
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.abstract
Background and Aims: Using decellularized scaffold to reengineer liver tissue is a promising alternative therapy for end-stage liver diseases. Though the decellularized human liver matrix is the ideal scaffold for reconstruction of the liver theoretically, the shortage of liver donors is still an obstacle for potential clinical application. Therefore, an appropriate alternative scaffold is needed. In the present study, we used a tissue engineering approach to prepare a rat decellularized spleen matrix (DSM) and evaluate the effectiveness of this DSM for primary rat hepatocytes culture. Methods: Rat decellularized spleen matrix (DSM) was prepared by perfusion of a series of detergents through spleen vasculature. DSM was characterized by residual DNA and specific extracellular matrix distribution. Thereafter, primary rat hepatocytes were cultured in the DSM in a 3-dimensional dynamic culture system, and liver cell survival and biological functions were evaluated by comparison with 3-dimensional sandwich culture and also with cultured in decellularized liver matrix (DLM). Results: Our research found that DSM did not exhibit any cellular components, but preserved the main extracellular matrix and the intact vasculature evaluated by DNA detection, histology, immunohistochemical staining, vessel corrosion cast and upright metallurgical microscope. Moreover, the method of DSM preparation procedure was relatively simple with high success rate (100%). After seeding primary hepatocytes in DSM, the cultured hepatocytes survived inside DSM with albumin synthesis and urea secretion within 10 d. Additionally, hepatocytes in dynamic culture medium had better biological functions at day 10 than that in sandwich culture. Albumin synthesis was 85.67 ± 6.34 μg/107cell/24h in dynamic culture in DSM compared to 62.43 ± 4.59 μg/107cell/24h in sandwich culture (P < 0.01) and to 87.54 ± 5.25 μg/107cell/24h in DLM culture (P > 0.05); urea release was 32.14 ± 8.62 μg/107cell/24h in dynamic culture in DSM compared to 20.47 ± 4.98 μg/107 cell/24h in sandwich culture (P < 0.05) and to 37.38 ± 7.29 μg/107cell/24h cultured in DLM (P > 0.05). Conclusion: The present study demonstrates that DSM can be prepared successfully using a tissue engineering approach. The DSM is an appropriate scaffold for primary hepatocytes culture.
Introduction
End-stage liver diseases, such as fulminant hepatitis, liver cirrhosis, and liver cancer, are serious health problems with high mortality. The most effective treatment for end-stage liver diseases is orthotopic liver transplantation. However, severe shortage of donor livers, high expense, and potential side effects of immunosuppression largely limit the application of this treatment.Citation1
In addition to organ transplantation, using a decellularized biological scaffold material to reconstruct a functional organ is a recent promising approach in the area of tissue engineering. The scaffold, which has good biocompatibility and reservation of the natural vascular network and extracellular matrix (ECM), is acquired by removing parenchymal cells from tissue and organ using decellular technology. A minimally immunogenic scaffold with a native intact structure for new tissue regeneration may be produced in that the ECM proteins are the most conserved proteins.Citation2 The initial applications of the decellularization matrix have been demonstrated only for the hollow organs or thin layer of tissues, such as bladder, artery, esophagus, trachea, and skin.Citation3–7 Ott et al. reported that they constructed a heart by using a rat's decellularized heart scaffold.Citation8 This study became the milestone in the field of reengineering whole organs using a decellularized scaffold. Since then, researchers have successfully constructed the primary functional engineering tissue of lung,Citation9,10 liverCitation11,12 and kidney Citation13 by using decellularized scaffolds.
Theoretically, the decellularized liver ECM matrix is the ideal scaffold for reconstruction of a liver.Citation11,12 However, the potential clinical application of the decellularized liver matrix is limited because of the shortage of liver donors. We intend to reconstruct functional liver tissue by using decellularized spleen matrix on the basis of theory analysis shown as below. Both the spleen and liver are parenchymal organs, and the hepatocytes, when seeded in spleen in vivo, could proliferate and reconfigure,Citation14 indicating that the microenvironment of spleen, especially the ECM and blood sinus, is suitable for proliferation of hepatocytes. In addition, the spleen can be obtained from patients with various diseases leading to splenectomy, such as splenic trauma, and immune thrombocytopenic purpura, as well as some patients with hypersplenism in cirrhosis and portal hypertension, who will undergo splenectomy. Moreover, spleen from donation after cardiac or brain death could also be used for scaffold preparing. Therefore, the spleen matrix may provide an appropriate alternative for a scaffold to cultivate hepatocytes with relatively easy availability.
Thus, we hypothesize that the decellularized splenic matrix (DSM) prepared in vitro may be an ideal scaffold for cultivating hepatocyte. However, the method by which a decellularized spleen scaffold is prepared has not yet been well established and the effectiveness of the DSM for hepatocyte cultures has not been evaluated. Therefore, the purpose of this study was to establish a simple and effective method to prepare the DSM, and then evaluate the survival and basic function of the hepatocytes cultured in DSM.
Results
Structure and components of DSM
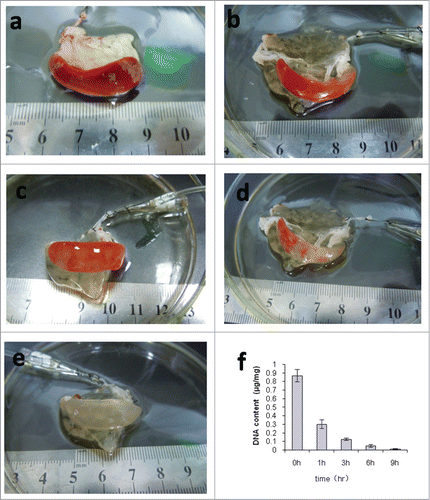
The color of the spleen changed gradually during the preparation of the DSM. After perfusion with 0.1% SDS, the red color of the spleen started to reduce as a result of the removal of cell components. Three to 6 hours later, the color changed to plaque-like and the branching tube structure was generally visible. The spleen showed a uniform white translucence after 9 h of perfusion. Meanwhile, the gross morphology was still intact (). These DSM were quite stretchable, which could be easily compressed and quickly re-expanded like sponges. When the spleen became white translucence, while there is no capsule breakage, precipitation or bubbles in the DSM, we defined it a successful case. Moreover, we had a 100% successful rate (40/40) of DSM preparation after 5 pre-experiments to optimize the procedures. Furthermore, the quantification of DNA showed that residual DNA was decreased with the prolongation of perfusion. The DNA of native spleen is about 0.8690 ± 0.0721 μg/mg. After 9 h of 0.1% SDS perfusion, the DNA in the spleen was about 0.0094 ± 0.0052 μg/mg, which was less than 2% of the native spleen (P < 0.01) ().
Figure 1. Decellularization of rat spleens. (a–e) Representative images of rat spleen during decellularization process at 0h (a), 1h (b), 3h (c), 6h (d), and 9h (e). (f) Representative the residual DNA content in the decellularized spleen matrix during the different time points.
Histological evaluation showed normal splenic organ structure and cell nuclei in the native isolated spleen (). The cells and the vessels can be seen clearly in the native spleen. Compared with the native isolated spleen, no cell nuclei and cytoplasm components were observed in the DSM, indicating that all the cellular components in the spleen were cleared out (). Many empty spaces, legacy for the prolapse of the cells, appeared in the matrix. In addition, the bases of the small blood vessels were still visible in the matrix components section ().
Figure 2. Comparison of native spleen (left) and DSM (right). Top to bottom: H&E staining, immunostaining of collagen I, collagen IV, fibronectin and laminin. Scale bar: 50μm.
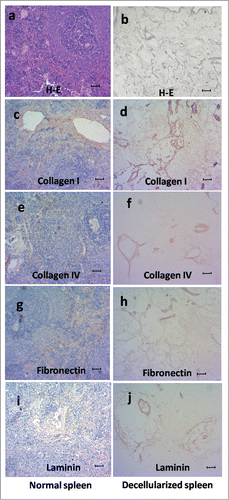
Immunohistochemical staining revealed that the presence of 4 major extracellular matrix proteins (collagen type I, collagen type IV, fibronectin, and laminin) in the DSM were retained similarly to those of the native spleens. It indicated that the structural and basement membrane components of the ECM were preserved (). The preservation of type I collagen was mainly within the blood sinus area (). Type IV collagen and fibronectin were observed mainly within the decellularized tissues (). The basement membrane of the vascular structures stained positive for laminin (). The distribution of these extracellular matrix proteins were maintained during decellularization, and it was quite close to that found in native spleen. Ultrastructural characterization of the DSM was evaluated by scanning electron microscopy (SEM) images. We found that DSM exhibited the complete spleen capsule (), continuous extracellular matrix, and the absence of cells ().
Figure 3. The spleen capsule (a) and the spleen extracellular matrix (b) of the DSM by SEM. Scale bars: 20μm.

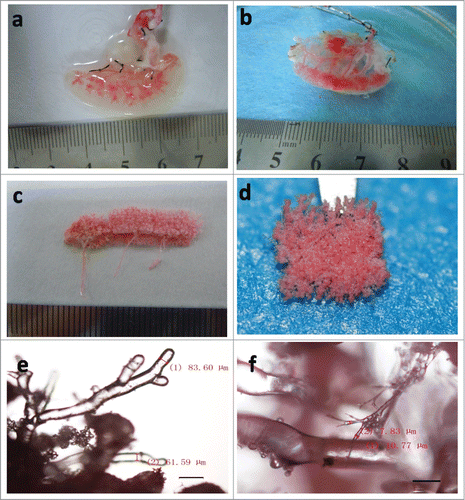
Red dye infusion through the splenic artery was used to assess the intact structure of the vascular network in the DSM. The main branch of the vascular system was seen first. With the advancement of the dye, the microvascular structures were gradually revealed from the larger vessels to small capillaries in the translucent matrix. Finally, the dye flowed out through the splenic vein. There was no leakage of red dye through the vascular walls, suggesting the maintenance of intact vasculature in DSM by the decellularization method used (). After the corrosion cast of the DSM was made, the structure and size of the splenic vasculature were evaluated by using upright metallurgical microscope. We observed fine branching structures, indicating that the architecture of micro-vasculature remained mostly intact in the DSM (). We found continuous small vessels with 69.07 ± 11.55 μm in diameter at 40× magnification, and also the smallest vessels with 7.63 ± 1.36 μm in diameter at 100× magnification ().
Figure 4. The vascular tree of DSM. (a and b) Representative red dye infusion into the DSM at initial stage (a) and the end stage (b). (c and d) Representative the entirety (c) and partial (d) of vascular tree cast. (e and f) Representative the vascular tree cast viewed by microscopy in 40x (e) and 100x (f) magnification. Scale bars: 200μm (e), 100μm (f).
Hepatocytes culture in DSM
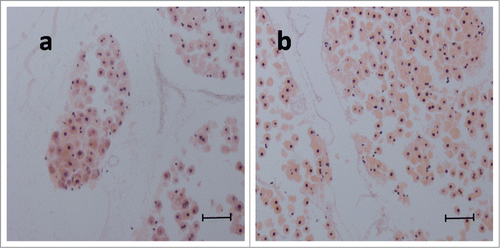
We introduced 1×107 primary adult rat hepatocytes into the DSM through the splenic artery. The engraftment efficiency with our protocol was 90.7% ± 4.2%.H-E staining of hepatocytes in DSM in the dynamic culture group clearly showed approximately 70% of normal cellular morphology. The results of histological examination indicated that the typical shape of hepatocytes with round nucleus (blue), prominent nucleoli, and polygonal cytoplasm (red) was well maintained in day 1 (68.2% ± 2.6%) and day 5 (70.6% ± 2.1%) in culture (). At day 1, the majority of the hepatocytes in the DSM was distributed in the network of blood vessels and only 10.8% ± 2.7% of cells dispersed into the perivascular area in the DSM. At day 5, about 83.7% ± 5.6% of the cells were found to distribute throughout the parenchymal space.
Figure 5. H&E staining of hepatocytes in dynamic culture in DSM at day 1 (a) and day 5 (b). Scale bar: 50μm.
Function of the hepatocytes
Albumin release in the dynamic culture in both DSM and DLM peaked at day 7 and slowly decreased thereafter. In the sandwich culture, albumin release reached the peak at day 5 (83.02 ± 4.13μg/107cell/24hr), and then slowly reduced. Albumin release from the dynamic culture in DSM at day 7 (102.24 ± 5.12 μg/107cell/24hr) and day 10 (85.67 ± 6.34 μg/107cell/24hr) was significantly higher than that from the sandwich culture (70.36 ± 6.22 μg/107cell/24hr at day 7 and 62.43 ± 4.59 μg/107cell/24hr at day 10, both p < 0.01, ), but there is no significant difference from that cultured in DLM (106.37 ± 8.26 μg/107cell/24hr at day 7 and 87.54 ± 5.25 μg/107cell/24hr at day 10, both P > 0.05, ).
Figure 6. Comparison of hepatocytes function among in the DSM, in the DLM, and the sandwich culture. (a) Representative albumin synthesis. (b) Representative urea release.* P < 0.05, **P < 0.01.
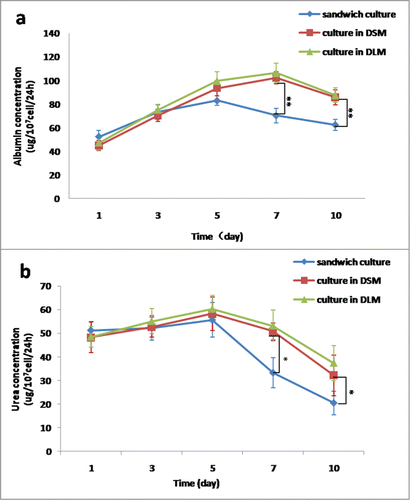
Urea release in the dynamic culture in DSM peaked at day 5 (58.26 ± 7.16 μg/107cell/24hr), and then gradually declined through 50.73 ± 3.57 μg/107cell/24hr at day 7 to 32.14 ± 8 .62 μg/107cell/24hr at day 10. Urea release in the sandwich culture reached the peak at day 5 (55.66 ± 7.29 μg/107cell/24hr), then gradually declined through 33.29 ± 6.37 μg/107cell/24hr at day 7 to 20.47 ± 4.98 μg/107cell/24hr at day 10. Urea release from the dynamic culture in DSM was significantly higher than that from the sandwich culture at days 7 and 10 (both P < 0.05, ). Urea release in the dynamic culture in DLM peaked at day 5 (60.35 ± 5.63 μg/107cell/24hr), and then gradually declined through 53.21 ± 6.57 μg/107cell/24hr at day 7 to 37.38 ± 7.29 μg/107cell/24hr at day 10. There was no significant difference in urea release between cultured in DSM and in DLM (both P > 0.05, ).
Discussion
There were 4 major findings in this study. Firstly, the procedure used to prepare DSM could preserve spleen matrix components, structure and intact vasculature. Secondly, isolated hepatocytes could be successfully seeded and dynamically cultured in the DSM prepared. Thirdly, hepatocytes cultured in the DSM were able to survive and secrete albumin and urea. Fourthly, the hepatocytes cultured in the DSM were functionally more active than those cultured in the sandwich culture, but it was comparable to that cultured in DLM assessed by albumin and urea secretion.
Native decellularized matrix is more feasible than the other bio- or synthetic scaffold for hepatocyte growth and proliferation because of its characteristics of retaining natural extracellular matrix components (e.g., collagen, glycoprotein, elastin, and a special 3-dimensional space frame structure). Additionally, the original vascular network in a decellularized matrix can ensure the provision of cell nutrients and removal of metabolites. However, the shortage of liver donors is still a big problem for liver tissue regeneration in a native liver scaffold. By contrast, the spleen exhibits a similar matrix structure to that of the liver. Liver cells implanted in the spleen in vivo have been proved to be able to survive and proliferate.Citation14 This in vivo method, however, may still cause severe problems, such as thrombosis. Therefore, in this study, we focused on the preparation of the DSM scaffold to determine if the decellularized procedure could maintain the tissue matrix structure, the microenvironment and the vascular network for cell proliferation. Furthermore, the DSM scaffold was used to see if hepatocytes could survive in the DSM and exhibit some physiological liver function.
The methodology of decellularization of an organ could be physical, chemical, enzymatic, or a combination of these.Citation15 The main purpose is to dissolve cell membranes and other components in order to cause cell disintegration and fragmentation for removal from the extracellular matrix. The decellular solution will significantly affect the decellularization, shape, and property of the scaffold. In the past, tissues were immersed into the decellular detergents for the removal of cells from the surface to the inside of the organ. To accelerate tissue decellularization, physical shock or stirring were also performed. However, these methods are ineffective for thicker tissues (>5 mm).Citation16 By contrast, enzymatic digestion disintegrates the cell membrane, and also damages the extracellular matrix of collagen and elastin, causing disruption of the decellularized matrix structure,Citation17,18 which is not beneficial for cell regeneration. Therefore, enzyme digestion was not considered for DSM preparation in this study.
Perfusion of detergents for decellularization of whole organ and tissue has been shown to achieve good results.Citation8 Similarly, in our study, ionic (0.1% SDS) and non-ionic (1% Triton X-100) detergents were perfused into the isolated spleen by a pump through the natural vascular network. SDS is an ionic detergent and a hydrophilic anionic surfactant with strong cellular removal effect that destroys the protein-protein interactions. Triton X-100, a non-ionic detergent, provides better retention of the extracellular matrix by disrupting fat-fat and fat-protein connections of cellular molecules.Citation19 In the procedures, PBS, low concentration (0.1%) SDS, deionized water, and 1% Triton X-100 were sequentially perfused into the spleen through the splenic artery to prepare the DSM scaffold. Deionized water was applied after SDS in order to rinse out residual SDS and cellular components. Subsequently, 1% Triton X-100 was perfused to further dispel residual cellular substances and SDS.
By macroscopic view, H-E staining, immunohistochemical staining, and scanning electron microscopy, we consistently found that spleen cells were completely removed in the DSM preparation. The preserved collagen, fibronectin, and laminin within the DSM were similar to the nature spleen and is comparable with the proteins preserved in dellcellularized liver matrix of other studies.Citation11 It showed that the process preserved the basic extracellular 3-dimensional architecture, the ultrastructure, and the native matrix components of the spleen.
Furthermore, our DSM preparation kept the intact vasculature structure. After red dye perfusion, splenic vessels were clearly shown in the DSM, and the dye flew from large blood vessels to the small vessels, and finally flew out through the splenic vein. The dye perfusion and vessel corrosion casts of the scaffold also suggested that the spleen vascular network remained intact. By microscopy measurement, cross-sectional splenic vascular was 69.07 ± 11.55 μm in diameter, and exhibited vascular continuity, and the minima vasculars observed were 7.63 ± 1.36 μm in diameter. Naturally, the diameter of the splenic vascular sinusoid is approximately 40 μm, and the capillaries is approximately 6–9 μm. When performing decellularization, endothelial cells on the vessel wall had been also destroyed; thus, the vessel lumen was larger than the original lumen.
As the prepared DSM retained the extracellular matrix, the 3-dimensional structure, and intact vascular network, it laid the foundation for the cultivation and long-term survival of the hepatocytes, and could also be connected to the blood vessels after implantation for oxygen and nutritional supply.
Many researches showed that the multiple step infusion was better than single step infusion and continually infusion.Citation12,16 In this study, the engraftment efficiency with 2-step infusion protocol was 90.7% ± 4.2%. This perfusion technique showed deposition of the primary hepatocytes within the parenchymal space and some in the vessels. The infusion speed of 1 ml/min and 10 min interval help to keep a high engraftment rate of cells.
After we seeded the primary hepatocytes in DSM, we found that these cells can survive for as long as 10 d. Meanwhile, albumin and urea can be detected from culture medium, indicating that the cultured liver cells exhibited basic hepatic function. DSM scaffolds have been shown to exhibit several advantages for hepatocyte cultivation: 1) the component of DSM is similar to that of the DLM, which can lead to hepatocyte adhesion and interaction by providing mechanical and biomechanical support; 2) the decellularized matrix maintains good biological and cellular compatibility; 3) the 3-dimensional structure in the DSM provides more adhesion and growth area for cell exchange of nutrients and oxygen and for formation of a spherical body; 4) Native vessel system provide a pathway for transportation of medium in thick tissue; and 5) the clinical source of the spleen is more widely available than that of the liver, not only from the donor, but also from the patients with splenectomy. Our findings provide novel evidence that DSM can be a potential scaffold alternative for liver tissue engineering.
The supply of oxygen and nutrients is especially important for hepatocyte survival. To better understand DSM compatibility for liver cell growth, we cultured hepatocytes in a dynamic culture condition in this study. We found that about 70% of cells maintained the normal morphology, suggesting that most of hepatocytes were survived in the SDM. Meanwhile, many hepatocytes were distributed in the perivascular space in the dynamic culture at day 5. The dynamic culture medium perfusion could force the engrafting of hepatocytes into the perivascular matrix, make hepatocytes to have a larger contact surface area with the splenic extracellular matrix, provide more oxygen and nutrients to the cultured liver cell, and remove metabolite waste faster.
Several protocols including plate cultivation, sandwich culture and co-culture have been attempted to acquire a more satisfying functional results. A large number of researches have come to a consensus that using sandwich culture for hepatocytes could create a 3 dimension environment close to that in vivo, and support hepatocytes growth and their specific function. So we choose the sandwich culture as the control group. For the better understanding of the influence of the DSM to the hepatocytes' function, we also used the DLM as another positive control group.
We demonstrated that the cultured primary hepatocytes in the DSM can perform the basic function. Albumin and urea were detected in the culture medium, which serve as specific indicators for the status of hepatic synthesis function. Furthermore, we found that albumin release peaked at day 5 in the sandwich culture group and at day 7 in the other 2 dynamic culture groups of DSM and DLM, and then gradually decreased afterward. The urea released peaked at day 3 in the sandwich culture group and at day 5 in the other 2 dynamic groups of DSM and DLM, and then gradually decreased afterward. There is no significant difference among the 3 groups at days 1∼5. At days 7∼10 of cultivation, albumin and urea in the 2 dynamic cultures of DSM and DLM were significantly higher than those in the sandwich culture, but there is no significant difference between cultured in DSM and in DLM. The structure of DSM provided a physical substratum for the attachment and spatial organization of cells. Proteins and other compositions preserved in scaffold were critical to keep cell survival and maintain cell biological functions, leading to significantly higher albumin and urea release from day 7 to day 10 in the dynamic culture than sandwich culture. Our data are consistent with another study showing that 3-dimensional cultures under constant perfusion can maintain the hepatocytes function.Citation20 Our data also demonstrated that DSM provide comparable environment for hepatocytes survive and function with DLM, since they had the similar mechanism.
We did notice that the functions of hepatocytes gradually declined after day 7 in the dynamic culture, which was also associated with the reduction of hepatocytes. The hepatocyte is the main cell type in the liver and makes up ∼80% of hepatic cells, and the other 20% are non-parenchymal cells, including endothelial cells, Kupffer cells, lymphocytes, and stellate cells. Studies have demonstrated that the non-parenchymal cells are important for maintaining the function of hepatocytes.Citation21–23 Consequently, our subsequent work is to co-culture non-parenchymal cells with hepatocytes to improve the hepatocytes' growth and functions.
As conclusion, our study suggests that DSM scaffold prepared by perfusion of detergents for decellularization preserves the matrix environment and intact vasculature. Furthermore, compared with hepatocytes in Sandwich culture, hepatocytes cultivated in this DSM scaffold had a significantly higher survival rate and better biological function. And compared with the hepatocytes cultured in DLM, there was no significant difference between their function. Therefore, it is suggested that the DSM may be a potential scaffold alternative for liver tissue regeneration.
Materials and Methods
Animals
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of Xi'an Jiaotong University Medical Center. The protocol was approved by the Committee on the Ethics of Animal Experiments of Xi'an Jiaotong University (Permit Number: 2011–309). All animals were housed in individual cages in a 12 h light/dark cycle, and fed with standard rat chow (Keaoxieli Inc.., Beijing, China). All surgery was performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering.
Preparation of DSM
Donor spleen harvest
Sprague-Dawley (SD) rats were anesthetized with an intraperitoneal injection (i.p.) of 3% pentobarbital sodium (45 mg/kg body weight) and heparin 1000 U i.p. was performed to prevent blood coagulation. After a midline laparotomy, the spleen and stomach were dissected to isolate the splenic artery and vein. An intravenous catheter (22 gauge) was inserted into the splenic artery followed by transection of the splenic vein. Thereafter, 100 U/ml heparinized PBS was infused via the splenic artery until the red blood cells were washed out completely. Lastly, the dissected spleen was frozen at −80°C to be used as a native spleen control or to be further decellularized after 24 h.
Spleen decellularization
The frozen spleen (n = 40) was slowly unfrozen in a chamber, and then the splenic artery was connected to the peristaltic pump. The isolated spleen was perfused with 100 U/ml heparinized PBS at 1.5 ml/min for 15 min. Subsequently, 0.1% SDS (Amresco, Solon, OH, USA) was perfused at 1.0 ml/min for 9 h, and the SDS solution was changed every 180 min. Thereafter, the spleen was perfused with distilled H2O for 15 min following 1% Triton X-100 (Solarbio, Beijing, China) for 30 min. Then the spleen was continuously perfused with distilled H2O for 15 min followed by PBS for 1 h. Finally, the DSM was sterilized in 0.1% peracetic acid PBS solution (Ruixing, Linyi, China) for 3 h and perfused extensively with sterile PBS for 48 h, then used to culture the hepatocytes following further process, or preserved in penicillin-streptomycin PBS solution at −80 ºC if it was not used immediately (). The DSM was observed for transparency change under macroscopic observation during the decellularization procedure.
Table 1. The decellularizing protocol of DSM
DNA in decellularized spleen
In order to evaluate the efficiency of cell removal by 0.1% SDS during the spleen decellularization, the spleen (n=12 ) was collected at 1h, 3h, 6h, and 9h during 0.1% SDS perfusion (n = 3 for each time point) for DNA measurement. First, native spleen and DSM were respectively cut into small pieces and lyophilized after perfused with distilled H2O for 15 min followed by PBS for 1hr. DNA was extracted using a Genomic DNA kit (TIANGEN Biotech, Beijing, China). Total DNA was quantified using ultramicro ultraviolet spectrophotometer (Quawell Technology, USA) according to the manufacturer's instructions. The remnants' base pair analysis of the DSM was performed by gel electrophoresis.
Characterization of DSM
DSM (n = 6) and native isolated spleens (n = 6) were fixed in 10% neutral buffered formalin, embedded in paraffin, and cut into 4-μm sections for hematoxylin and eosin (H-E) staining in order to compare cellular composition differences. Immunohistochemical staining for collagen type I (Abcam, Cambridge, MA USA), collagen type IV (Abcam, Cambridge, MA USA), fibronectin (Abcam, Cambridge, MA USA), and laminin (Abcam, Cambridge, MA USA) was used to determine the ECM distribution.
Allura Red dye (Marie's, Shanghai, China, 20%) was perfused to the DSM (n = 4) to evaluate the integrity and the structure of the vascular bed. Moreover, the vessels' corrosion casts of the DSM were generated, by using the methyl methacrylate (Kelong, Chengdu, China), in order to display the intricate vascular tree. The upright metallurgical microscope of the vessels' cast was used to evaluate the preservation of small vessel bed.
Hepatocyte culture in decellularized spleen matrix
Rat hepatocytes were harvested from SD rats by the Seglen 2-step perfusion method with modification.Citation24 Briefly, an intravenous catheter (22 gauge) was inserted into the portal vein of liver after anesthetization and midline laparotomy, and then the vena cava was sectioned. The liver was perfused with calcium-free buffer solution (37 ºC, pH 7.4) at 25 ml/min for 10 min. Once the liver became pale, it was excised and continuously perfused with 0.05% collagenase and 5 mmol/l of calcium chloride (37ºC, pH 7.6). Thereafter, the liver capsule was torn open, and the isolated hepatocytes were resuspended in a 4 ºC washing medium, which contained Williams'E medium (Sigma, USA), NaHCO3 0.35 mg/ml, HEPES 2.38 mg/ml, penicillin 100 U /ml, and streptomycin 100 U /ml. This cell suspension was filtered through a nylon mesh (grid size: 100 μm), and a cell pellet was collected by centrifugation at 300 rpm for 3 min. After the second resuspension and wash, the cell viability was tested by trypan blue exclusion; cells over 90% viability yield were seeded into the scaffolds.
Before the cell seeding, the DSM was perfused and kept immersed in culture medium for 20 min. The Williams'E culture medium(Sigma, USA) consisted of , 10% FBS (Sijiqing, Hangzhou, China), 0.5 U/ml insulin (Sigma, USA), 14 ng/ml glucagons (Sigma, USA), 0.5 μg/ml dexamethasone (Sigma, USA), 100 U/ml penicillin, and 100 μg/ml streptomycin (Dingguo, Beijing, China). Approximately a total of 1×107 primary adult rat hepatocytes was suspended in 2 ml of culture medium and then infused into the DSM by direct infusion into the splenic artery in 2 steps at 10-min intervals. An injection bump was used to infuse the cells at a rate of 1ml/min. After 1ml hepatocytes were infused, the graft would be let stand for 10min. At the end of the 20 min, the cells that flowed out from the splenic vein were collected and counted. The total number of cells retained in DSM was the difference between the initial number of cells seeded and the number of cell flowed out. The engraftment efficiency was defined as the rate of cell retainance in DSM which was obtained by the number of cells retained in DSM being divided by the initial number of cells infused.
After seeding, the recellularized spleen matrix (n = 12) were cultured for 10 d by continuously perfusing the culture medium at 1.5 ml/min through the vascular network of the DSM after statical culture for about 6 h. The culture medium was changed every 24 h. The perfusion culture system was composed of a peristaltic pump, a perfusion chamber, and tubing part. The system was then put into the carbon dioxide gas incubator. The chamber was a flask containing 50 ml perfusion culture medium. The tubing part was a long gas-permeable silicone tube. One end of the tube was penetrated into the flask and connected to splenic artery cannulation as the inlet. The other end of the tube was penetrated into the perfusion medium to suck the medium into the system. The culture medium intravenously flowed out from the spleen to the perfusion chamber. Before the perfusion culture, the perfusion medium was recirculated in the perfusion system. The DSM was kept immersed in the medium in the perfusion chamber while it was perfused through the splenic artery. The splenic vein was left open for outlet. The whole perfusing system was closed and changeable. The primary adult rat hepatocytes cultured in this system was named dynamic culture group.
The primary adult rat hepatocytes cultured in the sandwich configuration was used as control group. Tissue culture dishes were coated with a collagen solution that was composed of 9 portions of type I rat tail collagen (1.25 mg/ml, sigma, USA) and one portion of DMEM and incubated for 1 h at 37 °C to form a collagen gel. After gelation, cells were seeded at 1×106 cells/well when using 6-well cell culture cluster, in hepatocyte culture medium, and incubated in 90% air and 10% CO2 at 37 °C. The medium was changed daily. At day 2, after aspirating the culture medium from the dishes, the second collagen gel layer was overlaid on the hepatocytes and incubated for 1 h at 37°C before culture medium was added.
The primary adult rat hepatocytes cultured in the DLM was used as another control group. The DLM was obtained according to the modified protocol by Uygun, et al.Citation11 (). One hepatic lobule was used to culture the hepatocytes. The culture medium was infused from portal vein, and the procedure of the hepatocytes cultured in DLM is as same as in the DSM.
Table 2. The decellularizing protocol of DLM
Functional studies of hepatocytes in DSM
At days 1, 5, 7, and 10 after seeding, the grafts from the dynamic culture groups were fixed in 10% neutral buffered formalin, embedded in paraffin, and cut into 4-μm sections for H-E staining, to evaluate the hepatocytes' location and survival. The cell number was counted under 10 high fields, and the average values were calculated.
To assess the function of the hepatocytes in DSM, we quantified hepatocyte albumin synthesis and urea release. The supernatants of the culture medium were collected at days 1, 3, 5, 7, and 10. The rat albumin ELISA kit (R&D, USA) and QuantiChrom Urea Assay kit (Jian Cheng Biosciences, Nan Jing, China) were respectively used to determine the albumin and urea secreted into the culture medium. Compare three different culture methods, a mathematical formula shown as blow was utilized to calculate albumin synthesis and urea release by per unit cell.
c: albumin or urea (μg/ml) quantification; v: medium volume (ml); n: implantation cell number; q: cell engraftment rate (%), q = 100% when in sandwich culture system; m: albumin synthesis or urea release by per unit cells (μg/107cell/24h).
Statistical analysis
The values were expressed as means ± standard deviations. The data were analyzed by t-test. A value of p < 0.05 was considered to be statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This study was supported by grants from the Research Fund for the Doctoral Program of Higher Education of China (20110201130009) and the Science and Technology Research and Development Program of Shaanxi Province (2012KW-39–01).
REFERENCES
- Soltys KA, Soto-Gutierrez A, Nagaya M, Baskin KM, Deutsch M, Ito R, Shneider BL, Squires R, Vockley J, Guha C, et al. Barriers to the successful treatment of liver disease by hepatocyte transplantation. J Hepatol 2010 53:769; PMID:20667616; http://dx.doi.org/10.1016/j.jhep.2010.05.010.
- Hutter H, Vogel BE, Plenefisch JD, Norris CR, Proenca RB, Spieth J, Guo C, Mastwal S, Zhu X, Scheel J, et al. Conservation and novelty in the evolution of cell adhesion and extracellular matrix genes. Science 2000 287:989-94; PMID:10669422; http://dx.doi.org/10.1126/science.287.5455.989.
- Yoo JJ, Meng J, Oberpenning F, Atala A Bladder augmentation using allogenic bladder submucosa seeded with cells. Urology 1998 51:221-5; PMID:9495701; http://dx.doi.org/10.1016/S0090-4295(97)00644-4.
- Dahl SL, Koh J, Prabhakar V, Niklason LE Decellularized native and engineered arterial scaffolds for transplantation. Cell Transplant 2003 12:659-66; PMID:14579934; http://dx.doi.org/10.3727/000000003108747136.
- Nieponice A, Gilbert TW, Badylak SF Reinforcement of esophageal anastomoses with an extracellular matrix scaffold in a canine model. Ann Thorac Surg 2006 82; 2050-8; PMID:17126109; http://dx.doi.org/10.1016/j.athoracsur.2006.06.036.
- Macchiarini P, Jungebluth P, Go T, Asnaghi MA, Reese LE, Cogan TA, Dodson A, Martorell J, Bellini S, Parnigotto PP, et al. Clinical transplantation of a tissue-engineered airway. Lancet 2008 372:2023-30; PMID:19022496; http://dx.doi.org/10.1016/S0140-6736(08)61598-6.
- Schechner JS, Crane SK, Wang F, Szeglin AM, Tellides G, Lorber MI, Bothwell AL, Pober JS. Engraftment of a vascularized human skin equivalent. FASEB J 2003 17:2250-6; PMID:14656987; http://dx.doi.org/10.1096/fj.03-0257com.
- Ott HC, Matthiesen TS, Goh SK, Black LD, Kren SM, Netoff TI, Taylor DA. Perfusion-decellularized matrix: using nature's platform to engineer a bioartificial heart. Nat Med 2008 14:213-21; PMID:18193059; http://dx.doi.org/10.1038/nm1684.
- Ott HC, Clippinger B, Conrad C, Schuetz C, Pomerantseva L, Ikonomou L, Kotton D, Vacanti JP. Regeneration and orthotopic transplantation of a bioartificial lung. Nat Med 2010 16:927-33; PMID:20628374; http://dx.doi.org/10.1038/nm.2193.
- Petersen TH, Calle EA, Zhao L, Lee EJ, Gui, L, Raredon MB, Gavrilov K, Yi T, Zhuang ZW, Breuer C, et al. Tissue-engineered lungs for in vivo implantation. Science 2010 329:538-41; PMID:20576850; http://dx.doi.org/10.1126/science.1189345.
- Uygun BE, Soto-Gutierrez A, Yagi H, Izamis ML, Guzzardi MA, Shulman C, Milwid J, Kobayashi N, Tilles A, Berthiaume F, et al. Organ reengineering through development of a transplantable recellularized liver graft using decellularized liver matrix. Nat Med 2010 16:814-20; PMID:20543851; http://dx.doi.org/10.1038/nm.2170.
- Soto-Gutierrez A, Zhang L, Medberry C, Fukumitsu K, Faulk D, Jiang H, Reing J, Gramignoli R, Komori J, Ross M, et al. A Whole-Organ Regenerative Medicine Approach for Liver Replacement. Tissue Eng Part C Methods 2011 17:677-86; PMID:21375407; http://dx.doi.org/10.1089/ten.tec.2010.0698.
- Song JJ, Guyette JP, Gilpin SE, Gonzalez G, Vacanti JP, Ott HC., Regeneration and experimental orthotopic transplantation of a bioengineered kidney. Nat Med 2013 19:646-51; PMID:23584091; http://dx.doi.org/10.1038/nm.3154.
- Mito M, Ebata H, Kusano M, Onishi T, Saito T, Sakamoto S. Morphology and function of isolated hepatocytes transplanted into rat spleen. Transplantation 1979 28:499, 1979; PMID:516133; http://dx.doi.org/10.1097/00007890-197912000-00013.
- Gilbert TW, Sellaro TL, Badylak SF Decellularization of tissues and organs. Biomaterials 2006 27:3675-83; PMID:16519932.
- Badylak SF, Taylor D, Uygun K. Whole-organ tissue engineering: decellularization and recellularization of three-dimensional matrix scaffolds. Annu Rev Biomed Eng 2011 13:27-53; PMID:21417722; http://dx.doi.org/10.1146/annurev-bioeng-071910-124743.
- Badylak SF. The extracellular matrix as a biologic scaffold material. Biomaterials 2007 28:3587-93; PMID:17524477; http://dx.doi.org/10.1016/j.biomaterials.2007.04.043.
- Funamoto S, Nam K, Kimura T, Murakoshi A, Hashimoto Y, Niwaya K, Kitamura S, Fujisato T, Kishida A. The use of high-hydrostatic pressure treatment to decellularize blood vessels. Biomaterials 2010 31:3590-5; PMID:20149445; http://dx.doi.org/10.1016/j.biomaterials.2010.01.073.
- Hasna A, Banafshe L, Alicia A, Goni FM Detergent solubilization of phosphatidylcholine bilayers in the fluid state: Influence of the acyl chain structure. Biochim Biophys Acta 2006 1758:190-6; http://dx.doi.org/10.1016/j.bbamem.2006.01.016.
- Domansky K, Inman W, Serdy J, Dash A, Lim MH, Griffith LG. Perfused multiwell plate for 3D liver tissue engineering. Lab Chip 2010 10:51-8; PMID:20024050; http://dx.doi.org/10.1039/b913221j.
- Ding BS, Nolan DJ, Butler JM, James D, Babazadeh AO, Rosenwaks Z, Mittal V, Kobayashi H, Shido K, Lyden D, et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration, Nature 2010 468:310-5; PMID:21068842; http://dx.doi.org/10.1038/nature09493.
- LeCouter J, Moritz DR, Li B, Phillips GL, Liang XH, Gerber HP, Hillan KJ, Ferrara N. Angiogenesis-independent endothelial protection of liver: role of VEGFR-1, Science 2003 299:890-3; PMID:12574630; http://dx.doi.org/10.1126/science.1079562.
- Kyungsook KKO, Rie U, Kyoko K, Teruo O Preserved liver-specific functions of hepatocytes in 3D co-culture with endothelial cell sheets. Biomaterials 2012 33:1406-13; PMID:22118777; http://dx.doi.org/10.1016/j.biomaterials.2011.10.084.
- Seglen PO. Preparation of isolated rat liver cells. Methods Cell Biol 2008 13, 29, 1976.2023-30.