
Abstract
Advanced glycation end-products (AGEs) are involved in the development of vascular smooth muscle cell (VSMC) dysfunction and the progression of atherosclerosis. However, AGEs may indirectly affect VSMCs via AGEs-induced signal transduction between monocytes and human umbilical endothelial cells (HUVECs), rather than having a direct influence. This study was designed to elucidate the signaling pathway underlying AGEs-RAGE axis influence on VSMC dysfunction using a co-culture system with monocytes, HUVECs and VSMCs. AGEs stimulated production of reactive oxygen species and pro-inflammatory mediators such as tumor necrosis factor-α and interleukin-1β via extracellular-signal-regulated kinases phosphorylation and nuclear factor-κB activation in HUVECs. It was observed that AGEs-induced pro-inflammatory cytokines increase VSMC proliferation, inflammation and vascular remodeling in the co-culture system. This result implies that RAGE plays a role in AGEs-induced VSMC dysfunction. We suggest that the regulation of signal transduction via the AGEs-RAGE axis in the endothelium can be a therapeutic target for preventing atherosclerosis.
Introduction
Advanced glycation end products (AGEs) are formed by non-enzymatic glycation of proteins with other substances, such as carbohydrates or lipids (Basta et al. Citation2004). An increase in AGEs is one of the most important risk factors in the pathogenesis of accelerated atherosclerosis in patients with diabetes (Basta et al. Citation2004). In addition, the interaction of AGEs with their receptor (RAGE) on the cell surface causes upregulation of transcription factors such as nuclear factor (NF)-κB, leading to the expression of various pro-inflammatory cytokines and cell adhesion molecules that cause increased recruitment of monocytes into endothelial cells (ECs). This implies that AGEs-RAGE interactions are key steps in EC dysfunction and atherosclerosis (Davignon & Ganz Citation2004; Sitia et al. Citation2010; Mudau et al. Citation2012). In the pathogenesis of vessel disease, the proliferation of vascular smooth muscle cells (VSMCs) is an important event in the progression and rupture of atherosclerotic plaque, which subsequently contributes to heart disease (Ross Citation1993). Moreover, a recent study showed that VSMC cell cycle regulation is affected by inflammatory cytokines (Moon et al. Citation2003). Several studies have reported on vascular intercellular communication using co-culture systems. Monocytes co-cultured with ECs can promote the adhesion, activation, and transmigration of monocytes or lymphocytes, as well as increase the generation of cytokines including tumor necrosis factor (TNF), interleukin (IL), and monocyte chemoattractant protein (MCP)-1 (Lukacs et al. Citation1995; Tsouknos et al. Citation2003). However, the monocyte-derived mediators, which activate ECs have not yet been fully elucidated. Furthermore, monocytes alone or those treated with VSMC-conditioned media were found not to secrete matrix metalloproteinases (MMPs) (Lee et al. Citation1995). On the other hand, compared to when VSMCs were cultured alone, the secretion of MMPs was increased more than 20-fold when VSMCs were co-cultured with monocytes or treated with monocyte-conditioned media, suggesting that monocytes may play an important role in the pathogenesis of VSMCs. Although those authors provided a special cellular environment (Wu et al. Citation2008), the co-culture system with ECs and VSMCs could not exactly simulate an in vivo situation because of the absence of various extracellular matrix components, inflammatory cells, and some physical phenomena. Based on the results of previous studies, our research group established a co-culture model system through simultaneous co-cultivation of monocytes, HUVECs, and VSMCs to provide more in-vivo-like conditions (Nam et al. Citation2011). AGEs induced the production of IL-6 and TNF-α from monocytes and HUVECs in the co-culture system. Increased IL-6 and MCP-1 gene expression in VSMCs was induced by TNF-α, mediated primarily through stimulation by AGEs from HUVECs and monocytes.
Therefore, the present study was designed to investigate the effects of signaling pathways mediated by the AGEs-RAGE axis on the EC dysfunction observed during the initial period of atherosclerosis, as well as their contribution to VSMC proliferation and pathogenesis, using a co-culture system with THP-1 cells, HUVECs, and VSMCs.
Materials and methods
Chemicals
Endothelial cell basal medium-2 with supplemental growth factors (EGM-2) was purchased from Lonza Cambrex (Nottingham, UK), and Kaighn’s modification of Ham’s F-12K (F-12K) medium was purchased from Sigma Chemical (St. Louis, MO). RPMI 1640 medium and all other tissue culture reagents were obtained from GIBCO (Grand Island, NY). Endothelial cell growth supplement (ECGS), the multiwall insert system, and the trans-well insert system were purchased from BD Bioscience (Bedford, MA). Specific antibodies were obtained from Cell Signaling Technology Inc. (Danvers, MA) or Santa Cruz Biotechnology, Inc. (Heidelberg, Germany).
Cell culture
Human aortic VSMCs were obtained from ATCC (Manassas, VA), and cultured in F-12K medium containing 10% (v/v) FBS supplemented with insulin-transferrin-selenium, 10 mM HEPES, 0.05 g/L ascorbic acid, 0.03 g/L ECGS, 100 U/mL penicillin, and 100 U/mL streptomycin. VSMCs in passages 16 to 22 in active growth conditions were used for the experiments. Primary cultured human umbilical endothelial cells (HUVECs) were purchased from Lonza (Seoul, Korea). The cells were cultured in endothelial growth medium-2 (EGM-2) with 2% FBS. The human monocytic leukemia cell line THP-1 was grown in RPMI 1640 medium with 0.05 mM 2-mercaptoethanol and 10% (v/v) heat-inactivated fetal bovine serum (FBS). VSMCs were seeded at a density of 2 × 104 cells/well on the bottom of 6-well plates using the complete media and culture environment described above. HUVECs were seeded at a density of 5 × 104 cells/well in 0.4 μm transwell inserts and allowed to grow overnight in the above-mentioned conditions. The next day, the HUVECs cultured on the membrane transwell insert were placed into the 6-well plates containing the cultured VSMCs, and THP-1 monocytes were seeded at a density of 1 × 105 cells/well in the insert. All cultures were grown in EGM-2 medium and incubated for 24 h. The THP-1 monocytes and HUVECs were grown in the co-culture system for 24 h, and then the media was aspirated and replaced with fresh EGM-2 medium without FBS.
Preparation and treatment of glycated bovine serum albumin
Bovine serum albumin (BSA; 20 mg/mL) was incubated with 20 mM glycolaldehyde and 1 mM diethylene triamine penta acetic acid (DTPA) in potassium phosphate buffer (PBS; 0.01 M potassium phosphate buffer, pH 7.4, 0.138 M NaCl and 2.7 mM KCl) at 37 °C for 7 d. To prevent an unpredictable reversible glycation reaction, BSA was then directly reduced with 80 mM sodium borohydride for 30 min. Unincorporated glycolaldehyde and low molecular weight reactants were then removed through extensive dialysis with PBS. HUVECs and THP-1 cells were stimulated with 100 μg/mL of glycolaldehyde-derived AGEs (glycol-AGEs) with 1.5 mL of upper compartment media in the inner side of the insert, while VSMCs were treated with 2.5 mL of media alone in the bottom of the well.
Cell transfection with siRNA
Human umbilical endothelial cell and THP-1 cells were transfected at 40-60% confluence using Lipofectamine® 2000 (Invitrogen, Grand Island, NY), according to the manufactureŕs protocol. One day before transfection, approximately 5 × 104 cells/well were seeded in 6-well plates overnight. For transfection of each well, 20 pmol siRNA was diluted in 100 μL of Opti-MEM reduced serum medium and mixed gently. Next, 3 μL of Lipofectamine® 2000 was diluted in 100 μL of Opti-MEM reduced serum medium and mixed gently. The diluted mixtures were then combined and incubated for 20 min at room temperature, after which 200 μL of the siRNA-Lipofectamine complexes were added to each well. This produced a final volume of 2000 μL and a final RNA concentration of 10 nM. After 4 h, the complexes were removed and the cells were cultured in complete medium and incubated for 48 h at 37 °C in a CO2 incubator until ready to be assayed for gene knockdown. HUVECs were transfected with RAGE-specific siRNA (5'-TAGCTCCTGGTGGAACCGTAA-3”) or negative control siRNA (Qiagen, Hilden, Germany).
Measurement of intracellular reactive oxygen species production
The production of intracellular ROS was determined using 2',7'-dichlorofluorescein diacetate (DCF-DA) by fluorescence spectrophotometry. HUVECs were incubated with 10 μM DCF-DA in EGM-2 media for 30 min in a CO2 incubator, after which the cells were washed using 0.1 M PBS. The HUVECs were then treated with 100 μg/mL glycol-AGEs and incubated for a further 3 h. The HUVECs were pre-incubated with inhibitors (10 μM) for 1 h prior to treatment with glycol-AGEs. Cells were analyzed directly using a spectrofluorometer (VICTOR3™ Perkin-Elmer, Wellesley, MA) at an excitation wavelength of 485 nm/emission wavelength of 535 nm.
Real-time reverse transcription-PCR
mRNA expression was determined with quantitative real-time reverse transcriptase-polymerase chain reaction (qRT-PCR). Total RNA was extracted using TRIzol reagent, according to the manufacturer's protocol (TaKaRa Korea Biomedical Co, Seoul, Korea) and reverse-transcribed into cDNA using the LeGene Express 1st Strand cDNA Synthesis System (Legene, San Diego, CA). Real-time PCR was performed using TOPreal™ qPCR 2X PreMIX SYBR green (Enzynomics, Seoul, Korea) and an iQ5 thermal cycler (Bio-Rad, Hercules, CA). The primers were purchased from Cosmo Genetech (Seoul, Korea). The primers for amplification of specific human mRNA were as follows: RAGE (199 bp), 5'-GGAATGGAAAGGAGACCAAG-3' and 5'-CCCTTCTCATTAGGCACCAG-3'; TNF-α (80 bp), 5'-CCCAGGGACCTCTCTCTAATCA-3' and 5'-GCTACAGGCTTGTCACTC GG-3'; IL-1β (391 bp), 5'-AAACAGATGAAGTGCTCCTTCCAGG-3' and 5'-TGGAGA ACACCACTTGTTGCTCCA-3'; IL-6 (81 bp), 5′-GGTACATCCTCGACGGCATCT-3′ and 5′-GTGCCTCTTTGCTGCTTTCAC-3′; MCP-1 (161 bp), 5′-TCGCGAGCTATAGAAGAATCA-3′ and 5′-TGTTCAAGTCTTCGGAGTTTG-3′; Plasminogen activator inhibitor (PAI)-1 (112 bp), 5′-CAGAAAGTGAAGATCGAGGTGAAC-3′ and 5′-GGAAGGGTCTGTCCATGATGA-3′; GAPDH (250 bp), 5'- GAAGGTGAAGGTCGGAGT-3' and 5'-GAAGATGGTGATGGGATTTC-3' (sense and antisense, respectively). The levels of RAGE, TNF-α, IL-1β, IL-6, MCP-1, and PAI-1 mRNAs were normalized to the corresponding levels of GAPDH mRNA.
Preparation of nuclear extracts
After termination of the reactions by washing twice with ice-cold PBS, the cells were harvested and analyzed. HUVECs were collected in ice-cold PBS with protease inhibitors by scraping, and then pelleted by centrifugation at 12,000g for 5 min at 4 °C. The cells were then resuspended in 200 μL of 1X hypotonic buffer (10 mM Hepes, pH 7.8, 10 mM KCl, 0.1 mM EDTA, 1 mM DTT, and 0.8% NP-40). After moderate vortexing, the cells were centrifuged at 12,000g for 5 min at 4 °C. At this time, the supernatant contained the cytoplasmic fraction, while the pellet contained the nuclear fraction. The nuclear pellets were resuspended in 100 μL nuclear extraction buffer (50 mM Hepes, pH 7.8, 50 mM KCl, 300 mM NaCl, 0.1 mM EDTA, 1 mM DTT ,and 20% glycerol). After vortexing vigorously for 10 min at 4 °C, the cell pellet was centrifuged at 12,000g for 10 min, and the supernatant (nuclear extract) was collected, and stored at -80 °C for further experiments. Protein content in the nuclear extract was quantified by the BCA protein assay (Pierce Biotechnology, Rockford, IL).
Western blotting assay
After each experiment, cells were washed twice with ice-cold PBS, and resuspended in RIPA lysis buffer containing protease and/or phosphatase inhibitors. Cell homogenates were centrifuged at 10,000g for 20 min at 4 °C. The resulting supernatant was used as the cellular protein. Equal amounts of protein samples were subjected to 10% sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis, and transferred onto polyvinylidene fluoride membranes (Millipore, Billerica, MA). Afterwards, non-specific binding on the membranes was blocked with 5% non-fat milk in TBST (20 mM Tris–HCl, 500 mM NaCl, and 0.1% Tween 20) at room temperature for 1 h. The membranes were then incubated overnight at 4 °C with specific primary antibodies, including those for nuclear factor kappa B (NF-κB), proliferating cell nuclear antigen (PCNA), c-Jun NH2-terminal kinase (JNK), p-JNK (1:400) (SantaCruz Biotechnology, Santa Cruz, CA), p38 mitogen-activated protein kinase (p38-MAPK), p-p38, extracellular regulated kinase (ERK), p-ERK (1:1000) (Cell Signaling Technology, Boston, MA) and GAPDH (1:1000) (Millipore, Billerica, MA). The membranes were then probed with horseradish peroxidase-conjugated secondary antibodies at room temperature for 45 min. The protein–antibody complexes were detected with enhanced chemiluminescence reagents (iNtRON Biotechnology, Seongnam, Korea), and scanned with a luminescence image analyzer (Fuji Film LAS-4000, Tokyo, Japan). The bands were quantitated with ImageJ software (NIH, Bethesda, MD), and the protein signals were normalized to GAPDH or PCNA levels.
Immunofluorescence staining
Cells were washed with ice-cold PBS twice, then fixed with 3.7% formaldehyde in 1X PBS for 20 min at 4 °C. Cell membranes were then permeabilized with 0.1% Triton X-100 in PBS, and non-specific binding sites were blocked with 1% BSA in PBS for 1 h at RT. Cells were incubated with primary antibody (RAGE, NF-κB and Ki-67, 1:50, SantaCruz Biotechnology, Santa Cruz, CA) at 4 °C overnight. After incubation, the cells were rinsed with ice-cold PBS, and then incubated further with Alexa 488 green-fluorescent dye (1:1000) in PBS for 2 h at RT. Nuclei were stained with DAPI for 10 min at RT, and the cells were then observed under a confocal laser-scanning microscope (Carl-Zeiss, Oberkochen, Germany).
Gelatin zymography
MMP-2 and MMP-9 activities were detected by gelatin zymography on 8% polyacrylamide gels containing 0.2% gelatin using the media from the lower compartment of the glycol-AGEs-treated co-culture system. The gels were loaded with samples diluted in 1:1 non-reducing buffer (12.5% 0.5 M Tris–HCl pH 6.8, 10% glycerol, 4% SDS, and 0.05% bromophenol blue), and electrophoresis was carried out at 4 °C using a constant current of 40 mA for approximately 2 h. After electrophoresis, the gel was removed, and incubated with 2.5% Triton X-100 for 30 min at room temperature with gentle agitation. The gel was equilibrated for 30 min with 1 X Zymogram Developing Buffer (Koma Biotech, Seoul, Korea), and then incubated with fresh 1 X Zymogram Developing Buffer overnight at 37 °C. The bands were visualized by staining for 60 min with a solution containing 0.1% Coomassie R-250 in 40% ethanol and 10% acetic acid, followed by destaining for 2 h at room temperature in a solution containing 10% ethanol and 7.5% acetic acid. The gels were washed with distilled water and scanned. Band intensities were measured using ImageJ software (NIH, Bethesda, MD).
Statistical analysis
SAS software version 9.3 (SAS Institute, Cary, NC) was used for all statistical analyses. Data are expressed as mean ± standard deviation (SD) of three independent experiments performed in triplicate. Significant differences between means were determined by Duncan’s multiple range test.
Results
Gene silencing of RAGE and AGEs-induced intracellular ROS production
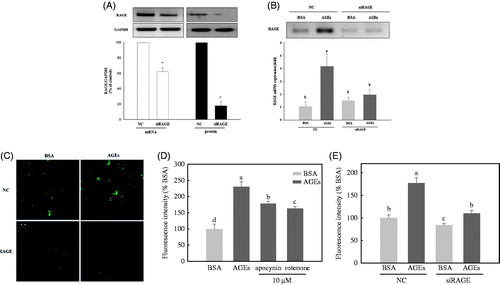
Human umbilical endothelial cells were transfected with either non-silencing control siRNA or RAGE-specific siRNA (20 nM), and RAGE expression was assessed by real-time PCR and Western blot analysis at 48 h post-transfection. Transfection of RAGE-specific siRNA caused a decrease in the mRNA and protein levels to about 40 and 80%, respectively, compared with those of the non-silencing control (). After exposure to 100 μg/mL of BSA or glycol-AGEs for 24 h at 48 h post-transfection, the levels of RAGE mRNA were again determined (). AGE treatment significantly increased RAGE expression prior to gene silencing of RAGE, but the effects of AGE exposure were no longer observed compared with the negative control-treated groups after silencing of RAGE. Immunofluorescence confocal microscopy was used to confirm that RAGE was expressed on the HUVEC cell membranes and that the siRNA reduced the expression levels of RAGE protein (). The effect of glycol-AGEs on endothelial ROS production was then investigated. To explore the sources of ROS, HUVECs were pretreated with apocynin (10 μM), a specific inhibitor of NADPH oxidase (Riganti et al. Citation2008), or rotenone (10 μM), a specific inhibitor of the mitochondrial electron transport chain (Zhao et al. Citation2014), for 1 h before treatment with glycol-AGEs (). The results showed that treatment with glycol-AGEs induced a significant increase in ROS generation, which could be inhibited by an addition of apocynin or rotenone. This suggests that glycol-AGEs-induced ROS generation was mainly derived from both mitochondria and NADPH oxidase. Binding of glycol-AGEs to RAGE induced the generation of intracellular ROS, which was also inhibited by RAGE-specific siRNA ().
Figure 1. Knockdown of the receptor for AGE (RAGE) inhibits advanced glycation end-products (AGEs)-induced intracellular reactive oxygen species (ROS) formation in HUVECs. (A) Examination of the receptor for advanced glycation end products (RAGE) expression upon knockdown with siRNA, assessed by real-time polymerase chain reaction (PCR) and Western blot. (B) The effect of AGEs on RAGE mRNA expression, with or without knockdown of RAGE. (C) HUVECs were transfected with non-silencing control siRNA or RAGE-specific siRNA for 48 h, then stimulated with BSA or glycol-AGEs for 24 h. The level of RAGE was analyzed by immunofluorescence staining. (D) Effect of an NADPH oxidase inhibitor (apocynin, 10 μM) and a mitochondrial respiratory chain complex inhibitor (rotenone, 10 μM) on glycol-AGEs-dependent ROS generation in HUVECs. (E) Examination of the effects of glycol-AGEs on ROS formation in RAGE knockdown HUVECs 48 h post-transfection. NC, non-silencing control; siRAGE, RAGE-specific siRNA. Bars with different letters (a, b, c, and d) differ significantly from each other (p < .001).
Effects of RAGE on mitogen-activated protein kinase phosphorylation
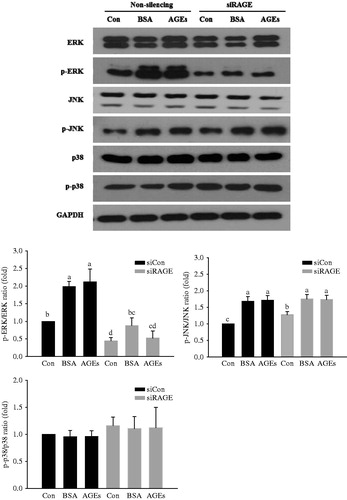
To determine the pathways involved in glycol-AGEs-induced signaling, the levels of phosphorylation of MAPK family proteins (ERK, JNK, and p38-MAPK) were measured in HUVECs stimulated with glycol-AGEs at 100 μg/mL for 10 min. As shown in , knockdown of RAGE expression with RAGE-specific siRNA attenuated glycol-AGEs-induced phosphorylation of ERK in HUVECs. However, JNK and p38 phosphorylation displayed no significant differences in the RAGE knock-down HUVECs. Taken together, these results imply that glycol-AGEs elevate RAGE expression mainly via the ERK pathway in HUVECs.
Figure 2. Activation of extracellular signal-regulated kinases (ERK), c-jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (p38 MAPK) downstream of RAGE. HUVECs were transfected with non-silencing control siRNA or RAGE-specific siRNA for 48 h before glycol-AGEs treatment. The phosphorylation levels and total protein levels of ERK, JNK, and p38 MAPK were examined by Western blot analysis. Knockdown of RAGE attenuated AGEs-induced phosphorylation of ERK, but not JNK or p38 MAPK. Levels of phospho-active kinases relative to total kinases are shown in the bar graphs. Values represent mean ± S.D. siCon, non-silencing control; siRAGE, RAGE-specific siRNA. Bars with different letters (a, b, c, and d) differ significantly from each other (p < .05).
Nuclear translocation of NF-κB
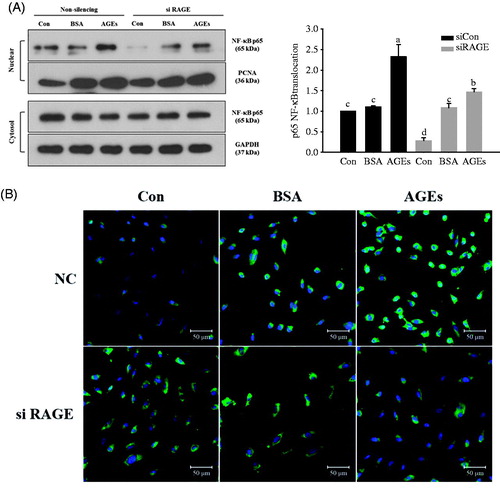
To verify the downstream action of glycol-AGEs-induced ERK in HUVECs, the nuclear translocation of NF-κB p65 was observed. Quiescent HUVECs (transfected with RAGE-specific siRNA) were treated with glycol-AGEs at 100 μg/mL for 1 h, after which NF-κB p65 activation was determined in the nuclear cell extracts by Western blotting. Stimulation of HUVECs with glycol-AGEs was shown to induce nuclear translocation of NF-κB, but translocation was significantly inhibited by RAGE knockdown (). NF-κB nuclear translocation was also monitored using immunofluorescence staining, and the nuclei were visualized with DAPI (blue). NF-κB (green) and DAPI were found to be co-localized in the nuclei of HUVECs stimulated with glycol-AGEs. However, RAGE knockdown blocked translocation of NF-κB, with the green stain remaining localized to the cytoplasm. These results indicate that blocking of RAGE expression in HUVECs prevents the translocation of NF-κB upon glycol-AGEs stimulation ().
Figure 3. Activation of nuclear factor kappa B (NF-κB) is responsible for AGEs-induced RAGE expression. (A) HUVECs were transfected with non-silencing control siRNA or RAGE-specific siRNA, and then treated with 100 μg/mL glycol-AGEs for 1 h. The nuclear fraction was used to analyze the concentration of p65, a subunit of NF-κB. Western blot showed that glycol-AGEs induced nuclear translocation of NF-κB, which was inhibited by RAGE knockdown in HUVECs. Proliferating cell nuclear antigen (PCNA), and GAPDH served as the internal controls. The bar graph shows the ratio of NF-κB localization from the cytoplasm to the nucleus, assessed using ImageJ software. (B) Examination of the translocation of p65 into nuclei via immunofluorescence staining. HUVECs were treated with 100 μg/mL glycol-AGEs for 1 h, and then stained with an antibody against p65 (green). Nuclei were stained with DAPI (blue). siCon, non-silencing control; siRAGE, RAGE-specific siRNA. Images were taken by confocal microscopy. Magnification: 100×.
Inflammatory cytokine expression in co-culture systems
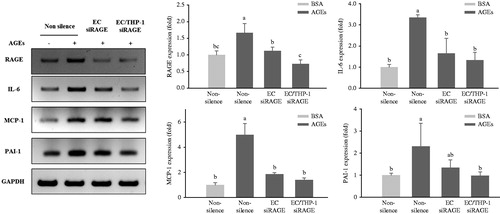
Based on our previous study (Nam et al. Citation2011) of a co-culture system with THP-1 monocytes, HUVECs and VSMCs are required to investigate the effects of the AGEs-RAGE axis on intercellular interactions occurring among these cells. As shown in , qRT-PCR revealed significant increases in the expressions of RAGE, TNF-α, and IL-1β mRNA in glycol-AGEs-induced HUVECs and THP-1 cells in the upper compartment of the insert during co-culture with VSMCs for 4 h. Under these experimental conditions, only HUVECs, but not THP-1 cells, were knocked down. However, only the group in which RAGE was knocked down in both HUVECs and THP-1 cells displayed a marked decrease in RAGE expression in the presence of glycol-AGEs (). In addition, we confirmed that the mRNA expression levels of TNF-α and IL-1β were decreased upon treatment with glycol-AGEs when both HUVECs and THP-1 cells were concurrently knocked down. Furthermore, it was observed that RAGE, IL-6, MCP-1, and PAI-1 mRNA expression was increased in VSMCs when the cells were co-cultured with THP-1 and HUVECs in the presence of glycol-AGEs for 24 h, whereas double knockdown in THP-1 and HUVECs caused a significant decrease in the expression levels of RAGE, MCP-1, IL-6, and PAI-1in VSMCs ().
Figure 4. Expression of pro-inflammatory cytokines in co-culture with HUVECs and THP-1 cells. HUVECs and THP-1 cells were co-cultured with VSMCs for 24 h after transfection with non-silencing control siRNA or RAGE-specific siRNA for 24 h. The cells were then stimulated with 100 μg/mL of glycol-AGEs for 4 h, and HUVECs and THP-1 cells were collected from the media in the upper insert. The bar graphs show the quantification of cytokine expression levels. The mRNA expressions of RAGE, tumor necrosis factor (TNF)-α and interleukin (IL)-1β are shown as fold changes (mean ± S.D.) compared with those of BSA-treated non-silenced controls. EC siRAGE, knock-down RAGE in HUVECs; EC/THP-1 siRAGE, knock-down RAGE in both HUVECs and THP-1 cells. Bars with different letters (a, b, c, and d) differ significantly from each other (p < .05).
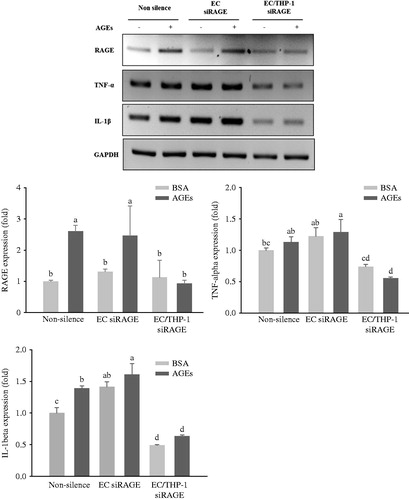
Figure 5. Expression of inflammatory cytokines in transfected HUVECs and THP-1 monocytic cells co-cultured with VSMCs. HUVECs and THP-1 cells were co-cultured with VSMCs for 24 h after transfection with non-silencing control siRNA or RAGE-specific siRNA for 24 h. The co-cultivated cells were then stimulated with 100 μg/mL of glycol-AGEs for 24 h, and then the VSMCs were collected from the lower sides of the co-cultures. The mRNA expression levels of RAGE, IL-6, monocyte chemoattractant protein (MCP)-1, and plasminogen activator inhibitor (PAI)-1 are shown as fold changes (mean ± S.D.) compared with those in BSA-treated non-silenced controls. The bar graphs show the quantification of cytokine expression, normalized to the corresponding value for GAPDH mRNA. EC siRAGE, knock-down RAGE in HUVECs; EC/THP-1 siRAGE, knock-down RAGE in both HUVECs and THP-1 cells. Bars with different letters (a, b, and c) differ significantly from each other (p < 0.05).
Impact of glycol-AGEs on VSMC function in the co-culture system
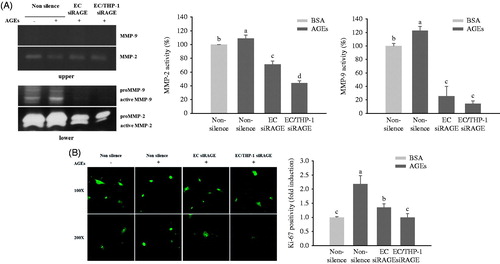
The lower compartment media from the co-culture system was next investigated for proteolytic activity. Treatment of HUVECs and THP-1 cells with glycol-AGEs stimulated proteolytic activity of MMP-2 and MMP-9 (). The mRNA levels of Pro-MMP-9, which has a molecular weight of 96 kDa and of activated MMP-9, which has a molecular weight of 88 kDa, were increased in the glycol-AGEs-treated group, whereas this was not the case in the RAGE knockdown groups. A slight induction of pro-MMP-2 and activated MMP-2, which have molecular weights of 72 kDa and 64 kDa, respectively, was shown in the glycol-AGEs-treated group but was significantly decreased in the RAGE knockdown groups. By contrast, the upper compartment media did not appear to change the activity of MMP-2, and did not produce a detectable MMP-9 band (), indicating that the MMPs did not arise from the upper insert layer (HUVECs or THP-1 cells). As shown in , treatment of HUVECs and THP-1 cells in the upper transwell insert with glycol-AGEs was found to trigger higher levels of VSMC proliferation (detected by Ki-67 staining in the nucleus) than did stimulation with BSA. The increased levels of Ki-67 expression were remarkably inhibited by RAGE knockdown in either HUVECs or both HUVECs and THP-1 cells.
Figure 6. Effects of AGEs on MMP expression and VSMC proliferation. HUVECs and THP-1 cells were co-cultured with VSMCs for 24 h after being transfected with non-silencing control siRNA or RAGE-specific siRNA. They were then treated with BSA or glycol-AGEs (100 μg/mL) for another 24 h. (A) Gelatin zymogram revealing the effects of glycol-AGEs and RAGE knockdown on inducible MMP-2 and MMP-9 activities in conditioned medium from co-cultures with HUVECs and THP-1 cells (upper), VSMCs (lower), the lower compartment was used to perform image analysis. (B) Effects of glycol-AGEs on VSMC proliferation (Ki-67 expression) induced by co-culture with HUVECs and THP-1 cells. The cells were stained with primary Ki-67 antibody and Alexa fluor® conjugated (green) secondary antibody. Magnification: 100 × (top), 200 × (bottom). Lower panels, statistical analysis of Ki-67 cell positivity as fold induction compared to the BSA-treated non-silenced control group. Each value represents the mean ± S.D. of three independent determinations. EC siRAGE, knock-down RAGE in HUVECs; EC/THP-1 siRAGE, knock-down RAGE in both HUVECs and THP-1 cells. Bars with different letters (a, b, c, and d) differ significantly from each other (p < .05).
Discussion
RAGE, existing in the plasma membrane, is a member of the immunoglobulin superfamily; it binds to AGEs on the cell surface, leading to a signaling cascade (Stern et al. Citation2002). The AGEs-RAGE interaction raises the permeability of ECs and promotes the invasion of molecules through the EC barrier (Basta et al. Citation2002). The AGEs-RAGE axis may induce intracellular ROS formation, stimulating NADPH oxidase (Wautier et al. Citation2001). In addition, other sources of ROS have been reported, including nitric oxide synthase, cytochrome P450 oxidase and several other enzymes, which may contribute to the generation of intracellular O2˙ and H2O2 in monocytes, ECs, and VSMCs (Puddu et al. Citation2008).
Our data showed that the glycol-AGEs-induced ROS generation was mainly derived from both the mitochondrial respiratory chain and NADPH oxidase in ECs (). The AGEs–RAGE interaction was previously reported to stimulate various signaling cascades involving MAPKs, such as ERK-1/2, JNK, and p38 (Ott et al. Citation2014).
However, in the present study, RAGE deletion markedly inhibited the phosphorylation of ERK, but not JNK or p38, in HUVECs, suggesting that the induction of pro-inflammatory cytokine expression by glycol-AGEs may be ERK-dependent (). Also, Yu et al. (Citation2013) reported that AGEs activated the time-dependent phosphorylation level of ERK, JNK, and p38. However, the ERK inhibitor PD98059 abolished AGE-RAGE axis-induced gene regulation. ERK phosphorylation is involved in the AGE-RAGE system. Glycol-AGEs enhanced ROS formation and activation of the ERK pathway could lead to activation of NF-κB. In our present study, control cells treated with siRAGE also showed NF-κB p65 staining (green), but p65 staining remained localized to the cytoplasm (). Addition of BSA alone to HUVECs transfected with non-silencing control siRNA stimulated translocation of NF-κB, as evidenced by immunofluorescence staining. It was reported that human serum albumin transactivated NF-κB in human proximal tubular HK-2 cells (Morigi et al. Citation2002). In that study, PKC-dependent ROS generation induced by albumin was related to the expression of the NF-κB-dependent MCP-1 gene.
The activation of NF-κB is followed by translocation into the nucleus for gene regulation. In addition, our in vivo study showed that ROS can stimulate a cascade leading to NF-κB activation, which elevated the expression of TNF-α in type 2 diabetic mice (Gao et al. Citation2008). Upon translocation to the nucleus, NF-κB increases the transcription of pro-inflammatory cytokine genes including IL-1β and TNF-α (Goldin et al. Citation2006).
In a previous study, we reported that all three cells types, including THP-1 monocytes, HUVECs, and SMCs, are required together to gain an understanding of the synergistic effects of AGEs on intercellular interactions (Nam et al. Citation2011). Co-culture of HUVECs and SMCs in the presence of glycol-AGEs and co-culture of HUVECs, SMCs, and THP-1 in the absence of glycol-AGEs, resulted in no significant increase in the proliferation of SMCs, whereas co-culture of all three cell types concurrently with glycol-AGEs treatment significantly enhanced SMC proliferation. In addition, significant induction of IL-6 and MCP-1 protein in the SMCs, which was stimulated by treatment with glycol-AGEs, was observed when cells were co-cultured with HUVECs and THP-1 cells. In the present study, glycol-AGEs were shown to be a potent stimulator of NF-κB p65 activation through ERK signaling and acted to regulate pro-inflammatory cytokines such as TNF-α or IL-1β when THP-1, HUVECs and VSMCs were co-cultured (). Moreover, NF-κB activation by AGEs has been reported to mediate the gene expression of cytokines such as IL-6 and MCP-1 in ECs and to induce VSMC activation (Hattori et al. Citation2002; Brasier Citation2010). These factors have important functions in the regulation of the inflammatory and proliferative responses in VSMCs, which are a major component of blood vessels (Chistiakov et al. Citation2015). In addition, VSMCs showed increased proliferation and production of fibronectin owing to AGEs-induced inflammatory cytokine production (Kume et al. Citation2000; Sakata et al. Citation2000; Lee et al. Citation2012). In our experiments, glycol-AGEs were found to increase the expressions of RAGE, MCP-1, IL-6, and PAI-1 in VSMCs for 24 h when cells were co-cultured with HUVECs and THP-1 cells (). Figure S1 shows that BSA in the media of the upper insert moved across the membrane, but glycer-AGEs in the same media hardly passed through the insert. Under these conditions, glycer-AGEs were less likely to directly affect VSMCs; however, increased expression of RAGE, TNF-α, and IL-1β in the glycol-AGEs-induced HUVECs and THP-1 cells in the upper compartment of the insert may trigger RAGE expression during co-culture with VSMCs in the lower compartment.
Abnormal conditions of inflammatory cells and cytokines are one of the pathological steps in the development of vascular lesions and play an important role in VSMC migration and control of the process of vascular wall remodeling (Siebenlist et al. Citation1994; Basta et al. Citation2002; Cullen et al. Citation2005). MCP-1 directs the migration of attached monocytes to the vascular wall, and was previously detected in atherosclerotic lesions, but not in normal arteries (Gu et al. Citation1998). IL-6 also plays an important role in the vessel wall, as a VSMC growth factor (Chistiakov et al. Citation2015). Moreover, PAI-1 is upregulated by various stimuli in cultured ECs, including TNF-α and IL-1β. Increased expression of PAI-1 attenuates apoptosis of VSMCs but is associated with significant proliferation after injury to arteries in rats (DeYoung et al. Citation2001). In addition, in the co-cultured system with HUVECs and VSMCs, a significant increase in both PAI-1 secretion from HUVECs and intracellular PAI-1 expression in VSMCs were observed (Gallicchio et al. Citation1994). Physiological fibrinolysis in VSMCs is controlled by specific molecules and affected by the presence of other cytokines secreted from adjacent cells (Lacolley et al. Citation2012).
On the other hand, THP 1 cells have been widely used to create a monocyte model system, because they are easily accessible without other blood contaminants and their use abolishes donor variability. Several reports have shown that the interaction between THP-1 and ECs is comparable to that between human primary monocytes and ECs (Kaplanski et al. Citation1998; Schildberger et al. Citation2013). However, THP-1 cells do not completely represent the phenotype of blood monocytes. Therefore, we investigated whether glycer-AGEs can also stimulate inflammatory mediators in ECs and human peripheral blood mononuclear cells (PBMCs) isolated from healthy donors. THP-1 and PBMCs showed comparable responses to inflammatory mediators such as TNF-α and IL-1β (Schildberger et al. Citation2013). Our findings show that glycer-AGEs clearly invoke the expression of inflammatory cytokines in HUVECs cultured with both THP-1 cells and PBMCs (Figure S2(A)), and treatment of PBMCs with glycer-AGEs released TNF-α and IL-1β to an even greater extent than did THP-1. Figure S2(B) shows that VSMCs upregulated inflammatory cytokines in the same manner when co-cultured with HUVECs and THP-1 or PBMCs.
Monocytes mature into macrophages in atherosclerotic lesions and secrete high concentrations of inflammatory cytokines and matrix metalloproteinases (MMPs), contributing to the progression of atherosclerotic plaque (Libby Citation2012). MMPs were implicated in ECM degradation and remodeling, which was another factor involved in vascular lesions, such as migration and intimal thickening (Lee & Moon Citation2005). Especially, MMP-9 is a crucial enzyme for regulating the migration and proliferation of VSMCs (Galis et al. Citation2002). The release of MMP-9, which was expressed upon treatment with TNF-α, was also reported to be mediated by transcriptional factors such as NF-κB and AP-1 via activation of the Ras/ERK pathways (Cho et al. Citation2000; Moon et al., Citation2003, Citation2004). MMP-2 and MMP-9 especially play pathogenic roles in the development of vascular remodeling by degrading various components of the ECM, and allowing invasion of collagen into the intimal layer (Rodriguez et al. Citation2007). Our results suggest that glycol-AGEs stimulate monocytes, ECs, and VSMCs in the vascular co-culture system, lead to activation of MMP-2 and 9, and can promote proliferation of VSMCs through regulation of inflammatory cytokines, NF-κB and ERK signaling (). ERK signaling promotes VSMC growth while inhibition of NF-κB decreases the proliferation of VSMCs (Maruyama et al. Citation1997). In addition, increased expression of PAI-1 inhibits apoptosis and promotes cell proliferation in VSMCs through downstream mediators (Chen et al. Citation2006). Our results clearly show that knockdown of RAGE inhibits ERK phosphorylation, NF-κB translocation to the nucleus, and consequent cytokine expression in HUVECs, as well as secretory MMP-2 and -9 activity and VSMC proliferation in our co-culture system. The results support that a co-culture system with monocytes, ECs, and VSMCs is effective for investigation of the involvement of AGEs in the intercellular signaling that occurs among these cells.
Conclusion
In conclusion, AGEs activate several signals in the endothelium, including ERK phosphorylation, NF-κB activation, and induction of inflammatory mediators, which affects VSMC proliferation and vascular remodeling. We showed that AGEs induced VSMC activation without direct treatment with inflammatory mediators such as TNF-α or IL-1β. These findings may advance the current understanding of vascular endothelial dysfunction, and may indicate a role of AGEs in the crosstalk among monocytes, ECs, and VSMCs. These results suggest that employment of a co-culture system is necessary to investigate the synergistic effects of the AGEs-RAGE axis on intercellular interactions, creating a more in-vivo-like environment to examine the implications of AGEs in atherosclerosis research.
Figures S1 and S2
Download PDF (233.1 KB)Acknowledgements
The authors thank Korea University Institute of Biomedical Science and Food Safety (Seoul, South Korea) for providing the equipment and facilities.
Disclosure statement
The authors report no conflicts of interest.
References
- Basta G, Lazzerini G, Massaro M, Simoncini T, Tanganelli P, Fu CF, Kislinger T, Stern DM, Schmidt AM, de Caterina R. 2002. Advanced glycation end products activate endothelium through signal-transduction receptor RAGE - a mechanism for amplification of inflammatory responses. Circulation. 105:816–822.
- Basta G, Schmidt AM, de Caterina R. 2004. Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc Res. 63:582–592.
- Brasier AR. 2010. The nuclear factor-kappaB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc Res. 86:211–218.
- Chen Y, Budd RC, Kelm Jr RJ, Sobel BE, Schneider DJ. 2006. Augmentation of proliferation of vascular smooth muscle cells by plasminogen activator inhibitor type 1. Arterioscler Thromb Vasc Biol. 26:1777–1783.
- Chistiakov DA, Orekhov AN, Bobryshev YV. 2015. Vascular smooth muscle cell in atherosclerosis. Acta Physiol (Oxf). 214:33–50.
- Cho A, Graves J, Reidy MA. 2000. Mitogen-activated protein kinases mediate matrix metalloproteinase-9 expression in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 20:2527–2532.
- Cullen JP, Sayeed S, Kim Y, Theodorakis NG, Sitzmann JV, Cahill PA, Redmond EM. 2005. Ethanol inhibits pulse pressure-induced vascular smooth muscle cell migration by differentially modulating plasminogen activator inhibitor type 1, matrix metalloproteinase-2 and -9. Thromb Haemost. 94:639–645.
- Davignon J, Ganz P. 2004. Role of endothelial dysfunction in atherosclerosis. Circulation. 109:III27–III32.
- DeYoung MB, Tom C, Dichek DA. 2001. Plasminogen activator inhibitor type 1 increases neointima formation in balloon-injured rat carotid arteries. Circulation. 104:1972–1977.
- Galis ZS, Johnson C, Godin D, Magid R, Shipley JM, Senior RM, Ivan E. 2002. Targeted disruption of the matrix metalloproteinase-9 gene impairs smooth muscle cell migration and geometrical arterial remodeling. Circ Res. 91:852–859.
- Gallicchio M, Argyriou S, Ianches G, Filonzi EL, Zoellner H, Hamilton JA, Mcgrath K, Wojta J. 1994. Stimulation of PAI-1 expression in endothelial cells by cultured vascular smooth muscle cells. Arterioscler Thromb. 14:815–823.
- Gao X, Zhang H, Schmidt AM, Zhang C. 2008. AGE/RAGE produces endothelial dysfunction in coronary arterioles in type 2 diabetic mice. Am J Physiol Heart Circ Physiol. 295:H491–H498.
- Goldin A, Beckman JA, Schmidt AM, Creager MA. 2006. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. 114:597–605.
- Gu L, Okada Y, Clinton SK, Gerard C, Sukhova GK, Libby P, Rollins BJ. 1998. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell. 2:275–281.
- Hattori Y, Suzuki M, Hattori S, Kasai K. 2002. Vascular smooth muscle cell activation by glycated albumin (Amadori adducts). Hypertension. 39:22–28.
- Kaplanski G, Marin V, Fabrigoule M, Boulay V, Benoliel AM, Bongrand P, Kaplanski S, Farnarier C. 1998. Thrombin-activated human endothelial cells support monocyte adhesion in vitro following expression of intercellular adhesion molecule-1 (ICAM-1; CD54) and vascular cell adhesion molecule-1 (VCAM-1; CD106). Blood. 92:1259–1267.
- Kume N, Moriwaki H, Kataoka H, Minami M, Murase T, Sawamura T, Masaki T, Kita T. 2000. Inducible expression of LOX-1, a novel receptor for oxidized LDL, in macrophages and vascular smooth muscle cells. Ann N Y Acad Sci. 902:323–327.
- Lacolley P, Regnault V, Nicoletti A, Li Z, Michel JB. 2012. The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res. 95:194–204.
- Lee B, Moon SK. 2005. Resveratrol inhibits TNF-alpha-induced proliferation and matrix metalloproteinase expression in human vascular smooth muscle cells. J Nutr. 135:2767–2773.
- Lee E, Grodzinsky AJ, Libby P, Clinton SK, Lark MW, Lee RT. 1995. Human vascular smooth muscle cell-monocyte interactions and metalloproteinase secretion in culture. Arterioscler Thromb Vasc Biol. 15:2284–2289.
- Lee IT, Shih RH, Lin CC, Chen JT, Yang CM. 2012. Role of TLR4/NADPH oxidase/ROS-activated p38 MAPK in VCAM-1 expression induced by lipopolysaccharide in human renal mesangial cells. Cell Commun Signal. 10: 33.
- Libby P. 2012. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 32:2045–2051.
- Lukacs NW, Strieter RM, Elner V, Evanoff HL, Burdick MD, Kunkel SL. 1995. Production of chemokines, interleukin-8 and monocyte chemoattractant protein-1, during monocyte endothelial-cell interactions. Blood. 86:2767–2773.
- Maruyama I, Shigeta K, Miyahara H, Nakajima T, Shin H, Ide S, Kitajima I. 1997. Thrombin activates NF-kappa B through thrombin receptor and results in proliferation of vascular smooth muscle cells: role of thrombin in atherosclerosis and restenosis. Ann N Y Acad Sci. 811:429–436.
- Moon SK, Cha BY, Kim CH. 2004. ERK1/2 mediates TNF-alpha-induced matrix metalloproteinase-9 expression in human vascular smooth muscle cells via the regulation of NF-kappaB and AP-1: involvement of the Ras dependent pathway. J Cell Physiol. 198:417–427.
- Moon SK, Cho GO, Jung SY, Gal SW, Kwon TK, Lee YC, Madamanchi NR, Kim CH. 2003. Quercetin exerts multiple inhibitory effects on vascular smooth muscle cells: role of ERK1/2, cell-cycle regulation, and matrix metalloproteinase-9. Biochem Biophys Res Commun. 301:1069–1078.
- Morigi M, Macconi D, Zoja C, Donadelli R, Buelli S, Zanchi C, Ghilardi M, Remuzzi G. 2002. Protein overload-induced NF-kappaB activation in proximal tubular cells requires H(2)O(2) through a PKC-dependent pathway. J Am Soc Nephrol. 13:1179–1189.
- Mudau M, Genis A, Lochner A, Strijdom H. 2012. Endothelial dysfunction: the early predictor of atherosclerosis. Cardiovasc J Afr. 23:222–231.
- Nam MH, Lee HS, Seomun Y, Lee Y, Lee KW. 2011. Monocyte-endothelium-smooth muscle cell interaction in co-culture: proliferation and cytokine productions in response to advanced glycation end products. Biochim Biophys Acta. 1810:907–912.
- Ott C, Jacobs K, Haucke E, Santos AN, Grune T, Simm A. 2014. Role of advanced glycation end products in cellular signaling. Redox Biol. 2:411–429.
- Puddu P, Puddu GM, Cravero E, Rosati M, Muscari A. 2008. The molecular sources of reactive oxygen species in hypertension. Blood Press. 17:70–77.
- Riganti C, Costamagna C, Doublier S, Miraglia E, Polimeni M, Bosia A, Ghigo D. 2008. The NADPH oxidase inhibitor apocynin induces nitric oxide synthesis via oxidative stress. Toxicol Appl Pharmacol. 228:277–285.
- Rodriguez JA, Orbe J, Paramo JA. 2007. Metalloproteases, vascular remodeling and atherothrombotic syndromes. Rev Esp Cardiol. 60:959–967.
- Ross R. 1993. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 362:801–809.
- Sakata N, Meng J, Takebayashi S. 2000. Effects of advanced glycation end products on the proliferation and fibronectin production of smooth muscle cells. J Atheroscler Thromb. 7:169–176.
- Schildberger A, Rossmanith E, Eichhorn T, Strassl K, Weber V. 2013. Monocytes, peripheral blood mononuclear cells, and THP-1 cells exhibit different cytokine expression patterns following stimulation with lipopolysaccharide. Mediators Inflamm. 2013:697972.
- Siebenlist U, Franzoso G, Brown K. 1994. Structure, regulation and function of NF-kappa B. Annu Rev Cell Biol. 10:405–455.
- Sitia S, Tomasoni L, Atzeni F, Ambrosio G, Cordiano C, Catapano A, Tramontana S, Perticone F, Naccarato P, Camici P, et al. 2010. From endothelial dysfunction to atherosclerosis. Autoimmun Rev. 9:830–834. ]
- Stern D, Yan SD, Yan SF, Schmidt AM. 2002. Receptor for advanced glycation end products: a multiligand receptor magnifying cell stress in diverse pathologic settings. Adv Drug Deliv Rev. 54:1615–1625.
- Tsouknos A, Nash GB, Rainger GE. 2003. Monocytes initiate a cycle of leukocyte recruitment when cocultured with endothelial cells. Atherosclerosis. 170:49–58.
- Wautier MP, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier J-L. 2001. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab. 280:E685–E694.
- Wu X, Zhou Q, Huang L, Sun A, Wang K, Zou Y, Ge J. 2008. Ageing-exaggerated proliferation of vascular smooth muscle cells is related to attenuation of Jagged1 expression in endothelial cells. Cardiovasc Res. 77:800–808.
- Yu L, Zhao Y, Xu S, Ding F, Jin C, Fu G, Weng S. 2013. Advanced glycation end product (AGE)-AGE receptor (RAGE) system upregulated Connexin43 expression in rat cardiomyocytes via PKC and Erk MAPK pathways. Int J Mol Sci. 14:2242–2257.
- Zhao W, Ma G, Chen X. 2014. Lipopolysaccharide induced LOX-1 expression via TLR4/MyD88/ROS activated p38MAPK-NF-κB pathway. Vascul Pharmacol. 63:162–172.