
ABSTRACT
The prevalence of end-stage liver disease (ESLD) in the US is increasing at an alarming rate. It can be caused by several factors; however, one of the most common routes begins with nonalcoholic fatty liver disease (NAFLD). ESLD is diagnosed by the presence of irreversible damage to the liver. Currently, the only definitive treatment for ESLD is orthotopic liver transplantation (OLT). Nevertheless, OLT is limited due to a shortage of donor livers. Several promising alternative treatment options are under investigation. Researchers have focused on the effect of liver-enriched transcription factors (LETFs) on disease progression. Specifically, hepatocyte nuclear factor 4-alpha (HNF4α) has been reported to reset the liver transcription network and possibly play a role in the regression of fibrosis and cirrhosis. In this review, we describe the function of HNF4α, along with its regulation at various levels. In addition, we summarize the role of HNF4α in ESLD and its potential as a therapeutic target in the treatment of ESLD.
Introduction
End-stage liver disease (ESLD) is a fatal condition with an increasing prevalence in the United States. ESLD is the final stage of chronic liver disease, characterized by irreversible damage to the cells, tissues, and hepatic functions.Citation1 From 1999 to 2016, the annual liver-associated deaths in the US increased by 65%, resulting in 34,174 deaths.Citation2 However, due to a high rate of underreported cases, the true mortality is likely much higher.Citation3
Currently, medical therapy of ESLD involves mitigation of risk factors, nutritional support coupled with the avoidance of harmful medications, and avoidance of high risk, low reward procedures.Citation4 Medical interventions can increase life expectancy, but OLT remains the only definitive treatment for ESLD.Citation5 However, liver transplantation is extremely limited due to a shortage of donor livers. According to data collected by the Organ Procurement and Transplantation Network, less than half (49.0%) of waiting list registrants listed in 2018 received a deceased donor liver transplant within one year. Additionally, from 2014 to 2016, only 56.0% of adult liver transplant candidates had undergone the necessary transplant.Citation6 This critical situation is further exacerbated by a forecasted increase in the cost of liver transplantation in the near future.Citation5
Since alternative treatment options for ESLD are urgently needed, different approaches to treat ESLD are currently under investigation.Citation7,Citation8 Moroni et al.Citation8 confirmed the safety of autologous macrophage transplantation for potential treatment of cirrhosis and other fibrotic diseases. One of the most promising methods is the exploration of liver-enriched transcription factors (LETFs). LETFs, along with transactivation factors, have a great influence on the maintenance of liver-specific gene transcription.Citation9 Liu et al.Citation10 reported the initial hypothesis, which described the role of hepatocyte-specific transcription factors in the development of ESLD.Citation10,Citation11 Interestingly, the expression of hepatocyte nuclear factor 4-alpha (HNF4α) was found to be directly correlated to liver disease in humans.Citation10,Citation12 HNF4α is a transcription factor that plays an important role in liver morphogenesis and maintenance of proper hepatocyte function in a mature liver.Citation13–15 Most liver diseases have been associated with altered HNF4α expression, isoform ratios, and localization.Citation16 Therefore, many studies have indicated that HNF4α may potentially be a target gene for the regression of fibrosis and cirrhosis.Citation17,Citation18 This review touches upon the functions and regulation of HNF4α in a healthy mature liver, as well as the role of HNF4α in liver disease, particularly in ESLD. Implementation of HNF4α as a key therapeutic target for future ESLD treatment is discussed as well.
ESLD
Patients diagnosed with ESLD often exhibit abnormalities of liver synthetic and excretory functions with the development of ascites, hepatic encephalopathy, and variceal hemorrhage, among other complications.Citation19 There are many causes of ESLD, including alcohol-related liver disease (ARLD)Citation20 and infection via hepatitis-inducing viruses, hepatitis B virus (HBV) and hepatitis C virus (HCV).Citation21,Citation22 However, one of the most common maladies that progresses to ESLD in the US is nonalcoholic fatty liver disease (NAFLD).Citation23 The histological spectrum of NAFLD can range from simple steatosis, characterized by lipid accumulation in the liver, to more severe nonalcoholic steatohepatitis (NASH).Citation24,Citation25 Patients with NASH are at an increased risk of developing fibrosis, cirrhosis, and potentially hepatocellular carcinoma of the liver.Citation24,Citation26
It is important to note that cirrhosis exists at two different stages: compensated and decompensated. Compensated cirrhosis is the early asymptomatic stage, while decompensated cirrhosis is synonymous with ESLD.Citation27,Citation28 Decompensated cirrhosis encompasses serious alterations at different levels, such as fibrosis with severe deposition of rigid extracellular matrix (ECM); disruption of the liver lobular architecture together with disruption of vascular organization, resulting in portal hypertension; and the inability of hepatocytes to perform adequate metabolic functions due to dedifferentiation or apoptosis of cells.Citation29,Citation30 Nishikawa T et al.Citation31 reported an adaptive metabolic shift in early stages of liver injury; hepatocytes start generating energy predominantly from glycolysis rather than oxidative phosphorylation. This adaptive shift results in hepatocytes that are unable to sustain high levels of energy production and subsequently leads to hepatocyte dysfunction.Citation31 Hepatocyte dysfunction in individuals with ESLD manifests in the liver with reduced hepatocyte mass, oxidative stress, impaired mitochondrial function, and limited regenerative capacity.Citation12 It is likely that the dysfunction observed can be attributed to cytokine release or injuries that, by extrinsic mechanisms, alter the expression of the hepatocyte transcription factor network.Citation18 Unfortunately, the precise molecular mechanisms responsible for the disruption of hepatic functions in liver injury remain enigmatic and require further investigation.
HNF4α function and regulation
There are six families of LETFs responsible for the maintenance of liver-specific gene transcription: HNF-1, HNF-3, HNF-4, HNF-6, CCAAT-enhancer-binding protein (C/EBP), and D-binding protein (DBP). The HNF-4 family is a subset of LETFs that includes HNF4β, HNF4γ, and most importantly HNF4α.Citation9 HNF4α is a transcription factor in the superfamily of nuclear receptorsCitation13,Citation32 encoded by the HNF4A gene on chromosome 20 in humans.Citation33,Citation34 Generally, nuclear receptors exhibit modular structure with six distinct regions, often referred to as regions A-F, which correspond to functional domains ().Citation35 As a nuclear receptor, HNF4α utilizes these regions to control the activity of its target genes directly through transactivation mechanisms.Citation36 Full transactivation activity results from the synchronization of HNF4α’s two activation domains, activation function-1 (AF-1) and activation function-2 (AF-2).Citation37 AF-1 and AF-2, located in the A/B and D/E regions, respectively, activate transcription in a cell-type independent manner. Region D in HNF4α acts as a hinge between the DNA binding domain in region C and the ligand-binding domain in region E.Citation35,Citation38 Additionally, region F has been indicated to express repressor activity.Citation39 Ultimately, region C contains a conserved zinc-finger DNA binding domain, which functions to bind HNF4α as a homodimer to the consensus DNA sequence ().Citation40,Citation41 Consensus DNA binding sequences have been found in the regulatory regions of many genes expressed in the liver and kidney.Citation42 Upon binding these sequences, HNF4α recruits transcriptional co-activators and the appropriate accessory proteins to positively regulate the expression of target genes.Citation43
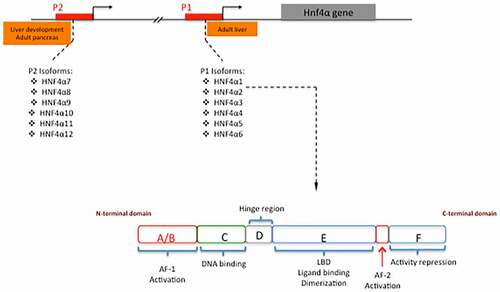
As the master regulator of liver homeostasis, HNF4α targets several genes essential in hepatocytes to maintain cellular processes. Traditionally, the target genes of HNF4α have been thought to encode apolipoproteins (apolipoproteins CIII, A-IV), involved in cholesterol homeostasis, transthyretin (TTR), involved in carrying thyroid hormone and Vitamin A in serum, pyruvate kinase, involved in glycolysis, and glutamate synthetase, involved in amino acid biosynthesis.Citation44,Citation45 Newer target genes are also associated with hepatic lipid homeostasis, xenobiotic detoxification, and drug metabolism.Citation46 HNF4α plays a crucial role in the expression of genes within Phase I and Phase II xenobiotic detoxification and drug metabolism.Citation46 Phase I is characterized by addition or exposure of a polar functional group to a lipophilic compound.Citation46 The most important Phase I gene that HNF4α regulates is CYP3A4, which is crucial to the body’s ability to metabolize drugs.Citation46 Phase II is characterized by conjugation reactions that help eliminate Phase I products from the body.Citation46 Genes in the SULT and UGT families account for the majority of Phase II metabolism and are regulated by HNF4α.Citation46 Bolotin et al., expanded on the classical roles of HNF4α by identifying additional target genes involved in signal transduction pathways (TAOK3, NGEF, FNTB) and in inflammation and immune responses (IL32, BRE, LEAP2).Citation47 Additionally, HNF4α has a role in controlling the expression of other LETFs such as HNF1α.Citation9
Regulation of HNF4α expression/activity occurs through multiple mechanisms and at different levels.Citation37, Citation48–54 The HNF4α gene is composed of 12 exons and alternative splicing of mRNA is a common way in which HNF4α expression is regulated at the transcriptional level. HNF4α protein variants are encoded by two separate promoters: P1 and P2. Transcription through the P1 or P2 promoters, combined with alternative splicing, produces up to 12 functionally distinct HNF4α isoforms.Citation55 Isoforms HNF4α1-6 are derived from the P1 promoter, while isoforms HNF4α7-12 are derived from the P2 promoter.Citation48 The P2-derived variants are in higher abundance within the adult pancreas and in the developing liver, whereas the P1-derived variants are in higher abundance within the adult liver.Citation49 Among the P1-products, the α1 and α2 isoforms are the most potent regulators of gene expression, while the α3 isoform exhibits significantly reduced activity.Citation56 The α4, α5 and α6 isoforms, which use an alternative first exon, display reduced transcriptional potential and inability to recognize the consensus response element of HNF4α.Citation56 P2 isoforms encode for a different amino-terminus on the 5ʹ end, which alters the AF-1 domain. This results in differential activation potential and target gene regulation compared to the P1-encoded isoforms.Citation57 These structural differences confer different capabilities in interacting with specific cofactors and in the transcriptional regulation of target genes in specific contexts.Citation56,Citation58The transactivation activity of HNF4α can be potentiated by the binding of specific co-activators at the AF-2 domain, such as steroid receptor co-activator-1 (SRC-1) and glutamate receptor interaction protein-1 (GRIP-1).Citation50 Additionally, enhanced activity can occur due to post-translational modifications, such as methylation and acetylation.Citation37 HNF4α methylation at the arginine 91 residue by protein arginine N-methyltransferase 1 (PRMT1) increases the efficiency of HNF4α binding to consensus DNA sequences.Citation51 Acetylation appears to be the most important post-translational modification in the regulation of HNF4α activity. Regarding this issue, acetylation of residues within the DNA binding domain by CREB-binding protein (CBP) has been shown to increase DNA binding activity as well.Citation52 However, acetylation of the lysine 458 residue in the repressor region of the F domain is responsible for normal repression and downregulation of HNF4α activity.Citation53 Conversely, phosphorylation modifications negatively regulate HNF4α activity. More specifically, phosphorylation of serine 304 by AMP-activated protein kinase (AMPK) serves to repress the activity of HNF4α by reducing its ability to form dimers and bind to DNA.Citation54
Through its network of transcriptional regulation, HNF4α plays a pivotal role in regulating hepatocyte functions, and it has been documented that HNF4α maintains hepatocyte differentiation and inhibits proliferation. In the adult mouse liver, hepatocyte-specific deletion of HNF4α resulted in increased hepatocyte proliferation.Citation59 Possible mechanisms by which HNF4α can inhibit cell proliferation involve epigenetic repression of pro-mitogenic genes, crosstalk with other cell cycle regulators including c-Myc and Cyclin D1, regulation of miRNAs, and post-translational modifications of HNF4α.Citation60,Citation61 These properties are key during initial and final phases of liver regeneration.Citation62
HNF4α in liver disease
The metabolic functions of hepatocytes in the early stages of liver disease are elevated by the upregulation of multiple networks of metabolism-related genes.Citation10 This elevated demand can only be sustained for a short period of time before hepatocellular failure.Citation10 Subsequently, it has been demonstrated that the molecular hallmarks in the events that lead to the progression of end-stage cirrhosis and terminal hepatic failure consist of a gradual down-regulation of LETFs, such as HNF4α, FOXA2, CEBPα, PPARα and HNF1α.Citation18 In an analysis of hepatocytes and liver tissue obtained from rats affected by liver disease, Nishikawa T et al.,Citation18 reported that the presence of HNF4α in hepatocytes with decompensated function was significantly reduced. Failure of the hepatic transcription system contributing to disease progression within rats, found by Nishikawa T et al.,Citation12 was later confirmed to occur in humans as well.
In response to acute liver injury, protective mechanisms appear to trigger a decline in HNF4α expression to redistribute transcriptional resources.Citation63 This decline in HNF4α subsequently contributes to a loss of cell-identifying gene expression.Citation63 However, HNF4α loss is temporary, and HNF4α expression can be restored with recovery from the injury.Citation63 On the other hand, prolonged liver injury persistently suppresses HNF4α expression and can contribute to the progression of liver disease.Citation63 In fact, evaluation of patient liver tissue samples at varying stages of decompensated liver function confirmed a correlation between HNF4α expression and degree of liver disease and fibrosis.Citation63
As a transcription factor, expression of HNF4α must be localized to the nucleus of the hepatocyte to properly control gene transcription associated with different hepatocellular processes (). In contrast to normal human hepatocytes, Florentino et al.Citation64 confirmed that, in addition to low protein expression, the expression of HNF4α in human hepatocytes isolated from livers with decompensated function is increasingly localized to the cytoplasm rather than the nucleus (). Markedly, they also showed that reduced HNF4α acetylation at lysine 106 was accompanied by a higher degree of liver dysfunction.Citation64 Signaling pathways possibly associated with HNF4α nuclear localization include AKT (protein kinase B) and cMET. AKT plays an important role in numerous cellular processes including development, proliferation, and survival.Citation65 Florentino et al. revealed a strong correlation among AKT and HNF4α levels; as AKT levels decreased in human hepatocytes from livers with decompensated function, HNF4α expression also decreased.Citation64 In addition, AKT directly influences cyclic adenosine monophosphate response element‐binding protein (CREB), a molecule controlling nuclear retention of transcription factors through acetylation activity.Citation52 It is suggested that AKT influences HNF4α nuclear localization through phosphorylation of threonine 308 by controlling the CREB-binding protein. Additionally, cMET, an inducer of AKT, was found to be reduced and directly correlated with decreased HNF4α nuclear localization.Citation64
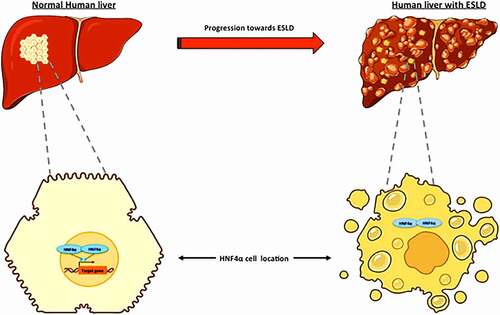
Furthermore, Yu et al.Citation66 identified that oxidative stress, which is elevated in fatty livers, fosters HNF4α cytoplasmic localization through activation of protein kinase C (PKC). PKC activation leads to phosphorylation of HNF4α and hinders its transfer into the nucleus by importin.Citation66 The decreased activity of HNF4α diminishes Apolipoprotein B (ApoB) and lessens very low-density lipoprotein (VLDL) secretion, leading to triglyceride (TAG) buildup in the liver. Consequently, this promotes the development of NAFLD.Citation66 Among the causes related to the decreased activity of HNF4α in liver disease, genetic alteration can be relevant. HNF4α mutations are associated with a large spectrum of diseases, including Crohn's disease,Citation67 Bowel disease,Citation68 and maturity-onset diabetes of the young (MODY).Citation69 Mutations in the HNF4α gene linked to maturity-onset diabetes in the young (MODY) are usually associated with a loss of HNF4α function.Citation69 The direct relationship between HNF4α mutations and liver disease progression has not been clearly established. Nonetheless, molecular mechanisms have been described. Mutations E276Q and R154X in the HNF4α gene lead to impaired HNF4α transcriptional activity through impaired physical interaction and functional cooperation between HNF4α and p300, a key HNF4α co-activator.Citation70 This decrease in HNF4α functional activity results in a decreased expression of its target genes, including HNF1α, which may finally alter the network of transcription factors in liver homeostasis.Citation41,Citation70,Citation71 These results support clinical findings that liver function can also be impaired in diabetic patients having HNF4α mutations.
Targeting HNF4α in molecular therapy
Liver transplantation remains the only curative treatment for patients with terminal liver failure. Recent advances in the understanding of the pathophysiology of cirrhosis, and the treatment of its complications, have resulted in improved management, leading to an increase in the quality of life and life expectancy of patients with liver disease.Citation72–74 Pharmacological treatments targeting fibrosis and cirrhosis have been investigated with some success, such as the utilization of antifibrotic medications focusing on hepatic stellate cells.Citation75,Citation76 Unfortunately, these therapeutic approaches fall short in achieving a significant restoration of vital liver function, as the potential side effects (e.g. hepatocarcinogenesis) outweigh the benefits.Citation72–74, Citation76
In the search for alternative treatments for ESLD, different approaches have revealed HNF4α as a potential target. Previously, HNF4α has been described as a potential therapeutic candidate for other liver diseases. Forced expression of HNF4α in animal models and cell lines of hepatocellular carcinoma (HCC) have shown to reverse invasiveness.Citation77 Downregulation of HNF4α promotes hepatic fibrosis and its re-expression alleviates hepatic fibrosis and improves liver function by suppressing the epithelial–mesenchymal transition (EMT). This evidence suggests that HNF4α could be an ideal option for the treatment of HCC or hepatic fibrosis.Citation78 Furthermore, it has been shown that the forced re-expression of HNF4α in rat models of end-stage liver disease corrected hepatocyte function at different levels, resetting target genes including liver-enriched transcription factors, serum proteins, coagulation factors and metabolic enzymes, as well as reversing impaired hepatic function.Citation18
Regarding the role of HNF4α as a possible candidate for transcriptional factor therapy, there are still many problems to be solved. In order to develop therapies based on target genes and bring them to the clinical trial stage, the type of therapy or delivery system must first be determined. The therapeutic potential of HNF4α appears very promising since mRNA-based therapeutics as an exogenous delivery system have been shown to be effective in replacing, reprogramming or modifying protein expression. The development of different anti-cancer therapiesCitation79 and the generation of vaccines against infectious diseases, such as SARS-CoV-2, have made apparent the power of this technology.Citation80–82
Interestingly, in a study closely modeling the pathophysiology of cirrhotic human livers, Tafaleng et al. showed that forced re-expression of HNF4α via mRNA treatment is effective in restoring, to some degree, endogenous expression of LETF and hepatocyte functional genes in human hepatocytes isolated from a cirrhotic liver. However, this study showed that this approach was not maximally effective in restoring normal HNF4α function, at least at the level of protein localization (Tafaleng et al., in press). Further investigation is necessary to increase the efficiency of HNF4α as a transcription factor in decompensated human hepatocytes. This may be done by targeting genes related to the function of HNF4α or those associated with its localization.
Currently, the concept of HNF4α as an ideal candidate for the treatment of end-stage liver disease is emerging, but to establish this potential molecular therapy, new challenges must be faced and questions must be answered, paving the way for clinical trials.
Abbreviations
ESLD, end-stage liver disease; NAFLD, nonalcoholic fatty liver disease; OLT, orthotopic liver transplantation; LETFs, liver-enriched transcription factors; HNF4α, hepatocyte nuclear factor 4 alpha; ARLD, alcohol-related liver disease; HBV, hepatitis B virus; HCV, hepatitis C virus; NASH, nonalcoholic steatohepatitis; ECM, extracellular matrix; EMT, epithelial–mesenchymal transition; HCC, hepatocellular carcinoma.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Hansen L, Lyons KS, Dieckmann NF, Chang MF, Hiatt S, Solanki E, Lee CS. Background and design of the symptom burden in end-stage liver disease patient-caregiver dyad study. Res Nurs Health. 2017;40(5):398–413. doi:https://doi.org/10.1002/nur.21807.
- Tapper EB, Parikh ND. Mortality due to cirrhosis and liver cancer in the United States, 1999-2016: observational study. Bmj. 2018;362:k2817. doi:https://doi.org/10.1136/bmj.k2817.
- Asrani SK, Larson JJ, Yawn B, Therneau TM, Kim WR. Underestimation of liver-related mortality in the United States. Gastroenterology. 2013;145(2):375–82.e1-2. doi:https://doi.org/10.1053/j.gastro.2013.04.005.
- Ge PS, Runyon BA, Campion EW. Treatment of patients with cirrhosis. N Engl J Med. 2016;375(8):767–77. doi:https://doi.org/10.1056/NEJMra1504367.
- Habka D, Mann D, Landes R, Soto-Gutierrez A. Future economics of liver transplantation: a 20-year cost modeling forecast and the prospect of bioengineering autologous liver grafts. PLoS One. 2015;10(7):e0131764. doi:https://doi.org/10.1371/journal.pone.0131764.
- Kwong AJ, Kim WR, Lake JR, Smith JM, Schladt DP, Skeans MA, Noreen SM, Foutz J, Booker SE, Cafarella M, et al. OPTN/SRTR 2019 Annual Data Report: liver. Am J Transplant. 2021;21(Suppl 2):208–315. doi:https://doi.org/10.1111/ajt.16494.
- Nishikawa K, Osawa Y, and Kimura K. Wnt/β-Catenin signaling as a potential target for the treatment of liver cirrhosis using antifibrotic drugs. Int J Mol Sci . 2018;10:19.
- Moroni F, Dwyer BJ, Graham C, Pass C, Bailey L, Ritchie L, Mitchell D, Glover A, Laurie A, Doig S, et al. Safety profile of autologous macrophage therapy for liver cirrhosis. Nat Med. 2019;25(10):1560–65. doi:https://doi.org/10.1038/s41591-019-0599-8.
- Schrem H, Klempnauer J, Borlak J. Liver-enriched transcription factors in liver function and development. Part I: the hepatocyte nuclear factor network and liver-specific gene expression. Pharmacol Rev. 2002;54(1):129–58. doi:https://doi.org/10.1124/pr.54.1.129.
- Liu L, Yannam GR, Nishikawa T, Yamamoto T, Basma H, Ito R, Nagaya M, Dutta-Moscato J, Stolz DB, Duan F, et al. The microenvironment in hepatocyte regeneration and function in rats with advanced cirrhosis. Hepatology. 2012;55(5):1529–39. doi:https://doi.org/10.1002/hep.24815.
- Haep N, Florentino RM, Squires JE, Bell A, Soto-Gutierrez A. The Inside-Out of End-Stage Liver Disease: hepatocytes are the Keystone. Semin Liver Dis. 2021;41(2):213–24. doi:https://doi.org/10.1055/s-0041-1725023.
- Guzman-Lepe J, Cervantes-Alvarez E, Collin De L HA, Wang Y, Wm M, Oda Y, Bekki Y, Shimokawa M, Wang H, Yoshizumi T, et al. Liver-enriched transcription factor expression relates to chronic hepatic failure in humans. Hepatol Commun. 2018;2(5):582–94. doi:https://doi.org/10.1002/hep4.1172.
- Babeu JP, Boudreau F, Boudreau F. Hepatocyte nuclear factor 4-alpha involvement in liver and intestinal inflammatory networks. World J Gastroenterol. 2014;20(1):22–30. doi:https://doi.org/10.3748/wjg.v20.i1.22.
- Battle MA, Konopka G, Parviz F, Gaggl AL, Yang C, Sladek FM, Duncan SA. Hepatocyte nuclear factor 4alpha orchestrates expression of cell adhesion proteins during the epithelial transformation of the developing liver. Proc Natl Acad Sci U S A. 2006;103(22):8419–24. doi:https://doi.org/10.1073/pnas.0600246103.
- Parviz F, Matullo C, Garrison WD, Savatski L, Adamson JW, Ning G, Kaestner KH, Rossi JM, Zaret KS, Duncan SA, et al. Hepatocyte nuclear factor 4alpha controls the development of a hepatic epithelium and liver morphogenesis. Nat Genet. 2003;34(3):292–96. doi:https://doi.org/10.1038/ng1175.
- Yeh MM, Bosch DE, Daoud SS. Role of hepatocyte nuclear factor 4-alpha in gastrointestinal and liver diseases. World J Gastroenterol. 2019;25(30):4074–91. doi:https://doi.org/10.3748/wjg.v25.i30.4074.
- Ji D, Chen GF, Wang JC, Cao LH, Lu F, Mu XX, Zhang X-Y, Lu X-J. Identification of TAF1,HNF4A, andCALM2 as potential therapeutic target genes for liver fibrosis. J Cell Physiol. 2019;234(6):9045–51. doi:https://doi.org/10.1002/jcp.27579.
- Nishikawa T, Bell A, Brooks JM, Setoyama K, Melis M, Han B, Fukumitsu K, Handa K, Tian J, Kaestner KH, et al. Resetting the transcription factor network reverses terminal chronic hepatic failure. J Clin Invest. 2015;125(4):1533–44. doi:https://doi.org/10.1172/JCI73137.
- Cox-North P, Doorenbos A, Shannon SE, Scott J, Curtis JR. The transition to end-of-life care in end-stage liver disease. Journal of Hospice & Palliative Nursing. 2013;15(4):209–15. doi:https://doi.org/10.1097/NJH.0b013e318289f4b0.
- Yadav DK, Zhang Q, Bai X, Li E, Liang T. Liver transplantation for alcohol-related liver disease (ARLD): an update on controversies and considerations. Can J Gastroenterol Hepatol. 2020;2020:8862152. doi:https://doi.org/10.1155/2020/8862152.
- D’Souza S, Lau KC, Coffin CS, Patel TR. Molecular mechanisms of viral hepatitis induced hepatocellular carcinoma. World J Gastroenterol. 2020;26(38):5759–83. doi:https://doi.org/10.3748/wjg.v26.i38.5759.
- Lin J, Wu JF, Zhang Q, Zhang HW, Cao GW. Virus-related liver cirrhosis: molecular basis and therapeutic options. World J Gastroenterol. 2014;20(21):6457–69. doi:https://doi.org/10.3748/wjg.v20.i21.6457.
- Calzadilla Bertot L, and Adams LA. The natural course of non-alcoholic fatty liver disease. Int J Mol Sci . 2016;5:17.
- Pierantonelli I. S-BG nonalcoholic fatty Liver disease: basic pathogenetic mechanisms in the progression from NAFLD to NASH. Transplantation. 2019;103(1):e1–e13. doi:https://doi.org/10.1097/TP.0000000000002480.
- Ma X, Li Z. Pathogenesis of nonalcoholic steatohepatitis (NASH). Chin J Dig Dis. 2006;7(1):7–11. doi:https://doi.org/10.1111/j.1443-9573.2006.00237.x.
- Sayiner M, Koenig A, Henry L, Younossi ZM. Epidemiology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis in the United States and the rest of the world. Clin Liver Dis. 2016;20(2):205–14. doi:https://doi.org/10.1016/j.cld.2015.10.001.
- Mazzarelli C, Prentice WM, Heneghan MA, Belli LS, Agarwal K, Cannon MD. Palliative care in end-stage liver disease: time to do better? Liver Transpl. 2018;24(7):961–68. doi:https://doi.org/10.1002/lt.25193.
- Potosek J, Curry M, Buss M, Chittenden E. Integration of palliative care in end-stage liver disease and liver transplantation. J Palliat Med. 2014;17(11):1271–77. doi:https://doi.org/10.1089/jpm.2013.0167.
- Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut. 2015;64(5):830–41. doi:https://doi.org/10.1136/gutjnl-2014-306842.
- Hernaez R, Solà E, Moreau R, Ginès P. Acute-on-chronic liver failure: an update. Gut. 2017;66(3):541–53. doi:https://doi.org/10.1136/gutjnl-2016-312670.
- Nishikawa T, Bellance N, Damm A, Bing H, Zhu Z, Handa K, Yovchev MI, Sehgal V, Moss TJ, Oertel M, et al. A switch in the source of ATP production and a loss in capacity to perform glycolysis are hallmarks of hepatocyte failure in advance liver disease. J Hepatol. 2014;60(6):1203–11. doi:https://doi.org/10.1016/j.jhep.2014.02.014.
- Fang B, Mane-Padros D, Bolotin E, Jiang T, Sladek FM. Identification of a binding motif specific to HNF4 by comparative analysis of multiple nuclear receptors. Nucleic Acids Res. 2012;40(12):5343–56. doi:https://doi.org/10.1093/nar/gks190.
- Azmi AS, Bao GW, Gao J, Mohammad RM, Sarkar FH. Network insights into the genes regulated by hepatocyte nuclear factor 4 in response to drug induced perturbations: a review. Curr Drug Discov Technol. 2013;10(2):147–54. doi:https://doi.org/10.2174/1570163811310020007.
- Gloyn AL, Ellard S, Shepherd M, Howell RT, Parry EM, Jefferson A, Levy ER, Hattersley AT. Maturity-onset diabetes of the young caused by a balanced translocation where the 20q12 break point results in disruption upstream of the coding region of hepatocyte nuclear factor-4alpha (HNF4A) gene. Diabetes. 2002;51(7):2329–33. doi:https://doi.org/10.2337/diabetes.51.7.2329.
- Hadzopoulou-Cladaras M, Kistanova E, Evagelopoulou C, Zeng S, Cladaras C, Ladias JA. Functional domains of the nuclear receptor hepatocyte nuclear factor 4. J Biol Chem. 1997;272(1):539–50. doi:https://doi.org/10.1074/jbc.272.1.539.
- Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83(6):841–50. doi:https://doi.org/10.1016/0092-8674(95)90200-7.
- Lu H. Crosstalk of HNF4α with extracellular and intracellular signaling pathways in the regulation of hepatic metabolism of drugs and lipids. Acta Pharm Sin B. 2016;6(5):393–408. doi:https://doi.org/10.1016/j.apsb.2016.07.003.
- Iyemere VP, Davies NH, Brownlee GG. The activation function 2 domain of hepatic nuclear factor 4 is regulated by a short C-terminal proline-rich repressor domain. Nucleic Acids Res. 1998;26(9):2098–104. doi:https://doi.org/10.1093/nar/26.9.2098.
- Suaud L, Formstecher P, Laine B. The activity of the activation function 2 of the human hepatocyte nuclear factor 4 (HNF-4alpha) is differently modulated by F domains from various origins. Biochem J. 1999;340(1):161–69. doi:https://doi.org/10.1042/bj3400161.
- Qu M, Duffy T, Hirota T, Kay SA. Nuclear receptor HNF4A transrepresses CLOCK:BMAL1 and modulates tissue-specific circadian networks. Proc Natl Acad Sci U S A. 2018;115(52):E12305–e12. doi:https://doi.org/10.1073/pnas.1816411115.
- Taniguchi H, Fujimoto A, Kono H, Furuta M, Fujita M, Nakagawa H. Loss-of-function mutations in Zn-finger DNA-binding domain of HNF4A cause aberrant transcriptional regulation in liver cancer. Oncotarget. 2018;9(40):26144–56. doi:https://doi.org/10.18632/oncotarget.25456.
- Drewes T, Senkel S, Holewa B, Ryffel GU. Human hepatocyte nuclear factor 4 isoforms are encoded by distinct and differentially expressed genes. Mol Cell Biol. 1996;16:925–31. doi:https://doi.org/10.1128/MCB.16.3.925.
- Gonzalez FJ. Regulation of hepatocyte nuclear factor 4 alpha-mediated transcription. Drug Metab Pharmacokinet. 2008;23(1):2–7. doi:https://doi.org/10.2133/dmpk.23.2.
- Sladek FM, Zhong WM, Lai E, Darnell JE Jr. Liver-enriched transcription factor HNF-4 is a novel member of the steroid hormone receptor superfamily. Genes Dev. 1990;4(12b):2353–65. doi:https://doi.org/10.1101/gad.4.12b.2353.
- Carrière V, Vidal R, Lazou K, Lacasa M, Delers F, Ribeiro A, Rousset M, Chambaz J, Lacorte JM. HNF-4-dependent induction of apolipoprotein A-IV gene transcription by an apical supply of lipid micelles in intestinal cells. J Biol Chem. 2005;280(7):5406–13. doi:https://doi.org/10.1074/jbc.M408002200.
- Hwang-Verslues WW, Sladek FM. HNF4α—role in drug metabolism and potential drug target? Curr Opin Pharmacol. 2010;10(6):698–705. doi:https://doi.org/10.1016/j.coph.2010.08.010.
- Bolotin E, Liao H, Ta TC, Yang C, Hwang-Verslues W, Evans JR, Jiang T, Sladek FM. Integrated approach for the identification of human hepatocyte nuclear factor 4alpha target genes using protein binding microarrays. Hepatology. 2010;51(2):642–53. doi:https://doi.org/10.1002/hep.23357.
- Lau HH, Ng NHJ, Loo LSW, Jasmen JB, Teo AKK. The molecular functions of hepatocyte nuclear factors - In and beyond the liver. J Hepatol. 2018;68(5):1033–48. doi:https://doi.org/10.1016/j.jhep.2017.11.026.
- Huang J, Levitsky LL, Rhoads DB. Novel P2 promoter-derived HNF4α isoforms with different N-terminus generated by alternate exon insertion. Exp Cell Res. 2009;315(7):1200–11. doi:https://doi.org/10.1016/j.yexcr.2009.01.004.
- Wang J-C, Stafford JM, Dk G. SRC-1 and GRIP1 coactivate transcription with hepatocyte nuclear factor 4*. Journal of Biological Chemistry. 1998;273(47):30847–50. doi:https://doi.org/10.1074/jbc.273.47.30847.
- Barrero MJ, Malik S. Two functional modes of a nuclear receptor-recruited arginine methyltransferase in transcriptional activation. Mol Cell. 2006;24(2):233–43. doi:https://doi.org/10.1016/j.molcel.2006.09.020.
- Soutoglou E, Katrakili N, Talianidis I. Acetylation regulates transcription factor activity at multiple levels. Mol Cell. 2000;5(4):745–51. doi:https://doi.org/10.1016/S1097-2765(00)80253-1.
- Yokoyama A, Katsura S, Ito R, Hashiba W, Sekine H, Fujiki R, Kato S. Multiple post-translational modifications in hepatocyte nuclear factor 4α. Biochem Biophys Res Commun. 2011;410(4):749–53. doi:https://doi.org/10.1016/j.bbrc.2011.06.033.
- Hong YH, Varanasi US, Yang W, Leff T. AMP-activated protein kinase regulates HNF4alpha transcriptional activity by inhibiting dimer formation and decreasing protein stability. J Biol Chem. 2003;278(30):27495–501. doi:https://doi.org/10.1074/jbc.M304112200.
- Ko HL, Zhuo Z, Ren EC. HNF4α combinatorial isoform heterodimers activate distinct gene targets that differ from their corresponding homodimers. Cell Rep. 2019;26(10):2549–2557.e3. doi:https://doi.org/10.1016/j.celrep.2019.02.033.
- É L, Babeu J-P, Simoneau J, Raisch J, Lavergne L, Lévesque D, Jolibois É, Avino M, Scott MS, Boudreau F, et al. Human hepatocyte nuclear factor 4-α encodes isoforms with distinct transcriptional functions. Mol Cell Proteomics. 2020;19(5):808–27. doi:https://doi.org/10.1074/mcp.RA119.001909.
- Briançon N, Weiss MC. In vivo role of the HNF4alpha AF-1 activation domain revealed by exon swapping. EMBO J. 2006;25(6):1253–62. doi:https://doi.org/10.1038/sj.emboj.7601021.
- Guo S, Lu H. Novel mechanisms of regulation of the expression and transcriptional activity of hepatocyte nuclear factor 4α. J Cell Biochem. 2019;120(1):519–32. doi:https://doi.org/10.1002/jcb.27407.
- Walesky C, Gunewardena S, Terwilliger EF, Edwards G, Borude P, Apte U. Hepatocyte-specific deletion of hepatocyte nuclear factor-4α in adult mice results in increased hepatocyte proliferation. Am J Physiol Gastrointest Liver Physiol. 2013;304:G26–G37. doi:https://doi.org/10.1152/ajpgi.00064.2012.
- Walesky C, Apte U. Role of hepatocyte nuclear factor 4α (HNF4α) in cell proliferation and cancer. Gene Expr. 2015;16(3):101–08. doi:https://doi.org/10.3727/105221615X14181438356292.
- Wu H, Reizel T, Wang YJ, Lapiro JL, Kren BT, Schug J, Rao S, Morgan A, Herman A, Shekels LL, et al. A negative reciprocal regulatory axis between cyclin D1 and HNF4α modulates cell cycle progression and metabolism in the liver. Proc Natl Acad Sci U S A. 2020;117(29):17177–86. doi:https://doi.org/10.1073/pnas.2002898117.
- Huck I, Gunewardena S, Espanol-Suner R, Willenbring H, Apte U. Hepatocyte nuclear factor 4 alpha activation is essential for termination of liver regeneration in mice. Hepatology (Baltimore, Md). 2019;70(2):666–81. doi:https://doi.org/10.1002/hep.30405.
- Dubois V, Staels B, Lefebvre P, Verzi MP, Eeckhoute J. Control of cell identity by the nuclear receptor HNF4 in organ pathophysiology. Cells. 2020;9(10):2185. doi:https://doi.org/10.3390/cells9102185.
- Florentino RM, Fraunhoffer NA, Morita K, Takeishi K, Ostrowska A, Achreja A, Animasahun O, Haep N, Arazov S, Agarwal N, et al. Cellular location of HNF4α is linked with terminal liver failure in humans. Hepatology Communications. 2020;4(6):859–75. doi:https://doi.org/10.1002/hep4.1505.
- Morales-Ruiz M, Santel A, Ribera J, Jiménez W. The role of Akt in chronic liver disease and liver regeneration. Semin Liver Dis. 2017;37:11–16. doi:https://doi.org/10.1055/s-0036-1597819.
- Yu D, Chen G, Pan M, Zhang J, He W, Liu Y, Nian, X, Sheng , L, and Xu, B, High fat diet-induced oxidative stress blocks hepatocyte nuclear factor 4α and leads to hepatic steatosis in mice. J Cell Physiol. 2018;233(6):4770–82. doi:https://doi.org/10.1002/jcp.26270.
- Marcil V, Sinnett D, Seidman E, Boudreau F, Gendron FP, Beaulieu JF, Menard D, Lambert M, Bitton A, Sanchez R, et al. Association between genetic variants in the HNF4A gene and childhood-onset Crohn’s disease. Genes Immun. 2012;13(7):556–65. doi:https://doi.org/10.1038/gene.2012.37.
- Chahar S, Gandhi V, Yu S, Desai K, Cowper-sal-lari R, Kim Y, Perekatt AO, Kumar N, Thackray JK, Musolf A, et al. Chromatin profiling reveals regulatory network shifts and a protective role for hepatocyte nuclear factor 4α during colitis. Mol Cell Biol. 2014;34(17):3291–304. doi:https://doi.org/10.1128/MCB.00349-14.
- Lausen J, Thomas H, Lemm I, Bulman M, Borgschulze M, Lingott A, Hattersley, A T, and Riffel, G U, Naturally occurring mutations in the human HNF4alpha gene impair the function of the transcription factor to a varying degree. Nucleic Acids Res. 2000;28(2):430–37. doi:https://doi.org/10.1093/nar/28.2.430.
- Jrm E, Formstecher P, Maturity-Onset LB. Diabetes of the young type 1 (MODY1)-Associated mutations R154X and E276Q in hepatocyte nuclear factor 4α (HNF4α) gene impair recruitment of p300, a key transcriptional coactivator. Molecular Endocrinology. 2001;15:1200–10.
- Oxombre B, Moerman E, Eeckhoute J, Formstecher P, Laine B. Mutations in hepatocyte nuclear factor 4α (HNF4α) gene associated with diabetes result in greater loss of HNF4α function in pancreatic β-cells than in nonpancreatic β-cells and in reduced activation of the apolipoprotein CIII promoter in hepatic cells. J Mol Med. 2002;80(7):423–30. doi:https://doi.org/10.1007/s00109-002-0340-8.
- Sebastiani G, Gkouvatsos K, Pantopoulos K. Chronic hepatitis C and liver fibrosis. World J Gastroenterol. 2014;20(32):11033–53. doi:https://doi.org/10.3748/wjg.v20.i32.11033.
- Pinzani M, Rombouts K, Colagrande S. Fibrosis in chronic liver diseases: diagnosis and management. J Hepatol. 2005;42(Suppl):S22–36. doi:https://doi.org/10.1016/j.jhep.2004.12.008.
- Schuppan D, Afdhal NH. Liver cirrhosis. Lancet. 2008;371(9615):838–51. doi:https://doi.org/10.1016/S0140-6736(08)60383-9.
- Chang Y, Li H. Hepatic antifibrotic pharmacotherapy: are we approaching success? J Clin Transl Hepatol. 2020;8(2):222–29. doi:https://doi.org/10.14218/JCTH.2020.00026.
- Rockey DC. Antifibrotic therapy in chronic liver disease. Clin Gastroenterol Hepatol. 2005;3(2):95–107. doi:https://doi.org/10.1016/S1542-3565(04)00445-8.
- Lazarevich NL, Cheremnova OA, Varga EV, Ovchinnikov DA, Kudrjavtseva EI, Morozova OV, Fleishman DI, Engelhardt NV, Duncan SA. Progression of HCC in mice is associated with a downregulation in the expression of hepatocyte nuclear factors. Hepatology. 2004;39(4):1038–47. doi:https://doi.org/10.1002/hep.20155.
- Yue HY, Yin C, Hou JL, Zeng X, Chen YX, Zhong W, Hu, PF, Deng, X, Tan, YX, and Zhang, JP, Hepatocyte nuclear factor 4alpha attenuates hepatic fibrosis in rats. Gut. 2010;59:236–46
- Verbeke R, Lentacker I, Wayteck L, Breckpot K, Van Bockstal M, Descamps B, Vanhove C, De Smedt SC, Dewitte H. Co-delivery of nucleoside-modified mRNA and TLR agonists for cancer immunotherapy: restoring the immunogenicity of immunosilent mRNA. J Control Release. 2017;266:287–300. doi:https://doi.org/10.1016/j.jconrel.2017.09.041.
- Richner JM, Himansu S, Dowd KA, Butler SL, Salazar V, Fox JM, Julander JG, Tang WW, Shresta S, Pierson TC, et al. Modified mRNA vaccines protect against Zika Virus Infection. Cell. 2017;168(6):1114–25.e10. doi:https://doi.org/10.1016/j.cell.2017.02.017.
- Corbett KS, Edwards DK, Leist SR, Abiona OM, Boyoglu-Barnum S, Gillespie RA, Himansu S, Schäfer A, Ziwawo CT, DiPiazza AT, et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature. 2020;586(7830):567–71. doi:https://doi.org/10.1038/s41586-020-2622-0.
- Anderson EJ, Rouphael NG, Widge AT, Jackson LA, Roberts PC, Makhene M, Chappell JD, Denison MR, Stevens LJ, Pruijssers AJ, et al. Safety and Immunogenicity of SARS-CoV-2 mRNA-1273 Vaccine in Older Adults. N Engl J Med. 2020;383(25):2427–38. doi:https://doi.org/10.1056/NEJMoa2028436.