
Abstract
PIWI-interacting RNAs (piRNAs) have long been associated with the silencing of transposable elements (TEs). However, over 20,000 unique species of piRNAs mapped to the human genome are more than the relatively few presumably required to regulate the known human transposon classes. Here, we present the results of the first genome-wide effort to study the effects of piRNAs on gene specific DNA methylation. We found that exon-derived piRNAs consist almost universally of species with 10 or fewer genomic copies, whereas piRNAs existing in high copies originate predominately from intronic and intergenic regions. Genome-wide methylation profiling following transfection of human somatic cells with piRNA mimics revealed methylation changes at numerous genic loci in single copy piRNA-transfected cells. Moreover, genomic regions directly adjacent to differentially methylated CpG sites were enriched for sequence matches to the transfected piRNAs. These findings suggest that a subset of single copy piRNAs may be able to induce DNA methylation at non-TE genic loci, a process that may be mediated in part by direct binding to either genomic DNA or nascent mRNA near target CpG sites.
Introduction
PIWI-interacting RNAs (piRNAs), together with small interfering RNAs (siRNAs) and microRNAs (miRNAs), form the trifecta of the endogenous small RNA-mediated silencing machinery found in metazoa. Originally identified in germ cells, piRNAs differ greatly from miRNAs and siRNAs in their structure and origin.Citation1-7 Roughly 26 to 31 nucleotides long in their mature forms, mammalian piRNAs can be divided into 2 groups characterized by distinct biogenic and functional pathways: pre-pachytene piRNAs, which are produced via a process known as ping-pong amplification and function to actively repress transposon activity at the pre-pachytene stage of meiosis in sperm development, and pachytene piRNAs, which are not produced by ping-pong amplification and whose precise functions remain enigmatic. Citation8-12
While both groups of piRNAs are believed to be processed from long single-stranded precursors,Citation5,7,Citation13 pre-pachytene piRNAs map primarily to repetitive genomic elementsCitation14-16 while, in mice, pachytene piRNAs appear to originate from ~3,000 genomic clusters.Citation17
Until recently, the predominance of the scientific focus on piRNAs has been on their transposon-silencing capacity in the germ line, a process that is mediated by their association with PIWI protein complexes, from which piRNAs derive their name, and which safeguards the genome against transposon-induced insertional mutations.Citation5,7,Citation12,18 In spite of this, there is mounting evidence to suggest that their range of functions extends beyond this role. Deep sequencing initiatives have revealed piRNA-like small RNA populations in multiple mammalian somatic tissues, including those of the central nervous system and the haematopoietic system.Citation19 There is also compelling evidence in lower level organisms to suggest that piRNAs may be capable of inducing histone modification and DNA methylation in a sequence-specific manner in the soma.Citation20-22 In addition, a plethora of studies now indicate that a specific population of piRNAs may be involved in the silencing of non-TE protein-coding genes in the Drosophila somatic tissues.Citation23-27 Likewise, Esposito et al. have recently demonstrated the capacity of a testis- and brain-specific piRNA to cis-regulate MTNR1A expression using human somatic cell models.Citation28 Together, these findings seem to suggest that piRNAs may be capable of regulating the expression of non-TE protein-coding genes outside the germ line.
Although there is a strong implication that the putative gene-regulatory capacity of piRNAs may be attributable, at least in part, to piRNA-induced epigenetic changes, searching the scientific literature reveals only two examples where piRNAs have been linked to gene-specific methylation changes in somatic tissue. In the first case, Rajasethupathy et al. demonstrated the endogenous capacity of a piRNA to alter CREB2 methylation in Aplysia neurons.Citation21 In the other, Cichocki et al. observed a correlation between expression of a piRNA encoded from the KIR3DL1 promoter and KIR3DL1 promoter methylatin in CD56+ NK cells.Citation20 This latter finding represents the only evidence of a piRNA-associated methylation change in human somatic tissue.
Given the limited but mounting evidence of a function beyond TE silencing and the relatively few piRNA species presumably required to regulate the known human transposon classes, we hypothesize that a portion of the 23,439 species of human piRNAs may have regulatory roles in other functional domains, including the regulation of non-TE gene-specific methylation. To test this hypothesis, we undertook the first genome-wide effort to map piRNAs and study their effect on gene methylation across the human genome. The distribution of the 667,944 piRNA loci in the human genome was analyzed in the context of their proximity to SINE and LINE transposable elements and non-TE protein-coding genes, as well as by piRNA genomic copy number. Somatic cancer cell lines were then transfected with piRNAs, chosen in accordance with their region of origin and copy number, and then analyzed for methylation changes at genic loci throughout the genome. Finally, genomic regions adjacent to differentially methylated CpG sites were interrogated for sequence matches to the transfected piRNAs to assess whether observed methylation changes could be mediated by direct piRNA binding near affected CpG sites.
Results
Distribution of piRNAs across the human genome
The 23,439 unique piRNA sequences map to 667,944 loci across the human genome. The frequency of these loci, along with loci for non-TE protein-coding genes, long interspersed nuclear elements (LINEs), and short interspersed nuclear elements (SINEs), was interrogated in 1 megabase blocks along the length of each chromosome. A frequency histogram for chromosome 1 is shown in ; frequency histograms for other chromosomes can be found in Figure S1. Proximity between piRNA loci and protein-coding genes, LINEs, and SINEs was approximated by the degree of linear correlation between chromosomal frequency distributions (). The distribution of piRNAs exhibited a high correlation with SINE distribution (Pearson's r = 0.79, p < 0.0001) and moderate correlations with gene (Pearson's r = 0.59, p < 0.0001) and LINE (Pearson's r = 0.34, p < 0.0001) distributions. Overall, these observations are consistent with findings in lower level organisms that indicate most piRNAs originate from repetitive elements,Citation13 but which have never been confirmed in humans.
Figure 1 (See previous page). piRNA distribution across chromosomes and by copy number. (A) A frequency map of piRNA loci on chromosome 1. The frequency of the 667,944 piRNA loci in the human genome, along with loci for non-TE protein-coding genes, long interspersed nuclear elements (LINEs), and short interspersed nuclear elements (SINEs), was interrogated in 1 megabase blocks along the length of each chromosome. (B) Proximity between piRNA loci and protein-coding genes, LINEs, and SINEs was approximated by the degree of linear correlation between chromosomal frequency distributions. piRNA loci exhibited the highest degree of overlap with SINE loci (Pearson's r = 0.79, p < 0.0001), and moderate overlap with gene (Pearson's r = 0.59, p < 0.0001) and LINE (Pearson's r = 0.34, p < 0.0001) loci. (C) Genomic distribution of piRNA loci by piRNA copy number. The 23,439 unique piRNA sequences were divided into 19,124 single copy piRNA loci, 11,881 loci corresponding to 3,261 piRNAs with 2 to 10 copies, 67,079 loci corresponding to 806 piRNAs with 11 to 1000 copies, and 564,111 loci corresponding to 61 piRNAs with greater than 1000 copies, and subsequently matched to one of 3 genomic regions (exons, introns, or intergenic regions). Overall, there was a tendency for lower copy piRNA loci to overlap exons of protein-coding genes and higher copy loci to overlap introns, where repetitive transposable elements are frequently found.
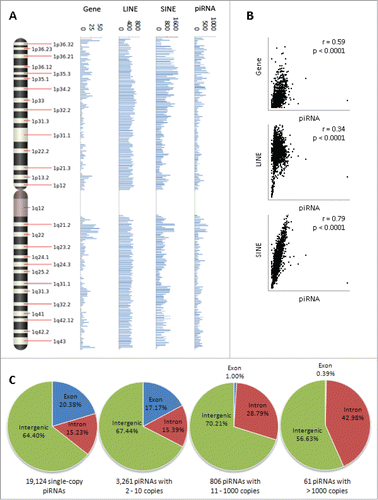
We further analyzed the genomic distribution of piRNA loci by piRNA copy number to test whether “repetitive” piRNAs are more likely to be encoded from repetitive regions, and “non-repetitive” piRNAs are more likely to originate from non-repetitive protein-coding regions. The 23,439 unique piRNA sequences were divided into 19,124 single-copy piRNAs, 3,261 piRNAs with 2 to 10 copies corresponding to 11,881 loci, 806 piRNAs with 11 to 1,000 copies corresponding to 67,079 loci, and 61 piRNAs with greater than 1,000 copies corresponding to 564,111 loci, and subsequently matched to one of 3 genomic regions (exon, intron, or intergenic). Of the single-copy piRNA loci, 20.38% overlap exons, 15.23% overlap introns, and 64.40% overlap intergenic regions (). Of loci corresponding to piRNAs with 2 to 10 copies, 17.17% overlap exons, 15.39% overlap introns, and 67.44% overlap intergenic regions. Of loci corresponding to piRNAs with 11 to 1000 copies, 1% overlap exons, 28.79% overlap introns, and 70.21% overlap intergenic regions. Finally, of loci corresponding to piRNAs with greater than 1000 copies, 0.39% overlap exons, 42.98% overlap introns, and 56.63% overlap intergenic regions. These findings are in directional agreement with our hypothesis that piRNA copy number is a function of the repetitiveness of the genomic elements they are derived from; while there did not appear to be a great degree of correlation between piRNA copy number and overlap with intergenic regions, there was a distinct tendency for lower copy piRNA loci to overlap exons of protein-coding genes and higher copy loci to overlap introns, where repetitive transposable elements are frequently found. Citation29,30 Overall, our findings suggest that human piRNAs can be broken down into one of 3 major categories by copy number and genomic distribution: single and low copy piRNAs encoded from exons, high number piRNAs encoded from introns and intergenic regions, and single and low copy piRNAs encoded from introns and intergenic regions.
Genome-wide effect of piRNAs on DNA methylation
The sequence diversity and distribution of single and low copy piRNAs, in conjunction with the established propensity of piRNAs to effect change at the epigenetic level, create a compelling case for an essential yet largely unexplored role for these piRNAs in the DNA methylation of non-TE protein-coding genes. A genome-wide DNA methylation analysis was performed to test the feasibility of this hypothesis. Four single-stranded piRNA mimics were individually transfected into the Farage human lymphoma and MCF-7 human breast cancer cell lines, in which we found detectable levels of PIWIL1, PIWIL2, and PIWIL4 mRNA expression (Figure S2). Endogenous expression of PIWIL2 and PIWIL4 in MCF-7 cells have also been documented elsewhere. Citation31-33
Genomic DNA was isolated and subsequently interrogated for genome-wide methylation changes using Illumina's Infinium HumanMethylation450 array. The piRNAs were chosen as one or more representative members of each copy number and distribution category. hsa_piR_020409 traces its origin to a single copy locus in the first exon of HIST1H4J, which encodes a histone subunit. hsa_piR_017183 derives from 17,043 loci, a significant portion of which are found to overlap LINE1 repetitive elements. hsa_piR_021285 and hsa_piR_023426 are both encoded from single copy loci in intergenic regions. Isolated genomic DNA from transfected cells were interrogated for methylation changes at 485,477 CpG sites. To minimize false positives and cell-specific changes, subsequent analyses were constrained to CpG sites exhibiting a unidirectional methylation change of 10% or greater in both Farage and MCF-7 cells and at nominal P-value of less than 0.05 in one or both cell lines. A total of 210 CpG sites met these criteria and 202 (96.2%) of these displayed increased methylation while only 8 (3.8%) displayed decreased methylation (). Interestingly, the single copy hsa_piR_020409, hsa_piR_021285, and hsa_piR_023426 transfections respectively accounted for 78, 56, and 41 of the differentially methylated CpG sites, while the high copy hsa_piR_017183 transfection accounted for just 17. Across the 4 piRNAs, a total of 117 unique genes were found to be differentially methylated at one or more CpG sites located within 1,500 bp of the transcriptional start site (“promoter”), the 5′ UTR, the first exon, the remaining gene body, or the 3′ UTR as designated by the HumanMethylation450 array. Information on methylation changes of individual CpG sites and their locations in the genome can be found in Figure S3.
Figure 2. Genome-wide methylation changes following piRNA transfection. (A) CpG site methylation changes following individual transfection of 4 single-stranded piRNA mimics (hsa_piR_020409, hsa_piR_017183, hsa_piR_021285, and hsa_piR_023426) into Farage human lymphoma and MCF-7 human breast cancer cells. Of the 223 CpG sites that exhibited a unidirectional methylation change of 10% or greater in both Farage and MCF-7 cells, 212 (95.1%) displayed increased (+) methylation while only 11 (4.9%) displayed decreased (−) methylation. (B) Validation of methylation changes using EpiTYPER. In order to validate array measurements, methylation at 4 CpG sites (cg25798413, cg06011925, cg21939303, and cg12072178) and corresponding ~400–600 bp flanking regions was re-evaluated using Sequenom's EpiTYPER platform. Results of the EpiTYPER measurement were in directional agreement with those of the Infinium array. (C) Tissue-specific enrichment in expression of differentially methylated genes. Genes that exhibited a unidirectional change of 10% or greater and a nominal P-value < 0.05 in both cell lines were examined for tissue enrichment using the UniProt tissue expression function as implemented in the DAVID Bioinformatic Resource (v6.7). Statistically significant or borderline significant expression enrichment was found only for the brain in any of the 4 transfections (Fisher Exact α = 0.05).
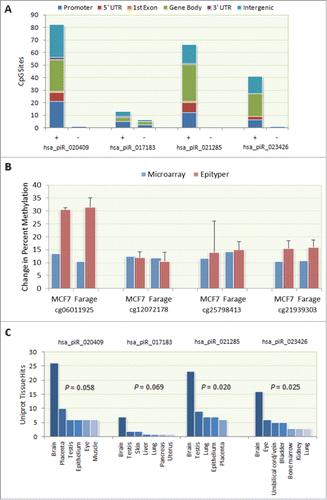
In order to validate our array measurements, methylation at 4 CpG sites (cg25798413, cg06011925, cg21939303, and cg12072178) and corresponding ∼400–600 bp flanking regions was re-evaluated using Sequenom's EpiTYPER platform. Results of the EpiTYPER measurement were in general agreement with those of the Infinium array (). Percent methylation values of individual CpG loci within the flanking region are given in Figure S4.
Genes that exhibited a unidirectional change of 10% or greater in both cell lines and a nominal P-value < 0.05 in one or both cell lines were examined for tissue enrichment using the UniProt tissue expression function as implemented in the DAVID Bioinformatic Resource (v6.7). Interestingly, expression of differentially methylated genes exhibited statistically significant or borderline significant enrichment (Fisher Exact α = 0.05) in only the brain for all of the piRNA transfections (), suggesting that the transfected piRNAs may play an as of yet unelucidated role there.
Correlation between piRNA mimic-associated gene methylation and mRNA expression
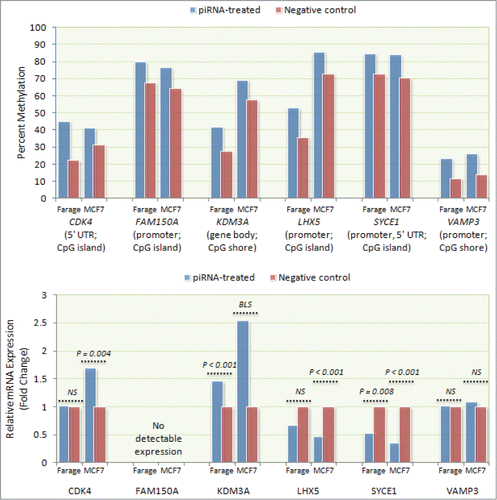
To check what effect piRNA mimic-associated gene methylation might have on gene expression, relative mRNA levels of 6 differentially methylated genes (CDK4, FAM150A, KDM3A, LHX5, SYCE1, and VAMP3) were measured by qPCR using RNA isolated from transfected Farage and MCF-7 cells. These genes were selected to examine the effect of differential methylation observed at a sample of CpG sites residing in the promoter region (FAM150A, LHX5, SYCE1, and VAMP3), 5′UTR (CDK4), or the gene body (KDM3A). Consistent with expectation, higher LHX5 and SYCE1 promoter methylation in hsa_piR_021285-transfected cells was concurrent with lower mRNA levels in both Farage (0.67-fold, NS and 0.53-fold, P = 0.008, respectively) and MCF-7 cells (0.47-fold, P < 0.001 and 0.35-fold, P < 0.001, respectively), although the expression difference for LHX5 did not achieve statistical significance in Farage cells (). Similarly, gene body methylation of KDM3A in hsa_piR_021285-transfected cells was concurrent with increased mRNA expression in both cell lines (Farage: 1.46-fold, P < 0.001; MCF-7: 2.54-fold, P = 0.057). Interestingly, despite higher 5′ UTR methylation in hsa_piR_023426-transfected cells, CDK4 exhibited higher expression in MCF-7 cells (1.69-fold; P = 0.004) and no expression difference in Farage cells. Expression differences were not observed for VAMP3 in either cell line, despite higher promoter methylation in hsa_piR_017183-transfected cells. No FAM150A expression was detected in either hsa_piR_020409-transfected Farage or MCF-7 cells.
Figure 3. Correlation between piRNA mimic-associated gene methylation and mRNA expression. mRNA expression levels of 6 differentially methylated genes (CDK4, FAM150A, KDM3A, LHX5, SYCE1, and VAMP3) were measured by qPCR using RNA isolated from transfected Farage and MCF-7 cells. Consistent with expectation, higher LHX5 and SYCE1 promoter methylation was associated with lower mRNA expression in both transfected Farage (0.67-fold, NS and 0.53-fold, P = 0.008, respectively) and MCF-7 (0.47-fold; P < 0.001 and 0.35-fold; P < 0.001, respectively) cells. Similarly, higher KDM3A gene body methylation was associated with increased mRNA expression in both cell lines (Farage: 1.46-fold, P < 0.001; MCF-7: 2.54-fold, P = 0.057 (borderline significant)). Despite exhibiting higher 5′ UTR methylation, CDK4 exhibited higher expression in MCF-7 cells (1.69-fold; P = 0.004) and no expression difference in Farage cells. Expression differences were not observed for VAMP3 in either Farage or MCF-7 cells, despite higher promoter methylation. No FAM150A expression was detected in either cell line.
Identification of possible piRNA binding sites
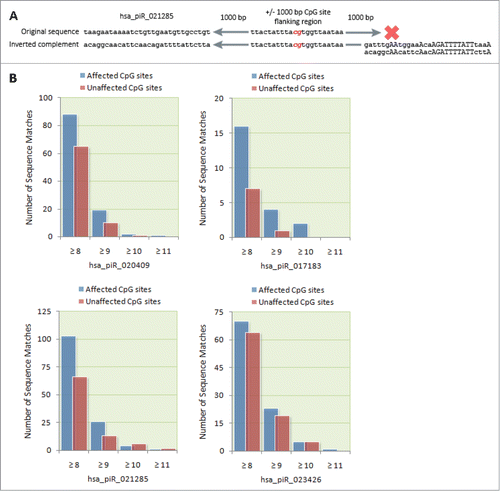
It is feasible that the mechanism by which piRNAs are able to affect DNA methylation involves direct binding of piRNAs to complementary genomic DNA or nascent mRNA transcripts. To investigate this possibility, we screened for ≥8 bp continuous sequence matches between the genomic region 1,000 bp upstream and downstream of differentially methylated CpG sites and the original and the inverted complementary sequences of the affecting piRNA(s). provides a schematic illustration of the screening process. The number of sequence matches to the flanking regions of differentially methylated CpG sites was then compared to the number of matches to the flanking regions of unaffected CpG sites. Overall, differentially methylated CpG sites were found to be enriched for adjacent ≥ 8 bp sequence matches to their affecting piRNA(s). Across the 4 piRNAs, 277 sequence matches to differentially methylated CpG sites were found compared to 202 matches to unaffected CpG sites (P = 0.021). This pattern was reflected at the level of each individual piRNA to varying degrees ().
Figure 4. Bioinformatic identification of possible piRNA binding sites. (A) Schematic of the algorithmic logic used to screen CpG site flanking regions for potential piRNA binding sites. A computer algorithm was designed to screen for ≥ 8 bp continuous sequence matches between a flanking region 1,000 bp upstream and downstream of affected CpG sites and the original and the inverted complement of corresponding piRNA sequences. (B) The number of sequence matches between transfected piRNAs and the flanking regions of differentially methylated CpG sites was compared to the number of matches to the flanking regions of unaffected CpG sites. For each of the transfected piRNAs, we found a higher number of ≥ 8 bp matches to differentially methylated CpG sites compared to unaffected CpG sites.
Discussion
Until now, the major thrust of piRNA research has focused on transposon silencing in the germ line of lower level organisms. Accordingly, the earliest evidence of a possible role for piRNAs in DNA methylation was found in the germ line of mice, where the murine Piwi proteins Mili and Miwi2 were shown to be linked to methylation of transposon loci. Citation14,16,Citation23 Only recently has evidence surfaced to suggest a role of piRNAs in the methylation of non-TE loci, as in the case of the imprinted Rasgrf1 locus in the murine male germ line.Citation34 More recent still is the finding that this function may extend beyond the germ line, most convincingly exemplified by the ability of a piRNA to alter the methylation of the CREB2 promoter in Aplysia neurons.Citation21 A similar function has never before been demonstrated in human somatic tissue, and it remains a mystery whether piRNA-associated methylation of non-TE loci can be characterized as a generalizable phenomenon. Moreover, due to the preponderance of piRNA research in lower level organisms, even rudimentary knowledge of piRNA distribution in the human genome has remained lacking.
The findings from our distribution analysis both reaffirm the traditional understanding of piRNA function and suggest a role for piRNAs beyond transposon silencing. The distinctly high degree of correlation between piRNA and SINE loci () is consistent with the role of piRNAs as transposon silencers, if we are to believe that piRNAs are able to regulate the same classes of transposable elements they derive from, as has been characterized in Drosophila.Citation9,13 Of potentially more interest is our finding that fewer than 1,000 of the 23,439 piRNA species in the human genome exist in more than 10 copies. These “high” copy piRNAs almost universally map to intronic and intergenic regions (), where the vast majority of transposable elements reside. The remaining ~22,000 are single and low copy piRNAs, which demonstrate a greater tendency to overlap non-TE exonic loci and presumably share less sequence similarity with transposons compared to their high copy number counterparts. The implication is that while high copy piRNAs may preferentially bind and regulate transposons, the sequence diversity of lower copy piRNAs may reflect the greater architectural complexity of protein-coding genes and demonstrate a propensity for these piRNAs in their regulation.
A cursory comparison of human single copy piRNAs and pachytene piRNAs found in mice reveals certain similarities. Most pachytene piRNAs in mice do not originate from repetitive genomic elementsCitation10,17 and overwhelmingly map to unique sequences within the genome.Citation2 Likewise, in humans, single copy piRNAs have a greater likelihood to map to non-repetitive genic loci relative to higher copy piRNAs (). Although the precise function of pachytene piRNAs remains a mystery, it is understood that they are required for post-meiotic genomic stability in mouse spermatocytes, which may not necessarily involve transposon repression.Citation11 The similarities in distribution and mapping frequency between mouse pachytene piRNAs and human single copy piRNAs suggest the possibility of a certain degree of overlap in function. Although it is premature to draw any conclusions, it should of interest whether at least a subset of single copy piRNAs are capable of protecting the genome against instability in human spermatocytes and whether this capacity could involve piRNA-dependent gene methylation.
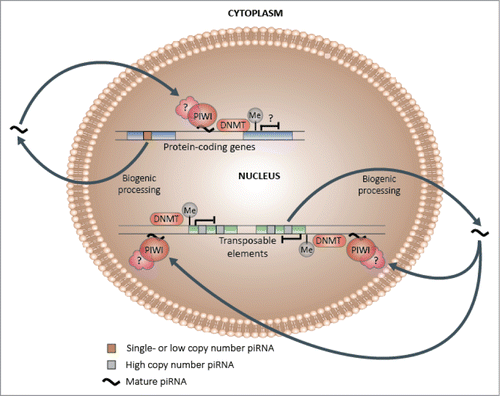
The idea that a subset of single and low copy piRNAs may have the capacity to alter methylation patterns at non-TE gene-coding loci is supported by the results of our methylation array. Despite displaying the greatest relative increase in nuclear expression following transfection, the high copy hsa_piR_017183 mimic affected just 14 of the 117 differentially methylated genes found across the 4 piRNA transfections (), which suggests that while both single and high copy piRNAs can potentially function as inductors of DNA methylation, a subset of single copy piRNAs may be more prone to target gene-associated CpG loci. Moreover, this targeting may be directed in part by piRNA binding to partially complementary genomic DNA or nascent mRNA transcripts adjacent to target CpG sites, as evident in the enrichment of complementary ≥ 8 bp sequence matches near differentially methylated CpG sites (). This observation is consistent with the previous finding of an 8-mer methylation-modulatory binding site between the 5′ end of the affecting piRNA and the Aplysia CREB2 promoter,Citation21 as well as the recent finding of likely piRNA binding to both genomic DNA and nascent mRNA in Drosophila.Citation35 summarizes the core tenets of our piRNA-induced DNA methylation model.
Figure 5. Hypothetical piRNA-dependent DNA methylation induction model. Following biogenic processing to their mature forms, a subset of single and low copy piRNAs may be capable of re-entering the nucleus and preferentially binding to complementary genic DNA or nascent mRNA, which may in turn lead to the induction of methylation at adjacent CpG sites and changes in transcriptional activity. In contrast, a higher proportion of high copy piRNAs, which are more likely to be derived from and share sequence similarity with transposable elements, may specialize in transposon regulation, which could encompass methylation-mediated transcriptional silencing.
piRNA-affected genes were found to be enriched for brain expression, which implies that the transfected piRNAs may be important for brain-related functions (). A function for piRNAs in the central nervous system is in congruence with the recent discovery of piRNAs in the hippocampus of mice, where they are theorized to play a role in spine morphogenesis, among other functions.Citation19 This is echoed by the finding of piRNAs in the Aplysia CNS and their ability to exert methylation-mediated control of the memory-relevant CREB2 gene.Citation21 Additionally, a brain- and testis-specific piRNA has demonstrated the ability to regulate the expression of MTNR1A in human somatic cells, the deregulation of which has been linked to neurodegenerative diseases, including Alzheimer's.Citation28 Taken in the context of these latter observations, our findings raise the intriguing possibility that certain piRNAs may play fundamental, but as yet unelucidated, roles in neurological development, neuroplasticity, and memory.
Of the 6 genes exhibiting piRNA mimic-associated hypermethylation interrogated for concurrent mRNA expression differences, 4 displayed statistically significant differences in mRNA levels in one or both transfected cell lines. While the directionality of expression differences for KDM3A, LHX5, and SYCE1 were consistent with the methylation differences found within their respective functional genic regions, CDK4 displayed increased expression in Farage cells and no expression difference in MCF-7 cells despite increased 5′ UTR methylation, an observation that is at odds with previous findings of 5′ UTR methylation and transcriptional activity being inversely correlative. Citation36-38 However, a previous investigation of the relationship between methylation in different genic regions and mRNA expression found an inverse correlation between 5′ UTR methylation and expression in only 37% of the 43 genes examined,Citation39 suggesting that while this relationship is common, it is not definitive. We also failed to observe expression differences in VAMP3 in either cell line, despite increased methylation of a CpG shore-affiliated CpG site within the proximal promoter. This was not unexpected, as the effect of CpG shore methylation on transcriptional activity is not clearly defined and may be highly gene- and tissue-dependent. Citation40,41 Additionally, it is also feasible that what tangible effects CDK4 and VAMP3 methylation may have had on transcription could have been confounded by crosstalk with histone or post-transcriptional modifiers, or that the methylation changes found for VAMP3 were below the biological threshold required to elicit a measurable downstream effect. Nonetheless, the observed expression changes in KDM3A, LHX5, and SYCE1 reinforce the idea that a subset of piRNAs may be capable of driving gene-associated methylation changes with biologically meaningful consequences.
It should be noted that a number of additional caveats limit the scope of our interpretations. First, the artificial increase in mature piRNA levels following the transient transfection of piRNA mimics bypasses the natural piRNA biogenic process and likely induces a higher level of expression than exists endogenously. However, the fact that these mimics aggregated in the nucleus in a time-dependent manner suggests that they were likely ferried there by one or more endogenously expressed PIWI proteins, which in turn suggests that the observed downstream methylation changes were achieved with input from piRNA mimic-PIWI complexes. Second, although our observation of piRNA-mediated gene methylation is in line with evidence from the soma of lower level organisms, care should be taken when attempting to extrapolate this finding beyond the 2 cell lines examined. The replicability of our findings in other somatic tissues and in untransformed tissues remains the topic of future investigations. Third, as the coverage of the Infinium HumanMethylation450 chip is inherently biased toward genic and CpG island regions, it is unclear whether it is monitoring non-genic elements of the genome where target regions of the piRNA mimics reside. In spite of this, the observation that transfection of the single copy piRNA mimics produced methylation changes at numerous genic loci whereas transfection of the high copy mimic did not is wholly consistent their hypothesized roles inferred from the relative genomic distribution of low and high copy piRNAs. So while the primary target base of the piRNA mimics may lie elsewhere within the genome, within the context of the cell lines tested, it is clear that the single piRNA mimics are capable of eliciting methylation changes at multiple genic loci and that this ability is attenuated in high copy piRNA mimic-transfected cells. Lastly, there are possibly statistically false positive findings from the whole epigenome-wide methylation screening. However, in addition to statistical criteria, we determined differential methylation sites according to the biological significance of >10 % change in both cell lines. Our bioinformatics search for enrichment of complementary sequences near the significantly altered sites further supports that the identified differentially methylated sites are not likely due to chance alone.
In summary, our findings suggest that a subset of single copy piRNAs may be capable of inducing DNA methylation at non-TE protein-coding genes. This capacity may be fulfilled in part by imperfect trans binding to partially complementary genomic DNA or nascent mRNA near target CpG sites. Overall, our investigation, although exploratory, highlights a potential role for a subset of piRNAs in the methylation of protein-coding genes within the human soma, a finding that warrants additional investigation in an expanded repertoire of piRNAs and normal somatic tissues.
Methods
Annotation and characterization of piRNAs
Data on all experimentally validated human piRNAs (n = 662,194) were downloaded from the piRNABank web resource (v1.0, accessed 10 October 2011),Citation42 including information on piRNA accession number, chromosomal position, strand, and sequence. The CNVision annotation tool (v1.73)Citation43 was utilized to characterize all piRNAs with respect to local genomic features (i.e., intron, exon, intergenic), gene name if intronic or exonic, and distance to the nearest genes in both the 5′ and 3′ directions if intergenic. An in-house Perl script (available upon request) was used to determine the copy number of each piRNA, defined as the number of times a piRNA sequence is perfectly mapped to the genome.
Distribution of piRNA sequences relative to other genomic elements
In order to visualize the positions of piRNA loci relative to key genomic elements, positions of piRNAs, non-TE protein-coding genes, and SINE and LINE elements were mapped along the length of each chromosome in 1 megabase blocks. Data on the positions of genes, SINE elements, and LINE elements were downloaded from the UCSC Genome Bioinformatics Table Browser Tool (http://genome.ucsc.edu/cgi-bin/hgTables). Proximity between piRNA loci and genomic elements was quantitated by the degree of linear correlation between mapped positions. Pearson's correlation coefficients and p-values were calculated using the SAS statistical software (v9.2) (SAS Institute, Cary, NC).
Confirmation of PIWIL1–4 expression in farage and MCF-7 cells
Farage, a human B cell lymphoma cell line, and MCF-7, a human breast cancer cell line, were purchased from the American Type Culture Collection (ATCC, Manassas, VA). Farage cells were maintained in Roswell Park Memorial Institute (RPMI) 1640 media supplemented with 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA). MCF-7 cells were maintained as monolayer cultures in 25 cm2 polystyrene culture flasks (Falcon, Becton Dickinson BioScience, Le Pont de Claix, France) in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% FBS and 1% penicillin-streptomycin (Invitrogen, Carlsbad, CA). Cells were incubated at 37°C in a humid atmosphere containing 5% CO2 and subcultured every 3–4 d. Total RNA from Farage and MCF-7 cells was isolated using the RNeasy Mini Kit (QIAGEN) per the instructions of the manufacturer. Isolated mRNA was subsequently converted to cDNA using the AffinityScript Multi-Temp cDNA Synthesis Kit (Agilent, Santa Clara, CA) per the instructions of the manufacturer. cDNA was then amplified using custom-designed gene-specific primers for PIWIL1, PIWIL2, PIWIL3, and PIWIL4 and the housekeeping gene GAPDH. All qPCR reactions were performed in technical triplicate using the SYBR Fast Universal qPCR Kit (Kapa Biosystems, Woburn, MA) on a 7500 Fast Real-Time PCR System (Life Technologies, Carlsbad, CA) with the following conditions: Denaturation at 95°C (3 minutes) followed by 40 cycles of denaturation at 95°C (3 seconds), annealing at 60°C (30 seconds), and elongation at 72°C (30 seconds). Cycle differences were calculated by subtracting the average Ct values for PIWIL1–4 from the average Ct value for GAPDH. Gene-specific primer sequences can be found in Table S1.
piRNA transfection into farage and MCF-7 cells
Single-stranded piRNA mimics (hsa_piR_020409: GGCUUGGUGAUGCCCUGGAUAUUGUCGC, hsa_piR_017183: CUGCAUAGUAUUCCAUGGUGUAUAUGUGC, hsa_piR_021285: UAAGAAUAAAAUCUGUUGAA UGUUGCCUGU, and hsa_piR_023426: UCAAAUAGCAAGUUCAGCCUUCCCAGG) were purchased from IDT (San Jose, CA) and individually reconstituted in nuclease-free water. MCF-7 and Farage cells were reverse transfected in 12-well plates (Falcon, Becton Dickinson BioScience, Le Pont de Claix, France) with either piRNA mimics or a single-stranded scrambled RNA control (QIAGEN, Valenica, CA) using the LipofectAMINE RNAiMAX transfection reagent (Invitrogen, Carlsbad, CA). ~25,000 cells suspended in either 10% FBS-supplemented DMEM or RPMI 1640 medium were added to a pre-incubated mixture of RNAiMAX (1 µl) and RNA oligo in each well to a final oligo concentration of 25 nM. Wells harboring cells intended for use in the methylation array were incubated at 37°C and 5% CO2 for 72 hours and then subjected to a second, forward, transfection at a final oligo concentration of 25 nM following aspiration of old media. Cells were then incubated for an additional 72 hours prior to DNA isolation. A number of wells were incubated for 48,72, and 96 hours without a second transfection for use in confirming piRNA delivery into the nucleus.
Confirmation of piRNA delivery into nucleus
Nuclear RNA was extracted from transfected Farage and MCF-7 cells at 48,72, and 96 hours using the SurePrep Nuclear or Cytoplasmic RNA Purification Kit (Fisher Scientific, Pittsburgh, PA), per the instructions of the manufacturer. RNA purity was measured using an Epoch Microplate Spectrophotometer (Biotek, Winooski, VT). Nuclear RNA was then poly-A tagged and converted to cDNA using the NCode miRNA First-Strand cDNA Synthesis Kit (Invitrogen, Carlsbad, CA). Relative abundance of hsa_piR_020409, hsa_piR_017183, hsa_piR_021285, and hsa_piR_023426 was quantitated using the 2−δδCt qPCR method with normalization to miR-16. piRNA- and miR-16-specific forward primers (hsa_piR_020409: GGCTTGGTGATGCCCTGGATATT GTCGC, hsa_piR_017183: CTGCATAGTATTCCATGGTGTATATGTGC, hsa_piR_021285: TAAGAATAAAATCTGTT GAATGTTGCCTGT, hsa_piR_023426: TCAAATAGCAAGTTCAGCCTTCCCAGG, miR-16: TAGCAGCACGTA AATATTGGCG) were purchased from IDT (San Jose, CA) and paired with a universal reverse primer (Invitrogen, Carlsbad, CA) in each reaction. All qPCR reactions were performed in triplicate using the SYBR Fast Universal qPCR Kit (Kapa Biosystems, Woburn, MA) on a 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA) with the following conditions: Denaturation at 95°C (3 minutes) followed by 40 cycles of denaturation at 95°C (3 seconds) and annealing/elongation at 60°C (30 seconds). Results of the confirmation can be found in Figure S5.
Genome-wide DNA methylation microarray
DNA was isolated from Farage and MCF-7 cells 72 hours following the second transfection using the DNeasy Blood & Tissue kit (QIAGEN, Valenica, CA), per the instructions of the manufacturer. DNA concentration and purity were measured using an Epoch Microplate Spectrophotometer (Biotek, Winooski, VT). Genome-wide DNA methylation measurements were performed using Illumina's (San Diego, CA) Infinium HumanMethylation450 array platform at Yale University's W.M. Keck Foundation Biotechnology Research Laboratory. 750 ng of DNA from each transfected cell was submitted to the facility for this purpose. The Infinium HumanMethylation450 array is designed to accommodate methylation measurements at 485,477 CpG sites distributed throughout the human genome.Citation44 31% (150,254 sites) of the array's CpG content are in CpG islands, 23% (112,072 sites) are in CpG shores, 10% (47,161) are in CpG shelves, and 36% (176,277 sites) are defined as “Open Sea.” 29% (140,004 sites) of CpG content are located in proximal gene promoters, defined as CpG sites within 200 bp (13% or 62,625 sites) or between 201 and 1,500 bp (16% or 77,379 sites) upstream of the transcriptional start site, 10% (49,525 sites) are in the 5′ UTR, 2% (10,810 sites) are in the first exon, 31% (150,212 sites) are in the gene body, 3% (15,383 sites) are in the 3′ UTR, and 25% (119,830 sites) correspond to intergenic regions. The Illumina Custom Model, as implemented in the Illumina GenomeStudio software, was used to assess methylation differences between piRNA mimic-transfected and negative control DNA samples at each CpG site as well as the statistical significance of these differences. This model generates P-values for differential methylation of each CpG site based on the average methylation indices β (a value ranging from 0 to 1, where 0 represents a completely unmethylated site and 1 a completely methylated site) and their variance across replicate probes. CpG sites exhibiting methylation differences of ≥ 10% in both Farage and MCF-7 cells as well as a nominal P-value < 0.05 in one or both cell lines were retained for further analysis. As an additional quality control measure, CpG sites with intensity values of less than 500 for both piRNA-transfected and control samples for both cells lines were filtered out to minimize the contribution from background signals.
Confirmation of DNA methylation
Four CpG sites exhibiting unidirectional methylation changes of 10% or greater in both cell lines were selected for validation using Sequenom's (San Diego, CA) EpiTYPER platform. One CpG site was chosen at random from each piRNA transfection (hsa_piR_020409: cg06011925, hsa_piR_017183: cg12072178, hsa_piR_021285: cg25798413, and hsa_piR_023426: cg21939303). As the EpiTYPER platform permits the measurement of DNA methylation in amplicons up to 600 bp in size, bisulfite sequencing PCR primers were designed using UCSF's MethPrimer program (http://www.urogene.org/methprimer) to target ∼400–600 bp regions flanking the primary CpG sites. The sequences of the bisulfite sequencing PCR primers may be found in the Table S2. 750 ng of DNA from each transfected cell and bisulfite sequencing PCR primer information were sent to Yale University's W.M. Keck Foundation Biotechnology Research Laboratory, where EpiTYPER measurements were performed in technical duplicates.
Tissue expression and gene function analysis
Transcripts associated with CpG sites exhibiting undirectional changes of 10% or greater in both cell lines and and a nominal P-value < 0.05 in one or both cell lines were interrogated for tissue expression using the UniProt tissue expression function as implemented in the DAVID Bioinformatic Resource (v6.7).Citation45,46 All transcripts meeting these criteria were uploaded individually by the species of piRNAs transfected as gene lists. An enrichment P-value < 0.05, as determined by the Fisher Exact test, was used to identify tissue enrichment over and above what might be expected due to random chance.
qPCR expression change confirmation
Total RNA from piRNA mimic- and scrambled RNA negative control-transfected Farage and MCF-7 cells was isolated concurrently with genomic DNA using the miRNeasy Mini Kit (QIAGEN) per the instructions of the manufacturer. Isolated mRNA was subsequently converted to cDNA using the AffinityScript Multi-Temp cDNA Synthesis Kit (Agilent, Santa Clara, CA) per the instructions of the manufacturer. cDNA was then amplified using custom-designed gene-specific primers for CDK4, FAM150A, KDM3A, LHX5, SYCE1, and VAMP3 and the housekeeping gene GAPDH. All qPCR reactions were performed in technical triplicate using the SYBR Fast Universal qPCR Kit (Kapa Biosystems, Woburn, MA) on a 7500 Fast Real-Time PCR System (Life Technologies, Carlsbad, CA) with the following conditions: Denaturation at 95°C (3 minutes) followed by 40 cycles of denaturation at 95°C (3 seconds), annealing at 58°C (30 seconds), and elongation at 72°C (30 seconds). Relative gene-specific cDNA abundance between transfections was assessed using the 2−δδCt method with normalization to GAPDH. Statistical differences in expression were assessed using the unpaired Student's t-test on δCt values. Gene-specific primer sequences can be found in Table S3.
Bioinformatic identification of possible piRNA binding sites
An in-house C++ script (available upon request) was used to identify continuous sequence matches of 8 bases or greater between a genomic flanking region 1000 bp upstream and downstream of differentially methylated CpG sites and the transfected piRNAs. Sequences of flanking regions were retrieved from the UCSC Genome Bioinformatics sequence browser (http://genome.ucsc.edu/cgi-bin/das/hg19/dna?segment=chr#:coord1,coord2) using hg19 (NCBI 37) coordinates. The script was designed to report sequence matches between the genomic flanking region and the original piRNA sequence and the inverted complement of the piRNA sequence in order to capture all possible binding orientations. Each transfected piRNA was matched only to those CpG sites that it affected, and an equal number of unaffected CpG sites, defined as the CpG sites exhibiting the least degree of change in methylation for each transfection, was screened for sequence matches to serve as a reference. Significance of enrichment in continuous ≥ 8 bp matches between transfected piRNAs and affected CpG sites was calculated using a paired Student's t-test, with the number of matches to affected and unaffected CpG sites for each piRNA as the paired comparators.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
suppl_tables_996091.docx
Download MS Word (1.7 MB)Acknowledgments
We would like to thank Christopher Castaldi and Irina Tikhonova at Yale University's W.M. Keck Biotechnology Resource Laboratory for performing the Illumina HumanMethylation450 and the Sequenom EpiTYPER arrays, respectively. In addition, we thank Yujuan Guo for helping to compose the C++ script.
Funding
This work was supported by funds from Yale University.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Aravin A, Gaidatzis D, Pfeffer S, Lagos-Quintana M, Landgraf P, Iovino N, Morris P, Brownstein MJ, Kuramochi-Miyagawa S, Nakano T, et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature 2006; 442:203-7; PMID:16751777
- Girard A, Sachidanandam R, Hannon GJ, Carmell MA. A germline-specific class of small RNAs binds mammalian piwi proteins. Nature 2006; 442:199-202; PMID:16751776
- Grivna ST, Beyret E, Wang Z, Lin H. A novel class of small RNAs in mouse spermatogenic cells. Genes Dev 2006; 20:1709-14; PMID:16766680; http://dx.doi.org/10.1101/gad.1434406
- Watanabe T, Takeda A, Tsukiyama T, Mise K, Okuno T, Sasaki H, Minami N, Imai H. Identification and characterization of two novel classes of small RNAs in the mouse germline: retrotransposon-derived siRNAs in oocytes and germline small RNAs in testes. Genes Dev 2006; 20:1732-43; PMID:16766679; http://dx.doi.org/10.1101/gad.1425706
- Bamezai S, Rawat VP, Buske C. Concise review: the piwi-piRNA axis: pivotal beyond transposon silencing. Stem Cells 2012; 30:2603-11; PMID:22996918; http://dx.doi.org/10.1002/stem.1237
- Kim VN. Small RNAs just got bigger: piwi-interacting RNAs (piRNAs) in mammalian testes. Genes Dev 2006; 20:1993-7; PMID:16882976; http://dx.doi.org/10.1101/gad.1456106
- Siomi MC, Sato K, Pezic D, Aravin AA. PIWI-interacting small RNAs: the vanguard of genome defence. Nat Rev Mol Cell Biol 2011; 12:246-58; PMID:21427766; http://dx.doi.org/10.1038/nrm3089
- Grimson A, Srivastava M, Fahey B, Woodcroft BJ, Chiang HR, King N, Degnan BM, Rokhsar DS, Bartel DP. Early origins and evolution of microRNAs and piwi-interacting RNAs in animals. Nature 2008; 455:1193-7; PMID:18830242; http://dx.doi.org/10.1038/nature07415
- Malone CD, Brennecke J, Dus M, Stark A, McCombie WR, Sachidanandam R, Hannon GJ. Specialized piRNA pathways act in germline and somatic tissues of the drosophila ovary. Cell 2009; 137:522-35; PMID:19395010; http://dx.doi.org/10.1016/j.cell.2009.03.040
- Nordstrand LM, Furu K, Paulsen J, Rognes T, Klungland A. Alkbh1 and Tzfp repress a non-repeat piRNA cluster in pachytene spermatocytes. Nucleic Acids Res 2012; 40:10950-63; PMID:22965116; http://dx.doi.org/10.1093/nar/gks839
- Zheng K, Wang PJ. Blockade of pachytene piRNA biogenesis reveals a novel requirement for maintaining post-meiotic germline genome integrity. PLoS Genet 2012; 8:e1003038; PMID:23166510; http://dx.doi.org/10.1371/journal.pgen.1003038
- Aravin AA, Hannon GJ, Brennecke J. The piwi-piRNA pathway provides an adaptive defense in the transposon arms race. Science 2007; 318:761-4; PMID:17975059; http://dx.doi.org/10.1126/science.1146484
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ. Discrete small RNA-generating loci as master regulators of transposon activity in drosophila. Cell 2007; 128:1089-103; PMID:17346786; http://dx.doi.org/10.1016/j.cell.2007.01.043
- Aravin AA, Sachidanandam R, Bourc'his D, Schaefer C, Pezic D, Toth KF, Bestor T, Hannon GJ. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell 2008; 31:785-99; PMID:18922463; http://dx.doi.org/10.1016/j.molcel.2008.09.003
- Kuramochi-Miyagawa S, Watanabe T, Gotoh K, Totoki Y, Toyoda A, Ikawa M, Asada N, Kojima K, Yamaguchi Y, Ijiri TW, et al. DNA methylation of retrotransposon genes is regulated by piwi family members MILI and MIWI2 in murine fetal testes. Genes Dev 2008; 22:908-17; PMID:18381894; http://dx.doi.org/10.1101/gad.1640708
- Aravin AA, Sachidanandam R, Girard A, Fejes-Toth K, Hannon GJ. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science 2007; 316:744-7; PMID:17446352; http://dx.doi.org/10.1126/science.1142612
- Gan H, Lin X, Zhang Z, Zhang W, Liao S, Wang L, Han C. piRNA profiling during specific stages of mouse spermatogenesis. RNA 2011; 17:1191-203; PMID:21602304; http://dx.doi.org/10.1261/rna.2648411
- Brennecke J, Malone CD, Aravin AA, Sachidanandam R, Stark A, Hannon GJ. An epigenetic role for maternally inherited piRNAs in transposon silencing. Science 2008; 322:1387-92; PMID:19039138; http://dx.doi.org/10.1126/science.1165171
- Lee EJ, Banerjee S, Zhou H, Jammalamadaka A, Arcila M, Manjunath BS, Kosik KS. Identification of piRNAs in the central nervous system. RNA 2011; 17:1090-9; PMID:21515829; http://dx.doi.org/10.1261/rna.2565011
- Cichocki F, Lenvik T, Sharma N, Yun G, Anderson SK, Miller JS. Cutting edge: KIR antisense transcripts are processed into a 28-Base PIWI-like RNA in human NK cells. J Immunol 2010; 185:2009-12; PMID:20631304; http://dx.doi.org/10.4049/jimmunol.1000855
- Rajasethupathy P, Antonov I, Sheridan R, Frey S, Sander C, Tuschl T, Kandel ER. A role for neuronal piRNAs in the epigenetic control of memory-related synaptic plasticity. Cell 2012; 149:693-707; PMID:22541438; http://dx.doi.org/10.1016/j.cell.2012.02.057
- Ross RJ, Weiner MM, Lin H. PIWI proteins and PIWI-interacting RNAs in the soma. Nature 2014; 505:353-9; PMID:24429634; http://dx.doi.org/10.1038/nature12987
- Peng JC, Lin H. Beyond transposons: the epigenetic and somatic functions of the piwi-piRNA mechanism. Curr Opin Cell Biol 2013; PMID:23465540; http://dx.doi.org/10.1016/j.ceb.2013.01.010
- Rouget C, Papin C, Boureux A, Meunier AC, Franco B, Robine N, Lai EC, Pelisson A, Simonelig M. Maternal mRNA deadenylation and decay by the piRNA pathway in the early drosophila embryo. Nature 2010; 467:1128-32; PMID:20953170; http://dx.doi.org/10.1038/nature09465
- Li MA, Alls JD, Avancini RM, Koo K, Godt D. The large maf factor traffic jam controls gonad morphogenesis in drosophila. Nat Cell Biol 2003; 5:994-1000; PMID:14578908; http://dx.doi.org/10.1038/ncb1058
- Qi H, Watanabe T, Ku HY, Liu N, Zhong M, Lin H. The yb body, a major site for piwi-associated RNA biogenesis and a gateway for piwi expression and transport to the nucleus in somatic cells. J Biol Chem 2011; 286:3789-97; PMID:21106531; http://dx.doi.org/10.1074/jbc.M110.193888
- Saito K, Inagaki S, Mituyama T, Kawamura Y, Ono Y, Sakota E, Kotani H, Asai K, Siomi H, Siomi MC. A regulatory circuit for piwi by the large maf gene traffic jam in drosophila. Nature 2009; 461:1296-9; PMID:19812547; http://dx.doi.org/10.1038/nature08501
- Esposito T, Magliocca S, Formicola D, Gianfrancesco F. piR_015520 belongs to piwi-associated RNAs regulates expression of the human melatonin receptor 1A gene. PLoS One 2011; 6:e22727; PMID:21818375; http://dx.doi.org/10.1371/journal.pone.0022727
- Zhang Y, Romanish MT, Mager DL. Distributions of transposable elements reveal hazardous zones in mammalian introns. PLoS Comput Biol 2011; 7:e1002046; PMID:21573203; http://dx.doi.org/10.1371/journal.pcbi.1002046
- Sela N, Kim E, Ast G. The role of transposable elements in the evolution of non-mammalian vertebrates and invertebrates. Genome Biol 2010; 11:R59; PMID:20525173; http://dx.doi.org/10.1186/gb-2010-11-6-r59
- Lee JH, Jung C, Javadian-Elyaderani P, Schweyer S, Schutte D, Shoukier M, Karimi-Busheri F, Weinfeld M, Rasouli-Nia A, Hengstler JG, et al. Pathways of proliferation and antiapoptosis driven in breast cancer stem cells by stem cell protein piwil2. Cancer Res 2010; 70:4569-79; PMID:20460541; http://dx.doi.org/10.1158/0008-5472.CAN-09-2670
- Lee JH, Schutte D, Wulf G, Fuzesi L, Radzun HJ, Schweyer S, Engel W, Nayernia K. Stem-cell protein piwil2 is widely expressed in tumors and inhibits apoptosis through activation of stat3/bcl-X-L pathway. Hum Mol Genet 2006; 15:201-11; PMID:16377660; http://dx.doi.org/10.1093/hmg/ddi430
- Giurato G, De Filippo MR, Rinaldi A, Hashim A, Nassa G, Ravo M, Rizzo F, Tarallo R, Weisz A. iMir: an integrated pipeline for high-throughput analysis of small non-coding RNA data obtained by smallRNA-seq. BMC bioinformatics 2013; 14:362; PMID:24330401; http://dx.doi.org/10.1186/1471-2105-14-362
- Watanabe T, Tomizawa S, Mitsuya K, Totoki Y, Yamamoto Y, Kuramochi-Miyagawa S, Iida N, Hoki Y, Murphy PJ, Toyoda A, et al. Role for piRNAs and noncoding RNA in de novo DNA methylation of the imprinted mouse rasgrf1 locus. Science 2011; 332:848-52; PMID:21566194; http://dx.doi.org/10.1126/science.1203919
- Huang XA, Yin H, Sweeney S, Raha D, Snyder M, Lin H. A major epigenetic programming mechanism guided by piRNAs. Dev Cell 2013; 24:502-16; PMID:23434410; http://dx.doi.org/10.1016/j.devcel.2013.01.023
- Yin H, Blanchard KL. DNA methylation represses the expression of the human erythropoietin gene by two different mechanisms. Blood 2000; 95:111-9; PMID:10607693
- Peng B, Hurt EM, Hodge DR, Thomas SB, Farrar WL. DNA hypermethylation and partial gene silencing of human thymine- DNA glycosylase in multiple myeloma cell lines. Epigenetics 2006; 1:138-45; PMID:17965616; http://dx.doi.org/10.4161/epi.1.3.2938
- Vincent A, Ducourouble MP, Van Seuningen I. Epigenetic regulation of the human mucin gene MUC4 in epithelial cancer cell lines involves both DNA methylation and histone modifications mediated by DNA methyltransferases and histone deacetylases. FASEB J 2008; 22:3035-45; PMID:18492726; http://dx.doi.org/10.1096/fj.07-103390
- Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet 2006; 38:1378-85; PMID:17072317; http://dx.doi.org/10.1038/ng1909
- Akalin A, Garrett-Bakelman FE, Kormaksson M, Busuttil J, Zhang L, Khrebtukova I, Milne TA, Huang Y, Biswas D, Hess JL, et al. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet 2012; 8:e1002781; PMID:22737091; http://dx.doi.org/10.1371/journal.pgen.1002781
- Hackett JA, Surani MA. DNA methylation dynamics during the mammalian life cycle. Philos Trans R Soc Lond B Biol Sci 2013; 368:20110328; PMID:23166392; http://dx.doi.org/10.1098/rstb.2011.0328
- Sai Lakshmi S, Agrawal S. piRNABank: a web resource on classified and clustered piwi-interacting RNAs. Nucleic Acids Res 2008; 36:D173-7; PMID:17881367; http://dx.doi.org/10.1093/nar/gkm696
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 williams syndrome region, are strongly associated with autism. Neuron 2011; 70:863-85; PMID:21658581; http://dx.doi.org/10.1016/j.neuron.2011.05.002
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et al. High density DNA methylation array with single CpG site resolution. Genomics 2011; 98:288-95; PMID:21839163; http://dx.doi.org/10.1016/j.ygeno.2011.07.007
- Dennis G, Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol 2003; 4:P3; PMID:12734009; http://dx.doi.org/10.1186/gb-2003-4-5-p3
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44-57; PMID:19131956; http://dx.doi.org/10.1038/nprot.2008.211