
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Edotecarin is an indolocarbazole class antitumor agent that has significant anticancer effects against various types of cancer, especially lung, breast, and stomach cancer.The conformation analysis of the edotecarin was performed using the PM3 method and six stable conformations were obtained.Afterwards the obtained lowest energy conformation was optimized at the DFT/B3LYP/6-31++G(d,p) level of theory. The vibrational wavenumbers, the highest occupied molecular orbital, the lowest unoccupied molecular orbital and molecular electrostatic potential of the most stable conformer of edotecarin were calculated at the DFT/B3LYP/6-31++G(d,p) level of theory.The molecular docking of the edotecarin molecule against DNA, Topoisomerase I, DNA-Topoisomerase I complex,α5β1 and αIIbβ3 integrins were performed to reveal its binding modes and binding affinities.
Introduction
Edotecarin, {6-N-(1-hydroxymethyl-2-hydroxy)ethylamino-12,13-dihydro-2,10-dihydroxy-13-(b-D-glucopyranosyl)-5H-indolo[2,3-a]-pyrrolo[3,4-c]-carbazole-5,7(6H)-dione}, was derived from the glucosyl derivative of indolo-carbazole (NB-506) [Citation1]. It is a topoisomerase I (TOPI) inhibitor, induces single-strand DNA cleavage more effectively than NB-506, thus having potential use in solid tumor treatment [Citation1]. In vitro research results indicate that edotecarin is a novel class of TOPI inhibitor with different and superior characteristics compared to other clinically used anticancer drugs [Citation2]. Studies of edotecarin against numerous human xenografts, murine tumors and their lethal toxicity in mice were performed in a study conducted by Arakawa et al. [Citation3]. The antitumor activity of edotecarin was tested in vitro and in vivo to observe the inhibition of tumor growth in models of breast, cervix, pharynx, lung, prostate, colon, stomach and hepatic cancer in a study conducted by Saif et al. Edotecarin was seen to be effective in cells with resistance acquired due to P-glycoprotein [Citation4] and has been shown to demonstrate significant antitumor's activity in human models of xenograft breast cancer and chemically and hormonally affected oncogene-derived breast tumors in a study published in 2007. Moreover, this compound has been shown to improve the effects of docetaxel and capecitabine in xenografts of human breast cancer. Therefore, it has been reported that the edotecarin molecule is an important therapeutic alternative for breast cancer patients as monotherapy or in conjunction with other treatment modalities [Citation5]. The DNA-topoisomerase I complex (DNA-TOPI complex) is stabilized by the edotecarin molecule. This culminated in the failure of cells to unwind DNA and hence preventing them from dividing [Citation3, Citation6]. A 2010 study by Saif et al. showed that in combination with 5-fluorouracil (5-FU) [Citation7], edotecarin, a novel topoisomerase I inhibitor, has an additional effect on colon cell lines (HCT-116) [Citation7]. Molecules with one or more degrees of freedom of rotation exhibit conformational isomerism leading to stable minimum-energy conformations. Conformation analysis plays an important role in biological activity since most mechanisms of action depend on the compatibility of the active site of the enzyme with the substrate, several methods of structure-activity relationship have been established to describe the three-dimensional structure of drug-like molecules [Citation8].
Literature survey reveals that, although having higly biological activity, no conformational analysis study on edotecarin molecule, and molecular docking simulations of edotecarin against DNA, Topoisomerase I, DNA-Topoisomerase I complex and integrins have been reported so far. In this study, vibration assignments, HOMO-LUMO, frontier molecular orbital (FMO) energies, MEP and molecular docking simulations were performed on the most stable conformer of the edotecarin molecule obtained by applying conformational analysis and then optimization at the DFT/B3LYP/6-31++G(d,p) theory level. Moreover, the drug likeness and ADMET properties of edotecarin were analyzed. These calculations are important to provide insight into molecular and pharmaceutical properties and the interaction mechanisms of edotecarin.
Methods and calculations
The conformational analysis of the edotecarin molecule was performed using Spartan06 program software [Citation9] with the PM3 method [Citation10–13] and six lowest energy conformers within 0-0.76 kcal/mol relative energies were determined. Among them, the lowest energy conformer (I) was then optimized using the Gaussian03 program [Citation14], DFT/B3LYP level of theory [Citation15], and 6-31++G(d,p) basis set. By the scaled quantum mechanical force field approach proposed by Pulay et al. (1983) [Citation16], the harmonic force field of edotecarin was defined using the MOLVIB program [Citation17–18]. The vibration modes' IR intensity, Raman activity and potential energy distribution (PED) were determined; the force fields of the Cartesian coordinate were translated into natural internal coordinates [Citation17, Citation18]. The calculated Raman activity of edotecarin was converted into Raman intensity by the simulation software Simirra [Citation19]. Lorentzian band shapes with a bandwidth (FWHM) of 10 cm−1 were used for the theoretical calculations. To provide the best fit for the experimental results, the following scale factors for DFT/B3LYP/6-31++G(d,p) level of theory calculations were selected:
Results and discussion
Structure
The comparison of the energies of the six lowest energy conformers of edotecarin () obtained as a result of the conformational analysis is given in . The differing dihedral angles of these six conformers are given in comparatively.
Figure 1. The six conformers with the lowest energy, obtained by conformational analysis of the edotecarin molecule.
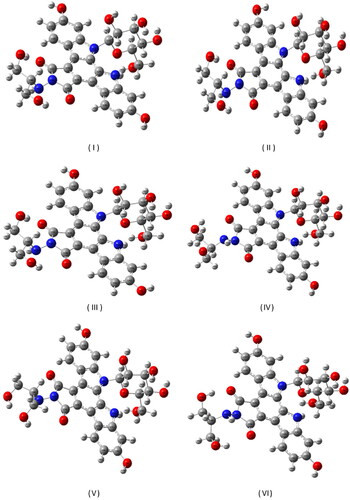
Table 1. The energies of the six most stable conformation obtained by conformation analysis.
Table 2. The dihedral angles differing in the six low energy conformers obtained by conformational analysis.
Following the conformation analysis, the lowest energy conformer (I) was optimized using the DFT/B3LYP level of theory and the 6-31++G(d,p) base set and the most stable molecular geometry was reached. The optimized geometry of edotecarin molecule obtained by DFT/B3LYP/6-31++G(d,p) level of theory was given in . In the optimized geometry parameters of edotecarin molecule were given and in some of the optimized parameters were compared with available related data.
Figure 2. The most stable geometric structure of the edotecarin molecule obtained with the DFT/B3LYP/6-31++G(d,p) level of theory.
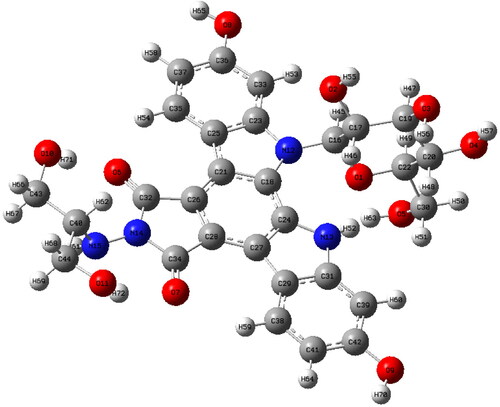
Table 3. Edotecarin's optimized geometry parameters * were calculated using 6-31++G(d,p) basis set at DFT/B3LYP level of theory.
Table 4. Comparison between the optimized parameters of the edotecarin molecule and the experimental [Citation20, Citation21] parameters of the carbazole molecule and also the theoretical [Citation22] parameters of the geometric 4-hydroxy carbazole molecule.
Molecular electrostatic potential
Dipole moment, electronegativity, and partial charges are associated by molecular electrostatic potential (MEP). It gives a simple way to understand a molecule's relative polarity. It is very beneficial in recognizing the electrophilic and nucleophilic reaction sites for the identification of interactions with hydrogen bonding. Negative electrostatic potential refers to the attracting of the proton by the molecular concentrated electron density (from lone pairs, pi-bonds, etc). Positive electrostatic potential corresponds to repulsion of the positive charge (proton) in regions where low electron density exists. The big differences in red and blue means the more the molecule is polar. MEP were calculated for the investigated molecule (red as negative, blue as positive) at the optimized geometry obtained using DFT method at B3LYP/6-31++G(d,p) level of theory. shows the molecular electrostatic potential surface of edotecarin. The red and blue colors regions represents the minimum electrostatic potential and the maximum of electrostatic potential, respectively.
Figure 3. Molecular electrostatic potential (MEP) of Edotecarin obtained by DFT/B3LYP/6- 31++G(d,p).
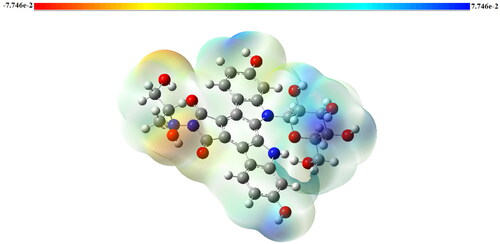
The color code of the edotecarin map changes in a range of −2.108 V (dark red) to 2.108 V (dark blue). shown that, the regions with positive potential value are predominantly positioned as possible nucleophilic attack sites above the hydrogen atoms belonging to the OH group. Also, as possible sites for electrophilic attack, the negative potential regions are located around oxygen atoms.
Vibrational spectral analysis
Edotecarin molecule (C29H28N4O11) has 72 atoms thus, 210 vibrational modes with C1 symmetry all modes are IR and Raman active. The wavenumbers of the vibrational modes of the title molecule, based on calculated potential energy distributions, IR and Raman intensities, are identified in . The simulated Raman and IR spectra of Edotecarin are shown in .
Figure 4. Calculated IR (a) and Raman (b) spectra of Edotecarin.
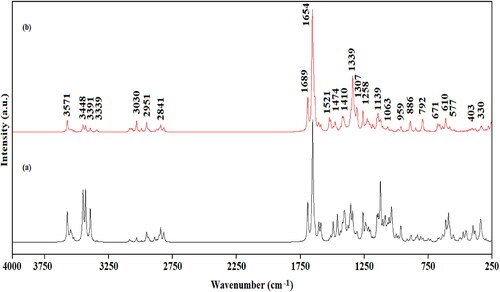
Table 5. Calculated wavenumbers (cm-1), calculated IR, Raman intensities (I) and the potential energy distribution of the vibrational modes of the Edotecarin.
The seven OH stretching modes (νOH) of the edotecarin were computed in the 3571-3428 cm−1 region (with PED contribution 94-100%). This mode for the 4-hydroxy carbazole molecule was observed at 3393 cm−1 in the FTIR spectrum and calculated at 3854 cm−1 at the DFT/B3LYP/6-311++G(d,p) level of theory [Citation22]. Morever in the Gly-Tyr molecule, the OH stretching modes were observed at 3566 and 3446 cm−1 in the FTIR spectrum and at 3577 and 3448 cm−1 in the Raman spectrum [Citation23].
The molecule has two N-H groups. These N-H stretching vibrations were calculated at 3391 and 3339 cm−1 as a pure mode (with PED contribution 100-97%). The N-H stretching mode for the 4- hydroxy carbazole molecule was observed at 3210 cm−1 in the FTIR spectrum and was calculated with the DFT/B3LYP/6-311++G(d,p) level of theory at 3670 cm−1 [Citation22]. This mode was computed at 3542 cm−1 for the 2,7-dimethoxy-9H-carbazole-3-carbaldehyde molecule using the B3LYP/LanL2Z level of theory [Citation24], for Gly-Tyr dipeptide the N-H stretching vibrations were observed at 3321 and 3210 cm−1 in the Raman and at 3326 cm−1 in the FTIR spectra [Citation23]. Our results are in Accord with the previous findings.
The CH stretching vibrations of edotecarin were computed in the range of 3086-2816 cm−1 with 92-99% PED contributions. The CH stretching vibrations of 4-hydroxy carbazole molecule were observed at 3053 and 3061 cm−1 in FTIR and Raman spectra, respectively and was calculated in the range of 3199-3149 cm−1 using the DFT/B3LYP/6-311++G(d,p) level of theory [Citation22]. In addition, the CH stretching vibrations of 2,7-dimethoxy-9H-carbazole-3-carbaldehyde molecule were found in the range of 3110-2905 cm−1 in calculations using the B3LYP/LanL2Z theory level [Citation24]. For 9-p-tolyl-9H-carbazole-3-carbaldehyde molecule, the CH stretching modes were observed in the 3056-2764 cm-1 range in the FTIR spectrum[Citation25], and for the CLTOAB molecule, the CH stretching modes were observed in the 2994-2850 cm−1 and 2993-2846 cm−1 ranges, in the ATR-IR and Raman spectra respectively [Citation26].
The vibrational wavenumbers of the C = O stretching modes of the edotecarin were computed at 1689 and 1633 cm−1 with 75% and 42% PED contributions, respectively. The same vibrations were calculated at 1619 and 1603 cm−1 for the 2,7-dimethoxy-9H-carbazole-3-carbaldehyde molecule using the B3LYP/LanL2Z level of theory [Citation24]. The C = O stretching vibration of the 9-p-tolyl-9H-carbazole-3-carbaldehyde molecule was observed at 1686 and 1692 cm−1 in the FTIR and Raman spectra, respectively [Citation25], and the C = O stretching modes of the Gly-Tyr dipeptide were observed in the 1698-1635 cm−1 and 1692-1630 cm−1 ranges, in the FT-IR and Raman spectra, respectively.
The computed 1468 and 1465 cm−1 wavenumbers were assigned to HCH in-plane angle bending vibrations of edotecarin, in accord to PED values. For the Val-Met dipeptide [Citation27] the HCH in plane bending vibrations were experimentally observed at 1451 and 1437 cm−1 and computed at 1455 and 1438 cm−1and for CLTOAB, were calculated in the range of 1487-1432 cm−1 and observed in the 1484-1416 cm−1.
The calculated N-N stretching vibrations of edotecarin were obtained as mixed vibrations at 1410 and 1139 cm−1, with 29% and 10% PED contributions, respectively. For the benzyl (imino (1H-pyrazol-1-yl) methyl) carbamate molecule, by using B3LYP/6-31G(d,p) level of theory, the N-N stretching modes were computed at 1269 (46% PED) and 1203 cm−1 (77% PED) [Citation28]. In the study on the 3-amino-1,2,4-triazole molecule, the N-N stretching vibrations were estimated at 1247; 1146; 1066; 1023 and 952 cm−1 with 10, 15, 12, 32 and 10% PED contributions, respectively, using with B3LYP/6-311++G(d,p) level of theory and the corresponding bands were observed at 1266; 1153; 1047; 1026 and 926 cm−1, respectively in the FTIR spectrum [Citation29].
Frontier molecular orbital analysis
The frontier molecular orbitals, namely, the highest energy occupied orbital (HOMO) and the lowest energy empty orbital (LUMO), are the molecular orbitals that participate in the chemical reactions or interactions with other species. Their energy difference (gap) gives information about the reactivity of the molecule and allow us to comprehend the wavenumbers they emit/reflect [Citation30].
By Time-Dependent Density Functional Theory (TD-DFT) using B3LYP/6-31++G(d,p) basis set, the energies of the frontier molecular orbitals of edotecarin molecule was determined. The energy gap between the ground energy level and the first excited state of edotecarin was found to be 2.769 eV, which indicated the presence of the charge transfer interaction in the molecule and reflected the biological activity of the molecule.
The percentage of contributions of atomic orbitals to HOMO and LUMO (frontier orbitals) was estimated using the program GaussSum (Version, 3.0) [Citation31]. gives the measured transition energies, oscillator strengths and main contributions. displays the schematic diagrams of the frontier HOMO and LUMO orbitals of edotecarin. The HOMO-LUMO energy difference is calculated to be 4.8 and 4.21 eV by DFT/B3LYP/6-31G(d) calculations in a report on carbazole and benzocarbazole, respectively [Citation32].
Figure 5. The atomic orbital HOMO-LUMO composition of the frontier molecular orbital for Edotecarin.
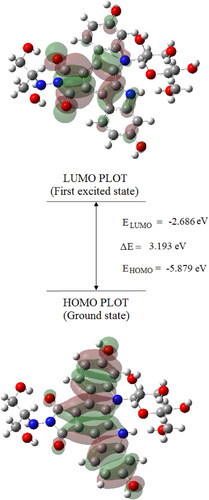
Table 6. Selected calculated positions of the pure electronic transitions, oscillator strengths (f) and major contributions.
In silico molecular docking studies of edotecarin as anticancer agent
Edotecarin (J-107088) is an DNA topoisomerase-1 inhibitor, and is known to interact with DNA and additionally stabilize the DNA-TOPI complex [Citation33, Citation34]. Edotecarin is a derivative of a compound called NB-506, an indolocarbazole antitumor agent, but edotecarin is a strong and selective TOPI inhibitor that is more effective at inducing single-stranded DNA cleavage than NB-506 [Citation4, Citation35, Citation36].
In order to investigate the interactions of edotecarin molecule with the target DNA, TOPI and TOPI-DNA complex, molecular docking simulations were performed. Moreover, the molecular docking simulations of edotecarin with the target proteins αIIbβ3 and α5β1 integrins were also performed to reveal its anticancer properties
The docking study of the edotecarin molecule was carried out using AutoDockVina [Citation37] and the crystal structures of DNA (PDB ID: 1BNA) and Topoisomerase I/DNA Complex (PDB ID: 1A36) were obtained with reference to the protein database [Citation38–40]. Before the docking analysis, DNA was modified for the docking analysis by extracting water molecules and adding polar hydrogens, and the DNA charges of Kollman were determined. Partial charges of the edotecarin molecule were calculated using the Geistenger method and the active region of DNA is defined as in size 40 Å × 40 Å × 40 Å.
The optimized structure of the edotecarin molecule, calculated in gas phase, was found to bind to the nucleic acids DC9, DG10, DC11, DG12, DG16 and DA17 by hydrogen bonds and Pi-Anion interaction (See ). The result shows that the binding affinity (ΔG) of the interaction of edotecarin with DNA is −8.1 kcal/mol. The identified active site of DNA was in accordance with the literature [Citation41–43]. The interactions between the edotecarin molecule and nucleic acids are described below:
Figure 6. Molecular docked models of edotecarin with DNA (a),The interactions between the edotecarin and DNA are labeled using colored dashed lines(b) (-8.1 kcal/mol).
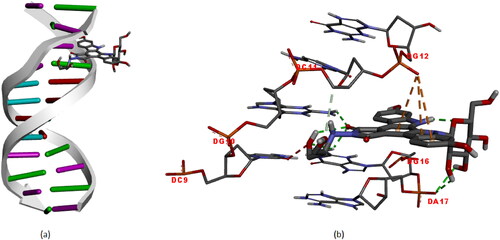
Unfavorable acceptor-acceptor interaction with a length of 2.71 Å between DC9 and the oxygen atom; Hydrogen bond interaction with a length of 2.44 Å between DC10 and oxygen atom; Carbon hydrogen bond interaction with a length of 3.67 Å between DC11 and oxygen atom; Pi-Anion interaction between DG12 and edotecarin molecule at lengths of 4.67, 4.31, 4.90 Å; Hydrogen bond interactions between DG16 and oxygen atom with lengths of 2.45, 2.51 and 2.19 Å; Hydrogen bond interaction between DA17 and hydrogen atom with lengths of 2.77 and 2.38 Å.
In a study performed by Das et al., the DNA docking of various piperidines that have antitumor activity were investigated by molecular dynamics method [Citation44]. The active site of DNA determined in DNA-piperidine complexes, is compatible with the active site of DNA where edotecarin interacts, found in this study. Hydrophobic interaction and hydrogen bonding have been determined to contribute to stabilizations of DNA-piperidine complexes [Citation44].
Before the docking analysis, the same preparation was made for Topoisomerase I, i.e. DNA and water molecules were extracted from the Topoisomerase I/DNA Complex, polar hydrogens were added and Topoisomerase I 's Kollman charges were calculated, and Topoisomerase I was prepared for docking analysis. And also the partial charges of the Edotecarin molecule were calculated using the Geistenger method. Using CAVER [Citation45], the active site of Topoisomerase I can be determined at max. Probe. The grid size is then specified as 40 Å × 40Å × 40Å for docking calculation. Optimized structure of the edotecarin molecule, determined with DFT/B3LYP/6-31++G(d,p) level of theory in the gas phase, binds to the amino acids of TOPI (ARG364, GLN421, ASN491, LYS493, THR498, ALA499, THR501, LYS532 ve ASP533) with −8.9 kcal/mol binding affinity(ΔG) ().
Figure 7. (a) and (b) Different representations of Edotecarin docking with TOPI. Dotted lines indicate interactions (-8.9 kcal/mol).
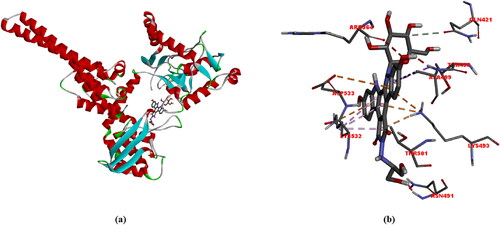
Interactions between the edotecarin molecule and Topoisomerase I are described below: Unfavorable acceptor-acceptor interaction with a length of 2.8 Å between ARG364 and the oxygen atom; Carbon hydrogen bond interaction with a length of 3.71 Å between GLN421 and edotecarin molecule; Unfavorable donor-donor interaction with a length of 1 Å between ASN491 and the Hyrogen atom; Between LYS493 and edotecarin molecule: Pi-Cation; Pi-Donor hydrogen bond interaction with a length 2.85 Å, Pi-Cation interaction with a 3.58 Å length, Pi-Cation interaction with a 4.02 Å length; Pi-Sigma interaction with a length of 3.99 Å between THR498 and edotecarin molecule; Unfavorable donor-donor interaction with a length of 1.41 Å between ALA499 and the edotecarin molecule and Pi-Alkyl interaction with a length of 5.09 Å; Hydrogen bond interaction with a length of 1.86 Å between THR501 and oxygen atom; Between LYS532 and edotecarin molecule: Pi-Cation interaction with a length 3.78 Å, Pi-Alkyl interaction with a 5.16 Å length, Pi-Alkyl interaction with a 4.37 Å length and Pi-Alkyl interaction with a 3.71 Å length; Pi-Anion interaction with a length of 4.82 Å between ASP533 and edotecarin molecule.
In a recent study (Dario et al.), the interaction of the vanadyl complex, an antitumor agent, with TOPI was investigated using molecular docking simulations and showed interactions of the vanadyl complex at different sites of TOPI [Citation46]. It was reported that the vanadyl complex interacted with the amino acids LEU485, ASN539, LEU629, ASN631, GLN713 and ILE714 in the external region of TOPI and interacted with LYS493, GLU495, GLY496, THR498, ALA499, ASP500, GLU561, ASP562 ve ASP563 residues in the lips region of TOPI. The complex interacted with GLU492, GLN421 and GLU494, in the center channel between the lips, where the DNA is bound for the activity of the protein. The GLN421 and ALA499 amino acids of TOPI where vanady complex interacted, in the center channel between lips and in lips regions, respectively, are the amino acids that edotecarin interacts. Thus docking simulations indicates that active site, where edotecarin docked, is the central channel between the lips close to lips region.
Before the docking analysis, the same process was made DNA-TOPI complex, i.e. water molecules were extracted, polar hydrogens were added, and DNA-TOPI complex 's Kollman charges were calculated, and DNA-TOPI complex was prepared for docking analysis. The active site of the DNA-TOPI complex was also determined according to the max probe value using the CAVER program [Citation45]. Then, grid size was defined as 60 Å × 60 Å × 60 Å for docking calculation.
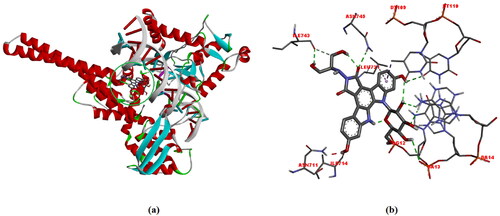
Optimized edotecarin molecule structure, determined by DFT/B3LYP/6-31++G(d,p) gas-phase theory, binds to DNA-TOPI amino acid complexes (ASN711, ILE714, LEU721, ILE743 ve ASN745) and also binds to DNA residues (DG12, DA13, DA14, DT109 ve DT110) (). It is quite consistent with literature studies [Citation47–50]. The binding affinity of this interaction (ΔG) is −10.7 kcal/mol.
Figure 8. (a) Structural model of DNA-TOPI complexes with ligand edotecarine binding site (green sphere), (b)The interactions between the edotecarin and DNA-TOPI complexes are labeled using colored dashed lines (-10.7 kcal/mol).
The results revealed that edotecarin is bound to the DNA with weak non-covalent interactions. More specifically it is bonded to DNA with conventional hydrogen bonding, carbon-hydrogen bond, pi-alkyl, pi-sigma and unfavorable donor-donor interactions. The details of the interactions are given below:
Unfavorable donor-donor interaction, 1.97 Å in length, between ASN711 and the edotecarin molecule; hydrogen bond interaction with ILE714 (2.49 Å); pi-alkyl interaction (5.46 Å) with LEU721; conventional hydrogen bond (2.42 Å) and carbon hydrogen bond (3.49 Å) interactions with ILE743; hydrogen bond interaction (2.33 Å) with ASN745; hydrogen bond interaction (2.39) Å with DG12; hydrogen bond interactions (2.59 Å and 3.02 Å) with DA13; hydrogen bond interaction (2.24 Å) with DA14; hydrogen bond (2.58 Å) and pi-sigma (3.77 Å) interactions with DT109; hydrogen bond interaction (2.75 Å) with DT110.
In order to examine the anti-proliferation effect of the edotecarin molecule for the anticancer function, molecular docking studies in target proteins α5β1 and αIIbβ3 integrins were performed.
After the α5β1 integrin (PDB ID: 4WK0) was prepared for molecular docking, the active side was determined at max probe by CAVER [Citation45]. The 3 D docked view of edotecarin in α5β1 integrin and the estimated interactions are shown in . The predicted binding affinity for α5β1 integrin is −9.1 kcal/mol (Kd = 0.212 µM). The interactions can be summarized as follows:
Figure 9. The 3 D docked view of the optimized structure of edotecarin in α5β1 integrin. The ligand interaction diagrams of receptor ligand complexes are Show (binding affinity -9.1 kcal/mol).
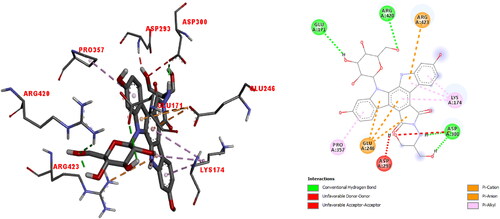
Unfavorable donor-donor interaction (1.48 Å) with ASP293; hydrogen bond (1.81 and 1.98 Å) and unfavorable acceptor-acceptor (2.7 Å) interactions with ASP300; pi-anion interactions (4.62, 4.01 and 3.85 Å) with GLU246; pi-alkyl interactions (5.39, 4.43 and 4.13 Å) with LYS174; hydrogen bond interaction (2.98 Å) with GLU171; pi-cation interaction (4.99 Å) with ARG423; hydrogen bond interaction (2.59 Å) with ARG420 and pi-alkyl interaction (5.33 Å) with PRO357.
In our previous study on the interaction of cationic pentapeptide, Glu-Gln-Arg-Pro-Arg, with α5β1 integrin [Citation51], it was found the the ligand interacted with PHE21,SER22,VAL23,ARG106, PHE168, GLU171, SER234, VAL235, LYS269, TYR287, LEU355, SER417 and ARG420, through salt bridge, attractive charge, hydrogen bond, carbon hydrogen bond, unfavorable positive-positive, unfavorable donor-donor interactions. The active site, where cationic pentapeptide docked [Citation51] was found to be highly compatible with the active site found by our calculations.
After the αIIbβ3 integrin (PDB ID: 3ZDX) was prepared for molecular docking, the active side was determined at max probe by CAVER [Citation45]. The 3 D docked view of edotecarin in αIIBβ3 integrin and the estimated interactions are shown in . The predicted binding affinity for αIIBβ3 integrin is −7.9 kcal/mol (Kd = 1.6 µM). The interactions can be summarized as follows:
Figure 10. The 3 D docked view of the optimized structure of edotecarin in αIIbβ3 integrin. The ligand interaction diagrams of receptor ligand complexes are Show (binding affinity -7.9 kcal/mol).
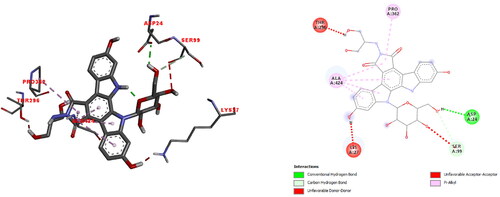
Unfavorable donor-donor interaction (1.89 Å) with LYS27; hydrogen bond interaction (2.2 Å) with ASP24; unfavorable acceptor-acceptor (3.0 Å) and carbon-hydrogen bond interactions (3.47 Å) with SER99; pi-alkyl interactions (4.36, 4.47, 4.55 ve 4.94 Å) with ALA424; pi-alkyl interaction (5.26 Å) with PRO362 and unfavorable donor- donor interaction (1.67 Å) with THR296.
In the previous study on the docking simulations of cationic pentapeptide, Glu-Gln-Arg-Pro-Arg, with αIIbβ3 integrin [Citation51], it was found the the ligand interacted with LEU23, ASP24, PHE25, ALA95, SER96, SER99, PHE171, SER173, TYR237, SER238, ARG261, HIS291, VAL293, ILE360, SER420, LEU421, ARG422 residues of integrin, through salt bridge, attractive charge, hydrogen bond, carbon hydrogen bond, unfavorable positive-positive, unfavorable donor-donor interactions. The active site of integrin, where cationic pentapeptide docked [Citation51] was found to be highly compatible with the active site found by our calculations.
The strong binding affinities of endotecarin molecule to the integrins α5β1 and αIIbβ3, with Kd values of 0.21 µM and 1.6 µM, respectively, shows its the anti-cancer activity.
Analysis of toxicological and physicochemical properties
The program can calculate lipophilicity (expressed as clogP), solubility in water (expressed as logS), molecular weight(MW), drug-likeness indices and drug scores. Moreover, the toxicological properties of the compounds may be shown also. CLogP is a central parameter in drug discovery and environmental toxicity research and this value must not be greater than 5.0 [Citation52, Citation53]. As increasing molecular weight decreases absorption, maintaining less molecular weight is an essential part of the drug development process. The solubility (logS) properties of a substance in aqueous solution influence the absorption and distribution characteristics. The solubility of a compound is predicted using the logS-based OSIRIS method. The preferred value is more than −4 [Citation52, Citation53].
TPSA is a molecular polar surface region and the transport properties of a drug are defined by it. This property is described by polar atoms and is predicted using the various classical 3 D PSA methods and the sum of tabulated surface contributions of polar fragments [Citation52, Citation53]. Drug-likeness is described on the basis of the molecular characteristics of the drugs advertised. The drug-likeness is the sum of the molecule score values of the fragments present. The positive drug-likeness value suggests that the molecule being tested mainly contains fragments that are commonly found in the products being marketed [Citation52, Citation53].
Using the OSIRIS Property Explorer (2010) [Citation53] software, which offers an overall risk prediction drug score, the approximate toxicity risk (Mutagenic, Tumorigenic, Irritant, Reproductive effect) values of Edotecarin and certain essential physicochemical properties (cLogP,Solubility,MW,TPSA,Druglikeness,Drug-score) have been calculated and given in .
Table 7. Osiris's estimation of title compounds toxicity hazards and physicochemical properties.
Absorption analysis of target molecule
The blood-brain barrier (BBB) is a barrier that separates circulating blood from the central nervous system (CNS) [Citation54], predicting BBB permeable compounds is critical to identifying toxic substances that can harm the brain and to design drug molecules that can reach its target in the CNS [Citation55]. Human intestinal absorption (HIA) estimation is an important goal for the production of oral drugs. The need for fast and effective models for predicting HIA and other biopharmaceutical properties has greatly increased in vitro parallel synthesis and screening of candidate compounds in drug development for pharmacological activity [Citation56–58]. Extensive studies have revealed that Caco-2 cell culture is widely used in vitro cell culture models for absorption screening assay of new drug and is a valuable index [Citation59–61]. P-glycoprotein (Pgp) is an essential protein in the cell membrane that removes from the cell several foreign substances [Citation62]. As such, the pharmacokinetic effects of drugs are a significant criterion of this. Chemotherapy resistance in cancer cells is specifically regulated by overexpression of P-glycoprotein (Pgp) [Citation63]. It is therefore of great importance to recognize substances that can either be transported out of the cell by Pgp (substrates) or hinder the function of Pgp (inhibitors) [Citation55]. Cytochrome P450 (CYP450) enzymes are important to the processing of cholesterol, steroids, etc. They are also used for the detoxification of foreign chemicals and for drug metabolism [Citation55, Citation64, Citation65]. Mutagens are chemicals that induce cancer-leading abnormal genetic mutations. A typical way of determining the mutagenicity of a chemical is the Ames test [Citation66]. This procedure has become the benchmark for chemical and drug safety evaluation and has been used to test thousands of molecules [Citation55]. In Ref [Citation67] probability values for Blood-Brain Barrier and Human Intestinal Absorption of Doxorubicin molecule with anticancer activity, were calculated as 0.9951, 0.8092, respectively. In this study, probability values were calculated as 0.6453 and 0.7758, respectively.
The silico module admetSAR [Citation68] was used to define various pharmacokinetic criteria, such as absorption, delivery, metabolism, excretion and toxicity, were examined for the scrutinized edotecarin molecule. Results from its ADMET analysis are presented in .
Table 8. Prediction of ADMET profiles of the edotecarin ADMET predicted profile – classification.
Conclusion
Conformational analysis of the edotecarin molecule was performed using a semi-experimental method PM3 and then obtained the lowest energy conformer (I) was optimized with the DFT/B3LYP/6-31++G(d,p) level of theory. Since protein-ligand interactions play a vital role in drug design, in this study, the docking simulations were performed for evaluation of the biological activity of the title molecule, using its most stable structure. The docking with DNA, TOP-I and DNA-TOPI complex was investigated in order to examine the inhibitory activity of the edotecarin molecule. The binding affinities of edotecarin to DNA, TOP-I and DNA-TOPI complex were obtainded −8.1, −8.9 and −10.7 kcal/mol, respectively, that corresponded to Kd = 1.24 µM, Kd= 0.3 µM and Kd= 0.015 µM, respectively. The predicted results support the exhibited antineoplastic activity of the edotecarin molecule. Moreover the predicted strong binding affinities of endotecarin molecule to the integrins α5β1 and αIIbβ3, with Kd values of 0.21 µM and 1.6 µM, respectively, are relevant to its the anti-cancer activity. The molecular docking predictions show that, the ligand edodecarin has good anti-tumor properties. The drug likeness and ADMET properties of edotecarin showed that molecule has good pharmacokinetic profiles.
This research indicates that the edotecarin can be further researched and evaluated for treatment and control techniques and for anticancer effects against different forms of cancer, in particular lung, breast, and stomach cancer.
Conflict of interests
The authors declare that they have no conflict of interests.
Additional information
Funding
References
- W. A. Denny, IDrugs 7 (2), 173 (2004).
- T. Yoshinari et al., Cancer Res. 59 (17), 4271 (1999).
- H. Arakawa et al., Jpn. J. Cancer Res. 90 (10), 1163 (1999). doi:10.1111/j.1349-7006.1999.tb00691.x
- M. W. Saif, and R. B. Diasio, Clin. Colorectal Cancer 5 (1), 27 (2005). doi:10.3816/ccc.2005.n.014
- M. Ciomei et al., Cancer Chemother. Pharmacol. 60 (2), 229 (2007). doi:10.1007/s00280-006-0365-8
- C. Sherer, and T. J. Snape, Eur. J. Med. Chem. 97, 552 (2015). doi:10.1016/j.ejmech.2014.11.007
- M. W. Saif et al., Anticancer Drugs 21 (7), 716 (2010). doi:10.1097/CAD.0b013e32833cb658
- M. P. D. Freitas, and T. D. C. Ramalho, Cienc. Agrotecnol. 37 (6), 485 (2013). doi:10.1590/S1413-70542013000600001
- Y. Shao et al., Phys. Chem. Chem. Phys. 8 (27), 3172 (2006). doi:10.1039/b517914a
- J. J. P. Stewart, J. Comput. Chem. 10 (2), 209 (1989). doi:10.1002/jcc.540100208
- J. J. P. Stewart, J. Comput. Chem. 10 (2), 221 (1989). doi:10.1002/jcc.540100209
- J. J. P. Stewart, J. Comput. Chem. 12 (3), 320 (1991). doi:10.1002/jcc.540120306
- J. J. P. Stewart, J. Mol. Model. 10 (2), 155 (2004). doi:10.1007/s00894-004-0183-z
- M. J. Frisch et al., 2004 Gaussian 03, Revision C. 02 (Gaussian, Inc: Wallingford, CT).
- A. D. Becke, J. Chem. Phys. 98 (7), 5648 (1993). doi:10.1063/1.464913
- P. Pulay et al., J. Am. Chem. Soc. 105 (24), 7037 (1983). doi:10.1021/ja00362a005
- T. Sundius, J. Mol. Struct. 218, 321 (1990). doi:10.1016/0022-2860(90)80287-T
- T. Sundius, Vib. Spectrosc. 29 (1–2), 89 (2002). doi:10.1016/S0924-2031(01)00189-8
- K. Istvan, 2002. Simirra, A Program for Simulation of IR and Raman Spectra (Chemical Research Center: Budapest). (It was obtained from Dr. Gabor Keresztury in Chemical Research Center in Budapest).
- I. P. Batra et al., Theoret. Chim. Acta 32 (4), 279 (1974). doi:10.1007/BF00526864
- S. Y. Lee, and B. H. Boo, J. Phys. Chem. 100 (37), 15073 (1996). doi:10.1021/jp960921p
- S. Selvaraj et al., Chem. Data Collect. 17–18, 302 (2018). doi:10.1016/j.cdc.2018.10.003
- S. Celik, S. Akyuz, and A. E. Ozel, Vib. Spectrosc. 92, 287 (2017). doi:10.1016/j.vibspec.2017.08.007
- T. Hasan, P. K. Singh, and N. Misra, Arch. Phys. Res. 3, 378 (2012).
- Y. Wang et al., Spectrochim. Acta A Mol. Biomol. Spectrosc. 135, 296 (2015). doi:10.1016/j.saa.2014.06.103
- S. Celik et al., J. Biomol. Struct. Dyn. 37 (10), 2515 (2019). doi:10.1080/07391102.2018.1495578
- S. Celik et al., Spectrosc. Lett. 53 (9), 648 (2020). doi:10.1080/00387010.2020.1821062
- Y. S. Rao et al., J. Mol. Struct. 1108, 567 (2016). doi:10.1016/j.molstruc.2015.12.008
- Y. Sert, Pamukkale J. Eng. Sci. 24 (7), 1272 (2018). doi:10.5505/pajes.2017.82195
- R. G. Pearson, 1973 Hard and Soft Acids and Bases (Van Nostrand Reinhold: New York, NY), Vol. 2.
- N. M. O'boyle, A. L. Tenderholt, and K. M. Langner, J. Comput. Chem. 29 (5), 839 (2008). doi:10.1002/jcc.20823
- Z. Geng, K. Shibasaki, and M. Kijima, Synth. Met. 213, 57 (2016). doi:10.1016/j.synthmet.2016.01.001
- S. K. Palanisamy, N. M. Rajendran, and A. Marino, Nat. Prod. Bioprospect. 7 (1), 1 (2017). doi:10.1007/s13659-016-0115-5
- S. Issa et al., J. Enzyme Inhib. Med. Chem. 34 (1), 1321 (2019). doi:10.1080/14756366.2019.1640692
- J. Moukharskaya, and C. Verschraegen, Hematol. Oncol. Clin. North Am. 26 (3), 507 (2012). doi:10.1016/j.hoc.2012.03.002
- K. Buzun et al., J. Enzyme Inhib. Med. Chem. 35 (1), 1781 (2020). doi:10.1080/14756366.2020.1821676
- O. Trott, and A. J. Olson, J. Comput. Chem. 31 (2), 455 (2010).
- H. R. Drew et al., Proc. Natl. Acad. Sci. U.S.A 78 (4), 2179 (1981). doi:10.1073/pnas.78.4.2179
- M. R. Redinbo et al., Science 279 (5356), 1504 (1998). doi:10.1126/science.279.5356.1504
- L. Stewart et al., Science 279 (5356), 1534 (1998). doi:10.1126/science.279.5356.1534
- S. Celik et al., J. Biomol. Struct. Dyn. 40 (2), 660 (2022). doi:10.1080/07391102.2020.1817150
- S. Celik et al., J. Biomol. Struct. Dyn. 38 (5), 1354 (2020). doi:10.1080/07391102.2019.1604263
- E. Akalin, S. Celik, and S. Akyuz, J. Appl. Spectrosc. 86 (6), 975 (2020). doi:10.1007/s10812-020-00926-2
- S. Das et al., J. Adv. Res. 9, 51 (2018). doi:10.1016/j.jare.2017.10.010
- A. Jurcik et al., Bioinformatics 34 (20), 3586 (2018). doi:10.1093/bioinformatics/bty386
- B. S. Dario et al., New J. Chem. 43 (45), 17831 (2019). doi:10.1039/C9NJ02480H
- G. S. Laco et al., Biochemistry 41 (5), 1428 (2002). doi:10.1021/bi011774a
- D. K. Neelima et al., Asian J. Sci. Technol. 8, 5563 (2017).
- A. Roy et al., Mol. Biol. Int. 2011, 1 (2011). (doi:10.4061/2011/854626
- A. Bhaumik, M. C. Eswaraiah, and R. Chakraborty, J. Pharm. Biol. Sci. 13, 61 (2018).
- O. K. Gasymov et al., J. Mol. Graph. Model.108, 107999 (2021). doi:10.1016/j.jmgm.2021.107999
- N. Balakrishnan, J. S. Raj, and N. Kandakatla, Int. J. Pharm. Pharm. Sci. 7, 295 (2015).
- Osiris. OSIRIS Property Explorer (Actelion Pharmaceuticals Ltd: Allschwil, Switzerland, 2010). http://www.organic-chemistry.org/prog/peo/
- N. J. Abbott, L. Rönnbäck, and E. Hansson, Nat. Rev. Neurosci. 7 (1), 41 (2006). doi:10.1038/nrn1824
- P. Schyman et al., Front. Pharmacol. 8, 889 (2017). doi:10.3389/fphar.2017.00889
- M. A. Gallop et al., J. Med. Chem. 37 (9), 1233 (1994). doi:10.1021/jm00035a001
- E. M. Gordon et al., J. Med. Chem. 37 (10), 1385 (1994). doi:10.1021/jm00036a001
- M. D. Wessel et al., J. Chem. Inf. Comput. Sci. 38 (4), 726 (1998). doi:10.1021/ci980029a
- H. Bohets et al., Curr. Top. Med. Chem. 1 (5), 367 (2001). doi:10.2174/1568026013394886
- P. Stenberg et al., J. Med. Chem. 44 (12), 1927 (2001). doi:10.1021/jm001101a
- M. Guangli, and C. Yiyu, J. Pharm. Pharm. Sci. 9 (2), 210 (2006).
- S. V. Ambudkar et al., Oncogene 22 (47), 7468 (2003). doi:10.1038/sj.onc.1206948
- P. Borst, and R. O. Elferink, Annu. Rev. Biochem. 71, 537 (2002). doi:10.1146/annurev.biochem.71.102301.093055
- C. M. Brown, B. Reisfeld, and A. N. Mayeno, Drug Metab. Rev. 40 (1), 1 (2008). doi:10.1080/03602530701836662
- M. Murray, J. Pharm. Pharmacol. 58 (7), 871 (2006). doi:10.1211/jpp.58.7.0001
- B. N. Ames et al., Proc. Natl. Acad. Sci. USA 70 (8), 2281 (1973). doi:10.1073/pnas.70.8.2281
- E. R. Kabir, M. K. S. Siam, and N. Mustafa, Proceedings of the Tenth International Conference on Computational Systems-Biology and Bioinformatics. Dec 2019, pp. 1–11.
- F. Cheng et al., J. Chem. Inf. Model. 52 (11), 3099 (2012). doi:10.1021/ci300367a