Abstract
The temporal dependence of cytoskeletal remodelling on cell–cell contact in HepG2 cells has been established here. Cell–cell contact occurred in an ultrasound standing wave trap designed to form and levitate a 2-D cell aggregate, allowing intercellular adhesive interactions to proceed, free from the influences of solid substrata. Membrane spreading at the point of contact and change in cell circularity reached 50% of their final values within 2.2 min of contact. Junctional F-actin increased at the interface but lagged behind membrane spreading, reaching 50% of its final value in 4.4 min. Aggregates had good mechanical stability after 15 min in the trap. The implication of this temporal dependence on the sequential progress of adhesion processes is discussed. These results provide insight into how biomimetic cell aggregates with some liver cell functions might be assembled in a systematic, controlled manner in a 3-D ultrasound trap.
INTRODUCTION
The temporal progression of cell-contact initiated cell morphology and F-actin cytoskeletal changes are of importance in processes such as tissue development (Carthew, Citation2005). Cell–cell adhesion is required for the formation of polarized aggregates (Carthew, Citation2005). Cadherin-mediated cell–cell adhesion initiates remodeling of the actin cytoskeleton through catenins (D'Souza-Schorey, Citation2005), leading to the development of a thick ring of F-actin at the cell periphery and termination of bundles of actin fibers at regions of cell–cell contact (Bamji, Citation2005; Zhang et al., Citation2005). Cell-substratum and cell-extracellular-matrix interactions through integrins can also direct cytoskeletal remodeling (Galler et al., Citation2006; Giancotti and Ruoslahti, Citation1999; Sawada et al., Citation2003). Significant attention has been paid to the temporal development of membrane spreading and F-actin cytoskeleton organization during integrin mediated cell-ECM adhesion of cells on artificial substrata (Cavalcanti-Adam et al., Citation2006; Chen et al., Citation2006; Kaido et al., Citation2004). The contribution of cell–cell interactions, independently of cell-ECM interactions, to adhesion processes has been more difficult to quantify due to the problem of identifying pairs of cells in suspension in a microscopic field that are about to interact and then maintaining those cells in focus during the development of membrane spreading. An ultrasound standing wave trap (USWT) that forms and levitates 2-D cell aggregates in suspension as a monolayer in a microscopic field has recently been described (Bazou et al., Citation2004, Citation2005a, Citation2006; Coakley et al., Citation2004; Morgan et al., Citation2004; Spengler and Coakley, Citation2003). The trap typically consists of a 0.5 mm layer of cell suspension that is driven at its half wavelength resonant frequency (1.5 MHz). Acoustic standing wave radiation forces drive the cells to form a single 2-D aggregate in the center of the resonating volume (). The aggregate can be continuously monitored microscopically through the quartz disc that forms the reflector of the standing wave device. Cell–cell adhesion and membrane spreading at the cell–cell contact interface in neural cell monolayers (Bazou et al., Citation2004, Citation2005; Coakley et al., Citation2004), the distribution of adhesion-related molecules NCAM and N-cadherin and F-actin in neural cells (Bazou et al., Citation2005a), and the development of the F-actin cytoskeleton and functioning gap junctions in chondrocytes (Bazou et al., Citation2006) in the USWT have been shown to change with time from aggregate formation. The cells remained viable throughout experiments. The times examined in the above studies looking at NCAM, N-cadherin, and F-actin were 1, 8, and 30 min (Bazou et al., Citation2005a) and 1 and 60 min for chondrocyte gap junction development (Bazou et al., Citation2006). It was shown that membrane spreading and molecular consequences following contact were beginning at 1 min, developing at 8 min and completed in 30–60 min (Bazou et al., Citation2005a, Citation2006). In the above cases, membrane spreading was defined as the increase in length of the cell–cell contact zone at the cell perimeter with time (Bazou et al., Citation2005a; Coakley et al., Citation2004; Coakley and Bazou, Citation2005).
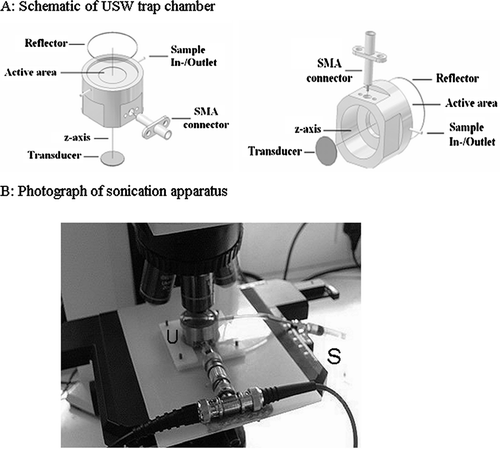
Figure 1 A: Schematic of the ultrasound trap (reproduced from Coakley et al., Citation2004, with permission). B: The ultrasound trap (U) on microscope stage and syringe used for loading and emptying the trap (S).
The work described here pays particular attention to establishing the initial rates of change over a 10-min period following adhesion. Rates of membrane spreading and cytoskeletal reorganization are established over those times. Adhesion of HepG2 cells is of interest in its own right and has additional significance because of the use of alginate-encapsulated HepG2 aggregates as a model for studying liver-specific biochemistry (Damelin et al., Citation2004). It is shown here that stable aggregates of HepG2 cells can be rapidly generated, without loss of viability, in the USWT. Cell morphology changes from an initial rounded form to a more cuboidal morphology, indicating that interactions between hepatocytes directly influence cell morphology. A significant redistribution of F-actin to the cell–cell interface occurs within 10 min of initiation of cell contact. This approach forms a basis for the development of a USWT hepatocyte culture system to rapidly form “microspheroids” as a tool for rapid biochemical analysis and functional characterisation.
MATERIALS AND METHODS
Cell Culture
HepG2 cells (European Collection of Cell Cultures, Health Protection Agency, Centre for Emergency Preparedness and Response, Salisbury, Wiltshire, UK), were maintained in Minimum Essential Medium (Eagle) (Sigma Aldrich, Poole, UK) supplemented with 10% v/v foetal bovine serum (Sigma Aldrich), 2 mM L-Glutamine (Invitrogen, Paisley, UK), 1% v/v non-essential amino acids (Invitrogen), 100 U/ml penicillin (Invitrogen), and 100 μ g/ml streptomycin (Invitrogen). Cultures were routinely passaged with 0.05% w/v trypsin-EDTA (Invitrogen) when 70% confluent (typically after 5 to 9 days). Single cell suspensions were obtained by washing monolayers in phosphate buffered saline (PBS, Sigma Aldrich) followed by incubation in Accutase (Sigma Aldrich) for 5 min. Cells were resuspended in serum-free medium, filtered through a 40-μ m Cell Strainer (BD Biosciences, Oxford, UK) and viability assessed by Trypan Blue (Sigma Aldrich) dye exclusion.
Ultrasound Exposure
The USWT employed here has been described previously (Bazou et al., Citation2004, Citation2005a). Its general structure () is based on a transducer with a nominal resonance frequency of 1.5 MHz (Ferroperm, Kvistgard, Denmark) attached to a steel plate, above which there is a sample space (to the depth of 0.5 mm, which equals λ /2, where λ represents the wavelength of sound in water) covered with a 1-mm-thick quartz glass reflector allowing optical access with a microscope. The transducer was driven by a sine wave generated by a Hewlett Packard 33120 A function generator. A 2-ml disposable syringe and small length of silicon tubing were employed to gently introduce cell suspensions into the USWT and to carefully remove aggregates from the trap at specified times after formation. The whole assembly was mounted on an Olympus BX41M reflection epifluorescence microscope (). The aggregation process was monitored using a CCD F-view camera mounted via a 0.3 × TV adapter. Images were captured with the analySIS 3.1 software package (Soft Imaging System GmbH, Münster, Germany). Sonication and thus levitation of cells commenced immediately following introduction of cells into the USWT and microscope focusing, which was taken as the zero timepoint in all experiments. The working acoustic pressure amplitude (110 kPa) was determined by trapping a single 25-μ m latex particle in the USWT and measuring the threshold voltage required to levitate the particle against gravity; this value was used to determine the working pressure on the basis of linear extrapolation (Coakley et al., Citation2004).
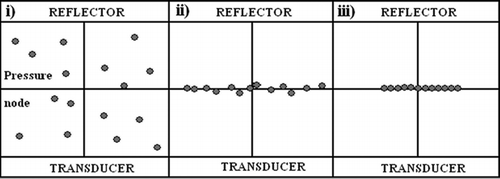
Figure 2 Schematic showing the formation of an aggregate of cells in the ultrasound trap (reproduced from Coakley et al., Citation2004, with permission). i: cells in suspension in trap with no ultrasound; ii: upon initiation of ultrasound exposure, cells congregate at the pressure node; and iii: the formation within a short time of a 2-D monolayer aggregate of cells.
Aggregate Area and Cell Death Assay
Ultrasound exposure has previously been used as a cell disruption tool resulting in cell death by rupturing cell membranes (Borthwick et al., 2006); hence, cells were incubated with Ethidium Homodimer, a DNA-binding fluorophore capable of passing across membranes with compromised integrity (Decherchi et al., Citation1997). Cells (3 × 105 cells · ml−1) in serum-free medium containing 1 μM Ethidium Homodimer (Invitrogen) were suspended in the USWT for 30 min, and aggregate micrographs were analyzed with Image-J 1.37b (Rasband, Citation1997–2006). Micrographs were converted from 8-bit grayscale brightfield to binary images by applying a threshold. Using the Wand tool, a region of interest (ROI) was selected around an aggregate. The area of this region (Aw) was measured. The cell-free area (Acf) within the region was determined (See Morphology and Void Analysis). The area of the aggregate (Aa) was given by Aw–Acf. Using the ‘ROI Manager,’ this ROI was applied to a fluorescence micrograph of the same aggregate, and the area of fluorescent cells (Afl) within the aggregate measured with the Analyze Particles tool. A measure of cell death was calculated as Afl/Aa and converted to a percentage. Differences in the area of fluorescing cells at 3 min and 30 min were determined with a one-tailed paired t-test in GraphPad Prism (GraphPad Software Incorporated, San Diego, CA); all experiments were carried out in triplicate unless otherwise stated and standard error of the mean was calculated. Membrane permeability of cells incubated in 1% v/v Triton X-100 at 4°C for 30 min and then suspended with Ethidium Homodimer in the USWT for 3 min was assessed as a positive control for reduced membrane permeability, and compared (t-test in GraphPad Prism) to untreated cells sonicated for 3 min.
Morphology and Void Analysis
Cells (3 × 105 cells · ml− 1) in serum-free medium were loaded into the USWT and aggregated. Using the Freehand tool, a ROI was selected around a cell and circularity was measured (defined in Image-J as 4Pi × [area/perimeter2]; a value of 1.0 represents a perfect circle) and differences were analyzed by analysis of variance (ANOVA) in Microsoft Excel. Interfacial length, i.e., the length of interfacial membrane spreading at the tangential contact area between two cells (Bazou et al., Citation2005a), was measured and differences were analyzed by ANOVA. To assess intercellular void space (Bazou et al., Citation2004), a ROI was selected around a binary image of an aggregate using the Wand tool, area was measured, and the Analyze Particles tool used to determine the void area within the ROI. A void index was established by dividing the void area within the ROI by the total ROI area (Bazou et al., Citation2004). The significance of correlation coefficients was assessed in GraphPad Prism using the F-test.
Fluorescent Localization of F-Actin
Cells (1 × 106 cells · ml−1) in serum-free medium were loaded into the USWT and exposed to ultrasound. While continuing sonication, aggregates were removed from the trap and placed on Histobond microscope slides (Raymond A Lamb Ltd., Eastbourne, UK), where they immediately sedimented. The supernatant was removed with a pipette, and the cells were fixed with 90% v/v ethanol within 60 s of removal from the USWT. After 20 min fixation, the cells were washed in PBS before being permeabilized for 30 min in PBS containing 0.1% v/v Triton X-100 (Sigma Aldrich), and then blocked for 30 min in PBS containing 1% w/v BSA. Actin was stained with 10 U/ml of Phalloidin-Alexa488 conjugate (Invitrogen) for 20 min in the dark. Slides were washed three times in PBS and mounted in VectaShield Mounting Medium (Vector Laboratories Ltd., Peterborough, UK). Images were collected as described previously.
A rectangular ROI was selected around a chosen area at the interface of a pair of interacting cells and used to generate a 3-D surface plot to visualize F-actin intensity (Barthel, Citation2004). A linear ROI was selected with the midpoint (0 μ m) over the center of the cell–cell boundary, halfway along the tangential length of the cell–cell interface, and fluorescence intensity values were obtained using the Plot Profile tool. Five fields of view were captured per replicate treatment. F-actin intensity was measured at five cell–cell interfaces from each field of view. Each dataset followed a Gaussian distribution (assessed in GraphPad Prism). These 25 measurements gave a mean value for each replicated experiment. The mean F-actin intensity over the distance 2 to 3 μ m on each side of the cell–cell interface was subtracted (as a background intensity level ‘correction’) from all intensity values over the range 0 to 2 μ m from the interface. The area under the resulting curves was calculated in GraphPad Prism. These measurements were repeated on unsonicated cells. The resulting ‘corrected’ values over 0–2 μ m from the cell rim were determined. A time zero control F-actin distribution was constructed from this data and its mirror image about the cell rim. To assess changes in the thickness of peripheral F-actin, the half-height of plot profile peak intensity was calculated and used to determine the half-width of F-actin at the cell–cell boundary.
RESULTS
Aggregate Formation and Strength
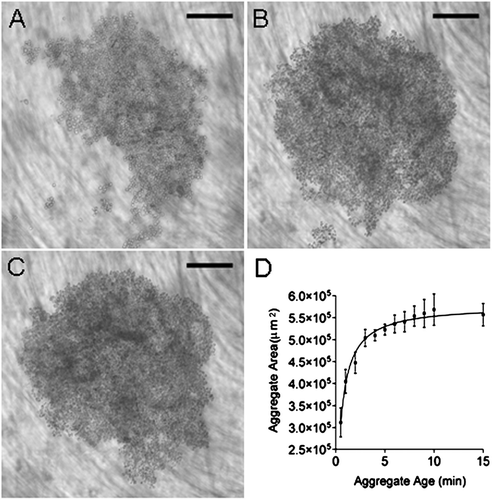
A cell aggregate was established from a suspension of HepG2cells at a concentration of 3 × 105 cells · ml− 1 within 30 s of commencing sonication (). The aggregate rapidly increased in size over 3 min () and had stabilizd by 15 min (). As the aggregate formed, its area showed a steep rise between 30 s and 3 min with a gradual increase beyond this point until 15 min (). Mean aggregate area at 15 min was 0.558 ± 0.003 mm2. The mean area of a HepG2 cell was 113 ± 12 μ m2 (n = 5), therefore aggregates contained approximately 5000 cells. To assess cell adhesion in the aggregate, levitation was terminated following 15 min and the aggregate allowed to sediment to the trap surface. The area of the sedimented aggregate, measured after 3 min on the trap base (0.568 mm2), was not significantly different (P = 0.83) from that of the suspended aggregate.
Figure 3 Development of HepG2 cell aggregate in the USWT at (A) 30 s, (B) 3 min, and (C) 15 min. Mean aggregate area increases with aggregate age until 3 min (D). Bar in micrographs represents 200 μ m.
Cell Death Assay
The membrane permeability cell death assay showed that 14.0 ± 0.24% (n = 3) and 15.0 ± 0.69% (n = 3) of the aggregate area contained fluorescing cells (i.e., those with compromised membrane integrity) at 3 and 30 min, respectively. A one-tailed paired-control t-test showed that this increase was not significant (P = 0.15). Following 3 min treatment with Triton X-100, 91.6 ± 3.0% of the aggregate contained fluorescing cells with compromised membrane integrity, which was significantly greater than the 14.0% for non-Triton-treated cells (P = 1.43 × 10− 5, n = 3).
Cell Morphology and Void Analysis
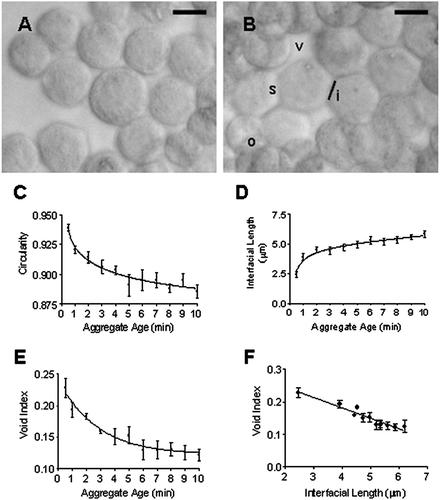
Cells entered the USWT with a rounded morphology characteristic of cells in suspension. Cells sonicated for 30 s () showed the same round morphology as the unsonicated cells (not shown). By 10 min, the loss of rounded morphology was distinguishable as a flattening of sides due to membrane spreading and overlapping (). Circularity was significantly reduced (P = 3.63 × 10− 8, n = 12) from 0.939 ± 0.003 at 30 s to 0.886 ± 0.006 at 10 min (). This occurred most extensively within the first 5 min of cell–cell contact during sonication. The interfacial length, an index of membrane spreading, increased significantly (P = 1.31 × 10− 6, n = 8) from 2.46 ± 0.25 μ m at 30 s to 5.86 ± 0.28 μ m at 10 min (). Void index measurements demonstrated an initial reduction in void area with time as cells formed a more closely packed aggregate (). This proceeded for 6 min until a plateau was reached. Void index was plotted against interfacial length at each time point (). The correlation index (r2 = 0.699) was significantly different from zero (P < 0.0001).
Figure 4 Representative images of (A) the circular morphology of HepG2 cells at 30 s following introduction into the USWT and (B) the departure from circular morphology following 10 min sonication in the trap. Cells show overlapping (o) and membrane spreading at points of contact (s) leading to an increase in the interfacial length (i) and an overall reduction of voids (v) within the aggregate. Aggregate age dependence of (C) Cell circularity; (D) Membrane spreading (as assessed by length of contact interface); and (E) void index. (F) Changes in circularity and void index correlate with membrane spreading (interfacial length). Bar in micrographs represents 10 μ m.
F-Actin Distribution
F-actin distribution was monitored as a function of time (the reported times were the time in the trap plus a 1 min delay for fixation). The distribution showed no difference at 2 min () from cells fixed and stained immediately following preparation (not shown). 3-D surface plots of F-actin intensity from fluorescence micrographs showed that F-actin was distributed towards the cell periphery after 2 min () but was localized at regions of cell–cell contact after 4 and 11 min (, ). This transition in staining at sites of contact was sharp as time increased from 2 to 4 min (, ) and became even more defined at 11 min (). F-actin distribution at the cell–cell boundary increased in peak intensity and decreased in width (). F-actin width perpendicular to the contact interface was 1.98 μ m at 2 min and 1.43 μ m at 11 min. The % change in the area under the curve in showed a rapid increase until 6 min, beyond which there was little further change (). The mean peak intensity of F-actin fluorescence increased more slowly than area under the curve as aggregate age increased ().
Figure 5 Fluorescence micrographs and 3-D surface intensity plots (inset shows magnified region of micrograph used for construction of surface plot) of F-actin stained with Phalloidin-Alexa488 in aggregates aged 2 min (A, B), 4 min (C, D), and 11 min (E, F), demonstrating that an increase in F-actin at regions of contact between HepG2 cells can be detected in aggregates aged 4 min. Bar in micrographs represents 20 μ m, and bar in magnified insets represents 2 μ m.
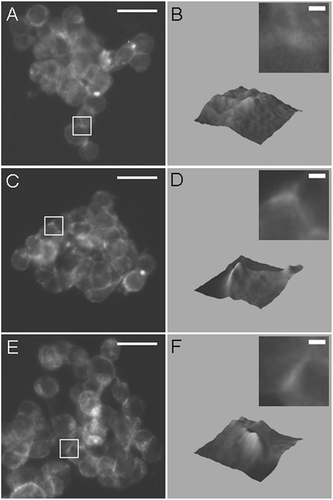
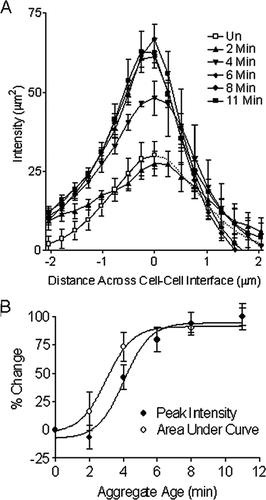
Figure 6 (A) Graphical representation of mean F-actin intensity plot profiles demonstrating a significant buildup of F-actin at the cell–cell boundary in aggregates aged greater than 3 min. A time zero control F-actin distribution across the periphery of a single cell was shown in mirror image (dotted line). (B) Area under curve and peak intensity of F-actin from A increase with aggregate age.
DISCUSSION
HepG2 cells introduced into the USWT had clearly established 2-D aggregates within 30 s, a time comparable to the 20 and 30 s periods previously reported for neural cells (Bazou et al., Citation2005a) and chondrocytes, respectively (Bazou et al., Citation2006). The speed of aggregate formation results in essentially synchronous initiation of cell–cell adhesive interactions across the aggregate. The cells retained viability, in agreement with previous results for neural cells following 60 min ultrasound exposure in the USWT (Bazou et al., Citation2005a). Indices of cell morphology demonstrated rapid changes following the initiation of cell–cell contact. Cells lost their rounded morphology within 5 min and exhibited membrane spreading (increased interfacial length at points of contact), which resulted in a reduction in the void index. While HepG2 integrin expression is required to facilitate cell adhesion, spreading, and loss of a round morphology on a substratum (Yin et al., Citation2003), the present study showed that spreading and morphological changes are also induced by cell–cell contact in suspension.
Actin reorganisation at points of cell–cell contact has previously been reported in a number of systems (Bazou et al., Citation2004, Citation2005a; Chu et al., Citation2004; Helwani et al., Citation2004; Huo et al., 2004; Ivanov et al., Citation2005; Zeggers et al., 1998; Zhang et al., Citation2005). Helwani et al. (Citation2004) demonstrated that E-cadherin colocalized with cortactin at points of adhesion between MDCK cells, which was required for F-actin recruitment to cell junctions. The result that cytoplasmic F-actin adjacent to the membrane had increased by 2 min () through cell–cell contact alone is comparable to the value of 2 min reported for a detectable increase in junctional F-actin using the ‘calcium switch’ approach to induce cell–cell contact in human keratinocyte cells grown on a monolayer of 3T3 fibroblasts treated with mitomycin-C (Zhang et al., Citation2005). Ivanov et al. (Citation2005) also employed the ‘calcium switch’ approach to study actin reorganization following the development of cell–cell contacts, namely during the assembly of the epithelial apical junctional complex, in T84 intestinal epithelial cells. Nascent E-cadherin-containing junctions developed by 30 min at the cell–cell boundary, which coincided with an increase in phalloidin-stained junctional F-actin (Ivanov et al., Citation2005). Peripheral F-actin can be detected within 4 min when mouse sarcoma cell doublets are formed using a pair of pipettes (Chu et al., Citation2004). By 8 min, F-actin can be detected at cell–cell junctions between neural cells in the USWT (Bazou et al., Citation2005a), and levels have stabilized between 8 min and 15 min of human keratinocyte cell contact (Zhang et al., Citation2005). Increased peripheral F-actin also occurs where HepG2 cells grown on quartz glass coverslips form cell–cell contacts (Huo et al., 2004; Zegers et al., Citation1998).
Morphological changes preceded the recruitment of F-actin to regions of cell contact. Membrane spreading (increased interfacial length) followed a reduction in circularity, but both showed a 50% change in 2.2 min (). Changes in cell morphology preceded F-actin redistribution by 1 min for actin area and by 2.2 min for actin peak intensity at the midpoint between cells (), as assessed by comparing the time required for a 50% change. On the assumption that membrane spreading arises from receptor–receptor interactions (Bazou et al., Citation2005a), these results demonstrate that cell–cell contact through aggregation leads to membrane spreading and a loss of circular morphology, followed by F-actin recruitment to regions of cell–cell contact. These early changes in F-actin distribution appear to be a response to changes in cellular morphology brought about by contact, rather than the morphological changes being driven by the redistribution of F-actin. A comparable relationship between morphology and cytoskeletal rearrangements can be observed in the early development and polarisation of embryos. Following cell division, daughter cells form adhesive junctions displaying membrane spreading at points of cell–cell contact where F-actin (Albertini et al., Citation1987) and adhesive molecules such as JAM-1 (Thomas et al., Citation2004) and F-actin (Albertini et al., Citation1987) become localized.
Table 1 Changes in indices of cell shape and F-actin with increasing aggregate age
HepG2 cells expressing GFP-actin rapidly adhere to fibronectin-treated surfaces, leading to an increase in cell area resulting from membrane spreading, reaching 50% of its steady-state value by 6 min (Feng et al., Citation2005). In the current cell–cell adhesion study, where membrane spreading was inferred from an increase in interfacial length rather than an increase in cell area, the duration of contact required to produce a 50% increase was shorter, at 2.2 min. Fluorescent staining demonstrated a 50% increase in F-actin area following 3.2 min and a 50% increase in peak intensity by 4.4 min ().
Changes in morphology and increased F-actin lead to the formation of a stable aggregate. Retention of aggregate integrity upon sedimentation following 15 min levitation demonstrated the acquisition of mechanical stability, a conclusion strengthened by the fact that large regions of the aggregate retained their structural integrity during the stress of removal from the trap for studying F-actin distribution at time points prior to 15 min. Both observations suggest that aggregates in suspension may be mechanically robust enough to withstand manipulation after 15 min. By using pairs of pipettes to hold doublets of mouse sarcoma cells together, it has been shown that strong adhesion develops between 4 min and 30 min, which is associated with an increase in actin at the cell–cell boundary (Chu et al., Citation2004).
Adhesive cell–cell interactions leading to cytoskeletal remodelling involve a range of cell-surface receptors, namely cell adhesion molecules (CAMs) and cadherins. It has previously been shown that wild-type HepG2 cells do not express the CAMs ICAM-1 (Qin et al., Citation2005), hepaCAM (Moh et al., Citation2005), and LI-CAM (Wong et al., Citation2003). The expression of E-cadherin in the HepG2 cell line is a contentious issue. On employing RT-PCR and immunofluorescence, E-cadherin was not detected in the HepG2 cell line by Yano and Yamasaki (Citation2001). Cui et al. (Citation2006) demonstrated that it could only be detected when cells were treated with the promoter demethylating agent 5-aza-2′-deoxycytidine. However, Lin et al. (Citation2006) detected E-cadherin in HepG2 cells by means of immunofluorescence and Western blotting, a result confirmed by the work of Liu et al. (Citation2006). During the current study, immunofluorescent detection of E-cadherin demonstrated a diffuse, punctuate cytoplasmic distribution rather than at cell–cell junctions, thus it was concluded that the HepG2 clone employed here did not express E-cadherin capable of forming functional adherens junctions (unpublished data).
Exposure to ultrasound is not likely to influence the properties of cell adhesion molecules. Aggregation occurs at a pressure node, where both local acoustic pressure and the axial direct radiation force that drives aggregation are zero (Kuznetsova and Coakley, Citation2004). Calculation of the acoustic interaction force between particles at such a pressure node are comparable to the Van der Waals force at membrane separations of 50 nm (Bazou et al., Citation2005b) and decreases relative to the van der Waals force at smaller cell separations. The Van der Waals force is in turn much smaller than the attractive force involved in surface receptor interactions at the membrane separations at which receptors engage (Bazou et al., Citation2006). We conclude that adhesion following cell–cell contact is due to normal receptor phenomena rather than to direct effects of experimental ultrasound exposure.
The formation of 2-D cell aggregates in the USWT allows the clear temporal optical microscopic examination of adhesion to be compared to detection of junctional F-actin in recovered aggregates. The ultrasound methodology also has the potential to rapidly form 3-D aggregates on a fast timescale. Like 3-D culture models, spheroids show enhanced liver-specific behavior in comparison to monolayers, making them an attractive model for in vitro toxicological studies (Ma et al., Citation2003). Current technology based on a rotating plate culture system provides spheroids following 7 days for studying liver-specific behavior and drug metabolism (Damelin et al., Citation2004) and for toxicological assessment (Ma et al., Citation2003; Xu et al., Citation2003). The data presented here provide guidance on the time required to rapidly form a 3-D aggregate of biologically interacting HepG2 cells in an appropriately designed ultrasound trap. The extent to which such ultrasonically generated 3-D aggregates formed from single-cell HepG2 suspensions develop a biochemistry comparable to spheroids will be investigated in future work.
ACKNOWLEDGMENTS
This work was supported by the Biotechnology and Biological Sciences Research Council, Grant No. BB/C515220/1.
REFERENCES
- Albertini D F, Overstrom E W, Ebert K M. Changes in the organisation of the actin cytoskeleton during preimplantation development of the pig embryo. Biol Reprod 1987; 37: 441–451
- Bamji S X. Cadherins: Actin within the cytoskeleton to form synapses. Neuron 2005; 47: 175–178
- Barthel K U. Interactive 3-D Surface Plot-Plugin for ImageJ, Internationale Medieninformatik. FHTW, Berlin 2004, http://rsb.info.nih.gov/ij/plugins/surface-plot-3d.html
- Bazou D, Coakley W T, Meek K, Yang M, Pham D T. Characterisation of the morphology of 2-D-particle aggregates in different electrolyte concentrations in an ultrasound trap. Colloids Surf. B: Physiochemical and engineering aspects 2004; 243: 97–104
- Bazou D, Dowthwaite G P, Khan I M, Archer C W, Ralphs J R, Coakley W T. Gap junctional intercellular communication and cytoskeletal organization in chondrocytes in suspension in an ultrasound trap. Mol. Membr. Biol 2006; 23: 195–205
- Bazou D, Foster G A, Ralphs J R, Coakley W T. Molecular adhesion development in a neural cell monolayer forming in an ultrasound trap. Mol Membr Biol 2005a; 22: 229–240
- Bazou D, Kuznetsova L, Coakley W T. Physical environment of 2-D animal cell aggregates formed in a short pathlength ultrasound standing wave trap. Ultrasound Med Biol 2005b; 31: 423–430
- Borthwick K A, Coakley W T, McDonnell M B, Nowotny H, Benes E, Groschl M. Development of a novel compact sonicator for cell disruption. J Microbiol Methods 2005; 60: 207–216
- Carthew R W. Adhesion proteins and the control of cell shape. Curr Opin Genet Dev 2005; 15: 358–363
- Cavalcanti-Adam E A, Micoulet A, Blümmel J, Auernheimer J, Kessler H, Spatz J P. Lateral spacing of integrin ligands influences cell spreading and focal adhesion assembly. Eur J Cell Biol 2006; 85: 219–224
- Chen M, Chen S C, Pallen C J. Integrin-induced tyrosine phosphorylation of protein-tyrosine phosphatase-α is required for cytoskeletal reorganization and cell migration. J Biol Chem 2006; 281: 11972–11980
- Chu Y S, Thomas W A, Eder O, Pincet F, Perez E, Thiery J P, Dufour S. Force measurements in E-cadherin-mediated cell doublets reveal rapid adhesion strengthened by actin cytoskeleton remodelling through Rac and Cdc42. J Cell Biol 2004; 167: 1183–1194
- Coakley W T, Bazou D. Particle and cell manipulation by radiation force in ultrasound standing waves. Bubble and particle dynamics in acoustic fields: modern trends and applications, A A Doinikov. Research Signpost, KeralaIndia 2005; 313–338
- Coakley W T, Bazou D, Morgan J, Foster G A, Archer C W, Powell K, Borthwick K AJ, Twomey C, Bishop J. Cell–cell contact and membrane spreading in an ultrasound trap. Colloids Surf B: Biointerfaces 2004; 34: 221–230
- Cui X, Wakai T, Shirai Y, Yokoyama N, Hatakeyama K, Hirano S. Arsenic trioxide inhibits DNA methyltransferase and restores methylation-silenced genes in human liver cancer cells. Hum Pathol 2006; 37: 298–311
- Damelin L H, Coward S, Choudhury S F, Chalmers S A, Cox I J, Robertson N J, Revial G, Miles M, Tootle R, Hodgson H JF, Selden C. Altered mitochondrial function and cholesterol synthesis influences protein synthesis in extended HepG2 spheroid cultures. Arch Biochem Biophys 2004; 432: 167–177
- Decherchi P, Cochard P, Gauthier P. Dual staining assessment of Schwann cell viability within whole peripheral nerves using calcein-AM and ethidium homodimer. J Neurosci Methods 1997; 71: 205–213
- D'Souza-Schorey C. Disassembling adherens junctions: breaking up is hard to do. Trends Cell Biol 2005; 15: 19–26
- Feng Z, Chen W N, Lee P VS, Liao K, Chan V. The influence of GFP-actin expression on the adhesion dynamics of HepG2 cells on a model extracellular matrix. Biomaterials 2005; 26: 5348–5358
- Galler A B, Arguinzonis M IG, Baumgartner W, Kuhn M, Smolenski A, Simm A, Reinhard M. VASP-dependent regulation of actin cytoskeleton rigidity, cell adhesion, and detachment. Histochem Cell Biol 2006; 125: 457–474
- Giancotti F G, Ruoslahti E. Integrin signalling. Science 1999; 285: 1028–1032
- Helwani F M, Kovacs E M, Paterson A D, Verma S, Ali R G, Fanning A S, Weed S A, Yap A S. Cortactin is necessary for E-cadherin-mediated contact formation and actin reorganization. J Cell Biol 2004; 164: 899–910
- Huo X, Xu X J, Chen Y W, Yang H W, Piao Z X. Filamentous-actins in human hepatocarcinoma cells with CLSM. World J Gastroenterol 2005; 10: 1666–1668
- Ivanov A I, Hunt D, Utech M, Nusrat A, Parkos C A. Differential roles for actin polymerization and a myosin II motor in assembly of the epithelial apical junction complex. Mol Biol Cell 2005; 16: 2636–2650
- Kaido T, Perez B, Yebra M, Hill J, Cirulli V, Hayek A, Montgomery A M. αv-Integrin utilization in human β-cell adhesion, spreading, and motility. J Biol Chem 2004; 279: 17731–17737
- Kuznetsova L, Coakley W T. Microparticle concentration in short path length ultrasonic resonators: role of radiation pressure and acoustic streaming. J Acoust Soc Am 2004; 116: 1956–1965
- Lin C Y, Lin C J, Chen K H, Wu J C, Huang S H, Wang S M. Macrophage activation increases the invasive properties of hepatoma cells by destabilization of the adherens junction. FEBS Lett 2006; 580: 3042–3050
- Liu J, Lian Z, Han S, Waye M M, Wang H, Wu M C, Wu K, Ding J, Arbuthnot P, Kew M, Fan D, Feitelson M A. Downregulation of E-cadherin by hepatitis B virus antigen X in hepatocellular carcinoma. Oncogene 2006; 25: 1008–1017
- Ma M, Xu J, Purcell W M. Biochemical and functional changes of rat liver spheroids during spheroid formation and maintenance in culture: I. Morphological maturation and kinetic changes of energy metabolism, albumin synthesis, and activities of some enzymes. J Cell Biochem 2003; 90: 1166–1175
- Moh M C, Lee L H, Shen S. Cloning and characterization of hepaCAM, a novel Ig-like cell adhesion molecule suppressed in human hepatocellular carcinoma. J Hepatol 2005; 42: 833–841
- Morgan J, Spengler J F, Kuznetsova L, Coakley W T, Xu J, Purcell W M. Manipulation of in vitro toxicant sensors in an ultrasound standing wave. Toxicol In Vitro 2004; 18: 115–120
- Qin P, Borges-Marcucci L A, Evans M J, Harnish D C. Bile acid signalling through FXR induces intracellular adhesion molecule-1 expression in mouse liver and human hepatocytes. Am J Physiol Gastrointest Liver Physiol 2005; 289: G267–G273
- Rasband W S. Image-J. U. S. National Institutes of Heath, Bethesda, MarylandUSA 1997, 2006 http://rsb.info.nih.gov/ij/
- Sawada S, Yoshimoto M, Odintsova E, Hotchin N A, Berditchevski F. The tetraspanin CD151 functions as a negative regulator in the adhesion-dependent activation of Ras. J Biol Chem 2003; 278: 26323–26326
- Spengler J F, Coakley W T. Ultrasonic trap to monitor morphology and stability of developing microparticle aggregates. Langmuir 2003; 19: 3635–3642
- Thomas F C, Sheth B, Eckert J J, Bazzoni G, Dejana E, Fleming T P. Contribution of JAM-1 to epithelial differentiation and tight-junction biogenesis in the mouse preimplantation embryo. J Cell Sci 2004; 117: 5599–5606
- Wong B W, Luk J M, Ng I O, Hu M Y, Liu K D, Fan S T. Identification of liver-intestine cadherin in hepatocellular carcinoma-a potential disease marker. Biochem Biophys Res Comm 2003; 311: 618–624
- Xu J, Ma M, Purcell W M. Biochemical and functional changes of rat liver spheroids during spheroid formation and maintenance in culture: II. Nitric oxide synthesis and related changes. J Cell Biochem 2003; 90: 1176–1185
- Yano T, Yamasaki H. Regulation of cellular invasion and matrix metalloproteinase activity in HepG2 cell by connexin 26 transfection. Mol Carcinog 2001; 31: 101–109
- Yin C, Liao K, Mao H Q, Leong K M, Zhuo R X, Chan V. Adhesion contact dynamics of HepG2 cells on galactose-immobilized substrates. Biomaterials 2003; 24: 837–850
- Zegers M MP, Zaal K JM, van Ijzendoorn S CD, Klappe K, Hoekstra D. Actin filaments and microtubules are involved in different membrane traffic pathways that transport sphingolipids to the apical surface of polarized HepG2 cells. Mol Biol Cell 1998; 9: 1939–1949
- Zhang J, Betson M, Erasmus J, Zeikos K, Bailly M, Cremer L P, Braga V MM. Actin at cell–cell junctions is composed of two dynamic and functional populations. J Cell Sci 2005; 118: 5549–5562