, significantly diminishes cellular proliferation, expression of osteogenic markers, and matrix mineralization (Citation16, Citation17). In contrast, ERK controls differentiation of uncommitted hMSC stimulated with dexamethazone (Citation18) but its role in driving the osteogenic commitment of hMSC in response to ECM binding is not known.
The transcriptional activity of runx2/cbfa1 is homologous to that of the drosophila transcription factor runt, and runx2/cbfa1 is absolutely essential for osteoblast differentiation and subsequent bone formation. Homozygous deletion of the runx2/cbfa1 gene in mice results in the complete absence of osteoblasts and bone (Citation19, Citation20). Runx2/cbfa1 activity is involved in controlling the expression of many osteogenic genes, including osteocalcin, osteopontin, alkaline phosphatase, and COLL-I (Citation21). This protein is part of a transcription complex that binds to osteoblast-specific cis acting element 2 (OSE2) in the promoter region of several osteogenic genes, where it may interact with other transcriptional complexes (e.g., SMADS, AP-1) to control osteogenesis (Citation22). Forced expression of runx2/cbfa1 in nonosteoblastic mesenchymal stem cells induces expression of osteoblastic phenotypes (Citation22, Citation23). One of the kinases known to target runx2/cbfa1 is ERK (Citation17, Citation24), although the specific amino acid residues responsible for this signaling event have not been identified. The role of ERK signaling in the initial activation of the runx2/cbfa1 transcription factor and its subsequent contribution to the commitment to osteogenic differentiation in hMSC are entirely unknown.
The mechanism by which hMSC translate adhesion to ECM into cell differentiation is poorly understood. We hypothesized that ERK signaling pathways provide a link between cell surface integrin activation by ECM and subsequent phosphorylation of the runx2/cbfa-1 transcription factor that controls osteogenic gene expression. To test this hypothesis, we cultured hMSC on purified COLL-I and VN in the presence or absence of PD98059, a specific inhibitor of ERK activation, and assayed for phosphorylation of runx2/cbfa-1 as well as expression of established osteogenic differentiation markers (osteopontin, osteocalcin, alkaline phosphatase, calcium deposition). Our results indicate that matrix-induced differentiation of hMSC primarily occurs through ERK signaling pathways when plated on purified COLL-I and VN.
EXPERIMENTAL PROCEDURES
Tissue culture media (DMEM) and penicillin G-streptomycin sulfate (GPS) were purchased from Mediatech (Cellgro, VA). Fetal bovine serum (FBS) was purchased from Gemini Bio-Products (Woodland, CA). Trypsin-EDTA and purified bovine type I collagen were obtained from Sigma Chemical Co. (St. Louis, MO). Purified human plasma vitronectin and human plasma fibronectin were from Chemicon International (Temecula, CA). The α vβ3 (catalog # MAB1976) and the α 1-6 and β 1-4 integrin function-blocking antibodies (Alpha integrin blocking and IHC kit, catalog # ECM 430; Beta integrin screening kit, catalog # ECM 440) were purchased from Chemicon International (Temecula, CA). Rabbit polyclonal IgG Anti-g-actin (catalog # AAM01-A) antibody was from Cytoskeleton (Denver, CO). Rabbit polyclonal IgG antibodies against anti-ERK1/2 (catalog # AB3053), osteopontin (catalog # AB1870), and phosphoserine (catalog # AB1603) were obtained from Chemicon International (Temecula, CA). Rabbit polyclonal IgG phosphospecific antibodies against anti-ERK 1/2 (pTpY185/187) (catalog # 44-680) was from Biosource International (Camarillo, CA). Mouse monoclonal IgG antibodies against anti-Runx2/Cbfa1 were purchased from MBL International (Watertown, MA). Horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG and HRP-conjugated goat anti-rabbit IgG secondary antibodies were obtained from Jackson Immuno Research (West Grove, PA). Protein A/G-agarose beads and the MEK1 inhibitor PD98059 were purchased from Calbiochem (San Diego, CA). Reverse transcriptase-polymerase chain reaction (RT-PCR) primers listed in were purchased from IDT Technologies (Coralville, Iowa). The protein assay kit was purchased from Pierce (Rockford, IL). Unless otherwise specified, the other standard reagents were obtained from Fisher Scientific (Fair Lawn, NJ).
TABLE 1 Primers used for RT-PCR
Cell Culture
Cryopreserved hMSC were purchased from Cambrex Inc. (Walkersville, MD) and were grown according to the manufacturers' instructions. Briefly, cells were plated at 5 × 103 cells/cm2 in a T75 flask (75 cm2) for continuous passaging in DMEM medium supplemented with 10% FBS, 1% L-glutamine [29.2 mg/mL], penicillin G [10,000 units/mL] and streptomycin sulfate 10,000 μg/mL]. Medium was changed twice weekly and cells were detached by trypsin-EDTA and passaged into fresh culture flasks at a ratio of 1:3 upon reaching confluence. Cultures were incubated at 37°C in a humidified atmosphere containing 95% air and 5% CO2.
For in vitro osteogenic assays, hMSC were passaged three times before they were inducted and plated at densities of 3.1 × 103 cells/cm2 in 0.2 mL/cm2 of medium on 100 mm Falcon dishes (78.5 cm2). The following day (day 0), we replaced the culture medium with fresh control medium in the absence or presence of osteogenic supplements (OS) containing 0.1 μ M Dex, 0.05 mM AsAP, and 10 mM β GP (Cambrex Inc). In each experiment, control and OS-treated cells were processed in parallel. For inhibition of MEK1, PD98059 in dimethyl sulfoxide (DMSO) was added to control and OS mediums at a final concentration of 50 μ M twice a week during media changes unless otherwise specified. The final concentration of DMSO never exceeded 0.1%, and the same amount of DMSO vehicle was added to control conditions. For all other assays, suspensions of cells were incubated with PD98059 (50 μ M) 15 min prior to plating. At days 14, 21, and 28, cultures were assayed as described below.
Immunoprecipitation of Runx2 and Western Blotting
Whole cell extracts were prepared by harvesting serum deprived cells overnight (DMEM + 0.1% FBS) in ice-cold RIPA buffer (150 mM NaCl, 50 mM Tris, 1% Triton-X, 0.3 mM Sodium vanadate, 1% Deoxycholic acid, 0.2% SDS, pH 7.4). Samples were precleaned twice with 50 μ l of protein A/G-agarose beads for 30 minutes followed by pelleting of beads. 5 μ l of mouse monoclonal anti-Runx2 was added and incubated for 2 h at 4°C with shaking. The immune complexes were collected on the addition of 30 μl of protein A/G-agarose beads and subsequently incubated for 1 h at 4°C, followed by centrifugation at 12,000 g for 10 min. Precipitates were washed thoroughly and suspended once in ice-cold RIPA buffer, centrifuged at 2,500 g for 10 sec, and resuspended in Laemmli's sample buffer. Proteins were denatured at 100°C for 5 min, resolved by 8% SDS-PAGE, and electrophoretically transblotted to Trans-Blot® nitrocellulose membranes (0.2 μ m) (Bio-Rad, Hercules, CA). The membranes were incubated with blocking solution (5% nonfat dried milk in 1X PBS + 0.2% Tween-20 (PBST)) for 1 h, then probed with various primary antibodies (1:500) overnight at 4°C. After three washes with PBST, membranes were incubated with HRP-conjugated secondary IgG (1:25,000) for 1 h, followed by another three washes with PBST. Immunoreactive bands were detected using the SuperSignal® Chemiluminescent reagent (Pierce) and quantitatively analyzed by normalizing band intensities to the controls (total ERK, total Runx2) on scanned films by IMAGEJ software.
Integrin-blocking western blots were performed according to the procedure above, but the cells were incubated with a functional integrin-blocking antibody for 30 min at 37°C, with vortexing every 5 min, prior to plating.
Adhesion Assays
Cell adhesion assays were performed as previously described using Sarstedt 96-well suspension cell culture plates (Citation25). Tissue culture plates were coated with purified ECM proteins (COLL-I or VN) at a concentration of 20 μ g/mL for 1 h at room temperature. Wells were washed twice with PBS and incubated with nd-blotto (5% nondiary creamer in PBS + 0.2% Tween 20) for 30 min prior to the assay. Suspensions containing cells were incubated with PD98059 at final concentrations of 0, 25, and 50 μ M, for 15 min prior to seeding. Cells were allowed to attach for 30 min at 37°C and were subsequently fixed with 3% paraformaldehyde, washed twice in PBS, and incubated in crystal violet dye for 15 min. Wells were washed thoroughly with water and the violet dye was extracted with 10% SDS solution. Absorbance was measured using a TECAN SPECTAFluor spectrophotometer at 595 nm and relative adhesion was compared to cells attached to nd-blotto.
Migration Assays
Cell migration assays were performed using 10 μm MIC plates from Millipore (Danvers, MA). Filters were coated with purified ECM proteins (COLL-I, VN, or fibronectin (FN)) at a concentration of 20 μ g/ml or nd-blotto for 1 h at room temperature prior to assay. Basal chambers for the nd-blotto wells were filled with migration medium (DMEM + 1% sodium pyruvate + 1X GPS) while the basal chambers for the ECM containing wells were filled with control medium. Cell suspensions in migration medium were incubated for 15 min with PD98059 at either 0, 10, 25, or 50 μ M concentrations and then seeded at a density of 5 × 103 cells per well. Migrations were allowed to run for 18 h at 37°C. Filters were then incubated for 30 min with 5 μ M calcein AM (Molecular Probes, Eugene, OR) and washed thoroughly with PBS. Residual cells were swabbed from the top of the wells to avoid false readings. To quantitate migration, plates were read at 485Ex/535Em with a TECAN SPECTAFluor spectrophotometer. Relative fluorescence values for each experimental condition were expressed relative to fibronectin and nd-blotto controls.
Reverse Transcipatase PCR
RNA was isolated from 10 × 108 hMSC cultured for 14 days in the presence or absence of PD98059 (50 μ M) on tissue culture plastic (±OS), or on COLL-I, or VN in control media. Total RNA was isolated using the RNeasy mini kit (Qiagen, Valencia, CA). RT-PCR was performed with the OneStep RT-PCR Kit (Qiagen) and a 96 well thermal cycler (MJ Research, Waltham, MA) using the primers listed in , which were designed by the Lasergene v5.0 program (DNASTAR, Madison, WI). One microgram of template RNA was used per reaction. The reverse transcription step ran for 30 min at 50°C, followed by PCR activation for 15 min at 95°C. Thirty amplification cycles were run, consisting of one min denaturation at 94°C, one min of annealing at 58°C, and one min of extension at 72°C. Final extension was allowed to run 10 min at 72°C. Reaction products were separated by gel electrophoresis using a 1% agarose gel. Bands were visualized by UV illumination of ethidium-bromide-stained gels and captured using a Chemilmager 4400 Gel imaging system (Alpha Innotech, San Leandro, CA). Band intensity was quantitatively analyzed by IMAGEJ software for each gene and was normalized to corresponding GAPDH values.
Immunohistochemistry
Osteopontin (OP) was localized in hMSC cultured on glass coverslips as previously described (Citation25). Briefly, primary OP antibodies (diluted 1:200 in 1X PBS + 1% BSA) were added for 1 h to cells fixed in 3% paraformaldehyde and blocked with PBS/1% BSA for 30 min. Secondary antibodies conjugated to rhodamine (Jackson Immuno Research) were added for 1 h, then coverslips were mounted using Prolong antifade medium (Molecular Probes). Cells were visualized with a Nikon TE2000-S inverted fluorescence/phase contrast microscope equipped with a digital SPOT camera.
Histological Staining of Alkaline Phosphatase Activity and Calcium Precipitation
For detection of alkaline phosphatase activity, a solution of naphthol AS-MX phosphate and fast blue RR dissolved in dH2O was poured into 100 mm dishes on day 14 according to the manufacturer's instructions contained in Sigma Kit #85 (Sigma). Cellular specimens were scored according to the quantity and intensity of the precipitated dye as determined by IMAGEJ software and was normalized to control values. For the detection of a calcium-phosphate-containing matrix after 28 days, cell layers were stained by the alizarin red-S method. Specimens were washed three times with PBS and fixed in ice-cold 70% ethanol for 1 h. Afterwards, cells were incubated with a 0.4% alizarin red-S solution in water (pH 4.2) for 15 min at room temperature. Cells layers were washed thoroughly with dH2O five times and left in PBS for 15 min. Cellular specimens were scored according to the quantity and size of precipitated granules.
Calcium Assay
Specimens were washed twice with PBS and extracted off the dishes in 0.5 N HCl. Accumulated calcium was removed from the cellular component by shaking for 5 h at 4°C, followed by centrifugation at 2,000 g for 10 min. The consequent supernatant was utilized for calcium determination according to the manufacturer's instructions contained in Sigma Kit #587 (Sigma). Total calcium was calculated from standard solutions prepared in parallel and expressed as ng/dish after absorbance at 575 nm was measured.
Statistical Analysis
All experiments were repeated a minimum of two times and the representative data were presented as mean ± S.E. Statistical analyses were preformed using Student's unpaired t test, and a p value less than 0.05 was considered significant.
RESULTS
Adhesion to ECM Proteins Stimulates ERK 1/2 Phosphorylation in hMSC
ERK 1 and ERK 2 were activated in hMSC plated on purified COLL-I or VN for 30 min, as evidenced by reactivity with an anti-ERK 1/2 (pTpY185/187) antibody in an immunoblot (, panel A). Specifically, cells plated on COLL-I and VN had 3.7 fold and 4.6 fold greater phosphorylation of ERK 1, respectively, than cells plated on the nonspecific substrate poly-L-lysine (, panel B). Similarly, ERK 2 phosphorylation levels were increased 1.7 fold and 1.8 fold, respectively, on these substrates (, panel C). Phosphorylation of ERK 1 and ERK 2 increased an additional 1.5 fold and 1.2 fold, respectively, in cells plated on COLL-I and VN for an additional 30 min (), but did not change in cells plated on fibronectin or poly-L-lysine. Control cells kept in suspension exhibited almost no detectable ERK phosphorylation (). As expected, cells plated on tissue culture plastic and stimulated with OS media supplements also exhibited activation of ERK 1 and ERK 2 over the same time course (Citation18) ().
Figure 1 Adhesion to COLL-I or VN activates ERK 1/2 in hMSC. (A) Cells were kept in suspension (Susp.) for 30 min, or plated for the same time on poly-L-lysine (Poly-L-L), tissue culture plastic in the presence of osteogenic supplements (OS), 20 μg/ml COLL-I or 20 μ g/ml VN in basal growth medium (no OS supplements), then lysed and probed for activated ERK 1/2 by western blot with phospho-ERK-specific antibodies (top row). Parallel cultures were treated with 50 μ M PD98059, as indicated by the + symbol. Total ERK was detected by western blot with a non-phospho-specific antibody. Immunoreactivity to G actin was used as a loading control (bottom row). Densitometric measures of band intensity for ERK 1 and ERK 2 are shown in (B) and (C), respectively. Gel shown is representative of three experiments.
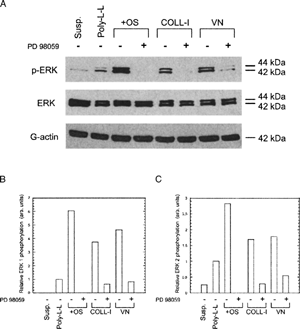
Figure 2 ERK phosphorylation increases with time in hMSC plated on COLL-I or VN compared to cells plated on fibronectin (FN) and poly-L-lysine (Poly-L-L) (A) Cells were plated for the indicated times on the indicated substrates, then lysed and probed for phosphorylated ERK as per . Densitometric measures of band intensity for ERK 1 and ERK 2 are shown in (B) and (C), respectively. ERK 1 bands were normalized to the intensity of ERK 1 in the 30 min/COLL-I lane. ERK 2 bands were normalized to the intensity of ERK 2 in the 30 min/Poly-L-L lane.
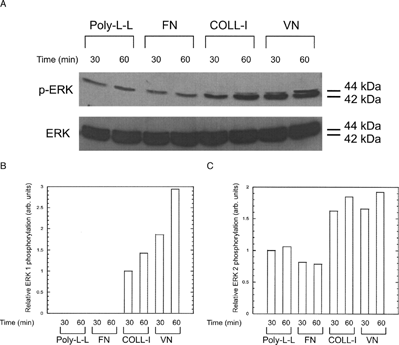
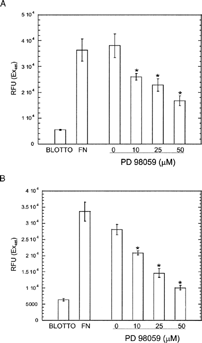
To assess the role of MEK signaling and specific integrins in this activation of ERK 1/2, we included the MEK inhibitor PD98059 and integrin function blocking antibodies in our assays. Addition of PD98059 significantly reduced ERK 1 and ERK 2 phosphorylation in all conditions (), demonstrating that ECM-induced ERK phosphorylation occurred through a MEK signaling pathway. Addition of PD98059 also reduced adhesion to COLL-I and VN in a dose dependent manner (). Addition of function blocking antibodies against the integrins α 1, α 2, and β 1 reduced phosphorylation of ERK 1 by 60–100% in cells plated on COLL-I (). Function blocking antibodies against the α v integrin subunit and the α vβ 3 integrin receptor eliminated ERK 1 phosphorylation in cells plated on VN. In contrast, these antibodies reduced phosphorylation of ERK 2 by only 40–60%, demonstrating that ERK 1, rather than ERK 2, is the predominant target of integrin-mediated COLL-I and VN binding in hMSC. Addition of PD98059 also inhibited integrin-mediated migration of these cells on COLL-I and VN ().
Figure 3 Adhesion of hMSC to COLL-I and VN is sensitive to ERK inhibition. Cells were plated on COLL-I or VN in the presence of the indicated concentrations of PD98059 and allowed to adhere for 30 min. As a control, cells were allowed to adhere to tissue culture plastic coated with nd-blotto (BLOTTO). Non adherent cells were removed by washing and adherent cells were stained with crystal violet, then solubilized in SDS and absorbance determined at 595 nm. Values represent mean ± standard deviation (n = 5). Asterisks indicate significant (p < 0.05) change from “0” values for each condition, using Student's unpaired t test.
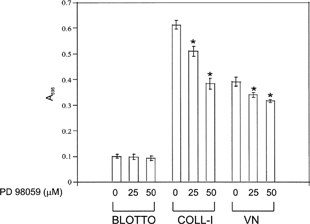
Figure 4 Integrin blocking reduces ERK 1/2 phosphorylation. (A) Cells were plated on COLL-I or VN and assayed for phosphorylated ERK as per . Prior to plating, cells were incubated with function blocking antibodies against the indicated integrin subunits. Control cells received no blocking antibodies (NB) or blocking antibodies against the α 3 subunit. (B) and (C) show densitometric measures of band intensity for ERK 1 and ERK 2 from panel A for cells plated on COLL-I and VN, respectively. Gel shown is representative of three experiments.
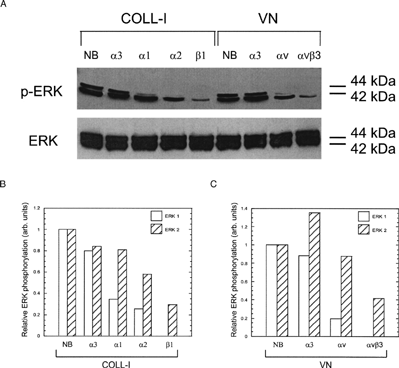
Figure 5 PD98059 inhibits hMSC migration on COLL-I (A) and VN (B). Cells were incubated in the indicated concentrations of PD98059 and plated in the upper chamber of a filter-based migration assay plate, then allowed to migrate towards the bottom of the filter for 18 h. Unmigrated cells were wiped from the upper surface of the filter and migrated cells were stained with calcein-AM. Migrated cells were detected with a fluorescence plate reader. To establish the dynamic range of the assay, cells were placed in filters coated with nd-blotto and fibronectin to establish the minimum and maximum number of cells that migrated, respectively. Values represent mean ± standard deviation (n = 5). Asterisks indicate significant (p < 0.05) change from “0” values for each condition, using Student's unpaired t test.
Inhibition of ERK Signaling Disrupts ECM-Stimulated Osteogenic Differentiation of hMSC
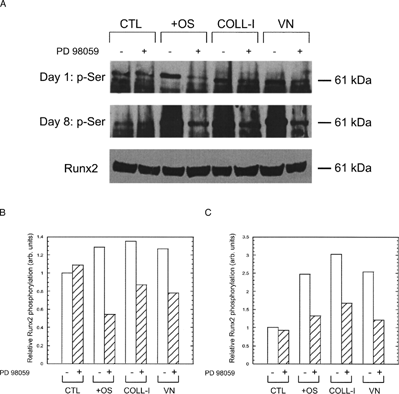
To assess the role of ERK activation on ECM-induced osteogenic differentiation of hMSC, we examined several hallmarks of this differentiation under conditions where the sole osteogenic stimulus was contact with ECM proteins, and the sole inhibitor was PD98059. To determine the effect of ECM adhesion on runx2/cbfa-1 activation, we assayed for runx2/cbfa-1 serine phosphorylation by immunoprecipitation and immunoblot analysis. shows that cells plated on COLL-I or VN exhibited increased serine phosphorylation of runx2/cbfa-1 compared to cells plated on tissue culture plastic, and that this increased phosphorylation occurred within one day of plating; the effect was exacerbated after eight days. Importantly, addition of PD98059 reduced serine phosphorylation of runx2/cbfa-1 in hMSC cultured for one and eight days under all conditions except those plated on tissue culture plastic. In control cells plated on tissue culture plastic and treated with OS media supplements, runx2/cbfa-1 phosphorylation was increased after one and eight days, and was reduced to basal levels by PD98059.
Figure 6 Adhesion to COLL-I and VN stimulates runx2/cbfa-1 phosphorylation. (A) Cells were plated on tissue culture plastic in basal media (CTL), on tissue culture plastic in media with OS supplements (OS), or in basal media on COLL-I or VN for one day (upper panel) or eight days (middle panel). Parallel cultures were treated with 50 μ M PD98059 or vehicle. At the end of the incubation period, cells were lysed and serine phosphorylated proteins were immunoprecipitated with a phospho-serine specific antibody. Immunopreciapiated proteins were separated by SDS-PAGE and blotted for runx2/cbfa-1, which is indicated by the 61 kDa band. Total runx2/cbfa-1 from each lysate was detected by western blot as a loading control (bottom panel). Quantitation of the 61 kDa band in each lane is shown for Day 1 and Day 8 samples in (B) and (C), respectively.
To measure the effect of ERK signaling on osteogenic gene expression, we performed RT-PCR analysis on cells plated on COLL-I or VN in the presence and absence of PD98059. shows that cells plated on these substrates for 14 days expressed increased levels of alkaline phosphatase, osteocalcin, and osteopontin genes compared to cells plated on tissue culture plastic, and that this increase was inhibited by PD98059. Control cells treated with OS media supplements also exhibited these increases, and were likewise sensitive to PD98059 treatment. Runx2/cbfa-1 expression was unchanged in all conditions tested. Direct measures of osteopontin protein deposition by fluorescence microscopy and immunoblot () and alkaline phosphatase expression by histological staining () confirmed our RT-PCR results.
Figure 7 Osteogenic differentiation of hMSC is ERK-dependent. (A) Cells were plated under the same conditions as in , and total RNA was isolated after 14 days in culture. RT-PCR was performed on each sample, using primers specific for amplification of the indicated genes (AP = alkaline phosphatase, OC = osteocalcin, OP = osteopontin). Cultures represented in the lanes marker “+” contained 50 μ M PD98059 throughout the culture period. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was amplified as a loading control. Densitometry of band intensities for AP, OC, OP, and CBFA1 are given in panels B–E, respectively.
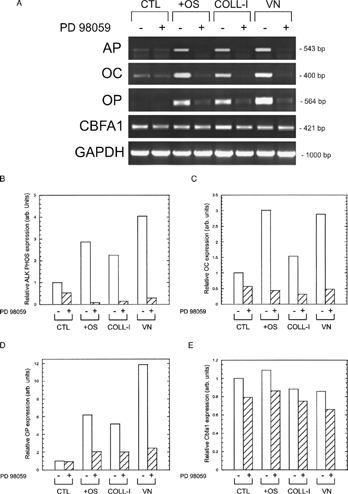
Figure 8 hMSC secretion of osteopontin is ERK dependent. Cells were plated for eight days under the same conditions as in , and osteopontin was detected by indirect immunofluorescence (Panel A) and immunoblot (Panel B). Arrows in panel A indicate osteopontin in perinuclear region, corresponding to secretory vesicles. Parallel cultures were treated with 50 μ M PD98059, as indicated by the + symbol. Photo “VN + PD98059” in Panel A was overexposed to reveal intracellular distribution of osteopontin in PD98059-treated cells, hence the bright nuclear staining.
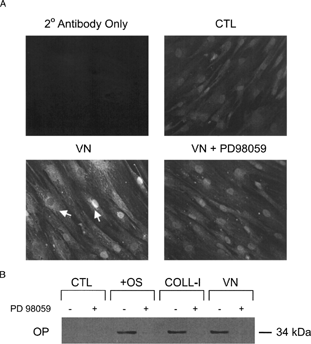
Figure 9 hMSC expression of alkaline phosphatase is ERK dependent. (A) Cells were plated for 14 days under the same conditions as in , then stained for alkaline phosphatase activity. The top and bottom rows indicate cultures without and with 50 μ M PD98059, respectively. Cells expressing alkaline phosphatase appear dark in phase contrast micrographs (10× magnification). (B) Densitometry of plates shown in panel A.
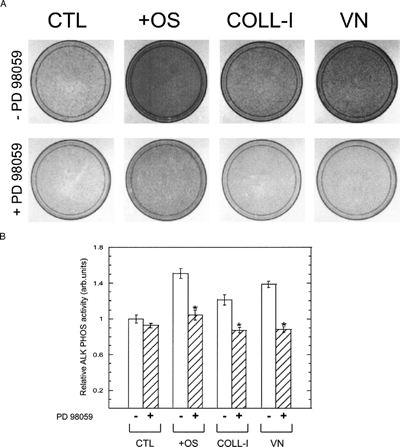
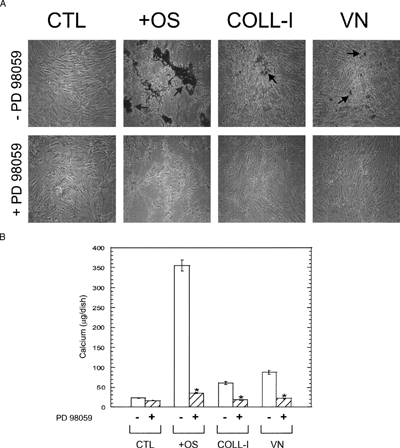
An additional measure of osteogenic differentiation is calcium deposition in the extracellular space. We measured calcium by alizarin red staining and by a colorimetric assay () and found that, whereas OS media supplements induced the greatest increase in calcium deposition, plating cells on COLL-I or VN induced significant increases in calcium deposition, and this increase was abolished by treatment with PD98059. Together these results, coupled with our functional assays described above, demonstrate that MEK-stimulated ERK signaling plays a central role in osteogenic differentiation of hMSC.
Figure 10 hMSC expression of calcified matrix is ERK dependent. (A) Cells were plated for 28 days under the same conditions as in , then stained for calcium with alizarin red. The top and bottom rows indicate cultures without and with 50 μ M PD98059, respectively. Calcified matrix appears dark in phase contrast micrographs (arrows, 10× magnification). (B) Colorimetric assessment of total calcium in hMSC cultures. Cells were plated for 28 as for panel A, then lysed and total calcium assayed as described in Materials and Methods. Asterisks indicate significant (p < 0.05) reductions in parallel untreated samples (white bars), using Student's unpaired t test.
DISCUSSION
Although activation of ERK signaling cascades has been associated with the proliferation, differentiation, and cellular function in a variety of cell systems, including osteoblasts, there is little evidence linking this pathway to the commitment of undifferentiated hMSC into becoming mineralized-matrix producing osteoblasts. We have previously reported that the interaction with COLL-I and VN matrix is sufficient to induce the osteogenic differentiation of hMSC (Citation10). In this investigation, we demonstrate that treatment of hMSC with the MEK1 inhibitor, PD98059, drastically inhibits the expression of osteogenic markers by blocking the phosphorylation of ERK 1/2. The inhibition of osteogenic differentiation is not a result of cellular apoptosis since only a small percentage of dead cells (< 0.5%) is detected in cultures treated with the DMSO vehicle or PD98059 as assessed by an MTT-viability assay (data not shown). This study presents the first evidence that ERK signaling pathways regulate the commitment step of hMSC differentiation on bone marrow ECM proteins.
The role of ERK signaling pathways in cellular proliferation and differentiation has been extensively investigated in a variety of cell systems, including osteoblasts (Citation26). Our data present the novel finding that ERK signaling plays a role in committing undifferentiated stem cells to a defined phenotype, rather than facilitating the differentiation of already-committed cells, and that this signaling is driven by adhesion to ECM. Specifically, our data show that ERK activation in uncommitted hMSC occurs within 30 min upon adhesion to COLL-I and VN and that this activity ultimately leads to the development of osteoblastic phenotypes days later. These observations are consistent with previous studies showing that COLL-I and VN stimulate ERK activity, and that this activity is required for differentiation (Citation11, Citation16). Previous studies in our laboratory have shown that osteogenic differentiation of hMSC does not occur on a fibronectin extracellular matrix, suggesting that specific ECM interactions with integrins (i.e., α 2β 1 or α vβ 3) is responsible for the signaling cascades responsible for osteogenesis (Citation10). While the total amounts of ERK 1 and ERK 2 proteins are expressed at similar levels in cells plated on all substrates tested, ERK 2 is strongly activated by phosphorylation compared to ERK 1 over an hr t course on COLL-I and VN. Furthermore, our data suggest that integrin blocking preferentially targets ERK 1. While the distinct roles played by ERK 1 and ERK 2 in integrin-mediated cell signaling are not well understood, our data suggest that the two ERK isoforms respond differently to adhesion on COLL-I and VN.
Since ERK phosphorylation is an integrin mediated event, several studies have implicated the role of FAK signaling as regulating matrix-induced differentiation (Citation11, Citation27, Citation28). Expression of antisense FAK mRNA in osteoblastic cells prevents alkaline phosphatase expression (Citation11), and thus differentiation. These studies provide a plausible link between integrin activation of ERK, by ECM induced tyrosine phosphorylation of FAK via the Grb2-Sos-Ras pathway. It is possible that FAK signaling represents one of the earliest decision making events controlling hMSC commitment to the osteogenic phenotype.
Our data demonstrate that matrix-induced ERK signaling pathways increase serine phosphorylation of runx2/cbfa-1 compared to control conditions. Xiao and colleagues have shown that runx2/cbfa-1 activity is controlled by MAPKs in differentiated osteoblasts (Citation17). Furthermore, its stimulation by constitutively active MEK enhances the expression of the bone marker protein osteocalcin, whereas expression of a dominant negative MEK or addition of PD98059 inhibits osteocalcin expression (Citation17). These results are consistent with our findings, that increased serine phosphorylation of runx2/cbfa-1 occurs within one day of plating and intensifies for eight days. Furthermore, our RT-PCR confirms that osteocalcin gene expression is significantly reduced upon the addition of PD98059 after 16 days. Importantly, addition of PD98059 reduces serine phosphorylation of runx2/cbfa-1, though not to basal levels found in cells plated on tissue culture plastic, suggesting that ERK 1/2 is one of several kinases that target this transcription factor. Protein kinase A has been implicated in the posttranslational activation of runx2/cbfa-1 in rat osteoblastic osteosarcoma cell lines (Citation29). However, the kinases and the specific amino acids responsible for the activation of runx2/cbfa-1 transcriptional activity have not been identified to date.
Although the present study demonstrates that the signaling pathway from integrins to runx2/cbfa-1 transcriptional activity via ERK phosphorylation is required for osteogenic commitment of hMSC, this alone is likely not the only signaling pathway that stimulates ECM-induced hMSC differentiation. For example, integrin binding of COLL-I activates the AP-1 transcription factor complex and stimulates bone morphogenetic protein (BMP) signaling cascades regulated by ERK 1/2 during osteoblast differentiation (Citation24), (Citation28), (Citation30), (Citation31). ERK 1/2 signaling pathways differentially regulate AP-1 activity by inducing c-fos expression, thus affecting early osteoblast-specific gene expression (Citation32). Genetic knockout of c-fos in mice results in abnormal bone development (Citation33). Suzawa and colleagues have shown that signals from BMPs and COLL-I are both important for osteoblastic differentiation (Citation28). They demonstrate that ERK phosphorylation enhances the transcriptional activity of Smads, which are in turn regulated by BMPs (Citation28). Furthermore, ERK is involved in BMP-2 induced osteoblastic differentiation of mesenchymal cell line C3H10T1/2 (Citation34). These observations suggest extensive cross-talk between various intracellular signaling pathways that may be differentially regulated by ERK signaling cascades. The relationships and roles of these pathways in the regulation of osteogenic differentiation of hMSC remain to be clarified.
ACKNOWLEDGEMENTS
This work was supported by Public Health Service Grant 5R01EB002197-02 from the National Institute of Biomedical Imaging and Bioengineering (NIBIB) (to GEP) and by SEED funds from Rensselaer Polytechnic Institute.
REFERENCES
- Franceschi R T. The developmental control of osteoblast-specific gene expression: Role of specific transcription factors and the extracellular matrix environment. Crit Rev Oral Biol Med 1999; 10: 40–57, [PUBMED], [INFOTRIEVE], [CSA]
- Franceschi R T, Iyer B S. Relationship between collagen synthesis and expression of the osteoblast phenotype in MC3T3-E1 cells. J Bone Miner Res 1992; 7: 235–246, [PUBMED], [INFOTRIEVE], [CSA]
- Franceschi R T, Iyer B S, Cui Y. Effects of ascorbic acid on collagen matrix formation and osteoblast differentiation in murine MC3T3-E1 cells. J Bone Miner Res 1994; 9: 843–854, [PUBMED], [INFOTRIEVE], [CSA]
- Stein G S, Lian J B. Molecular mechanisms mediating proliferation/differentiation interrelationships during progressive development of the osteoblast phenotype. Endocr Rev 1993; 14: 424–442, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Wenstrup R J, Witte D P, Florer J B. Abnormal differentiation in MC3T3-E1 preosteoblasts expressing a dominant-negative type I collagen mutation. Connect Tissue Res 1996; 35: 249–257, [PUBMED], [INFOTRIEVE], [CSA]
- Gajko-Galicka A. Mutations in type I collagen genes resulting in osteogenesis imperfecta in humans. Acta Biochim Pol 2002; 49: 433–441, [PUBMED], [INFOTRIEVE], [CSA]
- Schwartz M A, Ginsberg M H. Networks and crosstalk: Integrin signalling spreads. Nat Cell Biol 2002; 4: E65–E68, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Pittenger M F, Mackay A M, Beck S C, Jaiswal R K, Douglas R, Mosca J D, Moorman M A, Simonetti D W, Craig S, Marshak D R. Multilineage potential of adult human mesenchymal stem cells. Science 1999; 284: 143–147, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Gronthos S, Simmons P J, Graves S E, Robey P G. Integrin-mediated interactions between human bone marrow stromal precursor cells and the extracellular matrix. Bone 2001; 28: 174–181, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Salasznyk R M, Williams W A, Boskey A, Batorsky A, Plopper G E. Adhesion to Vitronectin and Collagen I promotes osteogenic differentiation of human mesenchymal stem cells. J Biomed Biotechnol 2004; 2004: 24–34, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Takeuchi Y, Suzawa M, Kikuchi T, Nishida E, Fujita T, Matsumoto T. Differentiation and transforming growth factor-beta receptor down-regulation by collagen-alpha2beta1 integrin interaction is mediated by focal adhesion kinase and its downstream signals in murine osteoblastic cells. J Biol Chem 1997; 272: 29309–29316, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Xiao G, Wang D, Benson M D, Karsenty G, Franceschi R T. Role of the alpha2-integrin in osteoblast-specific gene expression and activation of the Osf2 transcription factor. J Biol Chem 1998; 273: 32988–32994, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Mizuno M, Fujisawa R, Kuboki Y. Type I collagen-induced osteoblastic differentiation of bone-marrow cells mediated by collagen-alpha2beta1 integrin interaction. J Cell Physiol 2000; 184: 207–213, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Takeuchi Y, Nakayama K, Matsumoto T. Differentiation and cell surface expression of transforming growth factor-beta receptors are regulated by interaction with matrix collagen in murine osteoblastic cells. J Biol Chem 1996; 271: 3938–3944, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Schneider G B, Zaharias R, Stanford C. Osteoblast integrin adhesion and signaling regulate mineralization. J Dent Res 2001; 80: 1540–1544, [PUBMED], [INFOTRIEVE], [CSA]
- Lai C F, Chaudhary L, Fausto A, Halstead L R, Ory D S, Avioli L V, Cheng S L. Erk is essential for growth, differentiation, integrin expression, and cell function in human osteoblastic cells. J Biol Chem 2001; 276: 14443–14450, [PUBMED], [INFOTRIEVE], [CSA]
- Xiao G, Jiang D, Thomas P, Benson M D, Guan K, Karsenty G, Franceschi R T. MAPK pathways activate and phosphorylate the osteoblast-specific transcription factor, Cbfa1. J Biol Chem 2000; 275: 4453–4459, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Jaiswal R K, Jaiswal N, Bruder S P, Mbalaviele G, Marshak D R, Pittenger M F. Adult human mesenchymal stem cell differentiation to the osteogenic or adipogenic lineage is regulated by mitogen-activated protein kinase. J Biol Chem 2000; 275: 9645–9652, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Otto F, Thornell A P, Crompton T, Denzel A, Gilmour K C, Rosewell I R, Stamp G W, Beddington R S, Mundlos S, Olsen B R, Selby P B, Owen M J. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 1997; 89: 765–771, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson R T, Gao Y H, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997; 89: 755–764, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Franceschi R T, Xiao G. Regulation of the osteoblast-specific transcription factor, Runx2: Responsiveness to multiple signal transduction pathways. J Cell Biochem 2003; 88: 446–454, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Ducy P, Zhang R, Geoffroy V, Ridall A L, Karsenty G. Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation. Cell 1997; 89: 747–754, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Karsenty G. Transcriptional regulation of osteoblast differentiation during development. Front Biosci 1998; 3: D834–D837, [PUBMED], [INFOTRIEVE], [CSA]
- Xiao G, Gopalakrishnan R, Jiang D, Reith E, Benson M D, Franceschi R T. Bone morphogenetic proteins, extracellular matrix, and mitogen-activated protein kinase signaling pathways are required for osteoblast-specific gene expression and differentiation in MC3T3-E1 cells. J Bone Miner Res 2002; 17: 101–110, [PUBMED], [INFOTRIEVE], [CSA]
- Plopper G E, Domanico S Z, Cirulli V, Kiosses W B, Quaranta V. Migration of breast epithelial cells on Laminin-5: Differential role of integrins in normal and transformed cell types. Breast Cancer Res Treat 1998; 51: 57–69, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Cobb M H. MAP kinase pathways. Prog. Biophys Mol Biol 1999; 71: 479–500, [CSA], [CROSSREF]
- Tamura Y, Takeuchi Y, Suzawa M, Fukumoto S, Kato M, Miyazono K, Fujita T. Focal adhesion kinase activity is required for bone morphogenetic protein–Smad1 signaling and osteoblastic differentiation in murine MC3T3-E1 cells. J Bone Miner Res 2001; 16: 1772–1779, [PUBMED], [INFOTRIEVE], [CSA]
- Suzawa M, Tamura Y, Fukumoto S, Miyazono K, Fujita T, Kato S, Takeuchi Y. Stimulation of Smad1 transcriptional activity by Ras-extracellular signal-regulated kinase pathway: A possible mechanism for collagen-dependent osteoblastic differentiation. J Bone Miner Res 2002; 17: 240–248, [PUBMED], [INFOTRIEVE], [CSA]
- Selvamurugan N, Pulumati M R, Tyson D R, Partridge N C. Parathyroid hormone regulation of the rat collagenase-3 promoter by protein kinase A-dependent transactivation of core binding factor alpha1. J Biol Chem 2000; 275: 5037–5042, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Cowles E A, Brailey L L, Gronowicz G A. Integrin-mediated signaling regulates AP-1 transcription factors and proliferation in osteoblasts. J Biomed Mater Res 2000; 52: 725–737, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- McCabe L R, Banerjee C, Kundu R, Harrison R J, Dobner P R, Stein J L, Lian J B, Stein G S. Developmental expression and activities of specific fos and jun proteins are functionally related to osteoblast maturation: Role of Fra-2 and Jun D during differentiation. Endocrinology 1996; 137: 4398–4408, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Hipskind R A, Bilbe G. MAP kinase signaling cascades and gene expression in osteoblasts. Front Biosci 1998; 3: D804–D816, [PUBMED], [INFOTRIEVE], [CSA]
- Ruther U, Garber C, Komitowski D, Muller R, Wagner E F. Deregulated c-fos expression interferes with normal bone development in transgenic mice. Nature 1987; 325: 412–416, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Lou J, Tu Y, Li S, Manske P R. Involvement of ERK in BMP-2 induced osteoblastic differentiation of mesenchymal progenitor cell line C3H10T1/2. Biochem Biophys Res Commun 2000; 268: 757–762, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]