Abstract
The oncogenic tyrosine kinase, v-Src, phosphorylates connexin43 (Cx43) on Y247 and Y265 and inhibits Cx43 gap junctional communication (GJC), the process of intercellular exchange of ions and metabolites. To test the role of a negative charge on Cx43 induced by tyrosine phosphorylation, we expressed Cx43 with glutamic acid substitutions at Y247 or Y265. The Cx43Y247E or Cx43Y265E channels were functional in Cx43 knockout fibroblasts, indicating that introducing a negative charge on Cx43 was not likely the mechanism for v-Src disruption of GJC. Cells coexpressing v-Src and the triple serine to alanine mutant, Cx43S255/279/282A, confirmed that mitogen-activated protein (MAP) kinase phosphorylation of Cx43 was not required for v-Src-induced disruption of GJC and that tyrosine phosphorylation was sufficient. In addition, v-Src cells containing v-Src-resistant gap junctions, Cx43Y247/265F, displayed properties of cell migration, adhesion, and proliferation similar to Cx43wt/v-Src cells, suggesting that Cx43 tyrosine phosphorylation and disruption of GJC are not involved in these transformed cell properties.
INTRODUCTION
The exchange of small molecular weight metabolites and ions present in the cytoplasm of adjacent cells through intercellular channels, a process called gap junctional communication (GJC), has been associated with cellular growth control for decades (Loewenstein and Kanno Citation1966; Yamasaki and Naus Citation1996). Importantly, the disruption of GJC has been linked to the elevated proliferation of tumor cells and oncogene transformed cells (Hotz-Wagenblatt and Shalloway Citation1993). Many studies that modulated the expression level and functionality of the connexin channel proteins have revealed an inverse correlation between cell proliferation rates and the degree of GJC (Yamasaki et al. Citation1999). The observations that connexin genes introduced into certain cancer cell lines resulted in suppressed cell proliferation, and that normal cells treated with tumor promoters, such as 12-O-tetradecanoylphorbol 13-acetate, displayed a loss of GJC, have prompted the idea that connexins have a tumor suppressor function (Lee et al. Citation1992; Mesnil Citation2002). In addition, the establishment of a link between the activation of growth factor receptors and the downregulation of the connexin channel function has strengthened the evidence that GJC is involved in the regulation of cell proliferation (Maldonado et al. Citation1988).
The transforming oncogene of the Rous sarcoma virus, v-src, encodes a potent oncoprotein with unregulated tyrosine kinase activity. This kinase activity affects cell proliferation in fibroblasts by altering cell cycle control through suppression of the cyclin-dependent kinase inhibitor p27, induction of cyclins D1, E, and A, and the stimulation of the activity of the cyclin-dependent kinases CDK4/6 and CDK2 (Frame Citation2002). These stimulatory cell cycle effects are dependent on the PI 3-kinase and MAP kinase kinase (MEK) pathways (Riley et al. Citation2001; Yeatman Citation2004). In addition to proliferation, Src plays a primary role in altering cell adhesion, cell motility, and cell invasion, which play an important role in cancer development and progression.
The connexin43 (Cx43) gap junction protein is a v-Src substrate (Crow et al. Citation1990; Crow et al. Citation1992; Filson et al. Citation1990; Swenson et al. Citation1990), and in cells expressing the v-src gene, Cx43-mediated GJC is greatly reduced by a mechanism involving v-Src phosphorylation of tyrosine residues in the Cx43 C-terminal tail (Crow et al. Citation1990; Filson et al. Citation1990; Kanemitsu et al. Citation1997; Lin et al. Citation2001a, Citation2001b; Loo et al. Citation1995; Swenson et al. Citation1990). A model proposed for this mechanism suggests an initial interaction between the SH3 domain of v-Src and a proline-rich region of Cx43, which facilitates the phosphorylation of Cx43 on Y265 (Kanemitsu et al. Citation1997; Lin et al. Citation2001b). This phosphorylation at Y265 may provide a binding site for the SH2 domain of v-Src, and thus allow for the processive phosphorylation of Y247 (Lin et al. Citation2001b).
Our studies examining the mechanism by which v-Src disrupted Cx43 GJC involved the use of specific Cx43 mutants, the Y247F and Y265F single site mutant proteins, and a Y247/265F double tyrosine mutant (Cottrell et al. Citation2003; Lin et al. Citation2001b). In contrast to Cx43wt, these Cx43 mutant proteins created gap junctions that retained functionality in the presence of the v-Src kinase, indicating that tyrosine phosphorylation of Y247 and Y265 was necessary, and perhaps sufficient, to downregulate Cx43-mediated GJC (Cottrell et al. Citation2003; Lin et al. Citation2001b). Electrophysiological analysis of Cx43wt and the Cx43Y247/265F double tyrosine mutant coexpressed with v-Src indicated that the unitary conductance of the channels was not affected by tyrosine phosphorylation, but that the open probability, Po, of the channel was reduced and that channel selectivity may have been altered (Cottrell et al. Citation2003). In contrast, Zhou et al. (Citation1999) did not find a role for tyrosine phosphorylation in acute channel gating in Xenopus oocytes. How the tyrosine phosphorylation of the Y247 and Y265 sites affects the Po and selectivity of the Cx43 channel in our mouse fibroblast cell system has not been determined but may involve a conformational change in Cx43, perhaps caused by the negative charge of the tyrosine phosphorylation and/or an alteration in Cx43 interacting proteins.
Here we report on cell lines expressing Cx43 mutants harboring a glutamic acid substitution for the Y247 or Y265 residue. These phosphorylation-mimetic mutations permit the exploration of the possibility that a negative charge, similar to that introduced by tyrosine phosphorylation of Cx43 at the Y247 and Y265 sites, might be sufficient to alter channel function. In addition, because discrepancies have been reported on the involvement of MAP kinase phosphorylation of Cx43 serine residues in the v-Src-induced regulation of GJC (Lin et al. Citation2001b; Toyofuku et al. Citation2001; Zhou et al. Citation1999), we have generated cell lines expressing v-Src and Cx43 specifically engineered with triple alanine substitutions at the MAP kinase sites (S255/279/282A) to help clarify the role of MAP kinase serine phosphorylation in the v-Src-induced downregulation of Cx43 function.
The plasma membrane localization of Cx43 and the identification of several interacting or colocalizing partners in addition to Src, including ZO-1 and tubulin, have generated the notion of a role for Cx43 in the formation of protein complexes that may be important for linking scaffolding proteins and signaling enzymes to substrates and effectors linked to the cytoskeleton (Duffy et al. Citation2002; Herve et al. 2004). This possible link of Cx43 to the cytoskeleton has prompted our hypothesis that the tyrosine phosphorylation of Cx43 may play a role in v-Src's effects on cell adhesion and motility. The generation of novel cell clones and pools of selected cells that express v-Src and the double Y247/265F Cx43 tyrosine mutant has made it possible to evaluate the importance of the v-Src-induced disruption of GJC and tyrosine phosphorylation of Cx43 on cell adhesion, migration, and proliferation.
MATERIALS AND METHODS
Cell Culture and Generation of Cells Expressing Cx43 Y247E and Cx43 Y265E Mutants
Cx43 knockout (KO) cell clones (Martyn et al. Citation1997) stably expressing wild type rat Cx43wt (Beyer et al. Citation1987; Warn-Cramer et al. Citation1998; Lin et al. Citation2001b) or rat Cx43 with Phe mutations at the Y247 and Y265 sites (double tyrosine mutant, Cx43Y247/265F) were prepared by retroviral infection with pBabe-Cx43 and puromycin selection as described previously (Lin et al. Citation2001b; Martyn et al. Citation1997; Warn-Cramer et al. Citation1998). Communication-competent individual cell clones then were infected with pLxSH (vector alone) or with a v-src retrovirus pLvsrcSH and selected with hygromycin as described (Lin et al. Citation2001b). The isolated hygromycin-resistant cell clones and stable hygromycin-selected cell pools were used in these studies. The cells were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% FCS (fetal calf serum, BioWhittacker) under antibiotic selection (6–10 μg/ml puromycin and 200 U/ml hygromycin, Calbiochem). Rat-1 and Rat-1 v-Src cells used as controls were maintained in DMEM/10% FCS. All cells were grown at 37°C in 5% CO2 in a humidified incubator.
The rat cx43 gene (Beyer et al. Citation1987) encoding glutamic acid substitutions at the Y247 or Y265 site was prepared from a full-length cx43 gene using PCR site-directed mutagenesis, essentially as described for the generation of cx43Y247/265F (Lin et al. Citation2001b), using the following primers: Y247E, 5′AGAAGCGATCCTGAGCACGCCACCACT, 3′ AGTGGTGGCGTGCTCAGGATCGCTTCT; Y265E, 5′GGATCTCCAAAAGAGGCCTACTTC- AATGGC, 3′GCCATTGAAGTAGGCCTCTTTTG- GAGATCC.
DNA sequencing confirmed the fidelity of the cx43 PCR products and the introduction of the desired mutations. Cx43 KO fibroblasts were infected with the pBabe retrovirus containing either the cx43Y247E or cx43Y265E gene utilizing the PE501 and PA317 packaging cell lines as previously described for the generation of cell lines expressing the cx43Y247/265F gene (Lin et al. Citation2001b). See for a schematic representation of the various Cx43 mutants used in this study.
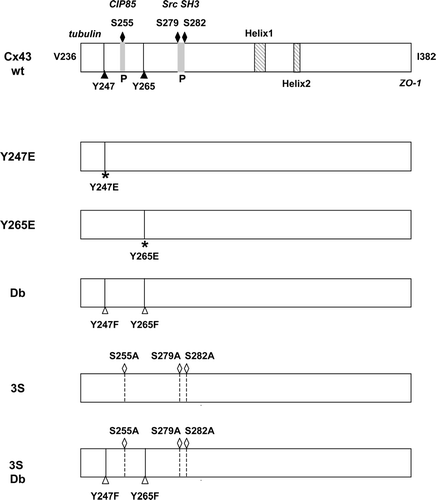
Figure 1 Schematic representation of the C-terminal domain of Cx43 showing the position of residues that were altered in these studies. Cx43wt is shown in the top panel with the Src tyrosine sites (Y247 and Y265) and the MAP kinase sites (S255, S279, and S282) indicated. The two helical regions in the C-terminal tail are also shown (Duffy et al. Citation2002), as well as the proline-rich (P) regions that overlap the MAP kinase sites and that provide interaction sites for the CIP85 protein (Lan et al. Citation2005) and the SH3 domain of Src (Kanemitsu et al. Citation1997). The C-terminal Ile382 residue involved in the interaction with ZO-1 and the juxtamembrane region near V236 that contains an interaction site for tubulin (Giepmans Citation2004) are also indicated. The phosphomimetic tyrosine (Y) to glutamic acid (E) mutations at Y247 or Y265 are shown in the second and third panels; the double (Db) tyrosine Src site mutant with phenylalanine (F) substitutions at Y247 and Y265 in the fourth panel; the triple serine to alanine (A) MAP kinase site mutant in the fifth panel (3S), and the MAP kinase/Src site mutant (3S Db) is shown in the final panel.
Cell Proliferation Assay
To examine the growth rates for the individual cell clones, cells were plated at a density of 1 × 104 cells/60 mm plate. Cells were fed every second day and counted on days 2, 4, 6, and 8 following plating. The data represent the average values obtained from single plates/cell type in three separate experiments (n = 3) for two different cell clones. To measure the growth rates of the antibiotic-selected cell pools, cells were plated at 2.4 × 104 cells/60 mm dish and were counted each day for a week. The cells were fed every second day. The data represent the average values from duplicate plates for each pool in three different experiments (n = 6).
Assays for Cell Migration, Adhesion, and Gap Junction Communication
To assess cell migration, cells were plated at a density of 1.6 × 105 on 35 mm dishes and grown overnight. The next day the cell monolayers were wounded by scraping with a P-200 plastic pipette tip. Then the cells were rinsed and fed with DMEM media lacking FCS, to limit cell proliferation. After 24 hours the cells were fixed in −20°C methanol and stained with 0.1% crystal violet. Cell monolayers were examined under the microscope and the number of cells that had moved into the wounded area was counted and used as a measure of the migratory ability of the cells. Two investigators counted two independent experiments with duplicate plates.
Calcium-dependent cell adhesion to the substratum was measured as described previously (Martyn et al. Citation1997). Equal numbers of cells were plated in a 96 well plate in six wells for each cell type. Cells were grown overnight in regular media and then treated with PBS buffer (Control, 3 wells) or with 5 mM EDTA/PBS (3 wells) for 5 min. The PBS or PBS/EDTA was then removed and the cells that remained attached to the plate were fixed with 10% TCA and stained with 0.4% SRB (sulforhodamine B) in 1% acetic acid. Protein-bound dye was extracted with 10 mM Tris base (unbuffered) and the dye concentration read at 570 nm in a Perkin Elmer HTS 7000 plate reader. The dye concentration in each well was taken to be proportional to the number of cells that remained adherent to the plate and was reported as a percentage of the control (PBS buffer treatment) for the same cell type. The data represent the average values obtained in triplicate wells in five independent experiments for the clones and six independent experiments for the pools.
GJC was measured by the microinjection of Lucifer yellow (LY) dye (10% w/v) into newly confluent cells, or cell patches, in 35 mm dishes. Single cells were injected with Eppendorf micropipettes using an Eppendorf Transjector 5246 and Eppendorf micromanipulator. Fluorescent images were visualized with a Zeiss phase-contrast inverted microscope equipped with eipfluorescence. The number of neighboring cells that became fluorescent due to dye transfer through gap junctions was counted at approximately one min following the injection of dye into a single cell.
Immunoblotting and Immunoprecipitation
Individual cell clones and selected cell pools were characterized for the levels of expression of Cx43 and Src and the levels of tyrosine phosphorylation on cellular proteins by SDS-PAGE analysis on 11% polyacrylamide gels, followed by immunoblotting. Cells were harvested and lysed in 20 mM Tris, pH 8.0, 150 mM NaCl, 1% NP-40 and 1 mM DTT with added protease and phosphatase inhibitors (1 mM PMSF, 10 mM NaF, and 160 μM Na3VO4), and equal amounts of clarified cell lysates (BioRad protein assay) were loaded onto the gels. Cx43 was detected with a rabbit polyclonal peptide antibody to the C-terminal 15 amino acids of Cx43 (aa 368-382); Src was detected with a monoclonal antibody that recognizes both c-Src and v-Src (antibody 2-17, generously provided by Dr. Sarah Parsons, Univ. of Virginia); and phosphotyrosine on cellular proteins was detected with a monoclonal antibody to phosphotyrosine (antibody PY99, Santa Cruz Biotechnology, Inc.). The immunoblots were developed with secondary antibodies and the enhanced chemiluminescence (ECL) immunoblotting kit (Amersham Pharmacia Biotech).
For metabolic labeling and Cx43 immunoprecipitation, newly confluent cells were labeled for 2 1/2 hr with [35S]-methionine (Amersham Pharmacia Biotech), 100 μCi/ml in methionine-free media supplemented with 4% dialyzed calf serum. Cells were treated with or without recombinant human epidemal growth factor (EGF; 100 ng/ml, Upstate Biotechnology) for the last 30 min of the labeling period. The cells were rinsed twice with cold PBS supplemented with protease and phosphatase inhibitors as described above and frozen at −20°C. Frozen cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 1% sodium deoxycholate, 1% Triton X-100, 0.1% SDS, 10 mM Tris, pH 7.2) containing protease and phosphatase inhibitors. Cx43 was immunoprecipitated from the clarified cell lysates using rabbit antiserum to the C-terminal peptide of Cx43 (aa 368-382) or normal rabbit serum and activated protein A-Staphylococcus aureus. The immune precipitates were washed 4 times with immunoprecipitation buffer and subjected to SDS-PAGE on 7.5%–15% acrylamide gradient gels. The resolved radiolabeled proteins were visualized by autoradiography of the dried fluorographed gels using Kodak X-OMAT film exposed at −70°C.
RESULTS
Characterization of Cx43Y247E and Cx43Y265E Expressing Cell Clones
To extend our understanding of the mechanism by which the tyrosine phosphorylation induced by v-Src inhibits Cx43-mediated GJC, the hypothesis that Cx43 channel function can be regulated by inducing a negative charge at either of two C-terminal tyrosine residues, Y247 and Y265, was tested (see schematic representation of Cx43 mutants in ). Cx43 mutant proteins with glutamic acid substitutions at one of these sites (Cx43Y247E or Cx43Y265E) were expressed in the Cx43 KO fibroblast cell line (Martyn et al. Citation1997). Immunofluorescence microscopy analysis revealed that Cx43 subcellular localization was not affected by substituting a glutamic acid for Y247 or Y265 (). Cells expressing the mutant Cx43Y247E or Cx43Y265E exhibited punctate gap junction plaques at the plasma membrane between cells, as well as within the cytoplasm, similar to the Cx43wt clone.
Figure 2 Characterization of Cx43Y247E and Cx43Y265E mutant proteins in Cx43 KO fibroblasts. (A) Cx43wt and the Cx43Y247E and Cx43Y265E mutants were examined by immunofluorescence microscopy using a Cx43 monoclonal antibody and Alexa-488. Parental Cx43 KO cells were used as a negative control. The level of GJC for each cell type is indicated in parentheses. (B) Immunoprecipitation of Cx43wt, Cx43Y247E, Cx43Y265E, and the Cx43S255/279/282A mutants from [35S]-labeled Cx43 KO fibroblasts. Connexin was immunoprecipitated with the Cx43 C-terminal peptide rabbit antiserum from cells treated (+) or untreated (−) with EGF (100 ng/ml for 30 min) and subjected to SDS-PAGE. The positions of the phosphorylated Cx43 isoforms (Cx43-P) are indicated at the left margin and the nonphosphorylated (NP) and P2 isoforms are marked at the right margin. An increase in phosphorylated Cx43 isoforms is visible for the EGF-treated cells compared with the control for each cell type (more Cx43 migrating in the P2 isoform and between the NP and P2 isoforms), except for the MAP kinase mutant (Cx43S255/279/282A) in the left-hand panel, which does not show this increase.
![Figure 2 Characterization of Cx43Y247E and Cx43Y265E mutant proteins in Cx43 KO fibroblasts. (A) Cx43wt and the Cx43Y247E and Cx43Y265E mutants were examined by immunofluorescence microscopy using a Cx43 monoclonal antibody and Alexa-488. Parental Cx43 KO cells were used as a negative control. The level of GJC for each cell type is indicated in parentheses. (B) Immunoprecipitation of Cx43wt, Cx43Y247E, Cx43Y265E, and the Cx43S255/279/282A mutants from [35S]-labeled Cx43 KO fibroblasts. Connexin was immunoprecipitated with the Cx43 C-terminal peptide rabbit antiserum from cells treated (+) or untreated (−) with EGF (100 ng/ml for 30 min) and subjected to SDS-PAGE. The positions of the phosphorylated Cx43 isoforms (Cx43-P) are indicated at the left margin and the nonphosphorylated (NP) and P2 isoforms are marked at the right margin. An increase in phosphorylated Cx43 isoforms is visible for the EGF-treated cells compared with the control for each cell type (more Cx43 migrating in the P2 isoform and between the NP and P2 isoforms), except for the MAP kinase mutant (Cx43S255/279/282A) in the left-hand panel, which does not show this increase.](/cms/asset/673b6a14-3366-4db2-aa96-7152f27ad387/icac_a_184784_uf0002_b.jpg)
To investigate the possibility that the introduction of glutamic acid at Y247 or Y265 in the Cx43 tail might alter other mechanisms of Cx43 regulation, which might interfere with the v-Src mechanism of downregulation of Cx43 GJC, Cx43wt and mutant proteins were immunoprecipitated from [35S]-labeled cells treated with or without EGF, and analyzed by SDS-PAGE and autoradiography. The results () revealed the expected increase in the levels of the phosphorylated Cx43 isoforms (Cx43-P) in the Cx43wt cells treated with EGF, particularly the P2 isoform and the intermediate P1 isoform that migrates between the nonphosphorylated (NP) and the P2 isoform, as compared with unstimulated control. In addition, both the Cx43Y247E and Cx43Y265E mutant proteins showed an increase in the phosphorylated isoforms, indicating that activated MAP kinases recognized these Cx43 mutants. The Cx43 triple serine mutant, Cx43S255/279/282A, lacking the MAP kinase serine phosphorylation sites, was used as a negative control and it did not display an increase in Cx43 phosphoisoforms. Therefore, the introduction of a negatively charged glutamic acid did not appear to affect the ability of the mutant Cx43 to be phosphorylated in response to EGF-induced signaling. The combined data suggested that the glutamic acid substitutions did not significantly alter protein structure in a way that interfered with Cx43 processing or growth factor regulation, and indicated that the mutant Cx43 could be used to test the effects of the negative charge on GJC.
To determine whether the introduction of the negative charge by the glutamic acid phosphotyrosine-mimetic at either the Y247 or Y265 site would affect GJC, we microinjected LY dye into the Cx43wt- and Cx43 mutant-expressing cells and counted the number of neighboring cells receiving dye. Both the Cx43Y247E and the Cx43Y265E cell clones were capable of transferring LY to neighboring cells to a degree similar to Cx43wt channels (, data in parentheses, and ). As demonstrated previously (Lin et al. Citation2001b), Cx43wt coexpressed with v-Src showed a marked decrease in GJC to around 1 to 1.3 cells (, clones Wt Src1 and Wt Src2). Although the levels of GJC were somewhat lower in the two E mutant clones, the levels were within the range of GJC that was observed with different Cx43wt clones, and were significantly higher than the GJC observed in the Cx43wt/v-Src expressing cells. Therefore, these results suggested that the introduction of a negative charge by the phosphorylation of Y247 or Y265 was likely not sufficient to downregulate Cx43 channel function and therefore not likely the v-Src mechanism for the closure of Cx43 channels.
TABLE 1 GJC of Cx43 KO cell clones expressing cx43wt or cx43 mutant genes with or without the v-src gene
Gap Junction Communication in Cells Expressing Cx43S255/279/282A and v-Src
In a previous study we showed that downregulating the activation of MAP kinase in v-Src expressing cells with the MEK inhibitor, PD98059, did not enhance the level of Cx43-mediated GJC, which suggested that MAP kinase-mediated serine phosphorylation was not required for the v-Src downregulation of Cx43 GJC (Lin et al. Citation2001b). Zhou et al. (Citation1999) however, using a different cell system, found that MAP kinase was involved in v-Src-induced downregulation of Cx43 GJC. In our study, the involvement of MAP kinase phosphorylation on S255, S279, and S282 could not be ruled out unequivocally since it was possible that not all the Cx43 phosphorylated by MAP kinase was degraded and removed from the cells during the MEK inhibitor treatment. Therefore, we extended the analysis of the role of MAP kinase by expressing v-Src in cells communicating through channels containing Cx43 with triple serine to alanine substitutions at the MAP kinase sites, Cx43S255/279/282A. The rationale for using cells expressing the Cx43S255/279/282A mutant was that they did not show enhanced Cx43 phosphorylation in the presence of EGF (), and thus activation of MAP kinase by v-Src in these cells could not induce additional Cx43 phosphorylation at these serine sites. Two v-Src-expressing clones, 3S S-1 and 3S S-2, were selected for further study, along with clones expressing the triple serine mutant (S255/S279/S282A) and the double tyrosine mutant (Y247/265F) with v-Src, 3S2Y S-1, and 3S2Y S-2. Immunoblotting of equal amounts of protein from whole cell lysates from these clones was used to evaluate the levels of the mutant Cx43, v-Src, and tyrosine phosphorylated proteins in the Src-transformed cells (). Comparison of the two clones expressing the triple serine substitutions and v-Src (3S S-1 and 3S S-2) showed similar levels of the mutant Cx43, v-Src, and tyrosine-phosphorylated proteins. Of the two clones expressing the triple serine substitutions and the double tyrosine substitution (3S2Y S-1 and 3S2Y S-2), one clone had a higher level of Cx43 and v-Src and an increased level of tyrosine phosphorylated proteins than the other. Immunoprecipitation of Cx43 and blotting back with an antibody to phosphotyrosine showed tyrosine phosphorylation on Cx43wt and the 3S Cx43 mutant with intact v-Src tyrosine sites and no detectable tyrosine phosphorylation on the Db and the 3S2Y Cx43 mutants that lacked the sites targeted by v-Src (data not shown).
Figure 3 Expression levels of Cx43, Src, and tyrosine phosphorylated proteins in Cx43 KO cell clones and cell pools. Immunoblotting analysis (IB) of equal amounts of protein from whole cell lysates was used to measure the relative levels of the Cx43, Src, or tyrosine phosphorylated proteins. (A) Cx43 KO cell clones expressing Cx43 mutants and v-Src (3S S-1 and 3S S-2 express Cx43S255/279/282A and v-Src, and 3S2Y S-1 and 3S2Y S-2 express Cx43S255/279/282A, Y247/265F and v-Src). Levels of GJC for the clones are indicated below the lanes. (B) cell clones, Wt Src1, and Wt Src2 (Cx43wt and v-Src), Db Src2, Db Src4 (Cx43Y247/265F and v-Src), and control clones, Wt 1 and Wt 2 (Cx43wt without v-Src). Cell pools, Wt, Wt Src, and Db Src. Rat-1 and Rat-1 v-Src fibroblasts were used as controls.
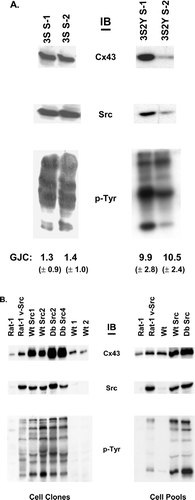
Lucifer Yellow dye transfer studies were performed to determine whether the lack of the MAP kinase serine phosphorylation sites would protect the cells from a loss of GJC caused by the effects of v-Src. The levels of GJC in the parental clones, 3S (Cx43S255/279/282A) and 3S 2Y (Cx43S255/279/282A, Y247/265F), were 12.9 and 9.1 cells, respectively (). As shown at the bottom of and in , changing the serine residues to alanine to prevent Cx43 phosphorylation at these sites did not prevent the v-Src-induced disruption of GJC (compare Cx43wt in clones Wt Src1 and Wt Src2 [1.3 and 1.0 communicating cells] to 3S S-1 and 3S S-2 [0.9 and 0.8 communicating cells]). Consistent with our previous results using the MEK inhibitor on the double tyrosine substitution mutants (Lin et al. Citation2001b), the GJC in the 3S2Y Src cell clones was protected from the effects of v-Src (see bottom of and ; 3S2Y S-1 and 3S2Y S-2, 9.9 and 10.5 communicating cells, respectively). These results confirmed that in the Cx43 KO fibroblast cell system, v-Src mediates the disruption of Cx43 GJC through phosphorylation of Y247 and Y265, and that additional MAP kinase-mediated serine phosphorylation on Cx43 is not required.
Characterization of Cx43, v-Src, and Tyrosine Phosphorylated Proteins in Cell Clones and Pools
Tyrosine phosphorylation creates SH2 and PTB binding motifs that are involved in the cell signaling that mediates various cell functions, including adhesion, migration, and proliferation. The tyrosine kinase activity of v-Src creates these binding motifs and transforms these cell properties (Yeatman Citation2004). Furthermore, Cx43 GJC can suppress cell proliferation. The communication-competent Cx43Y247/265F/v-Src cell lines and the noncommunicating Cx43wt/v-Src cells (Wt Src1 and Wt Src2; Lin et al. Citation2001b) allowed an investigation of the requirement for Cx43 tyrosine phosphorylation and the loss of GJC in v-Src-induced cell transformation. To accommodate possible bias introduced by using clonal v-Src-infected Cx43wt and Cx43Y247/265F cells, which may have unintentionally been selected because they grew more rapidly and displayed rounded morphologies indicative of v-Src expression, we also prepared antibiotic-selected cell pools of the control (Wt), Cx43wt and v-Src (Wt Src), and the Cx43Y247/265F double tyrosine mutant and v-Src (Db Src; Warn-Cramer et al. Citation2003). Two Cx43wt Src clones (Wt Src1 and Wt Src2), two Cx43Y247/265F Src clones (Db Src2 and Db Src4), and two Cx43 vector-only clones (Wt 1 and Wt 2) were used, along with the antibiotic-selected pools, in these studies. Immunoblotting of equal amounts of protein from whole cell lysates showed that the Cx43 levels were similar between the two control clones, but were significantly elevated in the four v-Src expressing clones, compared with the controls (, left-hand side), which suggested that v-Src protected Cx43 from degradation or enhanced its expression. Interestingly, the two Cx43Y247/265F/v-Src expressing clones (Db Src2 and Db Src4) had an additional increase in the connexin protein level over the Cx43wt/v-Src clones (Wt Src1 and Wt Src2). The Rat-1 and Rat-1 v-Src cells, used for comparison, showed a similar disparity in Cx43 levels. A similar expression pattern also was observed between the Wt, Wt Src, and Db Src cell pools, with the Db Src pool showing the highest levels of connexin protein (, right-hand side). In an earlier study, we demonstrated increased levels of Cx43 protein and mRNA in Rat-1 v-Src cells as compared with Rat-1 cells (approximately a two-fold increase in protein and mRNA [Goldberg and Lau, Citation1993]). In preliminary studies using the present cell system and a treatment with cycloheximide to block protein synthesis (30 μg/ml for 0, 3, and 6 hr) we found that the half-life for Cx43 was prolonged in the Wt Src cells (> 6 hr) as compared with the Wt cells (∼3 hr). This is in contrast to the previous study, where the half-lives for the Cx43 protein appeared to be similar in the Rat-1 and Rat-1 v-Src cells (∼2.5 hr). Taken together, these combined studies suggest that v-Src can increase the expression level of Cx43 and this may be regulated by different mechanisms in different cell types.
Immunoblot analysis of the v-Src protein levels showed equivalent levels of v-Src among all the v-Src expressing cells (). The levels and patterns of tyrosine phosphorylated proteins also were similar between the Cx43wt/v-Src (Wt Src1 and Wt Src2) and the Y247/265F/v-Src expressing cells (Db Src2 and Db Src4). The individual cell clones and cell pools not expressing v-Src showed very low levels of tyrosine phosphorylated proteins (). Furthermore, the parental cell clones expressing the Cx43Y247/265F tyrosine mutant showed very low levels of tyrosine phosphorylation on Cx43 (Lin et al. Citation2001b). These combined data indicated that these cell clones and cell pools were suitable for studies examining the role of Cx43 in v-Src-induced cell transformation.
GJC and Cx43 Tyrosine Phosphorylation Do Not Alter v-Src Effects on Cell-Substratum Adhesion and Cell Migration
The fact that v-Src reduces cell adhesion to the substratum through tyrosine phosphorylation of cytoskeletal proteins (Frame et al. Citation2002) and that Cx43 interacts with microtubules (Giepmans et al. Citation2001), which play a role in cell motility, prompted an analysis of the influence of Cx43 tyrosine phosphorylation and the downregulation of GJC on the v-Src-induced alteration of cell-substratum adhesion and cell migration. Ca2+-dependent cell adhesion to the substratum was assessed for cells plated on plastic dishes and allowed to attach and express matrix proteins overnight. We then determined the number of cells remaining attached to the tissue culture dish after a 5 min treatment with 5 mM EDTA/PBS by a protein-bound dye method (see Methods and [Martyn et al. Citation1997]). Compared with the cells treated with PBS alone, approximately 83% of the control Cx43 cells (Wt 1 and Wt 2) remained attached to the dish after treatment with EDTA (). In the clones expressing Cx43wt and v-Src (Wt Src1 and Wt Src2) the extent of cell attachment was reduced to approximately 75% of the untreated cells. Statistical analysis using Student's t test indicated that the difference between the Wt clones and the Wt Src clones was significant (p = 0.02). In the clones expressing the double tyrosine mutant Cx43Y247/265F (Db Src2 and Db Src4) there was an additional decrease in the number of cells remaining attached (∼64%). The difference in adhesion between the Wt Src and the Db Src also was statistically significant (p = 0.006). Similar patterns of attachment were observed between the Cx43wt and Cx43Y247/265F Src pools (Wt Src Pool, Db Src Pool, 73% and 67% of control, respectively, ), which were less than the control pool (Wt Pool, ∼90% of control). In the pools, the difference between the Wt and Wt Src was significant (p = 0.002), however, the difference between the Wt Src and Db Src was not statistically different (p = 0.36). These results showed that expression of v-Src in the KO fibroblast cell system caused the expected reduction in the adhesion of the cells to the substratum, but this effect was not blocked by removing the v-Src tyrosine phosphorylation sites on Cx43 and maintaining GJC. This data suggested that tyrosine phosphorylation of Cx43 and the loss of GJC induced by v-Src do not play a major role in the v-Src-induced reduction of cell-substratum adhesion.
Figure 4 Cell adhesion and migration for cell clones and cell pools of v-Src expressing cells were not affected by maintenance of GJC and lack of Cx43 tyrosine phosphorylation. Cell adhesion analysis: (A) cell clones and (B) cell pools. Equal numbers of cells were plated in 96 well culture dishes, grown overnight, and then treated for 5 min with 5 mM EDTA/PBS. Cells were washed and the remaining attached cells were trichloroacetic acid (TCA) fixed and then stained with SRB (see Materials and Methods). Protein determination at 570 nm was used as a measure of cell concentration and reported as a percent of the control (treated with PBS alone). An asterisk indicates values that are statistically different from the controls (Wt 1 and Wt 2) or groups statistically different from each other (the Wt Src clones versus the Db Src clones). Cell migration analysis: (C), cell clones and (D) cell pools. Cells were grown overnight and the monolayer was wounded by scraping with a plastic pipette tip. The cells then were rinsed and fed with medium lacking fetal calf serum (FCS) to reduce cell proliferation. After 24 hr, the cells were fixed in −20°C methanol and stained with 0.1% crystal violet. Wounded areas were microscopically examined, and cell migration was measured by counting the number of cells migrating into the wounded area within a microscopic field. Error bars represent the standard error of the mean (SEM).
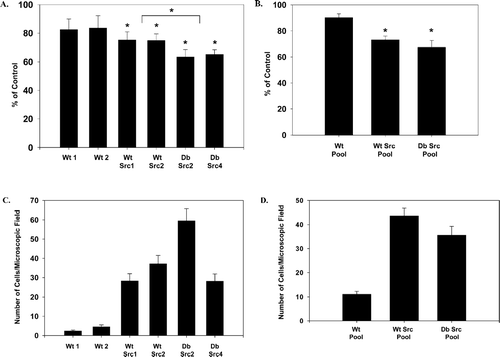
To evaluate the migration properties of v-Src expressing cells with and without Cx43-mediated GJC, a monolayer-wounding assay was utilized. Few of the control cells that lacked v-Src migrated into the wound area, as seen in , where ∼2–5 cells migrated/microscopic field for the Wt 1 and Wt 2 clones at 24 hr after wounding. When v-Src was expressed in these cells, however, an expected increase in cell migration was observed (to ∼28–37 cells migrated/microscopic field for the Wt Src1 and Wt Src2 clones). The two Cx43Y247/265F mutant clones (Db Src2 and Db Src4) also showed increased migration, but to different levels. The Db Src2 clone had 60 cells and the Db Src4 had on average 28 cells that migrated into the wound area per microscopic field. Because the lack of serum in the cell culture medium limited cell proliferation, the differences between these two Db Src clones is likely due to clonal differences. In addition, the increased number of cells in the wound area is due to increased migration rather than cell proliferation. The Cx43Y247/265F mutant Src pool (Db Src Pool, 36 cells) and the Cx43wt Src pool (Wt Src Pool, 44 cells) also migrated to a significantly greater extent than the Cx43wt vector pool (Wt Pool, 11 cells; see ). Although the two Y247/265F Src clones migrated on average to a higher level than the Cx43wt Src clones, this pattern was not observed in the cell pools, where the Cx43wt Src pool migrated to a greater extent than the Cx43Y247/265F Src pool. Taken together, these results suggested that the loss of GJC and the tyrosine phosphorylation of Cx43 are not involved in the v-Src-induced cell migration.
GJC and the Lack of Cx43 Tyrosine Phosphorylation Do Not Affect Rates of Proliferation in v-Src Transformed Cells
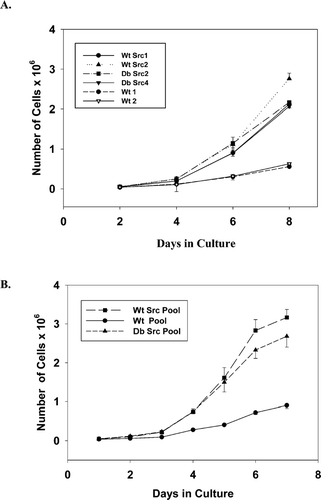
Reestablishment of Cx43-mediated GJC has been shown to reduce cell proliferation rates in our KO cell lines (Martyn et al. Citation1997). In this study, we asked whether maintaining GJC in the v-Src expressing KO cells would affect cell proliferation rates. In 10% serum, the cells in the Cx43wt vector clones and cell pools ( and , Wt 1, Wt 2, and Wt Pool) grew at significantly slower rates than the v-Src expressing cells. Over 8 days of growth, the v-Src expressing cell clones grew to an average density of 2.25 × 106 cells/60 mm plate while the control clones grew to a density of approximately 0.59 × 106 cells/60 mm plate. Analysis of the slopes of the growth curves during the exponential growth phase from day 2 to 6 suggested no significant differences in the rates of growth between the different v-Src expressing clones (Cx43wt versus the Cx43Y247/265F; see ). Similar rates of exponential growth were observed in the studies of the pools of cells for the Cx43wt and Cx43Y247/265F v-Src expressing cells, except that the pools grew very slowly at the plating density of 1 × 104, so for these experiments the cells were plated at 2.4 × 104/60 mm plate (). Thus, no significant differences were observed in the rates of cell proliferation when GJC was maintained and Cx43 was not tyrosine phosphorylated in cells expressing the Cx43Y265/247F mutant.
Figure 5 Cell proliferation rates for cell clones and cell pools expressing v-Src were not affected by maintenance of GJC and lack of Cx43 tyrosine phosphorylation. (A) growth curves for cell clones. Cells were plated at a density of 1 × 104 cells/60 mm plate, fed every second day, and then counted on days 2, 4, 6, and 8 following plating. Slopes of the exponential phase of the curves from day 4 to 8: Wt Src1, S = 0.481; Wt Src2, S = 0.626; Db Src2, S = 0.479; Db Src4, S = 0.467; Wt 1, S = 0.109; and Wt 2, S = 0.130. The data represent the average values obtained from single plates/cell type in three separate experiments (n = 3). (B) growth curves for cell pools. To measure the growth rates of the antibiotic-selected cell pools, cells were plated at 2.4 × 104 cells/60 mm dish and were counted each day for a week, with feeding on every second day. Slopes of the curves from day 2 to 6: Wt Src Pool, S = 0.685; Db Src Pool, S = 0.569; and Wt Pool, S = 0.164. The data represent the average values from duplicate plates for each cell pool in three different experiments (n = 6). Error bars represent the SEM.
DISCUSSION
Regulation of Cx43 function by phosphorylation is well documented and is a primary means of modulating GJC (Lampe and Lau Citation2004; Warn-Cramer and Lau Citation2004; Zhou et al. Citation1999). The mechanism for v-Src-induced phosphorylation of Cx43 on tyrosine residues in the C-terminal tail has been characterized by a number of investigators (Crow et al. Citation1990; Lin et al. Citation2001a, Citation2001b; Swenson et al. Citation1990; Toyofuku et al. Citation2001; Zhou et al. Citation1999) and involves v-Src SH3 and SH2 domain interactions with the proline-rich domain and phosphotyrosines, respectively, of Cx43 (Kanemitsu et al. Citation1997). In our previous study utilizing single phenylalanine substitutions in Cx43 at tyrosines 247 and 265 (Cx43Y247F and Cx43Y265F), we discovered that both mutants formed channels that were resistant to downregulation by v-Src. Tyrosine phosphorylation on Cx43 was reduced to ∼2% compared with Cx43wt in the Cx43Y247/265F double tyrosine mutant, indicating that v-Src induced phosphorylation primarily at these two tyrosine sites. Although the Y247F single site mutant displayed elevated tyrosine phosphorylation (∼57% compared with Cx43wt), presumably occurring at the Y265 site, this was not sufficient to inhibit Cx43 GJC. This observation led us to speculate that v-Src induces the processive phosphorylation of Cx43, where the first phosphorylation could occur on Y265, which may provide an SH2-interacting site for v-Src and lead to the second phosphorylation on Y247. We hypothesized that phosphorylation of Y247 either altered the structure of Cx43, and/or modified its interaction with a partner(s), in a way that elicited channel closure. Because the Cx43Y265F mutant was poorly phosphorylated on Y247 (∼10% of Cx43wt tyrosine phosphorylation), compared with the Y265 phosphorylation levels observed in the Cx43Y247F mutant (∼57% of Cx43wt; Lin et al. Citation2001b), we were unable to assess whether elevated phosphorylation on Y247 alone was sufficient to modulate channel function.
The phosphorylation of Y247 and Y265 by v-Src is necessary to inhibit Cx43 GJC in mouse Cx43 KO fibroblasts. Cx43wt is depicted as an open channel in the absence of v-Src in the model shown in (part 1) and a closed channel in v-Src cells (part 2). The channel remained open in v-Src cells that expressed a Cx43 mutant with phenylalanine mutations (F) at the Y247 and Y265 sites of Cx43 (the 2Y mutant shown in part 3). How the dual tyrosine phosphorylations of Cx43 close the channel has not been elucidated. To investigate the possibility that a negative charge introduced at these sites by tyrosine phosphorylation is a component of the regulatory mechanism, we have generated two Cx43 mutants with glutamic acid substitutions at either the tyrosine 247 or tyrosine 265 sites. Glutamic acid substitutions were employed because they introduce a negative charge that might induce conformational changes that mimic the effects of phosphorylation on tyrosine. It is well documented in the family of receptor tyrosine kinases that a phosphorylation at tyrosine can cause conformational changes that unmask active sites or relax negative regulatory controls (Cheetham Citation2004). In addition, it has been shown that a glutamic acid substitution for tyrosine 605 in the EphB2 receptor substituted for the required tyrosine phosphorylation at this site and caused a conformational change that activated the receptor's kinase activity (Zisch et al. Citation2000). It is important to note, however, that glutamic acid residues do not replace phosphotyrosine in binding to SH2 domains (Zisch et al. Citation2000).
Figure 6 Schematic model for the regulation of Cx43 channel function by v-Src in Cx43 KO cells that reexpress wt or mutant Cx43. In this model, Cx43wt is depicted as an open channel (part 1). In v-Src transformed cells, Cx43wt is directly phosphorylated by v-Src on Y247 and Y265 (Lin et al. Citation2001b), and activated MAP kinases can phosphorylate Cx43 on serine (S255, S279, and S282 [Cameron et al. Citation2003; Warn-Cramer et al. Citation1996)]. These events result in channel closure (part 2). Tyrosine phosphorylation of Cx43 was required for channel closure in the v-Src cells, since the channels remained open in a Cx43 mutant with phenylalanine (F) mutations at the Y247 and Y265 sites (2Y mutant, part 3). Phosphorylation of Cx43 at the MAP kinase sites was not required for channel closure, since the mutation of the serine sites to alanine (A) did not prevent v-Src-induced channel closure (3S mutant, part 4). The presence of a negative charge at either the Y247 or the Y265 site of Cx43 (introduced by mutation of tyrosine to glutamic acid (E)) was not sufficient to disrupt channel function (part 5). This suggested that a binding interaction between a phosphotyrosine site of Cx43 and a protein containing an SH2 or PTB domain or an alteration in Cx43 conformation might be required to mediate channel closure in the v-Src cells.
![Figure 6 Schematic model for the regulation of Cx43 channel function by v-Src in Cx43 KO cells that reexpress wt or mutant Cx43. In this model, Cx43wt is depicted as an open channel (part 1). In v-Src transformed cells, Cx43wt is directly phosphorylated by v-Src on Y247 and Y265 (Lin et al. Citation2001b), and activated MAP kinases can phosphorylate Cx43 on serine (S255, S279, and S282 [Cameron et al. Citation2003; Warn-Cramer et al. Citation1996)]. These events result in channel closure (part 2). Tyrosine phosphorylation of Cx43 was required for channel closure in the v-Src cells, since the channels remained open in a Cx43 mutant with phenylalanine (F) mutations at the Y247 and Y265 sites (2Y mutant, part 3). Phosphorylation of Cx43 at the MAP kinase sites was not required for channel closure, since the mutation of the serine sites to alanine (A) did not prevent v-Src-induced channel closure (3S mutant, part 4). The presence of a negative charge at either the Y247 or the Y265 site of Cx43 (introduced by mutation of tyrosine to glutamic acid (E)) was not sufficient to disrupt channel function (part 5). This suggested that a binding interaction between a phosphotyrosine site of Cx43 and a protein containing an SH2 or PTB domain or an alteration in Cx43 conformation might be required to mediate channel closure in the v-Src cells.](/cms/asset/639fdc2d-ff07-4a27-8a52-5ab356576540/icac_a_184784_uf0006_b.jpg)
Consistent with our previous observation that the Cx43Y247F mutant was functional and carried a negative charge on the phosphorylated Y265 site, the Cx43Y265E mutant was also functional, suggesting that a negative charge at the Y265 site was insufficient to close Cx43 channels (depicted as an open channel in the model in [part 5]). Thus, the introduction of a negative charge by the phosphorylation of Y247 or Y265 was insufficient in itself to downregulate Cx43 channel function and therefore not likely to be a mechanism for the v-Src induced closure of Cx43 channels. Although the Y265F Cx43 mutant was inefficiently phosphorylated in the v-Src cells, the stoichiometric introduction of a negative charge at the Y247 site in the Y247E mutant was not sufficient to disrupt GJC. These results best support a regulatory mechanism whereby phosphorylation of Y265 and Y247 by v-Src either establishes or disrupts interactions between Cx43 and other proteins containing SH2 or PTB domains and/or alters Cx43's conformation through a mechanism independent of the introduction of a negative charge, which may interfere with the open state of the Cx43 channel.
Contradictory results have been reported for the role of serine phosphorylation in the v-Src-induced downregulation of Cx43 GJC. In the Xenopus oocyte system utilized by Zhou et al. (Citation1999) v-Src-induced MAP kinase-mediated phosphorylation of Cx43 and not tyrosine phosphorylation was reported as the primary means for interrupting Cx43 communication (1999). Both MAP kinase serine phosphorylation and tyrosine phosphorylation were responsible for the inhibition of GJC produced by the overexpression of activated c-Src in HEK293 cells (Toyofuku et al. Citation2001). Using the MEK inhibitor, PD98059, we previously reported no involvement of MAP kinase phosphorylation in the v-Src-induced downregulation of Cx43 GJC in Cx43 KO fibroblast cells (Lin et al. Citation2001b). Although we confirmed that the MEK inhibitor effectively inhibited MAP kinase activation, we did not demonstrate, that at the time GJC was measured, that all of the Cx43 had turned over and the Cx43 in the channels was not phosphorylated at the MAP kinase sites. Therefore, we could not exclude the possibility that higher levels of GJC were not observed in the v-Src-expressing cells treated with the MEK inhibitor because the channels may have contained Cx43 that was still phosphorylated at the MAP kinase sites. To address this possibility we infected KO cells expressing a Cx43 triple MAP kinase serine to alanine (Cx43S255/279/282A) mutant (Warn-Cramer et al. Citation1998) with v-Src, and asked whether GJC in these v-Src-expressing cells was protected from downregulation, compared with the Cx43wt cells expressing v-Src. The lack of the Cx43 MAP kinase phosphorylation sites did not produce higher levels of GJC in cells expressing v-Src, compared with the Cx43wt (a closed channel is depicted for the Cx43 3S mutant in the model in , part 4), indicating that MAP kinase phosphorylation at S255, S279, and S282 was not involved in the v-Src downregulation of Cx43 GJC in the KO fibroblasts. This is in contrast to the results of Zhou et al. (Citation1999) in the Xenopus system. A very recent report suggests that v-Src requires Ras signaling for the suppression of Cx43-mediated GJC (Ito et al. Citation2005). Although the Ras signaling pathway activates MAP kinase, our results with the triple Cx43 serine mutant suggest that it is not the Ras downstream activation of MAP kinase and subsequent serine phosphorylation of Cx43 that mediates the v-Src-induced downregulation of Cx43 GJC.
Possible explanations for the differences observed in the role of serine phosphorylation in v-Src-induced downregulation of Cx43 GJC include chronic (Lin et al. Citation2001b) versus acute (Zhou et al. Citation1999) v-Src expression, as well as differences in Cx43 processing and regulation inherent in the cell systems utilized in the studies. In normal rat kidney (NRK) cells, the MEK inhibitor PD98059 protected Cx43 from downregulation of GJC by a temperature sensitive v-src after the shift to the permissive temperature and the acute expression of v-Src (Zhou et al. Citation1999). In contrast, chronic exposure to the activated v-Src kinase in our Cx43 KO cells expressing the Cx43 triple serine substitution, or cells treated with the MEK inhibitor, did not block the disruption of GJC. Perhaps chronic exposure to v-Src permanently changes the cell's signaling repertoire such that the MAP kinase serine phosphorylations become ineffective or do not occur. This may be due to changes in Cx43 protein interactions, since MAP kinase-mediated phophorylation of Cx43 is sufficient to disrupt GJC in other systems using acute stimulation of channel gating (Cameron et al. Citation2003; Warn-Cramer et al. Citation1998).
Inherent differences in the way Cx43 is regulated in different cell systems is apparent in the Xenopus oocytes where the half-life of Cx43 is 22 hours, as compared with 3 hours in the Cx43 KO fibroblasts used in our study. Fundamental differences between these two cell systems in the way Cx43 is regulated or processed could indirectly alter the effects of MAP kinase in the v-Src-induced regulation of Cx43. Given the differences reported for the role of MAP kinase in the downregulation of Cx43 GJC by v-Src from several thorough investigations, it seems likely that multiple signaling pathways and effectors could be activated by v-Src, that modulate the expression and functionality of Cx43 and that the particular pathway and effectors utilized are dependent on the status of the signaling components in a given cell type.
v-Src induces neoplastic transformation by stimulating signaling pathways that transmit proliferation and survival signals and by altering cytoskeletal networks that affect cell adhesion, migration, and invasion. Because Cx43 can negatively regulate cellular growth, and may serve as a nexus for assembling signaling and scaffolding proteins at the plasma membrane, which may connect with the cytoskeleton (Duffy et al. Citation2002), we considered the possibility that the downregulation of GJC by tyrosine phosphorylation of Cx43 might contribute to the mechanism of v-Src-induced cell transformation. In addition, Cx43 has a number of interacting partners that are involved in cell adhesion and migration including tubulin, actin, N-cadherin, and ZO-1 (Herve et al. Citation2004), which suggested the possibility of a link between v-Src's effects on cell adhesion and migration (Frame et al. Citation2002; Giepmans et al. Citation2001) and Cx43. No differences were detected, however, in cell-substratum adhesion and cell motility in cells expressing Cx43 mutations, that were impervious to Src's action. Because v-Src is a highly potent tyrosine kinase that affects numerous other signaling pathways that regulate adhesion and motility through modulation of the actin cytoskeleton, including integrins, p125 FAK, p190Rho/GAP, Rac/Rho GTPases and MEK/ERK (Frame Citation2004; Frame et al. Citation2002), these results are not surprising. Whether or not these cells expressing v-Src-resistant Cx43 mutants would behave differently in in vivo tumorigenicity studies is an open question.
In an earlier study we examined the density of Cx43wt/v-Src cells and Cx43Y247/265F/v-Src cells after a week of growth and observed no differences associated with GJC (Warn-Cramer et al. Citation2003). Here, we extended our studies to examine cell proliferation rates in these clones and cell pools. Differences in the exponential phase of cell growth might be apparent if Cx43 tyrosine phosphorylation and loss of GJC mediated the effects of v-Src on the cell cycle. No reduction in proliferation rates was observed, however, in the v-Src cells that remained communication competent and expressed Cx43 mutants lacking tyrosine phosphorylation. The lack of correlation between Cx43 GJC and growth regulation also recently was reported (Chandrasekhar et al. Citation2004) in v-Src-transformed mouse embryonic Cx43 KO brain cells that reexpressed Cx43wt or Cx32wt. Interestingly, in this study several of the Cx43-expressing clones established junction coupling despite Cx43 tyrosine phosphorylation, suggesting that this modification may not be sufficient for Src to inhibit GJC in this cell system and that Src may modulate other cellular effectors to downregulate Cx43 GJC.
Although the correlation between Src kinase activity and cell proliferation is well established in fibroblasts, colon cancer epithelial cell lines with elevated c-Src activity showed highly invasive behavior, but reduced cell proliferation (Frame Citation2002). It was postulated that activated Src may promote cell proliferation in early stages of tumor progression, but during later stages it may facilitate other properties of neoplasia, but not cell growth. It is possible that a similar situation exists for v-Src in our transformed cell model, and that changes occurring early in the transforming process, immediately after v-Src expression, create a unique signaling environment that determines v-Src's downstream effects. Therefore, to best evaluate a role for GJC in Src-induced growth promotion in cultured cell lines, it might be necessary to evaluate the connection between Src and GJC over specific time points after v-Src expression. In our study, we began with immortalized, but nontransformed and fully GJC competent cells that expressed a mutant Cx43 resistant to v-Src phosphorylation, and asked whether the presence of GJC and the lack of Cx43 tyrosine phosphorylation would inhibit the ability of v-Src to alter cell proliferation in cloned cells and cell pools. We discovered that the presence of GJC from the onset of v-Src expression did not reduce proliferation rates in these clones and pools of cells at time points shortly after they were isolated and expanded for the proliferation studies. In the Chandrasekhar et al. study (Citation2004), the role of GJC was examined in an immortalized cell line that was already fully transformed by v-Src prior to the reexpression of the connexin genes and the establishment of GJC, and, similarly, no effect on growth was observed. Taken together the results of these two studies would suggest that at time points beyond the first few hours of v-Src expression, the phosphorylation of Cx43 on tyrosine and subsequent loss of GJC do not play a significant role in the v-Src-induced cell proliferation.
In summary, we conclude that v-Src does not require serine phosphorylation of Cx43 to induce the downregulation of Cx43 GJC in the KO fibroblast cell system, and it is unlikely that v-Src induces Cx43 channel closure simply by introducing a potential conformation-altering negative charge at the Y247 or Y265 sites (see the model in ). Furthermore, our results suggest that v-Src does not alter cell adhesion or motility, or upregulate cell proliferation through the phosphorylation of Cx43 Y247 and Y265 and the inhibition of GJC.
ACKNOWLEDGMENTS
Supported by grants from the NIH, RR16453 (R. Shohet, PI; BWC, project PI) and CA 052098 (AFL) and support from the HS-BRIN (RR16467 to BWC). The authors thank Dr. Sarah Parsons for providing the Src antibody and Wendy Kurata, Anne Hernandez, and Chris Wallick for technical assistance.
REFERENCES
- Beyer E C, Paul D, Goodenough D A. Connexin43: A protein from rat heart homologous to a gap junction protein from liver. J Cell Biol 1987; 105: 2621–2629, [INFOTRIEVE], [CSA]
- Cameron S J, Malik S, Akaike M, Lerner-Marmarosh N, Yan C, Lee J D, Abe J, Yang J. Regulation of epidermal growth factor-induced connexin 43 gap junction communication by big mitogen-activated protein kinase1/ERK5 but not ERK1/2 kinase activation. J Biol Chem 2003; 278: 18682–18688, [INFOTRIEVE], [CSA], [CROSSREF]
- Chandrasekhar A, Merritt M, Huh S J, Nicholson B J, Zucker S N. Connexin expression and cell coupling fail to reverse the v-src transformed growth characteristics of a Cx43-/- cell line. Cell Commun Adhes 2004; 11: 103–119, [INFOTRIEVE], [CSA], [CROSSREF]
- Cheetham G M. Novel protein kinases and molecular mechanisms of autoinhibition. Curr Opin Struct Biol 2004; 14: 700–705, [INFOTRIEVE], [CSA], [CROSSREF]
- Cottrell G T, Lin R, Warn-Cramer B J, Lau A F, Burt J M. Mechanism of v-Src- and mitogen-activated protein kinase-induced reduction of gap junction communication. Am J Physiol Cell Physiol 2003; 284: C511–520, [INFOTRIEVE], [CSA]
- Crow D S, Beyer E C, Paul D L, Kobe S S, Lau A F. Phosphorylation of connexin43 gap junction protein in uninfected and Rous sarcoma virus-transformed mammalian fibroblasts. Mol Cell Biol 1990; 10: 1754–1763, [INFOTRIEVE], [CSA]
- Crow D S, Kurata W E, Lau A F. Phosphorylation of connexin43 in cells containing mutant src oncogenes. Oncogene 1992; 7: 999–1003, [INFOTRIEVE], [CSA]
- Duffy H S, Delmar M, Spray D C. Formation of the gap junction nexus: Binding partners for connexins. J Physiol Paris 2002; 96: 243–249, [INFOTRIEVE], [CSA], [CROSSREF]
- Filson A J, Azarnia R, Beyer E C, Loewenstein W R, Brugge J S. Tyrosine phosphorylation of a gap junction protein correlates with inhibition of cell-to-cell communication. Cell Growth Differ 1990; 1: 661–668, [INFOTRIEVE], [CSA]
- Frame M C. Src in cancer: Deregulation and consequences for cell behaviour. Biochim Biophys Acta 2002; 1602: 114–130, [INFOTRIEVE], [CSA]
- Frame M C. Newest findings on the oldest oncogene; how activated src does it. J Cell Sci 2004; 117: 989–998, [INFOTRIEVE], [CSA], [CROSSREF]
- Frame M C, Fincham V J, Carragher N O, Wyke J A. v-Src's hold over actin and cell adhesions. Nat Rev Mol Cell Biol 2002; 3: 233–245, [INFOTRIEVE], [CSA], [CROSSREF]
- Giepmans B N. Gap junctions and connexin-interacting proteins. Cardiovasc Res 2004; 62: 233–245, [INFOTRIEVE], [CSA], [CROSSREF]
- Giepmans B N, Verlaan I, Hengeveld T, Janssen H, Calafat J, Falk M M, Moolenaar W H. Gap junction protein connexin-43 interacts directly with microtubules. Curr Biol 2001; 11: 1364–1368, [INFOTRIEVE], [CSA], [CROSSREF]
- Goldberg G S, Lau A F. Dynamics of connexin43 phosphorylation in pp60v-src Transformed cells. Biochem J 1993; 285: 735–742, [CSA]
- Herve J C, Bourmeyster N, Sarrouilhe D. Diversity in protein-protein interactions of connexins: Emerging roles. Biochim Biophys Acta 2004; 1662: 22–41, [INFOTRIEVE], [CSA], [CROSSREF]
- Hotz-Wagenblatt A, Shalloway D. Gap junctional communication and neoplastic transformation. Crit Rev Oncog 1993; 4: 541–558, [INFOTRIEVE], [CSA]
- Ito S, Ito Y, Senga T, Hattori S, Matsuo S, Hamaguchi M. v-Src requires Ras signaling for the suppression of gap junctional intercellular communication. Oncogene 2005, Epub November 21[CSA]
- Kanemitsu M Y, Loo L W, Simon S, Lau A F, Eckhart W. Tyrosine phosphorylation of connexin 43 by v-Src is mediated by SH2 and SH3 domain interactions. J Biol Chem 1997; 272: 22824–22831, [INFOTRIEVE], [CSA], [CROSSREF]
- Lampe P D, Lau A F. The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol 2004; 36: 1171–1186, [INFOTRIEVE], [CSA], [CROSSREF]
- Lan Z, Kurata W E, Martyn K D, Jin C, Lau A F. Novel rab GAP-like protein, CIP85, interacts with connexin43 and induces its degradation. Biochem 2005; 44: 2385–2396, [CSA], [CROSSREF]
- Lee S W, Tomasetto C, Paul D, Keyomarsi K, Sager R. Transcriptional downregulation of gap-junction proteins blocks junctional communication in human mammary tumor cell lines. J Cell Biol 1992; 118: 1213–1221, [INFOTRIEVE], [CSA], [CROSSREF]
- Lin R, Warn-Cramer B J, Kurata W E, Lau A F. v-Src-mediated phosphorylation of connexin43 on tyrosine disrupts gap junctional communication in mammalian cells. Cell Commun Adhes 2001a; 8: 265–269, [INFOTRIEVE], [CSA]
- Lin R, Warn-Cramer B J, Kurata W E, Lau A F. v-Src phosphorylation of connexin 43 on Tyr247 and Tyr265 disrupts gap junctional communication. J Cell Biol 2001b; 154: 815–827, [INFOTRIEVE], [CSA], [CROSSREF]
- Loewenstein W R, Kanno Y. Intercellular communication and the control of tissue growth: Lack of communication between cancer cells. Nature 1966; 209: 1248–1249, [INFOTRIEVE], [CSA], [CROSSREF]
- Loo L W, Berestecky J M, Kanemitsu M Y, Lau A F. pp60src-mediated phosphorylation of connexin 43, a gap junction protein. J Biol Chem 1995; 270: 12751–12761, [INFOTRIEVE], [CSA], [CROSSREF]
- Maldonado P E, Rose B, Loewenstein W R. Growth factors modulate junctional cell-to-cell communication. J Membr Biol 1988; 106: 203–210, [INFOTRIEVE], [CSA], [CROSSREF]
- Martyn K D, Kurata W E, Warn-Cramer B J, Burt J M, TenBroek E, Lau A F. Immortalized connexin43 knockout cell lines display a subset of biological properties associated with the transformed phenotype. Cell Growth Differ 1997; 8: 1015–1027, [INFOTRIEVE], [CSA]
- Mesnil M. Connexins and cancer. Biol Cell 2002; 94: 493–500, [INFOTRIEVE], [CSA], [CROSSREF]
- Riley D, Carragher N O, Frame M C, Wyke J A. The mechanism of cell cycle regulation by v-Src. Oncogene 2001; 20: 5941–5950, [INFOTRIEVE], [CSA], [CROSSREF]
- Swenson K I, Piwinica-Worms H, McNamee H, Paul D L. Tyrosine phosphorylation of the gap junction protein connexin43 is required for the pp60v-src-induced inhibition of communication. Cell Regul 1990; 1: 989–1002, [INFOTRIEVE], [CSA]
- Toyofuku T, Akamatsu Y, Zhang H, Kuzuya T, Tada M, Hori M. c-Src regulates the interaction between connexin-43 and ZO-1 in cardiac myocytes. J Biol Chem 2001; 276: 1780–1788, [INFOTRIEVE], [CSA], [CROSSREF]
- Warn-Cramer B J, Cottrell G T, Burt J M, Lau A F. Regulation of connexin-43 gap junctional intercellular communication by mitogen-activated protein kinase. J Biol Chem 1998; 273: 9188–9196, [INFOTRIEVE], [CSA], [CROSSREF]
- Warn-Cramer B J, Lampe P D, Kurata W E, Kanemitsu M Y, Loo L WM, Eckhart W, Lau A F. Characterization of the mitogen-activated protein kinase phosphorylation sites on the connexin-43 gap junction protein. J Biol Chem 1996; 271: 3779–3786, [INFOTRIEVE], [CSA], [CROSSREF]
- Warn-Cramer B J, Lau A F. Regulation of gap junctions by tyrosine protein kinases. Biochim Biophys Acta 2004; 1662: 81–95, [INFOTRIEVE], [CSA], [CROSSREF]
- Warn-Cramer B J, Lin R, Martyn K, Guyette C V, Lau A F. Maintaining connexin43 gap junctional communication in v-Src cells does not alter growth properties associated with the transformed phenotype. Cell Commun Adhes 2003; 10: 299–303, [INFOTRIEVE], [CSA]
- Yamasaki H, Krutovskikh V, Mesnil M, Tanaka T, Zaidan-Dagli M L, Omori Y. Role of connexin (gap junction) genes in cell growth control and carcinogenesis. C R Acad Sci III 1999; 322: 151–159, [INFOTRIEVE], [CSA]
- Yamasaki H, Naus C C. Role of connexin genes in growth control. Carcinogenesis 1996; 17: 1199–1213, [INFOTRIEVE], [CSA]
- Yeatman T J. A renaissance for SRC. Nat Rev Cancer 2004; 4: 470–480, [INFOTRIEVE], [CSA], [CROSSREF]
- Zhou L, Kasperek E M, Nicholson B J. Dissection of the molecular basis of pp60(v-src) induced gating of connexin 43 gap junction channels. J Cell Biol 1999; 144: 1033–1045, [INFOTRIEVE], [CSA], [CROSSREF]
- Zisch A H, Pazzagli C, Freeman A L, Schneller M, Hadman M, Smith J W, Ruoslahti E, Pasquale E B. Replacing two conserved tyrosines of the EphB2 receptor with glutamic acid prevents binding of SH2 domains without abrogating kinase activity and biological responses. Oncogene 2000; 19: 177–187, [INFOTRIEVE], [CSA], [CROSSREF]