
ABSTRACT
Based on successes in preclinical animal transplant models, adoptive cell therapy (ACT) with regulatory T cells (Tregs) is a promising modality to induce allograft tolerance or reduce the use of immunosuppressive drugs to prevent rejection. Extensive work has been done in optimizing the best approach to manufacture Treg cell products for testing in transplant recipients. Collectively, clinical evaluations have demonstrated that large numbers of Tregs can be expanded ex vivo and infused safely. However, these trials have failed to induce robust drug-free tolerance and/or significantly reduce the level of immunosuppression needed to prevent solid organ transplant (SOTx) rejection. Improving Treg therapy effectiveness may require increasing Treg persistence or orchestrating Treg migration to secondary lymphatic tissues or places of inflammation. In this review, we describe current clinical Treg manufacturing methods used for clinical trials. We also highlight current strategies being implemented to improve delivered Treg ACT persistence and migration in preclinical studies.
Introduction
Regulatory T cells (Tregs) are a specialized subpopulation of CD4+ T cells that are crucial in maintaining immune homeostasis and preventing autoimmunity.Citation1 Tregs in humans are characterized by their prolific expression of the high-affinity interleukin IL-2 receptor α-chain (CD25), transcription factor forkhead box P3 (Foxp3), and low expression of IL-7 receptor α-chain (CD127).Citation2 The importance of Treg number and/or function in immune tolerance is illustrated by immunodysregulation polyendocrinopathy enteropathy X-lined (IPEX) syndrome which is caused by a mutation in the FoxP3 gene and can be cured by restoring normal Treg populations via hematopoietic cell transplant (HCT).Citation3,Citation4
Tregs function to suppress immune responses toward self and non-self by limiting the activation and proliferation of other T cells through multiple mechanisms. These mechanisms include the production of anti-inflammatory cytokines such as TGF-β (transforming growth factor beta) and IL-10 and the expression of membrane-bound molecules such as CTLA-4 (cytotoxic T cell-associated antigen), LAG-3 (lymphocyte activation gene 3), TIGIT (T cell immune receptor with Ig and ITIM domains), and CD39 (ectonucleoside triphosphate diphosphohydrolase 1).Citation1,Citation5 Tregs can also modulate antigen-presenting cells (APCs) through contact-dependent mechanisms that alter APCs capacity for co-stimulation and antigen presentation.Citation5 Additionally, high expression of CD25 enables them to sequester local IL-2 which limits effector T cell expansion and function by depriving them of IL-2.Citation6,Citation7
Interest in harnessing the suppressive capacity of Tregs for immunotherapy originates from early studies which demonstrated the importance of CD4+CD25+ Tregs in inhibiting the development of autoimmune disease.Citation8 It was then demonstrated that naturally occurring CD4+CD25+ Treg populations could be expanded ex vivo to treat autoimmune diseases in mice by suppressing and modulating effector T cells.Citation9 Thereafter, using adoptively transferred CD4+CD25+ Tregs in a series of preclinical sentinel model studies in solid organ transplantation (SOTx) and graft-versus-host disease (GvHD) after allogeneic hematopoietic cell transplantation (AlloHCT) further supported Treg therapeutic potential to induce immunological tolerance to non-self-antigens to prevent allograft rejection and GvHD. GvHD is a life-threatening complication of AlloHCT, which is used to treat hematological and non-hematological diseases.Citation10 GvHD is an alloreactive donor T cell-mediated response driven immunologically by differences between donor and recipient. In seminal studies completed by Taylor et al., they demonstrated that depletion of murine CD4+CD25+ cells from the BM donor increased GvHD, which was then reduced upon infusion of fresh donor CD4+CD25+ cells.Citation11 Since then, utilizing Treg to prevent/delay GvHD has been routinely demonstrated in preclinical murine models.Citation12–14
The therapeutic potential of Tregs to support allograft survival after SOTx has also been demonstrated using preclinical models of skin and heart Tx. For example, heart allograft survival was prolonged following transfer of ex vivo expanded CD4+CD25+ Tregs in wild-type mice.Citation15 Additionally, studies using humanized murine models demonstrated prolonged allograft survival in pancreatic islet and skin Tx following adoptive transfer of huTreg.Citation16–18 What would further translate to huTregs was the finding that murine Treg function was more potent than CD4+CD25+ Treg when sorted based on low expression of CD127.Citation19 Due to their immunological similarity to humans, the use of non-human primate models (NHP) has also been important in evaluating adoptive cell therapy (ACT) with Treg in SOTx. Ex vivo expanded Treg in NHP models are well characterized and adoptive transfer has been reported to suppress renal allograft rejection and prolong survival.Citation20–22
Recently, clinical studies have been completed or underway assessing Treg to improve outcomes after SOTx or with AlloHCT to prevent GVHD.Citation5, Citation23–25 These clinical studies have also shown that large numbers of CD4+CD25+ Treg can be expanded in the presence of high doses of IL-2 ex vivo and infused safely.Citation5, Citation26–34 The number of clinical trials utilizing Treg cell therapy in transplantation and other fields are expected to increase. In this review, we describe current clinical GMP methods used to isolate and expand Tregs ex vivo for clinical trials in solid organ Tx and treatment/prevention of GvHD. We then discuss in vivo and ex vivo strategies being used to improve adoptive Treg cell therapy persistence and migration to increase the efficacy of Treg therapy in preclinical studies. While numerous preclinical publications exist describing manufacturing methods of Treg cell products, few of these publications provide a correlation with completed clinical studies in SOTx. Therefore, to improve and enhance Treg cell therapy in the clinic, it is important to highlight manufacturing protocols specific to completed and recruiting clinical trials when possible. In this review, we aim to briefly introduce general concepts around Treg and then highlight clinical manufacturing methods used to generate Treg cell products used in SOTx referencing published clinical trial protocols, manuscripts, and preclinical protocol development. It is also noteworthy that even in preclinical animal transplant models Treg ACT has only provided prolonged SOTx survival. Thus, even at the preclinical level, Treg ACT is far from optimized. All clinical trials to date unfortunately have not reduced the levels of immunosuppressive drugs needed to prevent SOTx rejection.Citation5,Citation21,Citation28,Citation33 Thus, we also provide examples of ways investigators are working to improve Treg ACT and move the needle toward the field’s goal of maintenance of normal graft function without the use of immunosuppression, also known as drug-free tolerance or operational tolerance (OT).
Adoptive Treg therapy
Treg ACT has developed due to the need for alternative therapeutic agents to limit the need for immunosuppression (IS) after SOTx, and ideally support the routine induction and maintenance of immune transplant tolerance in recipients of allogeneic materials. SOTx remains the only effective lasting treatment for end-stage organ diseases. Strategies to increase short-term outcomes after SOTx have relied on better patient donor selection and improved immunosuppressive regimens.Citation24 For example, calcineurin inhibitors, antiproliferative agents, and mTOR (mammalian target of rapamycin) inhibitors can control the immune response early post-transplant, but have detrimental side effects such as cardiovascular diseases, kidney failure, and susceptibility to opportunistic infections.Citation24 Efforts to improve long-term outcomes and 10-year survival remain unsuccessful due to the failure of multi-drug immunosuppression to address chronic rejection despite their toxic side effects.Citation21,Citation35 Other therapeutic strategies such as co-stimulation-blockade (CoSB) or monoclonal antibody (Ab) therapy to target cytokines and other co-stimulatory molecules have only had limited success at reducing the use of immunosuppressive drugs.Citation21 Therefore, utilizing Tregs or other regulatory immune cells as a potential immune cell therapy may be a promising alternative to reduce the use of immunosuppressive drugs and even induce allograft tolerance based on data obtained from various preclinical murine and NHP studies.
Despite new prophylaxis strategies, GVHD is a common side effect of AlloHCT where alloreactive T cells destroy host tissues. Current treatments for GVHD involve nonspecific multi-drug immunosuppression, particularly corticosteroids, that often leads to morbidity and mortality due to cancer relapse or secondary infection.Citation10 Thus, alternative therapies such as Tregs are being investigated to prevent the development of GVHD after AlloHCT. If prophylaxes therapies can successfully prevent GVHD, this could also expand the use of AlloHCT beyond cancer treatment and for routine use as a means to induce transplant tolerance after SOTx.Citation36–38
Treg isolation
The donor source of Tregs is important when designing and describing Treg cell therapy products. Tregs are typically classified as thymic-derived Tregs (tTreg) and peripherally-derived Tregs (pTreg).Citation39 In contrast to tTreg cells that originate from the thymus, pTreg develop from conventional CD4+ T cells in the periphery after antigen encounter along with TGF-β, especially in the gut.Citation23 Similarly, investigators have “induced” Foxp3+ Treg (iTreg) in vitro from CD4+ Foxp3- populations by expanding CD4+CD25− cells in the presence of IL-2, rapamycin, and TGF-β.Citation40 To date, the majority of the Tregs used in clinical trials for SOTx, however, were isolated from autologous peripheral blood mononuclear cells (PBMC) or Umbilical cord blood (UCB). Unfortunately, there are no consistently reliable specific markers to distinguish between tTreg and pTreg in mice and humans, thus making specific origination of Treg isolated from these sources imprecise. There is evidence to support the use of allogenic Tregs in humans; however, that is beyond the scope of this review.Citation26,Citation27,Citation30 Therefore, this review will focus discussions on the detailed manufacturing of autologous Tregs from human subjects.
Human Tregs are relatively rare in the PBMCs and comprise only 2–10% of peripheral CD4+ T cells in healthy adults. Thus, Treg isolation and ex vivo expansion is required to reach the numbers of Treg predicted in animal studies to be therapeutic after adoptive transfer.Citation21,Citation41,Citation42 As introduced above, the Tregs for clinical trials have been most commonly isolated from PBMC or UCB.Citation24,Citation26,Citation27, Citation43–45 A recent novel strategy used pediatric thymuses which are routinely discarded during pediatric heart transplantation as a Treg source.Citation46,Citation47 Tregs develop in the thymus and thus the thymus represents an attractive alternative source to isolate Tregs for cellular therapies. In a study involving 11 different donors, roughly 12% of the CD4+ cells were CD3+ CD4+ CD8− CD25+ CD127− with 85% of these cells expressing the FoxP3.Citation46 This approach is an exciting alternative to isolate large numbers of Tregs; however, it is so far limited to pediatric heart transplant recipients or when third-party Tregs are utilized. The clinical feasibility to isolate and expand functional tTregs is currently being explored in an ongoing clinical trial (THYTECH; NCT0492449) where tTregs isolated from the thymus are being tested for their ability to prevent rejection after pediatric heart transplantation.Citation46
Treg isolation from leukapheresis has also been evaluated as a potential alternative source and offers increased starting cell density to improve Treg yields for subsequent ex vivo expansion.Citation48 Although not yet tested in clinical trials, the use of Granulocyte Colony-Stimulating Factor (G-CSF)-mobilized peripheral blood stem cells (G-PBSC) may be another potential source that could be used to isolate Tregs for patients receiving HCT.Citation49,Citation50 In this case, Treg isolation and expansion would be performed after CD34 selection utilizing the typically discarded CD34 negative fraction. Application of G-CSF significantly increased Treg yield while preserving suppressive function and phenotype.Citation50 To date, only PBMC and UBC have been clinically tested in SOTx and AlloHCT.
There has been rapid progress in the development of Good Manufacturing Practice (GMP) grade protocols to isolate and expand large numbers, and recent reviews have detailed the strategies used to isolate Tregs from various blood products for use in clinical trials.Citation24,Citation44,Citation51,Citation52 Of these, common isolation techniques include fluorescence-activated cell sorting (FACS), direct or untouched magnetic cell separation, or a combination of both. In the case of magnetic cell separation, these systems utilize magnetic microbeads to bind cells of interest (direct enrichment, or “positive” selection) or to selectively deplete unwanted cells (“negative” selection). Additionally, regardless of the methodology or source, high surface expression of CD25 remains the primary marker used to distinguish human Tregs from non-target cells along with low expression of CD127.Citation53 Unfortunately, there is no singular surface marker for Tregs that distinguish them from other immune cells, and Foxp3 is an intracellular transcription factor that cannot be utilized for the isolation of live cells. Therefore, multiple surface parameters are needed to isolate Treg from blood products. Many groups have utilized cell sorting based on parameters of CD3+CD4+CD25+CD127low.Citation28,Citation31, Citation54–56 While this process yields highly pure Tregs, it is less efficient as run times can take many hours depending on the purity and cell concentration of the starting product. Additionally, there can be considerable cell loss with traditional sorting via FACS or positive and negative bead selection. Such loss impacts the overall product yield and increases the need for robust ex vivo expansion. These shortcomings have driven advancements that have been made in streamlining GMP-compliant protocols to generate clinical grade Tregs.
One of the most notable advancements in this field has been with Miltenyi’s closed system magnetic cell separators CliniMACS Prodigy and CliniMACS Plus.Citation57 These systems utilize magnetic microbeads to enrich for Tregs or to selectively deplete non-Treg (CD19+, CD8+, or CD127+ cells). Like flow cytometric cell sorting, this platform allows for multiparameter selection, and numerous protocols specifically for Tregs have been developed and established. Currently, the approaches most cited are CD25 enrichment with or without prior selective depletion of CD8+ and/or CD19+ cells.Citation26,Citation27,Citation29,Citation30,Citation48, Citation58–63
Another notable strategy that is becoming more common for Treg isolation allowing for a high purity Treg product is CD25 enrichment utilizing Miltenyi’s CliniMACS systems as described above in combination with a purification based on CD4+CD25+ CD127low selection using the closed cartridge, low cost MACSQuant® Tyto® (Miltenyi Biotec: Bergisch Gladbach, Germany). This strategy works by using a clinical GMP grade CD25 reagent (like the CD25 Enrichment microbeads) that is also biotinylated which allows the user to fluorescently label CD25 positive cells with phycoerythrin (PE) and perform purification using the MACSQuant Tyto sorter following the CD25 enrichment step. In addition, other fluorescent markers in addition to CD25, such as CD4, CD127, or CD45RA, can be used for subsequent sorting if desired. This approach significantly reduces the processing time while also preserving the yield and purity of the product. If optimized, this type of processing approach could limit product variability and increase reliability. Investigators have demonstrated the feasibility of using the CliniMACS for CD4+CD25+ Treg enrichment; however, in this case, the purity was suboptimal.Citation64,Citation65 In theory, subsequent sorting for CD4+CD25+CD127low could significantly improve the purity and allow for robust ex vivo expansions of pure Treg populations.
Treg expansion
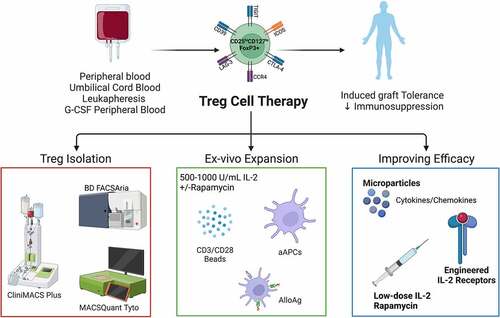
Based on the opinion, large doses of Tregs will be the most effective immunotherapy. CliniMACS-based and other Treg isolation protocols have been paired with ex vivo Treg expansion. There have been numerous different reagents and approaches used for generating clinical Treg products for the use in kidney transplantation () and liver and heart transplantation (). summarize completed and recruiting Phase I/II clinical trials in SOTx and the associated Treg manufacturing protocols. Using the isolation methods mentioned above, many groups have demonstrated that high numbers of Tregs can be isolated and subsequently expanded. Of note, it is evident in and 2 that there is a considerable lack of data on the purity and Treg number resulting from most isolation protocols. This is unfortunate as it is important to understand Treg viability, phenotype, and purity through isolation steps as it correlates to end product stability, function, as well as relevant clinical observations.
Table 1. Published manufacturing protocols for regulatory T cells assessed in kidney transplantation phase I/II clinical trials.
Table 2. Published manufacturing protocols for regulatory T cells assessed in liver and heart transplantation phase I/II clinical trials.
Human Tregs can be expanded in several ways all of which are dependent on IL-2 for proliferation and maintenance of the Treg phenotype (CD4+CD25+CD127lowFoxP3+). There is variation among protocols, however. IL-2 is typically added at day zero at 1000 U/mL and replenished as needed.Citation44,Citation52 There is no standard IL-2 concentration; however, a recent review has provided a detailed summary of reported concentrations (300–1000 U/mL) used for expansion in clinical trials.Citation44 The use of mTOR inhibitor rapamycin, an immunosuppressive drug used to prevent graft rejection, can also be used in clinical grade Treg expansion protocols.Citation29,Citation30,Citation45,Citation55,Citation57,Citation59,Citation66 Rapamycin selectively supports the ex vivo expansion of CD4+CD25+FoxP3+ Tregs while limiting the proliferation of contaminating non-Treg. Likewise, rapamycin promotes the stability and functional capacity of expanded Treg to suppress the proliferation of both autologous and allogenic CD4+ and CD8+ T cells in vitro.Citation67 This was also confirmed in preclinical rodent studies demonstrating that Tregs could be expanded ex vivo in the presence of rapamycin, and rapamycin delivery with Treg ACT can support tolerance to SOTx in rodents.Citation68–71 Therefore, in cases with less than ideal Treg purity following isolation, it may be beneficial to expand Tregs in the presence of rapamycin to block proliferation of unwanted CD4+CD25− cells.
Stimulating/activating strategies
T cell receptor (TCR) engagement is required for Treg cell differentiation and the induction of Foxp3.Citation72,Citation73 Recent studies in mice allowing the deletion of the Treg TCR have also revealed critical functions for TCR signaling in Treg lineage maintenance and their suppressive function.Citation74–77 In addition to TCR signaling, CD28 co-stimulation is necessary for Treg activation, proliferation, and function. Early experiments using mice deficient for CD28 or CD80/86 found reduced Treg populations, decreased CD25 expression, and suggested an important role for co-stimulation in Treg survival, FoxP3 expression, and suppressor function.Citation78,Citation79 The lack of CD28 or its ligands, CD80 and CD86, decreased Treg and exacerbates diabetes in nonobese diabetic (NOD) mice.Citation78 Additional observations in rodents and humans also demonstrate the importance of CD28 signaling for Treg proliferation.Citation80–83 As such, most clinical trials have expanded nonspecific polyclonal Tregs (polyTregs) using CD3/CD28 expander beads which provide both TCR and CD28 co-stimulation. There are several published GMP compatible methods used to ex vivo expand Tregs for cell therapy which have been previously described.Citation5,Citation24,Citation44,Citation52 The optimal way to stimulate Tregs remains unclear; however, there are two main approaches to ex vivo expand polyTregs for clinical trials. The most common approach to expand human polyTregs is to use polyclonal stimulation with anti-CD3 and anti-CD28 coated microbeads.Citation26, Citation28–31, Citation45,Citation54,Citation56,Citation84,Citation85 GMP grade anti-CD3 and anti-CD28-coated nanoparticles are available for clinical use from Miltenyi Biotec (ExpAct™ Treg kit) and Invitrogen (CTS Dynabeads™ CD3/CD28). These beads are typically added at the start of culture and are expanded over a period of 14–36 days. It is unclear if multiple restimulations are required. Of those reported, fold expansions have been variable and can range from 100 to 2000 with IL-2 alone.Citation26,Citation31,Citation54,Citation63 Tregs can also be activated using artificial antigen-presenting cells (aAPCs). These cells have been developed to replace natural antigen-presenting cells (APCs) which mediate T cell effector function. Using lentiviral vector technology, aAPCs can be generated to more closely replicate the features of DCs that deliver CD28 co-stimulation.Citation86 For example, aAPCs expressing CD64 and CD86 were used to expand human UCB-derived Tregs.Citation27 These cells are advantageous because they do not require removal post expansion as cells are lethally irritated prior to use. K562 cells expressing CD86 and CD64 that are loaded with soluble anti-CD3 have been used in clinical trials.Citation27,Citation40 It has been reported that these aAPCs are highly effective at expanding Treg.Citation27,Citation60 For example, Brunstein et al. were able to achieve a 10,000-fold expansion of Tregs from UBC in 2 weeks using the aAPCs that expressed CD64 and CD86. In the presence of rapamycin, 3000-fold expansion was achieved.Citation87
In addition to manufacturing polyclonal Tregs through the process discussed above, generating donor-antigen-specific/reactive Tregs (darTreg) has been gaining traction in recent attempts to induce tolerance after SOTx. In comparison to polyTregs, darTregs are expanded in the presence of donor cells and exhibit donor specificity after alloantigen exposure by selectively targeting alloreactive effector T cells that are detrimental to the allograft.Citation88 Generally, darTreg are generated by isolating recipient Tregs and culturing them with donor alloantigen-expressing APCs from the transplant donor, such as monocyte-derived DC and B cells.Citation89,Citation90 The global Treg pool contains a small fraction (10–20%) of darTregs, and generating them in large quantities has been a barrier for successful implementation.Citation5,Citation16,Citation21, Citation91–94 Despite manufacturing challenges due to low frequency of darTregs, experimental models of transplantation have demonstrated that donor antigen-specific Tregs (darTregs) are more potent than polyTregs in promoting allograft survival.Citation16,Citation71,Citation88,Citation95 Therefore, due to increased specificity for alloreactive effector T cells and increased potency, it is likely that fewer darTreg are required to induce tolerance compared to polyTregs.
The first report of successful drug-free tolerance with a donor antigen specific cell therapy product in patients undergoing organ transplantation was published by Todo et al. In this study, investigators isolated recipient and donor lymphocytes and co-cultured recipient lymphocytes with irradiated donor lymphocytes with monoclonal antibodies to CD80/86 without IL-2 or rapamycin.Citation85 Patients who underwent living donor liver Tx (LDLT) were lymphodepleted 8–10 days post liver Tx and received a single infusion of the darTreg-enriched product. Of the 10 patients infused, 7 met the trial-defined endpoint of tolerance and were successfully weaned off immunosuppression within the study period.Citation85 Overall, infusions of 100–300 million cells were well tolerated without significant adverse event and the mean number of Tregs (CD4+CD25+FoxP3+) infused were 24.8% of the CD4+cells, which is a low Treg concentration compared to other clinical studies ( and 2).Citation85 The investigators measured suppressive function of the expanded cells in vitro using the mixed lymphocyte reaction (MLR) and inhibited recipient T cell proliferation by donor antigen stimulation. Yet, it is unclear what cell populations in the darTreg-enriched product were indeed responsible for tolerance induction in vivo and suppressive function in vitro. Nonetheless, these were impressive and exciting studies. As the Tregs made up a very small fraction of the total product, this would suggest that lower numbers of darTreg are needed to induce tolerance or at the least weaning of immunosuppressive regimens. Additionally, it suggests that possibly other regulatory cells in addition to darTregs may be beneficial, contributing to tolerance induction or reduction of immunosuppression. Todo et al.’s study has facilitated the development of protocols to expand antigen-specific or darTregs from humans.Citation9,Citation89,Citation93,Citation96,Citation97 Several clinical trials are testing darTregs in SOTx ( and 2). For example, the ONE Study was a massive multicenter study that included two polyTreg products and two darTreg products.Citation98
Recently, results of the Artemis Phase I/II clinical trial (NCT02474199) have been published. The objective of this study was to determine safety and efficacy of a single dose of darTreg to facilitate reduction of immunosuppression in patients 2–7 years post liver Tx. Nine participants initiated immunosuppression reduction and were eligible for a single darTreg infusion; however, only 5 of the 9 products manufactured met release criteria.Citation94 The products that were not infused failed release criteria due to insufficient dose highlighting the challenge to manufacture enough darTregs for clinical trials. Investigators aimed to infuse a total of 100–500 million darTreg. Two patients received >300 million and the other three received 100–200 million darTregs. In comparison to the study by Todo et al., their product manufactured from starting population Tregs was more pure meeting release criteria of >95% CD4+, >60% FoxP3+, and <5% CD4−CD8+.Citation94 Of the 5 participants who received darTregs, all infusions were well tolerated with no reported significant adverse events. Two participants met the primary endpoint of 75% reduction of calcineurin inhibition or discontinuation of a second drug, but none of the participants attempted complete immunosuppression withdrawal.Citation94 Efficacy could not be assessed due to too few treated participants. However, the investigators’ mechanistic studies offer insights to darTreg function in this patient population and current challenges to overcome. Their studies suggest darTreg are dysfunctional in patients 2–7 years after liver Tx. Mechanistic and phenotypic studies suggest an upregulation in activation, exhaustion, senescence, or progressive deletion after liver transplantation.Citation94 To investigate Treg donor reactivity after liver TX, the authors performed in vitro studies using blood samples from the AWISH study (AWISH; NTC00135694). Tregs were assessed longitudinally in 16 patients at pre-Tx, 6 months post-Tx, and 2 years post-Tx to assess darTreg and conventional T cell reactivity to donor antigen. Overall, authors showed reduced proliferation and selective reduction of donor reactivity in all T cells after liver Tx starting 6 months post liver Tx persisting until 2 years after transplant.Citation94 The differences in epigenetic regulation, transcription, and donor reactivity of darTregs in liver Tx highlight important insights to the difficulties of generating successful Treg products and underpin challenges faced when manufacturing enough darTregs. Future longitudinal studies should investigate both darTreg and polyTreg transcriptional changes and donor reactivity pre- and post-Tx when possible, which may offer solutions to improve clinical manufacturing of Treg products that are functionally potent and persist after infusion.
Alternative strategies to improve efficacy and Treg in vivo persistence
Multiple clinical trials have demonstrated the safety and tolerability of adoptive Treg cell therapy after AlloHCT or SOTx.Citation5,Citation23,Citation24,Citation31,Citation44 However, clinical trials attempting to limit recipient anti-AlloAg responses have yet failed to translate to reduced use of immunosuppressants after SOTx.Citation5,Citation28,Citation32,Citation33 One factor potentially leading to the limited efficacy of Treg ACT may be due to their reduced persistence in vivo. Results from clinical trials demonstrate that Tregs expanded in culture with high doses of IL-2 decrease rapidly in vivo and methods of tracking infused Tregs are limited.Citation54,Citation85,Citation94,Citation99 Thus, alternative methods such as administering low-dose IL-2 (ld-IL-2) to increase their persistence in vivo are being investigated. As discussed in earlier sections, Tregs require the growth factor IL-2 for proliferation, survival, and functional activity.Citation43 Additionally, Treg ACT requires extensive ex vivo expansion with high doses of IL-2 which may affect their stability, phenotype, and survival once transferred in vivo. It is thought that using IL-2 at low doses in vivo in combination with adoptive Treg cell therapy could promote Treg survival and improve therapeutic efficacy. Use of IL-2 in combination with adoptive Treg cell therapy has not been approved and clinical trials are underway investigating safety, dosing, and efficacy of low-dose IL-2 and Treg infusion for GvHD and T1D.Citation99–104 More recently, it has also been clinically tested in stable liver transplant recipients patients 2–6 years post-transplant.Citation105 Unfortunately, it is clear that using ld-IL-2 to increase Treg persistence is not without its risks, as there is clear evidence for activation of effector cells such as cytotoxic CD8+ T cells and NK cells in the recipient.Citation99,Citation105 In the context of SOTx, recently published results from a clinical trial (NCT02949492) that used ld-IL-2 to expand endogenous Tregs in LTx recipients reported rejection episodes in 4 of 5 participants who initiated immunosuppression withdrawal.Citation105 These episodes were mild to moderate and tended to resolve after initiating immunosuppression. One participant, however, developed T cell mediated rejection that was unresponsive to steroids and the individual required re-transplantation.Citation105 Due to these negative outcomes, it is very unlikely that ld-IL-2 can be easily developed as an adjuvant to Treg ACT supporting tolerance induction. As discussed below, however, several groups are developing IL-2 orthologs to target transferred Tregs more precisely without impact on other immune cells responsive to the natural endogenous cytokine.Citation106
Microparticles delivering chemokine CCL22
The rapid loss of infused Treg may also suggest that they undergo generalized and unfocused migration into the tissues or are lost in the absence of signals supporting their survival and required functions. As such, harnessing mechanisms that can orchestrate Treg ACT recruitment and function in vivo may be a promising new approach to improve clinical outcomes. In a recent proof-of-principle study, it was demonstrated that poly lactic-co-glycolic acid (PLGA) microparticles (MP) could generate a chemokine gradient of C-C-Motif Chemokine 22 (CCL22) to recruit CCR4-expressing Tregs in vivo.Citation107 These CCL22 MP prolonged hindlimb allograft survival and promoted donor-specific tolerance.Citation108 Additionally, synthetic human CCL22 MP induced human Treg migration in vitro demonstrating that this technology has the potential to improve Treg cell therapy efficacy by delivering chemokines, like CCL22, that directs them to the graft, or by providing other stimuli (i.e., IL-2, TGF-β) that support their survival or functions in the graft.
The use of IL-2, TGF-β, and rapamycin has been shown to favor Treg suppressive function and development.Citation109 Currently, the use of low dose IL-2 in combination with Treg cell therapy is being evaluated to promote Treg persistence and endogenous Treg expansion.Citation99 A limitation to this is that this approach is nonspecific and can have systemic off target effects. Additionally, a recently published study found that administration of low-dose IL-2 in liver Tx patients did increase the number of Tregs but failed to induce transplantation tolerance.Citation105 An alternative to this would be to provide local extended release of these cytokines and drugs at the graft site to promote tolerance. In one study, Treg inducing microparticles (TRI-MP) were engineered to release TGF-β1, IL-2, and rapamycin to induce differentiation from naïve T cells.Citation110 Using a rat hindlimb vascular composite allotransplantation (VCA) model, TRI-MP prolonged allograft survival without the use of immunosuppression. This system also enriched Treg and reduced inflammatory Th1 populations. The studies above suggest that microparticles can be engineered to support Treg localization, survival, and persistence after adoptive transfer to prevent allograft rejection and promote tolerance. Microparticles delivering supportive or stimulatory signals are an attractive potential method to add value to Treg ACT by boosting the efficacy of a product that, to date, has shown minimum impact.
Engineering IL-2 cytokine-cytokine receptors
As described earlier, low dose IL-2 in combination with adoptive Treg transfer is an approach currently being evaluated in clinical trials to promote Treg survival and persistence. However, low dose IL-2 may support an inflammatory environment consisting of activated CD4+ and CD8+ cytotoxic effectors as well as NK cells making it difficult for Tregs to persist. Therefore, cytokine-cytokine receptor engineering such as the IL-2 cytokine receptor complex on Tregs may be an innovative approach to induce allograft tolerance and Treg survival without supporting effector T cell and NK cells that may contribute to GvHD response.
In a murine mixed chimerism model, FoxP3+GFP+-BALB/c Tregs (green fluorescent protein under the control of the mouse Foxp3 promotor) were transduced to express an orthogonal IL-2 (oIL-2) receptor β chain (oIL-2Rβ). Transduced Tregs were adoptively transferred along with C57BL/6 bone marrow cells (BMC) into wild type BALB/c recipients. Recipients that received oIL-2Rβ Tregs and treated with oIL-2 had significantly improved engraftment and increased percentage of FoxP3+GFP+ in CD4+ T cells without increasing CD8+ T cells. In addition, those same recipients had improved acceptance of heart allografts from C57BL/6 donor mice demonstrating donor-specific tolerance.Citation111
Another study used a murine major mismatch acute GvHD model to investigate the suppressive function of this Treg orthogonal IL-2/IL-2 receptor complex. Briefly, irradiated BALB/c recipients received T cell-depleted BMC from C57Bl/6 mice with or without fresh C57Bl/6 FoxP3 Treg cells or with oIL-2Rβ Treg. On Day 2, C57Bl/6 T responder cells were injected to induce GvHD. Increased survival was observed in recipients that received oIL-2Rβ+ Tregs with oIL-2. Additionally, oIL-2 selectively expanded oIL-2Rβ Treg in vivo and retained their ability to migrate to the gastrointestinal tract and lymph nodes.Citation112 These preclinical data support the potential of using cytokine-cytokine receptor engineering to improve adoptive Treg therapy efficacy for the treatment of Tx and GvHD. These data demonstrate the specificity of cytokine-cytokine receptor interactions avoiding interactions with the natural cytokine response, thus reducing potentially harmful off target expansion of effectors, improving allograft tolerance.
Conclusions/future directions
Overall, the steps for human Treg isolation and ex vivo expansion for clinical use have been outlined here and in other reviews.Citation24,Citation44,Citation52,Citation113 However, it is clear from this discussion that there is still work required to get to the generation of a functionally therapeutic product for clinical use that matches the effectiveness observed in preclinical studies. Clinical trials using Treg-based therapeutics for the prevention of GvHD after HCT or rejection after SOTx have been described and consistently shown that infusion of ex vivo expanded Tregs is well tolerated and safe.Citation24,Citation44,Citation52 Technical advancements in clinical manufacturing of Treg cell products for ACT have improved the isolation of these cells, with many coming online that allow isolation in closed systems as part of GMP processes. This includes positive selection for CD25 followed by subsequent sorting for CD4+CD127loCD25hi using Miltenyi’s closed system magnetic cell separator (CliniMACS Plus) and sorter (MACSQuant Tyto) (). Variations in the approach and reagents used for ex vivo expansion indicate that optimization and standardization are still needed to enable consistent generation of functionally stable Tregs for clinical use, and are summarized in . Additionally, variation in manufacturing and reporting characteristics of Treg cell products may be a barrier to effectively compare study results between institutions and hamper future product development. Therefore, efforts have been made in this review to outline the minimum information required to interpret and compare experimental findings.Citation113 This may help standardize future studies allowing for more effective comparison and reproducibility. Lastly, results from clinical trials have driven novel preclinical studies aiming to improve the clinical application of Tregs. Studies include developing modified cytokine-cytokine receptor complexes to promote in vivo persistence and migration to target graft sites or lymphatic tissues (see ).
Figure 1. Overview of clinical manufacturing of regulatory T cell (Treg) products. Tregs can be isolated from peripheral blood, umbilical cord blood, leukapheresis, or G-CSF mobilized peripheral blood by magnetic cell separation and/or flow cytometric sorting using GMP grade closed systems. Isolated Tregs are then ex vivo expanded using anti-CD3/CD28 magnetic expander beads or artificial antigen-presenting cells (K562 64/86 aAPCs) in the presence of interleukin (IL)-2 with or without rapamycin. AlloAg-specific Tregs can be generated by culturing recipient Tregs with donor AlloAg-expressing APCs. Current in vivo and ex vivo strategies are being used to improve Treg ACT persistence and migration. Figure created with BioRender.com.
Acknowledgments
KB and HRT together generated and revised the text. KB generated the figures. Both authors approve of the submitted version.
Disclosure statement
HT receives remuneration as a scientific advisor to Slate Biotech and research funding from ECM Therapeutics. HT is a listed inventor on patent application PCT/US2019/030547 [Matrix Bound Vesicles (MBVs) Containing IL-33 and Their Use].
Additional information
Funding
References
- Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133(5):775–17. doi:10.1016/j.cell.2008.05.009.
- Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, Gottlieb PA, Kapranov P, Gingeras TR, de St. Groth BF, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ Treg cells. J Exp Med. 2006;203(7):1701–11. doi:10.1084/jem.20060772.
- Barzaghi F, Amaya Hernandez LC, Neven B, Ricci S, Kucuk ZY, Bleesing JJ, Nademi Z, Slatter MA, Ulloa ER, Shcherbina A, et al. Long-term follow-up of IPEX syndrome patients after different therapeutic strategies: an international multicenter retrospective study. J Allergy Clin Immunol. 2018;141(3):1036–49 e5. doi:10.1016/j.jaci.2017.10.041.
- Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27(1):20–21. doi:10.1038/83713.
- Raffin C, Vo LT, Bluestone JA. Treg cell-based therapies: challenges and perspectives. Nat Rev Immunol. 2020;20(3):158–72. doi:10.1038/s41577-019-0232-6.
- Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T cells and human disease. Annu Rev Immunol. 2020;38(1):541–66. doi:10.1146/annurev-immunol-042718-041717.
- Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, Gasteiger G, Feng Y, Fontenot JD, Rudensky AY. An essential role for the IL-2 receptor in treg cell function. Nat Immunol. 2016;17(11):1322–33. doi:10.1038/ni.3540.
- Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155(3):1151–64. doi:10.4049/jimmunol.155.3.1151.
- Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, Masteller EL, McDevitt H, Bonyhadi M, Bluestone JA. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. 2004;199(11):1455–65. doi:10.1084/jem.20040139.
- Hefazi M, Bolivar-Wagers S, Blazar BR, Romero MP. Regulatory T cell therapy of graft-versus-host disease: advances and challenges. Int J Mol Sci. 2021;23(1):22. doi:10.3390/ijms23010022.
- Taylor PA, Lees CJ, Blazar BR. The infusion of ex vivo activated and expanded CD4(+)CD25(+) immune regulatory cells inhibits graft-versus-host disease lethality. Blood. 2002;99(10):3493–99. doi:10.1182/blood.V99.10.3493.
- Cohen JL, Trenado A, Vasey D, Klatzmann D, Salomon BL. CD4(+)CD25(+) immunoregulatory T cells: new therapeutics for graft-versus-host disease. J Exp Med. 2002;196(3):401–06. doi:10.1084/jem.20020090.
- Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, Negrin RS. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat Med. 2003;9(9):1144–50. doi:10.1038/nm915.
- Hoffmann P, Ermann J, Edinger M, Fathman CG, Strober S. Donor-type CD4(+)CD25(+) regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. J Exp Med. 2002;196(3):389–99. doi:10.1084/jem.20020399.
- Xia G, He J, Leventhal JR. Ex vivo-expanded natural CD4+CD25+ regulatory T cells synergize with host T-cell depletion to promote long-term survival of allografts. Am J Transplant. 2008;8(2):298–306. doi:10.1111/j.1600-6143.2007.02088.x.
- Sagoo P, Ali N, Garg G, Nestle FO, Lechler RI, Lombardi G. Human regulatory T cells with alloantigen specificity are more potent inhibitors of alloimmune skin graft damage than polyclonal regulatory T cells. Sci Transl Med. 2011;3(83):83ra42. doi:10.1126/scitranslmed.3002076.
- Xiao F, Ma L, Zhao M, Huang G, Mirenda V, Dorling A, Lechler R, Lombardi G. Ex vivo expanded human regulatory T cells delay islet allograft rejection via inhibiting islet-derived monocyte chemoattractant protein-1 production in CD34+ stem cells-reconstituted NOD-scid IL2rgammanull mice. PLoS One. 2014;9(3):e90387. doi:10.1371/journal.pone.0090387.
- Issa F, Hester J, Goto R, Nadig SN, Goodacre TE, Wood K. Ex vivo-expanded human regulatory T cells prevent the rejection of skin allografts in a humanized mouse model. Transplantation. 2010;90(12):1321–27. doi:10.1097/TP.0b013e3181ff8772.
- Nadig SN, Wieckiewicz J, Wu DC, Warnecke G, Zhang W, Luo S, Schiopu A, Taggart DP, Wood KJ. In vivo prevention of transplant arteriosclerosis by ex vivo-expanded human regulatory T cells. Nat Med. 2010;16(7):809–13. doi:10.1038/nm.2154.
- Bashuda H, Kimikawa M, Seino K, Kato Y, Ono F, Shimizu A, Yagita H, Teraoka S, Okumura K. Renal allograft rejection is prevented by adoptive transfer of anergic T cells in nonhuman primates. J Clin Invest. 2005;115(7):1896–902. doi:10.1172/JCI23743.
- Ezzelarab MB, Thomson AW. Adoptive cell therapy with tregs to improve transplant outcomes: the promise and the stumbling blocks. Curr Transplant Rep. 2016;3(4):265–74. doi:10.1007/s40472-016-0114-9.
- Ma A, Qi S, Song L, Hu Y, Dun H, Massicotte E, Dupuis M, Daloze P, Chen H. Adoptive transfer of CD4+CD25+ regulatory cells combined with low-dose sirolimus and anti-thymocyte globulin delays acute rejection of renal allografts in Cynomolgus monkeys. Int Immunopharmacol. 2011;11(5):618–29. doi:10.1016/j.intimp.2010.11.001.
- Elias S, Rudensky AY. Therapeutic use of regulatory T cells for graft-versus-host disease. Br J Haematol. 2019;187(1):25–38. doi:10.1111/bjh.16157.
- Romano M, Tung SL, Smyth LA, Lombardi G. Treg therapy in transplantation: a general overview. Transpl Int. 2017;30(8):745–53. doi:10.1111/tri.12909.
- Terry LV, Oo YH. The next frontier of regulatory T Cells: promising immunotherapy for autoimmune diseases and organ transplantations. Front Immunol. 2020;11:565518. doi:10.3389/fimmu.2020.565518.
- Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, DeFor T, Levine BL, June CH, Rubinstein P, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. 2011;117(3):1061–70. doi:10.1182/blood-2010-07-293795.
- Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, Curtsinger J, Verneris MR, MacMillan ML, Levine BL, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood. 2016;127(8):1044–51. doi:10.1182/blood-2015-06-653667.
- Chandran S, Tang Q, Sarwal M, Laszik ZG, Putnam AL, Lee K, Leung J, Nguyen V, Sigdel T, Tavares EC, et al. Polyclonal regulatory T cell therapy for control of inflammation in kidney transplants. Am J Transplant. 2017;17(11):2945–54. doi:10.1111/ajt.14415.
- Mathew JM, Hv J, LeFever A, Konieczna I, Stratton C, He J, Huang X, Gallon L, Skaro A, Ansari MJ, et al. A phase I clinical trial with ex vivo expanded recipient regulatory T cells in living donor kidney transplants. Sci Rep. 2018;8(1):7428. doi:10.1038/s41598-018-25574-7.
- Theil A, Tuve S, Oelschlagel U, Maiwald A, Dohler D, Ossmann D, Zenkel A, Wilhelm C, Middeke JM, Shayegi N, et al. Adoptive transfer of allogeneic regulatory T cells into patients with chronic graft-versus-host disease. Cytotherapy. 2015;17(4):473–86. doi:10.1016/j.jcyt.2014.11.005.
- Trzonkowski P, Bieniaszewska M, Juscinska J, Dobyszuk A, Krzystyniak A, Marek N, Myśliwska J, Hellmann A. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin Immunol. 2009;133(1):22–26. doi:10.1016/j.clim.2009.06.001.
- Koyama I, Bashuda H, Uchida K, Seino KI, Habu S, Nakajima I, Fuchinoue S, Okumura K, Teraoka S. A clinical trial with adoptive transfer of ex vivo-induced, donor-specific immune-regulatory cells in kidney transplantation – a second report. Transplantation. 2020;104(11):2415–23. doi:10.1097/TP.0000000000003149.
- Sanchez-Fueyo A, Whitehouse G, Grageda N, Cramp ME, Lim TY, Romano M, Thirkell S, Lowe K, Fry L, Heward J, et al. Applicability, safety, and biological activity of regulatory T cell therapy in liver transplantation. Am J Transplant. 2020;20(4):1125–36. doi:10.1111/ajt.15700.
- Tang Q, Vincenti F. Transplant trials with Tregs: perils and promises. J Clin Invest. 2017;127(7):2505–12. doi:10.1172/JCI90598.
- Juneja T, Kazmi M, Mellace M, Saidi RF. Utilization of treg cells in solid organ transplantation. Front Immunol. 2022;13:746889. doi:10.3389/fimmu.2022.746889.
- Safinia N, Grageda N, Scotta C, Thirkell S, Fry LJ, Vaikunthanathan T, Lechler RI, Lombardi G. Cell therapy in organ transplantation: our experience on the clinical translation of regulatory T cells. Front Immunol. 2018;9:354. doi:10.3389/fimmu.2018.00354.
- Leventhal JR, Ildstad ST. Tolerance induction in HLA disparate living donor kidney transplantation by facilitating cell-enriched donor stem cell infusion: the importance of durable chimerism. Hum Immunol. 2018;79(5):272–76. doi:10.1016/j.humimm.2018.01.007.
- Spitzer TR, Sykes M, Tolkoff-Rubin N, Kawai T, McAfee SL, Dey BR, Ballen K, Delmonico F, Saidman S, Sachs DH, et al. Long-term follow-up of recipients of combined human leukocyte antigen-matched bone marrow and kidney transplantation for multiple myeloma with end-stage renal disease. Transplantation. 2011;91(6):672–76. doi:10.1097/TP.0b013e31820a3068.
- Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, Jiang S, Kuchroo VK, Mathis D, Roncarolo MG, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol. 2013;14(4):307–08. doi:10.1038/ni.2554.
- MacMillan ML, Hippen KL, McKenna DH, Kadidlo D, Sumstad D, DeFor TE, Brunstein CG, Holtan SG, Miller JS, Warlick ED, et al. First-in-human phase 1 trial of induced regulatory T cells for graft-versus-host disease prophylaxis in HLA-matched siblings. Blood Adv. 2021;5(5):1425–36. doi:10.1182/bloodadvances.2020003219.
- Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25high regulatory cells in human peripheral blood. J Immunol. 2001;167(3):1245–53. doi:10.4049/jimmunol.167.3.1245.
- Dieckmann D, Plottner H, Berchtold S, Berger T, Schuler G. Ex vivo isolation and characterization of CD4(+)CD25(+) T cells with regulatory properties from human blood. J Exp Med. 2001;193(11):1303–10. doi:10.1084/jem.193.11.1303.
- Abbas AK, Trotta E, Simeonov DR, Marson A, Bluestone JA. Revisiting IL-2: Biology and therapeutic prospects. Science Immunology. 2018;3(25). doi:10.1126/sciimmunol.aat1482.
- MacDonald KN, Piret JM, Levings MK. Methods to manufacture regulatory T cells for cell therapy. Clin Exp Immunol. 2019;197(1):52–63. doi:10.1111/cei.13297.
- Safinia N, Vaikunthanathan T, Fraser H, Thirkell S, Lowe K, Blackmore L, Whitehouse G, Martinez-Llordella M, Jassem W, Sanchez-Fueyo A, et al. Successful expansion of functional and stable regulatory T cells for immunotherapy in liver transplantation. Oncotarget. 2016;7(7):7563–77. doi:10.18632/oncotarget.6927.
- Bernaldo-de-Quiros E, Cozar B, Lopez-Esteban R, Clemente M, Gil-Jaurena JM, Pardo C, Pita A, Pérez-Caballero R, Camino M, Gil N, et al. A novel GMP protocol to produce high-quality treg cells from the pediatric thymic tissue to be employed as cellular therapy. Front Immunol. 2022;13:893576. doi:10.3389/fimmu.2022.893576.
- Dijke IE, Hoeppli RE, Ellis T, Pearcey J, Huang Q, McMurchy AN, Boer K, Peeters AMA, Aubert G, Larsen I, et al. Discarded human thymus is a novel source of stable and long-lived therapeutic regulatory T cells. Am J Transplant. 2016;16(1):58–71. doi:10.1111/ajt.13456.
- Golab K, Grose R, Trzonkowski P, Wickrema A, Tibudan M, Marek-Trzonkowska N, Matosz S, Solomina J, Ostrega D, Millis JM, et al. Utilization of leukapheresis and CD4 positive selection in Treg isolation and the ex-vivo expansion for a clinical application in transplantation and autoimmune disorders. Oncotarget. 2016;7(48):79474–84. doi:10.18632/oncotarget.13101.
- Patel P, Mahmud D, Park Y, Yoshinaga K, Mahmud N, Rondelli D. Clinical grade isolation of regulatory T cells from G-CSF mobilized peripheral blood improves with initial depletion of monocytes. Am J Blood Res. 2015;5:79–85.
- Ukena SN, Velaga S, Goudeva L, Ivanyi P, Olek S, Falk CS, Ganser A, Franzke A. Human regulatory T cells of G-CSF mobilized allogeneic stem cell donors qualify for clinical application. PLoS One. 2012;7(12):e51644. doi:10.1371/journal.pone.0051644.
- Safinia N, Scotta C, Vaikunthanathan T, Lechler RI, Lombardi G. Regulatory T cells: serious contenders in the promise for immunological tolerance in transplantation. Front Immunol. 2015;6:438. doi:10.3389/fimmu.2015.00438.
- Mamo T, Hippen KL, MacMillan ML, Brunstein CG, Miller JS, Wagner JE, Blazar BR, McKenna DH. Regulatory T cells: a review of manufacturing and clinical utility. Transfusion. 2022;62(4):904–15. doi:10.1111/trf.16797.
- Eggenhuizen PJ, Ng BH, Ooi JD. Treg enhancing therapies to treat autoimmune diseases. Int J Mol Sci. 2020;22(1):21. doi:10.3390/ijms22010021.
- Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, Herold KC, Lares A, Lee MR, Li K, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. 2015;7(315):315ra189. doi:10.1126/scitranslmed.aad4134.
- Canavan JB, Scotta C, Vossenkamper A, Goldberg R, Elder MJ, Shoval I, Marks E, Stolarczyk E, Lo JW, Powell N, et al. Developing in vitro expanded CD45RA + regulatory T cells as an adoptive cell therapy for Crohn’s disease. Gut. 2016;65(4):584–94. doi:10.1136/gutjnl-2014-306919.
- Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Techmanska I, Juscinska J, Wujtewicz MA, Witkowski P, Młynarski W, Balcerska A, et al. Administration of CD4+CD25highCD127− regulatory T cells preserves β-Cell function in type 1 diabetes in children. Diabetes Care. 2012;35(9):1817–20. doi:10.2337/dc12-0038.
- Marin Morales JM, Munch N, Peter K, Freund D, Oelschlagel U, Holig K, Böhm T, Flach A-C, Keßler J, Bonifacio E, et al. Automated clinical grade expansion of regulatory T cells in a fully closed system. Front Immunol. 2019;10:38. doi:10.3389/fimmu.2019.00038.
- Alsuliman A, Appel SH, Beers DR, Basar R, Shaim H, Kaur I, Zulovich J, Yvon E, Muftuoglu M, Imahashi N, et al. A robust, good manufacturing practice-compliant, clinical-scale procedure to generate regulatory T cells from patients with amyotrophic lateral sclerosis for adoptive cell therapy. Cytotherapy. 2016;18(10):1312–24. doi:10.1016/j.jcyt.2016.06.012.
- Fraser H, Safinia N, Grageda N, Thirkell S, Lowe K, Fry LJ, Scottá C, Hope A, Fisher C, Hilton R, et al. A rapamycin-based GMP-compatible process for the isolation and expansion of regulatory T cells for clinical trials. Mol Ther Methods Clin Dev. 2018;8:198–209. doi:10.1016/j.omtm.2018.01.006.
- McKenna DH Jr., Sumstad D, Kadidlo DM, Batdorf B, Lord CJ, Merkel SC, Koellner CM, Curtsinger JM, June CH, Riley JL, et al. Optimization of cGMP purification and expansion of umbilical cord blood-derived T-regulatory cells in support of first-in-human clinical trials. Cytotherapy. 2017;19(2):250–62. doi:10.1016/j.jcyt.2016.10.011.
- Parmar S, Liu X, Najjar A, Shah N, Yang H, Yvon E, Rezvani K, McNiece I, Zweidler-McKay P, Miller L, et al. Ex vivo fucosylation of third-party human regulatory T cells enhances anti-graft-versus-host disease potency in vivo. Blood. 2015;125(9):1502–06. doi:10.1182/blood-2014-10-603449.
- Peters JH, Preijers FW, Woestenenk R, Hilbrands LB, Koenen HJ, Joosten I. Clinical grade treg: GMP isolation, improvement of purity by CD127 depletion, treg expansion, and treg cryopreservation. PLoS One. 2008;3(9):e3161. doi:10.1371/journal.pone.0003161.
- Voskens CJ, Fischer A, Roessner S, Lorenz C, Hirschmann S, Atreya R, Neufert C, Atreya I, Neurath MF, Schuler G, et al. Characterization and expansion of autologous GMP-ready regulatory T cells for TREG-based cell therapy in patients with ulcerative colitis. Inflamm Bowel Dis. 2017;23(8):1348–59. doi:10.1097/MIB.0000000000001192.
- Hoffmann P, Boeld TJ, Eder R, Albrecht J, Doser K, Piseshka B, Dada A, Niemand C, Assenmacher M, Orsó E, et al. Isolation of CD4+CD25+ regulatory T cells for clinical trials. Biol Blood Marrow Transplant. 2006;12(3):267–74. doi:10.1016/j.bbmt.2006.01.005.
- Wichlan DG, Roddam PL, Eldridge P, Handgretinger R, Riberdy JM. Efficient and reproducible large-scale isolation of human CD4+ CD25+ regulatory T cells with potent suppressor activity. J Immunol Methods. 2006;315(1–2):27–36. doi:10.1016/j.jim.2006.06.014.
- Wiesinger M, Stoica D, Roessner S, Lorenz C, Fischer A, Atreya R, Neufert CF, Atreya I, Scheffold A, Schuler-Thurner B. Good manufacturing practice-compliant production and lot-release of ex vivo expanded regulatory T cells as basis for treatment of patients with autoimmune and inflammatory disorders. Front Immunol. 2017;8:1371. doi:10.3389/fimmu.2017.01371.
- Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol. 2006;177(12):8338–47. doi:10.4049/jimmunol.177.12.8338.
- Lu L, Qian XF, Rao JH, Wang XH, Zheng SG, Zhang F. Rapamycin promotes the expansion of CD4(+) Foxp3(+) regulatory T cells after liver transplantation. Transplant Proc. 2010;42(5):1755–57. doi:10.1016/j.transproceed.2009.10.008.
- Ogino H, Nakamura K, Iwasa T, Ihara E, Akiho H, Motomura Y, Akahoshi K, Igarashi H, Kato M, Kotoh K, et al. Regulatory T cells expanded by rapamycin in vitro suppress colitis in an experimental mouse model. J Gastroenterol. 2012;47(4):366–76. doi:10.1007/s00535-011-0502-y.
- Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood. 2005;105(12):4743–48. doi:10.1182/blood-2004-10-3932.
- Raimondi G, Sumpter TL, Matta BM, Pillai M, Corbitt N, Vodovotz Y, Wang Z, Thomson AW. Mammalian target of rapamycin inhibition and alloantigen-specific regulatory T cells synergize to promote long-term graft survival in immunocompetent recipients. J Immunol. 2010;184(2):624–36. doi:10.4049/jimmunol.0900936.
- Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22(3):329–41. doi:10.1016/j.immuni.2005.01.016.
- Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, Rudensky AY. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007;445(7129):771–75. doi:10.1038/nature05543.
- Kim JK, Klinger M, Benjamin J, Xiao Y, Erle DJ, Littman DR, Killeen N. Impact of the TCR signal on regulatory T cell homeostasis, function, and trafficking. PLoS One. 2009;4(8):e6580. doi:10.1371/journal.pone.0006580.
- Vahl JC, Drees C, Heger K, Heink S, Fischer JC, Nedjic J, Ohkura N, Morikawa H, Poeck H, Schallenberg S, et al. Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity. 2014;41(5):722–36. doi:10.1016/j.immuni.2014.10.012.
- Schmidt AM, Lu W, Sindhava VJ, Huang Y, Burkhardt JK, Yang E, Riese MJ, Maltzman JS, Jordan MS, Kambayashi T. Regulatory T cells require TCR signaling for their suppressive function. J Immunol. 2015;194(9):4362–70. doi:10.4049/jimmunol.1402384.
- Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol. 2014;15(11):1070–78. doi:10.1038/ni.3004.
- Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12(4):431–40. doi:10.1016/S1074-7613(00)80195-8.
- Tai X, Cowan M, Feigenbaum L, Singer A. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat Immunol. 2005;6(2):152–62. doi:10.1038/ni1160.
- Golovina TN, Mikheeva T, Suhoski MM, Aqui NA, Tai VC, Shan X, Lui R, Balcarcel RR, Fisher N, Levine BL. CD28 costimulation is essential for human T regulatory expansion and function. J Immunol. 2008;181(4):2855–68. doi:10.4049/jimmunol.181.4.2855.
- He X, Smeets RL, van Rijssen E, Boots AM, Joosten I, Koenen HJ. Single CD28 stimulation induces stable and polyclonal expansion of human regulatory T cells. Sci Rep. 2017;7(1):43003. doi:10.1038/srep43003.
- Hombach AA, Kofler D, Hombach A, Rappl G, Abken H. Effective proliferation of human regulatory T cells requires a strong costimulatory CD28 signal that cannot be substituted by IL-2. J Immunol. 2007;179(11):7924–31. doi:10.4049/jimmunol.179.11.7924.
- Lin CH, Hunig T. Efficient expansion of regulatory T cells in vitro and in vivo with a CD28 superagonist. Eur J Immunol. 2003;33(3):626–38. doi:10.1002/eji.200323570.
- Kellner JN, Yvon E, Parmar S. Ex vivo generation of umbilical cord blood T regulatory cells expressing the homing markers CD62L and cutaneous lymphocyte antigen. Oncotarget. 2018;9(72):33694–701. doi:10.18632/oncotarget.26097.
- Todo S, Yamashita K, Goto R, Zaitsu M, Nagatsu A, Oura T, Watanabe M, Aoyagi T, Suzuki T, Shimamura T, et al. A pilot study of operational tolerance with a regulatory T-cell-based cell therapy in living donor liver transplantation. Hepatology. 2016;64(2):632–43. doi:10.1002/hep.28459.
- Suhoski MM, Golovina TN, Aqui NA, Tai VC, Varela-Rohena A, Milone MC, Carroll RG, Riley JL, June CH. Engineering artificial antigen-presenting cells to express a diverse array of co-stimulatory molecules. Mol Ther. 2007;15(5):981–88. doi:10.1038/mt.sj.6300134.
- Hippen KL, Merkel SC, Schirm DK, Sieben CM, Sumstad D, Kadidlo DM, McKenna DH, Bromberg JS, Levine BL, Riley JL. Massive ex Vivo expansion of human natural regulatory T Cells (T regs) with minimal loss of in Vivo functional activity. Sci Transl Med. 2011;3(83):83ra41. doi:10.1126/scitranslmed.3001809.
- Sanchez-Fueyo A, Sandner S, Habicht A, Mariat C, Kenny J, Degauque N, Zheng XX, Strom TB, Turka LA, Sayegh MH, et al. Specificity of CD4+CD25+ regulatory T cell function in alloimmunity. J Immunol. 2006;176(1):329–34. doi:10.4049/jimmunol.176.1.329.
- Putnam AL, Safinia N, Medvec A, Laszkowska M, Wray M, Mintz MA, Trotta E, Szot GL, Liu W, Lares A, et al. Clinical grade manufacturing of human alloantigen-reactive regulatory T cells for use in transplantation. Am J Transplant. 2013;13(11):3010–20. doi:10.1111/ajt.12433.
- Lee LM, Zhang H, Lee K, Liang H, Merleev A, Vincenti F, Maverakis E, Thomson AW, Tang Q. A comparison of ex vivo expanded human regulatory T cells using allogeneic stimulated B cells or monocyte-derived dendritic cells. Front Immunol. 2021;12:679675. doi:10.3389/fimmu.2021.679675.
- Golshayan D, Jiang S, Tsang J, Garin MI, Mottet C, Lechler RI. In vitro-expanded donor alloantigen-specific CD4+CD25+ regulatory T cells promote experimental transplantation tolerance. Blood. 2007;109(2):827–35. doi:10.1182/blood-2006-05-025460.
- Noyan F, Lee YS, Hardtke-Wolenski M, Knoefel AK, Taubert R, Baron U, Manns MP, Jaeckel E. Donor-specific regulatory T cells generated on donor B cells are superior to CD4+CD25high cells in controlling alloimmune responses in humanized mice. Transplant Proc. 2013;45(5):1832–37. doi:10.1016/j.transproceed.2013.01.073.
- Trenado A, Fisson S, Braunberger E, Klatzmann D, Salomon BL, Cohen JL. Ex vivo selection of recipient-type alloantigen-specific CD4(+)CD25(+) immunoregulatory T cells for the control of graft-versus-host disease after allogeneic hematopoietic stem-cell transplantation. Transplantation. 2004;77:S32–4. doi:10.1097/01.TP.0000106470.07410.CA.
- Tang Q, Leung J, Peng Y, Sanchez-Fueyo A, Lozano JJ, Lam A, Lee K, Greenland JR, Hellerstein M, Fitch M, et al. Selective decrease of donor-reactive T regs after liver transplantation limits T reg therapy for promoting allograft tolerance in humans. Sci Transl Med. 2022;14(669):eabo2628. doi:10.1126/scitranslmed.abo2628.
- Tsang JY, Tanriver Y, Jiang S, Xue SA, Ratnasothy K, Chen D, Stauss HJ, Bucy RP, Lombardi G, Lechler R, et al. Conferring indirect allospecificity on CD4+CD25+ Tregs by TCR gene transfer favors transplantation tolerance in mice. J Clin Invest. 2008;118(11):3619–28. doi:10.1172/JCI33185.
- Mathew JM, Voss JH, McEwen ST, Konieczna I, Chakraborty A, Huang X, He J, Gallon L, Kornbluth RS, Leventhal JR. Generation and characterization of alloantigen-specific regulatory T cells for clinical transplant tolerance. Sci Rep. 2018;8(1):1136. doi:10.1038/s41598-018-19621-6.
- Trenado A, Charlotte F, Fisson S, Yagello M, Klatzmann D, Salomon BL, Cohen JL. Recipient-type specific CD4+CD25+ regulatory T cells favor immune reconstitution and control graft-versus-host disease while maintaining graft-versus-leukemia. J Clin Invest. 2003;112(11):1688–96. doi:10.1172/JCI17702.
- Sawitzki B, Harden PN, Reinke P, Moreau A, Hutchinson JA, Game DS, Tang Q, Guinan EC, Battaglia M, Burlingham WJ, et al. Regulatory cell therapy in kidney transplantation (The ONE Study): a harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet. 2020;395(10237):1627–39. doi:10.1016/S0140-6736(20)30167-7.
- Dong S, Hiam-Galvez KJ, Mowery CT, Herold KC, Gitelman SE, Esensten JH, Liu W, Lares AP, Leinbach AS, Lee M. The effect of low-dose IL-2 and treg adoptive cell therapy in patients with type 1 diabetes. JCI Insight. 2021;6(18):e147474. doi:10.1172/jci.insight.147474.
- Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP, Armand P, Cutler C, Ho VT, Treister NS, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med. 2011;365(22):2055–66. 3rd. doi:10.1056/NEJMoa1108188.
- Kennedy-Nasser AA, Ku S, Castillo-Caro P, Hazrat Y, Wu MF, Liu H, Melenhorst J, Barrett AJ, Ito S, Foster A. Ultra low-dose IL-2 for GVHD prophylaxis after allogeneic hematopoietic stem cell transplantation mediates expansion of regulatory T cells without diminishing antiviral and antileukemic activity. Clin Cancer Res. 2014;20(8):2215–25. doi:10.1158/1078-0432.CCR-13-3205.
- Hartemann A, Bensimon G, Payan CA, Jacqueminet S, Bourron O, Nicolas N, Fonfrede M, Rosenzwajg M, Bernard C, Klatzmann D. Low-dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2013;1(4):295–305. doi:10.1016/S2213-8587(13)70113-X.
- Whangbo J, Nikiforow S, Kim HT, Wahl J, Reynolds CG, Chamling Rai S, Kim S, Burden A, Alho AC, Lacerda JF, et al. A phase 1 study of donor regulatory T-cell infusion plus low-dose interleukin-2 for steroid-refractory chronic graft-vs-host disease. Blood Adv. 2022;6(21):5786–96. doi:10.1182/bloodadvances.2021006625.
- Whangbo JS, Kim HT, Nikiforow S, Koreth J, Alho AC, Falahee B, Kim S, Dusenbury K, Fields MJ, Reynolds CG. Functional analysis of clinical response to low-dose IL-2 in patients with refractory chronic graft-versus-host disease. Blood Adv. 2019;3(7):984–94 doi:10.1182/bloodadvances.2018027474.
- Lim TY, Perpinan E, Londono MC, Miquel R, Ruiz P, Kurt AS, Codela E, Cross A, Berlin C, Hester J, et al. Low dose interleukin-2 selectively expands circulating regulatory T cells but fails to promote liver allograft tolerance in humans. J Hepatol. 2022;77:S809. doi:10.1016/S0168-8278(22)01920-1.
- Sockolosky JT, Trotta E, Parisi G, Picton L, Su LL, Le AC, Chhabra A, Silveria SL, George BM, King IC, et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science. 2018;359(6379):1037–42. doi:10.1126/science.aar3246.
- Jhunjhunwala S, Raimondi G, Glowacki AJ, Hall SJ, Maskarinec D, Thorne SH, Thomson AW, Little SR. Bioinspired controlled release of CCL22 recruits regulatory T cells in vivo. Adv Mater. 2012;24(35):4735–38. doi:10.1002/adma.201202513.
- Fisher JD, Zhang W, Balmert SC, Aral AM, Acharya AP, Kulahci Y, Li J, Turnquist HR, Thomson AW, Solari MG, et al. In situ recruitment of regulatory T cells promotes donor-specific tolerance in vascularized composite allotransplantation. Sci Adv. 2020;6(11):eaax8429. doi:10.1126/sciadv.aax8429.
- Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25(3):455–71. doi:10.1016/j.immuni.2006.07.011.
- Jhunjhunwala S, Balmert SC, Raimondi G, Dons E, Nichols EE, Thomson AW, Little SR. Controlled release formulations of IL-2, TGF-beta1 and rapamycin for the induction of regulatory T cells. J Control Release. 2012;159:78–84.
- Hirai T, Simonetta F, Su LL, Picton L, Baker J, Seo K, Lohmeyer JK, Mavers M, Blazar BR, Garcia C, et al. Engineered IL-2 cytokine-cytokine receptor complex enables selective expansion of regulatory T cells and facilitates establishment of organ transplantation tolerance. Biol. Blood Marrow Transplant. 2020;26(3):S59–S60. doi:10.1016/j.bbmt.2019.12.226.
- Hirai T, Ramos TL, Lin PY, Simonetta F, Su LL, Picton LK, Baker J, Lin J, Li P, Seo K. Selective expansion of regulatory T cells using an orthogonal IL-2/IL-2 receptor system facilitates transplantation tolerance. J Clin Invest. 2021;131(8): e139991. doi:10.1172/JCI139991.
- Fuchs A, Gliwinski M, Grageda N, Spiering R, Abbas AK, Appel S, Bacchetta R, Battaglia M, Berglund D, Blazar B, et al. Minimum information about T regulatory cells: a step toward reproducibility and standardization. Front Immunol. 2017;8:1844. doi:10.3389/fimmu.2017.01844.
- Atif M, Conti F, Gorochov G, Oo YH, Miyara M. Regulatory T cells in solid organ transplantation. Clin Transl Immunol. 2020;9(2):e01099. doi:10.1002/cti2.1099.
- Gedaly R, De Stefano F, Turcios L, Hill M, Hidalgo G, Mitov MI, Alstott MC, Butterfield DA, Mitchell HC, Hart J, et al. mTOR inhibitor everolimus in regulatory T cell expansion for clinical application in transplantation. Transplantation. 2019;103(4):705–15. doi:10.1097/TP.0000000000002495.