
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Splice-site selection is controlled by secondary structure through sequestration or approximation of splicing signals in primary transcripts but the exact role of even the simplest and most prevalent structural motifs in exon recognition remains poorly understood. Here we took advantage of a single-hairpin exon that was activated in a mammalian-wide interspersed repeat (MIR) by a mutation stabilizing a terminal triloop, with splice sites positioned close to each other in a lower stem of the hairpin. We first show that the MIR exon inclusion in mRNA correlated inversely with hairpin stabilities. Employing a systematic manipulation of unpaired regions without altering splice-site configuration, we demonstrate a high correlation between exon inclusion of terminal tri- and tetraloop mutants and matching tri-/tetramers in splicing silencers/enhancers. Loop-specific exon inclusion levels and enhancer/silencer associations were preserved across primate cell lines, in 4 hybrid transcripts and also in the context of a distinct stem, but only if its loop-closing base pairs were shared with the MIR hairpin. Unlike terminal loops, splicing activities of internal loop mutants were predicted by their intramolecular Watson-Crick interactions with the antiparallel strand of the MIR hairpin rather than by frequencies of corresponding trinucleotides in splicing silencers/enhancers. We also show that splicing outcome of oligonucleotides targeting the MIR exon depend on the identity of the triloop adjacent to their antisense target. Finally, we identify proteins regulating MIR exon recognition and reveal a distinct requirement of adjacent exons for C-terminal extensions of Tra2α and Tra2β RNA recognition motifs.
Abbreviations
| TE | = | transposable element |
| SINE | = | Short INterspersed Element |
| MIR | = | mammalian-wide interspersed repeat |
| SSO | = | splice-switching oligonucleotides |
| FGB | = | gene for fibrinogen |
| ESE | = | exonic splicing enhancer, ESS, exonic splicing silencer. |
Introduction
Introns are removed from eukaryotic genes by a large and dynamic RNA-protein complex termed the spliceosome.Citation1 Spliceosomes identify splice sites in mRNA precursors (pre-mRNAs) with single-nucleotide precision and often in a cell type-, developmental stage- or gender-specific manner, contributing to transcriptomic and proteomic diversity and organismal complexity through alternative splicing.Citation1 Splice site choice is controlled by conserved but degenerate pre-mRNA signals at intron-exon boundaries and also by auxiliary motifs in introns and exons that activate or inhibit splicing, termed enhancers and silencers.Citation2-9 These motifs are preferentially located in single-stranded regions of pre-mRNAs and have been under selection in evolution,Citation10 however, their structural correlates are largely unknown.
Numerous examples in the literature show that so-called ‘splicing code’, or information required for accurate RNA processing decisions, is influenced by a sequestration or approximation of pre-mRNA splicing signals.Citation11-23 For example, alternative splice sites are enriched for conserved RNA secondary structures and GC-content, which may promote looping out of intervening segments.Citation24-26 Long-range RNA structures and intramolecular base pairing have been associated with alternative splicing in Drosophila Citation27 and cDNA segments ultraconserved between rodents and humans were found to be AT-rich and resistant to folding.Citation28 However, no universal exon recognition rules have been uncovered even for the most common RNA building blocks.
Transposable elements (TEs) are repetitive sequences that can move or transpose themselves to new positions within the genome of a single cell.Citation29,30 They constitute up to 80% of eukaryotic genomes, substantially shaping genome evolution, species divergence, exon-intron structure and gene expression.Citation29,30 Although each TE family has contributed to protein diversity and can be ‘exonized’, Short INterspersed Elements (SINEs) have been particularly active in evolution in this respect.Citation31-35 The overrepresentation of SINEs among exonized TEs was found also for mutation-induced cryptic exons that led to genetic disease.Citation36 SINEs occupy ∼13% of the human genome and comprise primate-specific Alus and more ancient mammalian-wide interspersed repeats (MIRs). Each SINE subfamily contributed both traditionalCitation34-38 and auxiliaryCitation3,34,39-41 splicing motifs to exonized sequences, thus enhancing proteomic diversity and gene expression control through alternative splicing, but molecular pathways leading to their exonization are only partially understood.
SINEs are overrepresented in introns where they are often present in the opposite orientation within a short distance from each other, which can enhance formation of very stable stem-loop structures and influence RNA processing.Citation34,42 Stem-loops (or hairpins) are the most common RNA secondary structure motifs that play an important role in transcription, RNA processing and mRNA export, mRNA stability, subcellular localization, translation and viral replication (reviewed in ref.Citation43). Well defined hairpins, such as an iron responsive element (IRE), have been implicated in important cellular functions, including ion uptake and storage, energy metabolism, hypoxic regulation, cytoskeletal and neuronal organization.Citation43-45 Highly predictable hairpin structures formed by inverted SINEs may thus provide useful models for studying their importance in splice-site recognition. However, TEs have rarely been in the spotlight of structural models of pre-mRNA splicing, which were focused largely on traditional rather than auxiliary splicing signals.Citation20
Here, we have developed a new structural splicing model, consisting of a single-hairpin MIR exon activated by A > G mutation in intron 1 of the human gene for fibrinogen (FGB). Using a combination of biochemical and biophysical methods, we show an unexpected autonomy of an IRE-like MIR hairpin in exon recognition and its long-range interaction with an upstream, Tra2-induced exon. Terminal tri- and tetra-loops of the MIR hairpin were sufficient to maintain this autonomy even in the context of a distinct stem but only if coupled with loop-closing base pairs of the original MIR hairpin. We also identify splicing enhancer and silencer categories that best predicted exon inclusion of terminal tri- and tetra-loop mutants. In contrast to terminal loops, splicing activities of internal triloops of the MIR hairpin were predicted by their intramolecular interactions with the antiparallel strand of the helix rather than frequencies of matching trinucleotides in splicing enhancers/silencers. Finally, we identify proteins that control usage of the MIR exon.
Results
Splicing activity and stability of a single-hairpin exon
To develop a structural model of auxiliary splicing sequences, we searched previously published cases of exonized TEs in genetic diseaseCitation36 for mutations within SINEs. RNA secondary structure predictions with multiple pre-mRNA segments overlapping new SINE exons revealed that an exonized antisense MIR in FGB intron 1 was fully encompassed in a very stable hairpin structure (). As compared to the MIR consensus sequence, this MIR copy sustained a 4-nt deletion that created a purine-rich exonic splicing enhancer (ESE), which was further optimized by the disease-causing A > G mutation (Fig. S1), leading to afibrinogenemia.Citation46 In this structure, the 5′ and 3′ splice sites of the new exon were positioned close to each other in a stable lower stem whereas the mutation stabilized a closing base-pair of a terminal triloop ().
Figure 1. MIR hairpin stabilization is associated with increased exon skipping. (A) Predicted RNA secondary structure of the MIR exon. The 5′ and 3′ splice sites are shown as blue and red arrows, respectively. Intronic sequences are in lower case, exon positions (upper case) are numbered. A > G mutation is shown by a star. Alignments of FGB intron 1 MIR elements with the MIR consensus is in Figure S1; 4-bp sequence deleted in the antisense MIR copy is shown to the left. Putative branch points are indicated in Figure S1. Target sequences of splice-switching oligoribonucleotides (SSOs) are shown as red lines (5′ and 3′ stem) and a blue curve (loop SSO). (B) Far UV-CD spectrum of the 24-mer hairpin at 25°C showing a strong positive ellipticity at 265 nm. (C) 800 MHz 1H NMR spectrum of the RNA oligo (inset) showing signals for Watson-Crick hydrogen bonded A-U and G-C base pairs between 12–13.5 ppm. (D) FGB minigene reporter. Exons are shown as boxes, introns as lines; their scale is at the top in base pairs. The MIR and T exons are shown as red and blue boxes; the MIR exon-activating mutation is denoted by a star. Dotted lines show canonical (top) and aberrant (bottom) RNA products. MIRs are denoted by blue arrowheads, cloning primers by black arrows and amplification primers by gray arrows. Primer sequences are shown in Table S3. A FGB segment cloned into a hybrid in vitro splicing reporter () is denoted by a black rectangle. (E) Stabilization of the MIR hairpin increases exon skipping. Free energies were computed for the hairpin shown in panel A. A predicted structure of a hairpin destabilising double mutant 13C/14C, which led to full exon inclusion, is shown in the inset. (F) MIR exon inclusion levels for 38 FGB mutations (numbered as in panel A). WT, wildtype minigene; MUT, minigene carrying the disease-causing A > G substitution. All mutations were introduced in the MUT minigene; the same mutations in the WT reporter failed to activate the exon (data not shown). Spliced RNA products (bottom left) are shown as minigene exons throughout; their color corresponds to that in (D). Error bars are SDs of duplicate experiments. Gel panel was merged from separate gels as indicated; cryptic splice sites within or adjacent to the MIR exon were not observed for any construct.
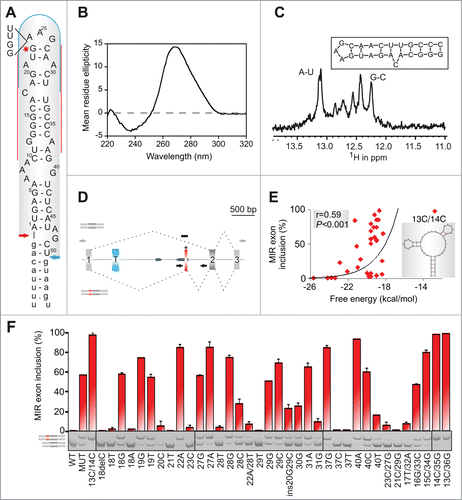
To support formation of this structure experimentally and test if its stability correlates with inclusion of the MIR exon in mature transcripts, we carried out biophysical studies using a synthetic, MIR-derived RNA () and mutagenesis with minigene constructs containing FGB exons 1–3 (). The far-UV circular dichroism (CD) spectrum of a 3 μM buffered solution (pH 7.0) of the 24-mer representing exon positions 13–36 gave a strong positive ellipticity at 265 nm (), consistent with a double-stranded stem-loop structure in solution. The NMR spectrum at 800 MHz of the same oligonucleotide sample showed imino proton signals in the region 12 to 13.5 ppm (), confirming the formation of stable Watson-Crick hydrogen bonded A-U and G-C base pairs. Structure prediction calculations suggested that the oligo hairpin structure () has a high stability (ΔG = −6.6 kcal mol−1) which we examined using CD melting studies. We observed a clear, but broad, melting profile (consistent with mis-matched or unpaired bases) with a mid-point unfolding transition corresponding to a Tm = 74.0 ± 3.5°C (data not shown).
Mutagenesis of single- and double-stranded regions of minigene reporters transfected into human embryonic kidney (HEK) 293 cells showed that substitutions of unpaired nucleotides altered exon inclusion levels more than mutations of paired residues (median difference 54.5% vs 31.6%, P < 0.05) and that inclusion of the MIR exon in the mRNA correlated positively with predicted free energy of mutated constructs (). Most notably, the MIR exon inclusion was eliminated by deletion or mutation of a cytosine bulge at position 18, which separates a stable lower helix from a more dynamic upper stem of the hairpin. C or G at this position was associated with exon inclusion and A or T with exon skipping, suggesting that ligand interactions are mediated by hydrogen bonding. Exon inclusion was also diminished by mutations of other single-nucleotide bulges at positions 21, 29 and 37.
Collectively, computational, CD, NMR and mutagenesis studies supported the structural model of MIR exon activation shown in . This model permits systematic manipulation of terminal loops without altering splice-site configuration of the new exon, thus providing an insight into structural correlates of auxiliary splicing sequences.
MIR exon inclusion levels of 64 RNA triloops
Next, we cloned 64 minigenes representing all possible nucleotide combinations in the terminal triloop (exon positions 24–26, ) and determined inclusion levels of mutated MIR exons after transfection (). The GAN triloops (where N is any base) produced the highest exon inclusion levels, accounting for over a third of cryptic splicing, while the GA-and AG-containing loops contributed >50% to the total. The A > G > C > U hierarchy of triloop nucleotide splicing activities (, inset) was the same as base frequencies found for most splicing enhancer categories (). Silencers showed the opposite order (data not shown; cf. Fig. 2C). Notably, our triloop mutants exhibited almost identical hierarchy in exon inclusion levels in 4 different primate cell lines although epithelial HeLa cells produced systematically less cryptic splicing than the remaining tested cells (Fig. S2).
Figure 2. RNA triloops as structural correlates of splicing enhancers and silencers. (A) Mean inclusion levels of 64 terminal triloop mutants of the MIR hairpin exon (top right; mutated residues are in red). Inclusion levels are on a ln scale. Inset shows weighted triloop nucleotide-specific contribution to exon inclusion levels (see Materials and Methods). (B, C) Nucleobase fraction (%) of the indicated categories of splicing enhancers and silencers. Splicing enhancers (B) and silencers (C) were published previously as RESCUE-ESEs,Citation4 exon and intron identity elements (EIEs and IIEs),Citation7 ESEs and ESSs derived by neighborhood inference (trusted NI ESEs and ESSs) Citation77 and QUEPASA ESEs and ESSs.Citation9 (D, E) MIR exon inclusion levels of MIR triloop mutants were best predicted by frequencies of corresponding trinucleotides in QUEPASA hexamers.Citation9 Pearson correlation coefficients and associated P values are shown at the top. Trinucleotide frequencies in QUEPASA enhancers and silencers are shown in Figure S3. Correlation matrix for all enhancer and silencer classes is in Table S1.
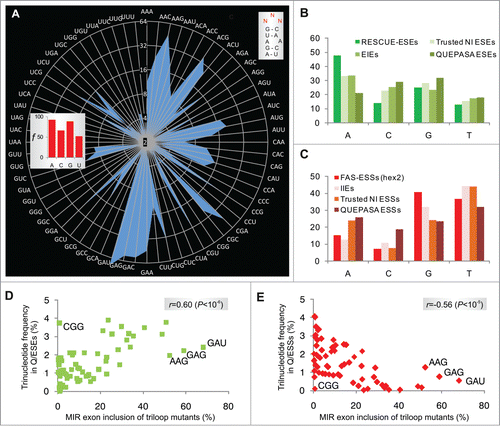
The most stable secondary structures predicted for each triloop mutant maintained the identical hairpin in 60/64 (94%) cases, altering only the terminal triloop display. The four remaining loop trinucleotides (CCC, GGG, AGG and GGC) and stable alternative secondary structures predicted for CCU, CUC, UCC and UGC triloops lacked the cytosine bulge; remarkably, they all failed to activate the MIR exon ().
Taken together, splice-switching properties of single nucleotide bulges of the hairpin were complemented by a remarkably wide range of splicing activities of terminal triloops, in which GAN/NAG trinucleotides contributed most to exonization. The order of triloop splicing activities was very similar across primate cell lines.
Trinucleotide frequencies in splicing enhancers and silencers correlate with splicing activities of corresponding terminal triloops
To determine the extent to which each triloop mutant contributes to universal exon recognition, we correlated their exon inclusion levels with trinucleotide frequencies in previously defined categories of splicing enhancers and silencers. A significant positive correlation was found for most enhancer categories and negative correlation for most silencer classes (). The highest correlation was found for trinucleotide frequencies in the most recently derived class of enhancers (termed QUEPASA), particularly when normalized for their previously defined ESEseq scoresCitation9 (Table S1, ). The pattern of splicing activities of 64 terminal MIR triloops strikingly resembled the radar chart showing frequencies of matching trinucleotides in QUEPASA enhancers (cf. and Fig. S3); this pattern was dramatically distinct for QUEPASA silencers. Thus, the terminal triloop identity contributed to a great extent to enhancer/silencer activities of the MIR exon.
Triloop identity controls splicing outcomes of an adjacent antisense target
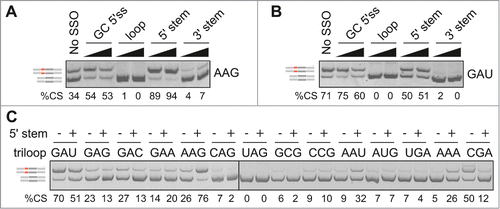
To further test the relative importance of single and double-stranded regions in MIR exon recognition, we employed splice-switching oligonucleotides (SSOs) that targeted the 5'stem, terminal loop and 3'stem of the hairpin (). All SSOs were 2'-O-methyl-modified at each sugar residue and uniformly labeled with phosphorothioates (Table S3). Cotransfection of loop and 3'stem SSOs with FGB reporters produced invariably exon skipping. Interestingly, the effect of the 5'stem SSO depended on the loop identity (). This SSO promoted exon inclusion of terminal loop mutants that contained diadenosines (except for CAA and AAC) but increased exon skipping or had no effect for the remaining loops (). Thus, the splicing outcome of the SSO that targeted a more accessible face of the MIR hairpin was controlled by the adjacent single-stranded sequence.
Figure 3. Triloop-dependent promotion and repression of the MIR exon by SSOs. (A, B) Identification of SSOs that target the MIR exon and promote its inclusion in mature transcripts. SSOs are shown at the top, percentage of cryptic splicing (%CS) at the bottom, spliced products schematically to the left and tested triloop sequence to the right. SSO sequences are shown in Table S3 and their hairpin targets in . Final concentrations of each SSO in cell cultures were 40 and 80 nM. GC 5'ss denotes a 2′-O-methyl SSO used as one of the controls; this SSO targets the 5′ splice site of a cryptic exon in the BTK gene and induces exon skipping Citation74 and is also partially complementary to the 5'stem MIR at exon positions 8–10 and 13–17 (). (C) Triloops that promote or repress MIR exon inclusion upon binding of the 5′ stem SSO. A final concentration of the SSO in the culture medium was 50 nM. The panel was merged from 2 separate gels.
MIR hairpin acts as an autonomous exon selection module
To test generality of the observed triloop hierarchies of exon usage () and the role of their sequence context in exon recognition, we examined splicing activities of the same 64 hairpins inserted in central exons of 3 additional minigenes. MIR hairpins were cloned in F9 exon 3, SMN2 exon 7 and at 2 insertion sites of L1CAM exon 18 ( and S4). If exon recognition is facilitated largely by autonomous hairpin interactions through the terminal loop, we should observe a similar order of exon inclusion levels for triloop mutants in FGB and hybrid transcripts and a significant correlation of exon inclusion levels between FGB and hybrid mutants. In contrast, if the inclusion of central exons relies more on other interactions, we would expect a more variable ranking of their triloop-specific splicing activities.
Figure 4. Loop- and stem-dependencies of MIR exon recognition. (A) Hybrid reporter constructs. Exons are shown as boxes (numbered and to a scale shown at the top); introns (not to a scale) are shown as lines. Splicing to a cryptic 5′ splice site in F9 (cr5ss-104) Citation73 is shown by dotted lines. MIR hairpins are in red; r1 and r2 denote their positions in 2 L1CAM constructs. (B) Spliced products of wild-type (WT) reporters and their hybrid counterparts with (MIR MUT) and without (MIR WT) the A > G transition. hd, heteroduplexed PCR products. F9 BP-G constructs had substitution of the branch point adenine for guanine Citation73 to obtain sufficient expression of transcripts skipping exon 3. (C) Exon inclusion levels (%) of terminal triloop mutants in FGB (y-axes) and of corresponding triloop mutants in the indicated hybrid pre-mRNAs (x-axes). Outliers/purine-rich triloops are highlighted. r, Pearson correlation coefficient. (D) Splicing activities of FGB MIR and SMN2 TSL2 triloop mutants. Secondary structure of the native TSL2 hairpin (TSL2/N) Citation21,47 is shown schematically to the left; mutated nucleotides are in red. The 5′ splice site of SMN2 exon 7 is denoted by an arrow. The number of tested triloops is shown at the bottom. (E) Loop-closing base-pairs significantly contribute to the context-dependency of splicing reactions. x-axis shows splicing activities of corresponding triloop mutants in SMN2 TSL2/H. Heptamers shared between MIR () and hybrid (TSL2/H) triloop mutants are denoted by a blue curve; a shared sequence between TSL2/N and MIR is limited to the triloop, as in panel D. (F) A lack of correlation between exon inclusion levels of matching internal and terminal triloop mutants in FGB. Triloops generating high exon inclusion are indicated. (G) MIR exon inclusion levels of internal triloop mutants inversely correlate with the number of hydrogen bonds formed between mutated triloops and the antiparallel strand. The number of hydrogen bonds (x-axis) was calculated from Mfold predictions for each mutant (Table S2). Inset shows that MIR exon inclusion levels (%, y-axis) significantly increased with the number of unpaired bases between the internal loop and the antiparallel strand of the MIR hairpin (P <0.001).
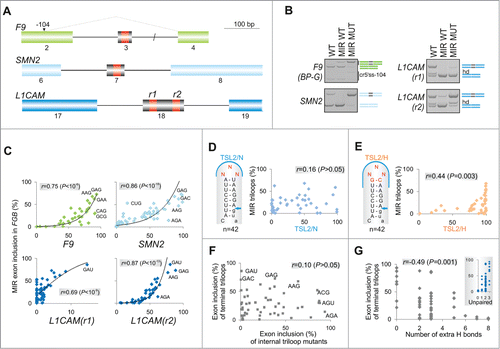
shows that the MIR hairpin containing the disease-causing A > G substitution (MUT) increased inclusion of the central exon in each hybrid minigene. By contrast, the hairpin lacking this substitution and unable to activate the MIR exon (WT) induced exon skipping in SMN2 and L1CAM, nevertheless it could still promote inclusion of a very small exon in the F9 minigene. We then transfected 256 sequence-verified constructs (i.e., 64 triloops for each hybrid) into HEK293 cells and determined inclusion of central exons in their mRNAs. As shown in , exon inclusion of 64 triloop mutants in the context of FGB pre-mRNAs positively correlated with exon inclusion of the same triloop mutants in each hybrid minigene. We conclude that the terminal triloop identity of the MIR hairpin was a major determinant of exon inclusion in each hybrid transcript, acting autonomously through its ligand interactions.
Loop-closing base-pairs as a principal contributor to stem-specific splicing activities
To determine the relative importance of stem identity of this potentially universal exon-promoting structure, we investigated the TSL2 hairpin in SMN2 exon 7, which terminates with the AAU triloopCitation21,47 (Fig. S4A). We randomly mutated this triloop in the SMN2 minigene, measured exon 7 inclusion of the resulting 42 TSL2 triloop variants and correlated their inclusion levels with those established for terminal triloops in the FGB minigene (). In the native SMN2 context (TSL2/N), exon 7 inclusion levels was not significantly correlated with the MIR exon inclusion of FGB constructs, suggesting that the triloop identity alone does not predict splicing activity. However, when the A-U closing base pair of TSL2 was replaced by G-C in each mutant, thereby extending the shared motif between SMN2 and FGB hairpin caps to heptamers, the hybrid (TSL2/H) or ‘MIR-onized’ reporters produced significant correlation ().
We next tested the importance of the loop-closing guanine for MIR exon recognition. We exchanged the loop-closing bases in a randomly chosen subset of FGB triloop mutants and examined their exon inclusion levels. The GC pair yielded higher exon inclusion than the CG pair in 8 of 10 tested constructs. The splicing enhancement was particularly pronounced in pre-mRNAs in which the 5′ guanine in the base-pair was followed by an unpaired adenine at the first triloop position (Fig. S5).
Taken together, these data showed that the stem dependency of MIR exon activation was to a significant extent determined by the identity of loop-closing base-pairs. They also explain previous observations of higher predictive values of hexa- or octamer splicing enhancers for exon inclusion than for shorter oligomers. Finally, the higher exon inclusion of G(NNN)C triloops as compared to their C(NNN)G counterparts (Fig. S5) may reflect a previously described lower thermodynamic stability of the formerCitation48 and contribute to the observed positive correlation between splicing activity of the hairpin and free energy ().
Intramolecular base-pairing interactions of the internal triloop predict MIR exon inclusion
Apart from the terminal triloop, the MIR hairpin contains a predicted internal triloop (or trinucleotide bulge) located closer to the 3′ splice site at exon positions 7–9 (). Unlike the terminal triloop, however, systematic mutagenesis of the internal triloop would introduce new base-pairing contacts with the antiparallel strand of the hairpin. To explore their significance in MIR splicing, we randomly mutagenized this unpaired segment in the FGB reporter, measured exon inclusion levels of the resulting mutants and compared them to the number of predicted hydrogen bonds between triloop mutants and the antiparallel strand of the hairpin (Table S2). Interestingly, inclusion levels of internal triloop mutants correlated neither with corresponding terminal loop mutants () nor with frequencies of matching trinucleotides in splicing silencers and most enhancers (Table S1). By contrast, exon inclusion levels of internal triloop mutants significantly correlated with the number of hydrogen bonds between mutated trinucleotide and the antisense sequence (). Together, these data further support a critical role of hairpin structures and their stabilities in exon recognition and suggest that activities of splicing enhancers and silencers, particularly motifs that show the highest correlation with splicing outcomes in our minigenes (Table S1, ), are better predicted by exposed rather than less accessible short RNA loops.
Triloops frequent in previously determined RNA secondary structures tend to confer low inclusion of the MIR exon
Are inclusion levels of 64 MIR triloop mutants related to triloop frequencies in natural RNAs? To begin to answer this question, we compared frequencies of 823 triloops in previously determined 1349 RNA secondary structures and the MIR exon inclusion of matching triloop mutants. The triloop database was derived from structures of 123 small subunit rRNAs, 223 large subunit rRNAs, 309 5S rRNAs, 484 tRNAs, 91 signal recognition particles, 16 RNase P RNAs, 100 group I introns and 3 group II intronsCitation48 (Brent Znosko, personal communication). Interestingly, triloops frequent in natural RNAs tended to have low exon inclusion values and vice versa (Fig. S6A). The only outlier was the GAA triloop: although relatively frequent in nature, it was associated with a high inclusion of the MIR exon, both in FGB and hybrid transcripts. In contrast, we observed no significant correlation for the internal MIR loop mutants, in which the GAA triloop outlier was not dominant (Fig. S6B). These data suggest that triloops most frequent in nature (including UAA, GCA, UUU and AUA) tend to have splicing silencer activitites, except for GAA, probably the most potent splicing enhancer motif.Citation36,49 With the notable exception of GAA, triloops common in these natural RNAs thus appear to contribute more than expected to repression of pseudo-splice sites, which outnumber authentic splice sites by an order of magnitude.Citation3 A key assumption of this hypothesis is that the number and type of triloops in the database are representative of triloops formed in naturally occurring pre-mRNAs immediately upon transcription, which remains to be tested.
RNA tetraloops that promote inclusion of the MIR exon
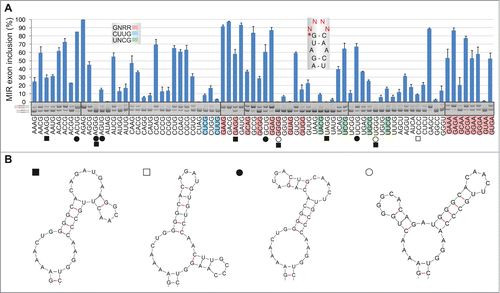
The most common terminal loops in natural RNAs have 4 nucleotides.Citation50 To test their exon-enhancing activities, we converted our terminal triloop mutants into tetraloops, including stable UNCG-, CUUG- and GNRA-type tetraloops that are preferred in nature,Citation50 and examined exon inclusion of mutated reporter constructs in the FGB mRNA. As with terminal triloops, GA-containing tetraloops gave the highest level of exon inclusion (). The GNRR-type tetraloops tended to be overrepresented among splice-proficient pre-mRNAs (median exon inclusion 58% vs 24%), whereas 2 CUYG constructs were found among splicing spoilers. Secondary structure predictions of each mutated transcript suggested that as many as 252/256 (98.4%) tetraloops would maintain the canonical structure of the MIR hairpin, including the cytosine bulge at exon position 18 and the internal AAA triloop. Stable alternative structures, which were predicted for 11 of the tested 76 FGB pre-mRNAs, lacked the cytosine bulge (); interestingly, these constructs were significantly associated with skipping of the MIR exon (32.9% vs 9.5%, P < 0.05; Mann-Whitney rank sum test). Similar to triloops, correlation between exon inclusion levels of tetraloop mutants and corresponding tetranucleotide frequencies in enhancers/silencers was highly significant (Table S1, cf. and S7A–C).
Figure 5. RNA tetraloops that promote and repress MIR exon splicing. (A) MIR exon inclusion levels of 76 constructs with the indicated tetraloops in COS7 cells. RNA products are to the left, merged gels are denoted by vertical lines. Error bars represent SDs from 2 transfections experiments. Stable tetraloops are colored as indicated. A default option of Mfold identified 11 transcripts (marked by symbols at the bottom) with stable alternative RNA secondary structures (shown in panel B); predicted structure of the remaining pre-mRNAs is shown in the inset of panel A.
Splicing activities of terminal tetraloops and stem identity
To determine the extent to which the tetraloop sequence can predict exon inclusion levels in the presence of a distinct stem, we created a library of random tetraloops that cap the TSL1 hairpin in SMN2 exon 7 (Fig. S4A). In the native context (TSL1/N; Fig. S7D), the TSL1 hairpin is formed by 6 Watson-Crick base-pairs terminating by the AGAC tetraloop.Citation21 The 5′ loop-closing base is uracil at exon position 6 of SMN2; in contrast, cytosine at this position in the splice-proficient SMN1 counterpart eliminates the loop-closing base-pairing, increases the loop size and shortens the stem to 5 base-pairs,Citation21 the size of the upper stem of the MIR hairpin (). Exon inclusion levels measured for 61 different TSL1/N tetraloops in SMN2 reporters showed significant correlation with frequencies of matching tetranucleotides in splicing enhancers (Fig. S7E) and silencers (Fig. S7F). When the native loop-closing base pair U-A in TSL1 was replaced by G-C (TSL1/H, ), exon inclusion levels increased but the correlation was preserved even in the less informative context of the MIR-onized hairpin. Thus, as for triloops, tetraloop-closing base pairs accounted for a significant fraction of stem-mediated splicing activities.
Figure 6. Tra2-induced activation of cryptic exons in FGB intron 1. (A) Activation of the MIR exon is promoted by Tra2. siRNAs targeting the indicated SR or SR-like proteins are at the top, spliced products to the right, FGB reporters are at the bottom. SC1, SC2, scrambled controls. MIR and T exons are denoted throughout as red and blue boxes, respectively. Their position in the pre-mRNA is shown in . SRSF1 and Tra2 immunoblots are shown in panels E and G; depletion levels of endogenous proteins were as described.Citation74,75 (B) T exon activation in cells overexpressing Tra2β. Expression plasmids are at the top; EV, empty vector DNA. (C) Nucleotide sequence of transcripts with T and MIR exons. Sequenced RT-PCR products are color coded as in panel B. (D) Downregulation of SRSF1 promotes MIR exon inclusion. Final concentration and sequence of SRSF1-targeting siRNAs (termed K and N) was as described.Citation75,82 (E) Western blot analysis with anti-SRSF1 antibodies. (1/4), a quarter of the lysate loaded to facilitate estimates of depleted proteins. (F) Distinct role of Tra2α and Tra2β residues R190 and Y/F193 in activation of adjacent exons. The upper panel shows relative positions of mutated residues in the indicated β-sheets of Tra2 RRMs, based on previously published alignments.Citation52,53 The relative abundance of cryptic exons in cells overexpressing wild-type and mutated Tra2 proteins is in the lower panel. Tra2 substitutions are shown at the top. Spliced products (shown to the right) are quantified at the bottom. Concentration of reporter and expression plasmid DNA was 250 and 500 ng/mL, respectively. (G) Immunoblotting of HEK293 cell lysates using antibodies against β-actin and Xpress. Tra2 mutations are at the top. C, no plasmid DNA control. (H) Dose-dependent activation of T- and MIR exons in cells expressing Tra2α R190A and Tra2β R190A. Final DNA concentrations of plasmid expressing Tra2α and Tra2β were 30, 100, 300 and 600 ng/mL, and 6, 20, 60 and 180 ng/mL, respectively.
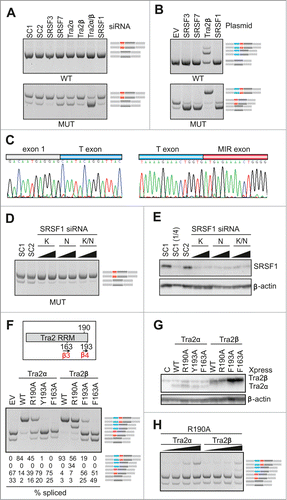
Figure 7. Tra2-induced cryptic exon activation is controlled by the MIR hairpin located >1.5 kb downstream. FGB and hybrid SMN2-MIR constructs were cotransfected with Tra2 expression plasmids and controls (top). The FGB pre-mRNA is shown in and the hybrid SMN2-FGB pre-mRNA in . Spliced products are shown schematically to the right; exons are color -coded as in the pre-mRNAs. Mutated splicing reporters are to the left; mutations were introduced in the terminal triloop (exon position 24–26)/bulge (exon position 18) of the MUT construct, as indicated. WT, wild construct without the 23A > G substitution; EV, empty vector; C, reporter-only controls.

Tra2-induced activation of cryptic exons in FGB intron 1
To identify ligands that control MIR exon inclusion and their interactions with the hairpin, we first examined MIR exon splicing in cells depleted of known splicing factors, including serine/arginine-rich (SR) proteins as important regulators of exon inclusion.Citation51 Screening with siRNAs targeting >20 factors (data not shown) revealed significant exon skipping in cells depleted of the human homolog of Drosophila transporter (Tra2) and in cells transfected with siRNAs targeting SR protein SRSF7 (also known as 9G8; ). Interestingly, overexpression of Tra2, but no other tested splicing factors or controls, activated an upstream exon (termed T, for Tra2-activated; ). The T exon contained optimal Tra2β binding sites Citation52,53 and was about twice as long as the MIR exon (Fig. S8). In contrast to Tra2, down-regulation of SR proteins SRSF3 (SRp20) and SRSF1 (SF2/ASF) promoted exon inclusion while their overexpression increased exon skipping (). For a subset of SR proteins, we also found significant correlation between MIR exon inclusion and scores for their putative exonic binding sites, including SRSF1 and SRSF2 (Fig. S9).
The RNA recognition motif (RRM) of Tra2β binds GAA- and CAA-containing sequences through hydrogen-bond formation with several amino acids as well as through stacking interactions mediated by unusually aligned rings on the β-sheet surface.Citation52,53 In a solution structure of RRM-Tra2β/(GAA)2, the protein contacted the A3G4A5A6 tetramer through arginine 190 (A3G4) and phenylalanines 193 (A5) and 163 (A5A6).Citation52 Cotransfection of our FGB MUT reporter with expression plasmids encoding Tra2α or Tra2β that had R190A, Y/F193A and F163A substitutions revealed that exon T activation was completely abrogated by mutation F163A (), indicating that this β3 residue is critical for T exon inclusion in each protein. However, mutations of β4 amino acids in the 2 proteins produced a distinct splicing pattern. Most notably, Tra2β R190A was capable of activating transcripts that contained the T exon but lacked the MIR exon, whereas Tra2α R190A promoted only transcripts with both exons. This was further supported by addition of increasing amounts of each R190A plasmid to cell cultures to exclude that the distinct activation of adjacent exons is due to a higher expression of Tra2β over Tra2α and by immunoblots confirming expression of each mutated construct (). We conclude that MIR and T exons have distinct requirements for the C-terminal extension of Tra2 RRMs.
To test if the triloop identity in the MIR hairpin can influence T exon activation, we transfected WT and MUT versions of FGB and hybrid SMN2-FGB reporter plasmids into cells overexpressing Tra2α and/or Tra2β and compared inclusion levels of their mid-exons (). Interestingly, Tra2-induced T-exon activation was observed in the WT FGB clone but ‘T-exon only’ transcripts disappeared in the context of the AAG and other tested triloops, except for ACG. Even more remarkably, the Tra2-induced activation of the T exon was completely abrogated in FGB by deletion of the bulged cytosine at position 18 of the MIR exon and was also reduced by substitutions of this residue. The same changes also diminished activation of SMN2-FGB exon 7 ().
Collectively, these data identify proteins that control inclusion of the MIR exon in mature transcripts and reveal a Tra2-activated cryptic exon >1.5 kbp upstream whose selection was dramatically affected by single-stranded regions of the MIR hairpin. Finally, they show a distinct role of C-terminal residues in Tra2α and Tra2β RRMs in activation of the 2 adjacent exons.
Tra2β, SRSF1 and hnRNPs A1 and H bind to hybrid FGB RNAs
To identify proteins that bind to the MIR hairpin, we subcloned a 99-nt amplicon containing the MIR exon and flanking sequences into a PY7 reporter that is efficiently spliced in nuclear extractsCitation54 (). Similar to the canonical FGB minigene (), the MUT version of the hybrid PY7-FGB reporter subcloned into pCR3.1 was spliced more efficiently in cellulo than the WT counterpart (). In vitro splicing of WT and MUT PY7-FGB transcripts showed that both alleles were spliced at optimized conditions at 2 mM Mg2+/60 mM KCl, but the MUT PY7-FGB construct was spliced more efficiently than the WT PY7-FGB hybrid (). Additional 11 mutated FGB constructs that were subcloned into PY7 revealed positive correlation between the percentage of splicing in nuclear extracts and MIR exon inclusion levels upon transfection into HEK293 cells (r = 0.55, P < 0.05, t-test, data not shown), indicating that the MIR hairpin contributes significantly to splicing efficiency of the hybrid pre-mRNA substrate.
Figure 8. Identification of proteins that bind to PY7-FGB transcripts (A) Schematics of hybrid PY7-FGB reporter construct for in vitro splicing. PY7 Citation54 and MIR () exons are represented by white and black rectangles (to scale at the top), respectively. The A > G mutation is shown by an asterisk. Cloning primers are in Table S3. (B) Splicing of pCR-PY7-FGB hybrid constructs in HEK293 cells. Spliced products are shown to the right. (C) In vitro splicing of WT and MUT PY7-FGB hybrids in HeLa nuclear extracts. 32P-UTP labeled RNAs were incubated in HeLa nuclear extracts for 1 hr. Concentrations of Mg2+ and KCl are at the top, spliced products are to the right. The size of the pre-mRNA/mRNA was 327/201 bp. (D, E) UV-crosslinking of proteins to 32P-UTP-labeled WT and MUT transcripts and their mutated versions. Asterisks correpond to proteins identified by immunoprecipitation in panel F. Panel E shows differential crosslinking to 32P-CTP-labeled bulge mutants at MIR exon position 18. (F) Immunoprecipitation of the crosslinked proteins with the indicated antibodies.
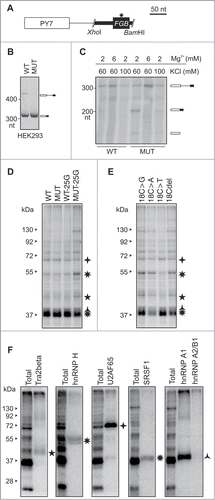
Disappointingly, UV-RNA crosslinking of PY7-FGB reporters under splicing conditions did not reveal a distinct pattern of bound proteins between WT and MUT transcripts with either 32P-UTP or 32P-CTP (lane 1 and 2, , data not shown). As compared to controls, however, we observed lower signal intensity from ∼38–40 kD bands and stronger signal from ∼45 and ∼55 kD proteins for transcripts that contained an A > G substitution at exon position 25 of the MUT construct (lane 4, , data not shown). This AAG > AGG triloop substitution diminished inclusion of the MIR exon (, Table S4). Proteins of the same size range also differentially bound transcripts derived from cytosine bulge mutants (). Immunoprecipitation of crosslinked proteins revealed that apart from a control U2AF65/35, the transcript was bound by Tra2β, SRSF1 and heterogenenous ribonucleoproteins (hnRNPs) H and A1 ().
Discussion
Using a novel, single-hairpin exon selection model, we have shown that short and exposed RNA loops are key components of the structural splicing code and account for most exon-repressing/-promoting activities of splicing silencers/enhancers, demonstrating a pervasive role of the most common RNA structural motifs in exon recognition. Our results strongly indicate that these structures act as principal exon selection modules (, Fig. S7) that control activation of adjacent exons (), with exposed loops and bulges providing key exon recognition contacts. They also show that intramolecular base-pairing contributes significantly to the mysterious context dependency of auxiliary splicing signals ().
The observed correlation between free energies of MIR hairpin mutants and their exon inclusion levels () is consistent with a higher exon inclusion by stem-loop destabilization previously noticed for MAPT,Citation55 SMN2,Citation17 FN1,Citation13 PSEN2 Citation56 and TNNT2 Citation57 and also with decreased stability of secondary structures of exonized Alus as compared to their non-exonized counterparts.Citation42 The size of the MIR hairpin corresponds to the most common range of human RNA folds predicted using genome-wide approaches, with many thousands estimated to exist in introns.Citation58 The MIR upper stem shows similarities to structures previously found to promote exon inclusion, including a 4-nt upper stem interrupted by a single uridine bulge and terminating in a tetraloop in PS2 exon 5 Citation56 or a 5-nt upper stem with a single adenine bulge, terminating in a well-characterized purine-rich ESE in FN1 exon ‘EDA’.Citation13 The MIR hairpin is also similar to the IRE, a regulatory conserved stem-loop increasingly found in mRNAs encoding important proteins with diverse funct-ions Citation44,45,59 (Fig. S10). IREs also have a 5-nt upper stem and a flexible cytosine-type bulge, which was shown to bind domain 4 of iron regulatory protein 1.Citation60,61
Secondary structure predictions are generally more accurate for inverted repeats than average pre-mRNAs. Inverted repeats are significantly enriched in eukaryotic genomes, suggesting that they are functionally important.Citation43,62,63 The majority of human inverted repeats are derived from SINEs.Citation43 These elements should therefore provide the most attractive structural models of mammalian pre-mRNA splicing, alleviating uncertainties in accuracy of RNA secondary structure predictions encountered with typical pre-mRNAs. This notion is consistent with a low fraction of predicted alternative foldings with a similar energy, both for triloop (8/64, 12.5%) and tetraloop (11/76, 14.5%) FGB mutants. Such models should also facilitate identification of RNA-protein interactions across their exonized sequences. Proteins that influenced MIR exon inclusion () may be engaged in interactions across the hairpin, perhaps similar to a cooperative assembly of Tra2/SRSF7 on a purine-rich enhancer in Drosophila doublesex.Citation64 also suggests that Tra2 is a stronger splicing activator than other SR protein family members, confirming earlier observations Citation65; in addition, optimal binding sites for Tra2β Citation52,53 were enriched among high-inclusion triloops (). Future studies should therefore explore the role of proteins that recruit Tra2 to MIR RNA, including hnRNP G, which was recently shown to contact diadenosines in single-stranded regions.Citation66
As shown for SMN2 exon 7,Citation67 SSOs that bind exonic sequences and promote their inclusion in mRNA are relatively uncommon. For example, exon-promoting antisense targets were found only for 2/10 (20%) tested oligos in the initial walk across SMN2 exon 7.Citation67 By analogy, we found only 2 intron retention-repressing SSOs among 20 (10%) initially tested across INS intron 1.Citation68 Our study revealed that exon-promoting and -repressing effect of the SSO targeting a more accessible side of the MIR hairpin is controlled by the flanking triloop sequence (). This observation has important implications for potential clinical use of antisense reagents and for personalized medicine because even optimized SSOs may produce unintended outcomes if the target is surrounded natural variants in the vicinity that have not been tested. Also, distance between such natural variants and the antisense target might be much longer than that observed for the MIR hairpin, highlighting the requirement for structural design of SSOs.
Finally, clone resources developed in this work will be helpful for systematic studies that investigate ligand preferences and their requirements for splicing outcomes. Reference exon inclusion levels for tri- and tetra-loop sequences capping a stable stem (, S2A and Table S4) will be useful for studying auxiliary splicing motifs in other hairpins and their long-range interactions. Identification of distant contacts between the MIR element and upstream exon ()Citation27,69,70 will facilitate our understanding of tertiary interactions in splicing regulation. Splicing-proficient triloops were rich in diadenosines () and could thus be engaged in more extensive RNA tertiary interactions than splicing spoilers. Diadenosines are frequently involved in formation of the most common 3 dimensional motifs.Citation71 This hypothesis is also supported by the observed promotion of the MIR exon inclusion by the 5′ stem SSO, which required AA-containing triloops (). As shown for group II self-splicing introns,Citation72 adoption of complex tertiary structures could remain very important also for splicing catalysis of their likely evolutionary descendants, i.e., human introns. Our model should therefore prove useful for evaluating the relative importance of diadenosine binding proteins Citation66 and intramolecular tertiary contacts in RNA processing. Ultimately, high resolution structures will help us understand how hairpins facilitate exon selection and alternative splicing, which should substantially improve future iterations of exonic and intronic splicing code, accurate prediction of phenotypic consequences of disease-causing splicing mutations and their correction by antisense strategies.
Materials and Methods
Splicing reporter constructs
To facilitate mutagenesis, the BamHI-XhoI fragment of pcDNA-FGB-V5-His-TOPO (a gift of Dr Ryan Davis, University of Otago)Citation46 was subcloned into pCR3.1 (Invitrogen). The HindIII site was removed from the pCR3.1 polylinker using a NheI/BamHI digest and Klenow enzyme treatment. Mutagenesis of the resulting minigene (termed pCR3.1-FGB) was carried with 673-bp products amplified by cloning primers C-F and C-R (, Table S3). Mutagenic PCR primers for tri- and tetra-loops were degenerated at each loop position. PCR products were digested by KpnI/HindIII, subcloned into pCR3.1-FGB and propagated in a E. coli strain DH5α. Each plasmid DNA was completely sequenced using primer S (Table S3) to confirm desired mutations and exclude PCR-introduced errors. Reporter constructs with and without the disease-causing A > G substitution are referred as MUT and WT, respectively, throughout the text.
Hybrid reporter constructs SMN2-FGB, F9-FGB and L1CAM-FGB were prepared by introducing a 24-bp FGB hairpin into central exons of previously published minigenes.Citation17,73 The SMN2 minigeneCitation17 was a gift from Drs Ravi and Nathalia Singh, Iowa University. The F9 minigene had a substitution of the branch point adenineCitation73 to obtain similar levels of exon inclusion and skipping. The L1CAM minigeneCitation73 contained the MIR hairpin at 2 different positions of the central exon to create reporters L1CAM(r1)-FGB and L1CAM(r2)-FGB. Cloning primers for hybrid reporters were SMN2-FGB-F and SMN2-FGB-R, F9-FGB-F and F9-FGB-R, L1CAM(r1)-FGB-F1 and L1CAM(r1)-FGB-R1, and L1CAM(r2)-FGB-F2 and L1CAM(r2)-FGB-R2, while triloop mutants were obtained using primers SMN2-NNN, F9-NNN, L1CAM(r1)-NNN and L1CAM(r2)-NNN (Table S3).
To prepare constructs for in vitro splicing and UV crosslinking (PY7-FGB), we amplified a 99-bp segment encompassing the cryptic exon and flanking MIR sequences using primers PY7-FGB-F-XhoI and PY7-FGB-R-BamHI (Table S3). PCR products were digested with XhoI/BamHI and subcloned into an in vitro splicing-proficient clone PY7Citation54 (a gift from Professor Christopher Smith, University of Cambridge). Minigene pCR-PY7-FGB (pCR3.1, LifeTechnologies) for in vivo splicing was prepared with primers pCR-PY7F-NheI and PY7-FGB-R-BamHI (Table S3) using the PY7-FGB plasmid DNA as a template. Spliced products were amplified using primers PL1 and PL2 (Table S3).
Mammalian expression plasmids encoding Tra2 proteins were obtained by PCR employing the Pfu polymerase, primers shown in Table S3 and cDNA as a template. PCR products were first cloned between BamHI and NotI sites of pcDNA3.1-HisC (Invitrogen). To increase the intron-mediated expression of Tra2 proteins and move the Xpress tag to the N-terminal, plasmids were digested using NheI and NotI and inserted into the pCI-neo vector (Promega). Mutated Tra2 versions were prepared by overlap-extension PCR using wild-type pCI-neo Tra2 DNA and mutagenic primers Tra2α-F163A, Tra2β-F163A, Tra2α-Y193A, Tra2β-F193A, Tra2α-R190A and Tra2β-R190A (Table S3). All clones were sequenced to confirm the identity of intended mutations and exclude undesired changes.
Cell cultures and transfections
HEK293, HeLa, HepG2 and COS7 cell lines were cultured under standard conditions as described previously.Citation74 Transient transfections were performed in 12- or 24-well plates using FuGENE (Roche) according to manufacturer's recommendations. Final concentration of each plasmid DNA in the culture medium was 500 ng/mL. Cells were harvested 24 hours post-transfection for RNA extraction, as described.Citation74
Detection and measurements of spliced products
Total RNA was isolated using TRI-reagent (Ambion) according to manufacturer's recommendations and reverse-transcribed using the Moloney murine leukemia virus reverse transcriptase (Invitrogen) and the oligo-d(T) primer, essentially as described.Citation74 For RT-PCR, we used vector primers PL1 and PL2 (Table S3). To amplify spliced products of SMN2, we used primers P1 and P2, as reported previously.Citation47 Amplifications were for 28 cycles. PCR products were separated on polyacrylamide gels and exon inclusion levels were measured as described.Citation39,74
Splice-switching oligonucleotides (SSOs) and small interfering RNAs (siRNAs)
The WT and MUT reporters were individually cotransfected with SSOs targeting the loop, 5'stem and 3'stem in the MIR hairpin. SSOs (MWG Biotech) were 2′-O-methyl-modified at each sugar residue and uniformly labeled with phosphorothioates. Oligoribonucleotides targeting loops (SSO-AAG and SSO-GAU) and the 5′ and 3′ stem are in Table S3. Spliced products were detected and measured as described above. Sequences of siRNAs targeting splicing factors were described previously.Citation75
Western blot analysis
Cells (mock)-transfected with siRNA and expression plasmids were washed twice with PBS and immediately lysed in the RIPA buffer (New England Biolabs).Citation75 Protein lysates were loaded on to 10% SDS-PAGE and transferred to polyvinyl difluoride membranes (Amersham) using electroblotting. Membranes were incubated with the following antibodies: Xpress (Invitrogen, R910-25), SFRS7 (9G8; Sigma, SAB1101226), Tra2β (Abcam, ab31353), SRSF1 (Abcam, ab38017), actin (Abcam, ab37063) and horseradish peroxidase-conjugated secondary antibodies. Proteins were detected using the Immun-Star WesternC kit (Biorad).
In vitro splicing assay
The PY7-FGB substrates were transcribed using the MAXIscript kit (Ambion) with BamHI-linearized plasmids as templates. In vitro transcription reaction contained 1x transcription buffer, 1 μg of linear DNA, 60 μCi of [α-32P]UTP or [α-32P]CTP (800 Ci/mmol, PerkinElmer), 0.5 mM ATP, 0.5 mM CTP or 0.5 mM of UTP, 0.1 mM GTP, 0.4 mM m7G(5′)ppp(5′)G, 1 U/μL of ribonuclease inhibitor (Promega), and 2U/μL of SP6 RNA polymerase. Transcription reactions were incubated at 37°C for 1 and 3 hrs. Internally labeled transcripts were gel purified using PAGE.
Each splicing reaction contained 0.1 nM of RNA (∼10ng), 30% of HeLa nuclear extract (4C Biotech), 1 mM DTT, 60 mM KCl, 12 mM Hepes–KOH, pH 7.9, 0.5 mM ATP, 20 mM creatine phosphate, 2 mM MgCl2, 0.25 U/μL of ribonuclease inhibitor, and 2.6 % of polyvinylalcohol. Following incubation at 30°C for 2 hours, 100-μL splicing reactions were digested with proteinase K (100 mM Tris, pH 7.5, 1% SDS, 150 mM NaCl, 10 mM EDTA, 0.05 μg/μl tRNA, and 0.25 mg/mL proteinase K) at 37°C for 15 min, extracted with phenol and chloroform, precipitated with ethanol and separated using PAGE. Gels were dried and exposed to a Typhoon PhosphorImager (Molecular Dynamics).
UV-RNA crosslinking and immunoprecipitation
Binding reactions were the same as in vitro splicing, except for omission of polyvinylalcohol. Reactions were irradiated with 254-nm UV light (Ultralum) on ice for 10 min and treated with a mixture of RNase A (0.05 U/1 μl) and RNase T1 (2U/μl; Ambion) at 30°C for 20 min. An equal volume of 2x Laemmli buffer was added and samples were boiled for 5 min before loading on to 10% SDS-PAGEs. The gels were fixed, dried and exposed to PhosphorImager screens. For immunoprecipitation, 50 μl of cross-linked reactions were mixed with 400 μl of the IP buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM EDTA, 0.05% NP-40), 50 μl of protein G-coupled Sepharose beads and the indicated antibodies. Antibodies were purchased from Sigma (U2AF65, hnRNP A2/B1), ABcam (Tra2β, SRSF1) or were a generous gift from Professors Douglas Black (hnRNP H), UCLA, and Gideon Dreyfuss (hnRNP A1), University of Pennsylvania. After incubation at 4°C for 2 hrs, the samples were washed 5 times with the IP buffer. Beads were resuspended in 20 ul of the Laemmli buffer, boiled for 5 min and loaded on a 10% SDS-PAGE.
Tri- and tetra-nucleotide frequencies in splicing silencers and enhancers
These data were computed for previously defined RESCUE exonic splicing enhancers (ESEs),Citation4 FAS exonic splicing silencers (FAS-ESSs),Citation5 putative exonic splicing enhancers and silencers (PESSs and PESEs),Citation76 exon identity elements (EIEs),Citation7 QUEPASA ESEs/ESSs,Citation9 neighborhood inference (NI) ESEs/ESSsCitation77 and exon regulatory sequences (ERSs)Citation6 (Table S1). Frequencies of tri- and tetra-nucleotides in these motifs were computed using Microsoft Excel functions or VBA scripts (available on request).
Circular dichroism (CD) and nuclear magnetic resonance spectroscopy (NMR)
RNA oligo for CD and NMR were purchased from Thermo Scientific, deprotected according to manufacturer's instructions, lyophilised and stored at −20°C. Stock solutions were prepared from the desalted, lyophilised samples by resuspending in milliQ water or KCl buffer (100 mM KCl, 10mM K2HPO4/KH2PO4, pH 7.0, milliQ water) to a final concentration of 2–4 μM.
CD spectra were acquired using a PiStar-180 spectrophotometer (Applied Photophysics Ltd, Surrey, UK), equipped with a LTD6G circulating water bath (Grant Instruments, UK) and thermoelectric temperature controller (Melcor, USA). Samples were heated in the cell to 95°C for 15 minutes and samples were then annealed by cooling to room temperature over a period of 4 h. CD spectra were recorded over a wavelength range of 215–340 nm using a 1 cm path length strain-free quartz cuvette and at the temperatures indicated. Data points were recorded at 1 nm intervals. A bandwidth of 3 nm was used and 5,000 counts acquired at each point with adaptive sampling enabled. Each trace is shown as the mean of 3 scans (±SD ). CD temperature ramps were acquired at 265 nm corresponding to the band maxima of the folded hairpin structure. Ranges between 5 and 95°C were used, with points acquired at 0.5°C intervals with a 120–180 second time-step between 0.5°C increments. Points were acquired with 10,000 counts and adaptive sampling enabled. Heating and cooling studies were compared to check for hysteresis and overall reversibility.
NMR spectra (1H) were collected at 800 MHz using a Bruker Avance III spectrometer with a triple resonance cryoprobe. Standard Bruker acquisition parameters were used. Data were collected using Topspin (v. 3.0) and processed in CCPN Analysis (v. 2.1).
Triloop database
Triloop frequencies of previously determined RNA stem-loop structures were determined for a total of 823 triloops in 123 small subunit rRNAs, 223 large subunit rRNAs, 309 5S rRNAs, 484 tRNAs, 91 signal recognition particles, 16 RNase P RNAs, 100 group I introns and 3 group II introns Citation48 (and Brent Znosko, personal communication). The triloop database was considered to be representative of triloops found in naturally occurring RNAs.Citation48
RNA secondary structure predictions
Overlapping segments of FGB intron 1 and all mutated transcripts were used as an input into RNAstructure (v. 5.2, default options),Citation78 which accommodates a dynamic programming algorithm on the principle of minimizing free energy.Citation79 We used the same input also for Mfold Citation80 and RNAfold from the Vienna RNA suite Citation81 to confirm secondary structure predictions.
Statistical analysis
Pearson correlation coefficients and Mann-Whitney rank sum tests were computed using SigmaStat (v. 3.5; Systat Software). Kruskall-Wallis and Friedman tests were computed using SigmaStat or Stat200 (Biosoft, UK). Correlation coefficients were compared using Fisher r-to-z transformations. Weighted contribution of each base (N) to cryptic splicing (f, ) for n triloops was calculated as
where k is the mean exon inclusion level of 2 independent transfections.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Douglas Black, UCLA, Ryan Davis, University of Otago, Javier Cáceres, University of Edinburgh, Gideon Dreyfuss, University of Pennsylvania, Adrian Krainer, Cold Spring Harbor, Gavin Screaton, Imperial College, Ravindra and Natalia Singh, Iowa University, and Christopher Smith, University of Cambridge, for reagents. We also thank Huw Williams, Nottingham Center for Biomolecular Sciences, for technical support and Ian Eperon, University of Leicester, and Sam Griffiths-Jones, University of Manchester, for manuscript comments.
Supplemental Materials
Download PDF (1.7 MB)Funding
This work was supported by grants from JDRF and LLR to IV and from VEGA to J.K.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell 2009; 136:701-18; PMID:19239890; http://dx.doi.org/10.1016/j.cell.2009.02.009
- Burge CB, Tuschl T, Sharp PA. Splicing of precursors to mRNAs by the spliceosome. In: Gesteland RF, Cech TR, F. AJ, eds. The RNA World. New York: Cold Spring Harbor Laboratory Press, 1999:525-60.
- Fairbrother WG, Chasin LA. Human genomic sequences that inhibit splicing. Mol Cell Biol 2000; 20:6816-25; PMID:10958678; http://dx.doi.org/10.1128/MCB.20.18.6816-6825.2000
- Fairbrother WG, Yeh RF, Sharp PA, Burge CB. Predictive identification of exonic splicing enhancers in human genes. Science 2002; 297:1007-13; PMID:12114529; http://dx.doi.org/10.1126/science.1073774
- Wang Z, Rolish ME, Yeo G, Tung V, Mawson M, Burge CB. Systematic identification and analysis of exonic splicing silencers. Cell 2004; 119:831-45; PMID:15607979; http://dx.doi.org/10.1016/j.cell.2004.11.010
- Goren A, Ram O, Amit M, Keren H, Lev-Maor G, Vig I, Pupko T, Ast G. Comparative analysis identifies exonic splicing regulatory sequences–The complex definition of enhancers and silencers. Mol Cell 2006; 22:769-81; PMID:16793546; http://dx.doi.org/10.1016/j.molcel.2006.05.008
- Zhang C, Li WH, Krainer AR, Zhang MQ. RNA landscape of evolution for optimal exon and intron discrimination. Proc Natl Acad Sci USA 2008; 105:5797-802; PMID:18391195; http://dx.doi.org/10.1073/pnas.0801692105
- Chasin LA. Searching for splicing motifs. Adv Exp Med Biol 2007; 623:85-106; PMID:18380342; http://dx.doi.org/10.1007/978-0-387-77374-2_6
- Ke S, Shang S, Kalachikov SM, Morozova I, Yu L, Russo JJ, Ju J, Chasin LA. Quantitative evaluation of all hexamers as exonic splicing elements. Genome Res 2011; 21; PMID:21659425; http://dx.doi.org/10.1101/gr.119628.110
- Hiller M, Zhang Z, Backofen R, Stamm S. Pre-mRNA secondary structures influence exon recognition. PLoS Genet 2007; 3:e204; PMID:18020710; http://dx.doi.org/10.1371/journal.pgen.0030204
- Eperon LP, Graham IR, Griffiths AD, Eperon IC. Effects of RNA secondary structure on alternative splicing of pre-mRNA: is folding limited to a region behind the transcribing RNA polymerase? Cell 1988; 54:393-401; PMID:2840206; http://dx.doi.org/10.1016/0092-8674(88)90202-4
- Blanchette M, Chabot B. A highly stable duplex structure sequesters the 5′ splice site region of hnRNP A1 alternative exon 7B. RNA 1997; 3:405-19; PMID:9085847
- Muro AF, Caputi M, Pariyarath R, Pagani F, Buratti E, Baralle FE. Regulation of fibronectin EDA exon alternative splicing: possible role of RNA secondary structure for enhancer display. Mol Cell Biol 1999; 19:2657-71; PMID:10082532
- Chen Y, Stephan W. Compensatory evolution of a precursor messenger RNA secondary structure in the Drosophila melanogaster Adh gene. Proc Natl Acad Sci USA 2003; 100:11499-504; PMID:12972637
- Buratti E, Baralle FE. Influence of RNA secondary structure on the pre-mRNA splicing process. Mol Cell Biol 2004; 24:10505-14; PMID:15572659; http://dx.doi.org/10.1128/MCB.24.24.10505-10514.2004
- Graveley BR. Mutually exclusive splicing of the insect Dscam pre-mRNA directed by competing intronic RNA secondary structures. Cell 2005; 123:65-73; PMID:16213213; http://dx.doi.org/10.1016/j.cell.2005.07.028
- Singh NN, Singh RN, Androphy EJ. Modulating role of RNA structure in alternative splicing of a critical exon in the spinal muscular atrophy genes. Nucleic Acids Res 2007; 35:371-89; PMID:17170000; http://dx.doi.org/10.1093/nar/gkl1050
- Barash Y, Calarco JA, Gao W, Pan Q, Wang X, Shai O, Blencowe BJ, Frey BJ. Deciphering the splicing code. Nature 2010; 465:53-9; PMID:20445623; http://dx.doi.org/10.1038/nature09000
- Jin Y, Yang Y, Zhang P. New insights into RNA secondary structure in the alternative splicing of pre-mRNAs. RNA Biol 2010; 8:450-7; PMID:21558794; http://dx.doi.org/10.4161/rna.8.3.15388
- Warf MB, Berglund JA. Role of RNA structure in regulating pre-mRNA splicing. Trends Biochem Sci 2010; 35:169-78; PMID:19959365; http://dx.doi.org/10.1016/j.tibs.2009.10.004
- Liu W, Zhou Y, Hu Z, Sun T, Denise A, Fu XD, Zhang Y. Regulation of splicing enhancer activities by RNA secondary structures. FEBS Lett 2010; 584:4401-7; PMID:20888818; http://dx.doi.org/10.1016/j.febslet.2010.09.039
- McManus CJ, Graveley BR. RNA structure and the mechanisms of alternative splicing. Curr Opin Genet Dev 2011; 21:373-9; PMID:21530232; http://dx.doi.org/10.1016/j.gde.2011.04.001
- Gahura O, Hammann C, Valentova A, Puta F, Folk P. Secondary structure is required for 3' splice site recognition in yeast. Nucleic Acids Res 2011; 39:9759-67; PMID:21893588; http://dx.doi.org/10.1093/nar/gkr662
- Shepard PJ, Hertel KJ. Conserved RNA secondary structures promote alternative splicing. RNA 2008; 14:1463-9; PMID:18579871; http://dx.doi.org/10.1261/rna.1069408
- Zhang J, Kuo CC, Chen L. GC content around splice sites affects splicing through pre-mRNA secondary structures. BMC Genomics 2011; 12:90; PMID:21281513; http://dx.doi.org/10.1186/1471-2164-12-90
- Miriami E, Margalit H, Sperling R. Conserved sequence elements associated with exon skipping. Nucleic Acids Res 2003; 31:1974-83; PMID:12655015; http://dx.doi.org/10.1093/nar/gkg279
- Raker VA, Mironov AA, Gelfand MS, Pervouchine DD. Modulation of alternative splicing by long-range RNA structures in Drosophila. Nucleic Acids Res 2009; 37:4533-44; PMID:19465384; http://dx.doi.org/10.1093/nar/gkp407
- Sathirapongsasuti JF, Sathira N, Suzuki Y, Huttenhower C, Sugano S. Ultraconserved cDNA segments in the human transcriptome exhibit resistance to folding and implicate function in translation and alternative splicing. Nucleic Acids Res 2010; 39:1967-79; PMID:21062826; http://dx.doi.org/10.1093/nar/gkq949
- Jurka J, Kapitonov VV, Kohany O, Jurka MV. Repetitive sequences in complex genomes: structure and evolution. Ann Rev Genomics Hum Genet 2007; 8:241-59; PMID:17506661; http://dx.doi.org/10.1146/annurev.genom.8.080706.092416
- Feschotte C, Jiang N, Wessler SR. Plant transposable elements: where genetics meets genomics. Nat Rev Genet 2002; 3:329-41; PMID:11988759; http://dx.doi.org/10.1038/nrg793
- Makalowski W, Mitchell GA, Labuda D. Alu sequences in the coding regions of mRNA: a source of protein variability. Trends Genet 1994; 10:188-93; PMID:8073532; http://dx.doi.org/10.1016/0168-9525(94)90254-2
- Nekrutenko A, Li WH. Transposable elements are found in a large number of human protein-coding genes. Trends Genet 2001; 17:619-21; PMID:11672845; http://dx.doi.org/10.1016/S0168-9525(01)02445-3
- Sorek R, Ast G, Graur D. Alu-containing exons are alternatively spliced. Genome Res 2002; 12:1060-7; PMID:12097342; http://dx.doi.org/10.1101/gr.229302
- Sela N, Mersch B, Gal-Mark N, Lev-Maor G, Hotz-Wagenblatt A, Ast G. Comparative analysis of transposed element insertion within human and mouse genomes reveals Alu's unique role in shaping the human transcriptome. Genome Biol 2007; 8:R127; PMID:17594509
- Krull M, Petrusma M, Makalowski W, Brosius J, Schmitz J. Functional persistence of exonized mammalian-wide interspersed repeat elements (MIRs). Genome Res 2007; 17:1139-45; PMID:17623809; http://dx.doi.org/10.1101/gr.6320607
- Vorechovsky I. Transposable elements in disease-associated cryptic exons. Hum Genet 2010; 127:135-54; PMID:19823873; http://dx.doi.org/10.1007/s00439-009-0752-4
- Lev-Maor G, Sorek R, Shomron N, Ast G. The birth of an alternatively spliced exon: 3′ splice-site selection in Alu exons. Science 2003; 300:1288-91; PMID:12764196; http://dx.doi.org/10.1126/science.1082588
- Sorek R, Lev-Maor G, Reznik M, Dagan T, Belinky F, Graur D, Ast G. Minimal conditions for exonization of intronic sequences: 5′ splice site formation in Alu exons. Mol Cell 2004; 14:221-31; PMID:15099521; http://dx.doi.org/10.1016/S1097-2765(04)00181-9
- Lei H, Vorechovsky I. Identification of splicing silencers and enhancers in sense Alus: a role for pseudo-acceptors in splice site repression. Mol Cell Biol 2005; 25:6912-20; PMID:16055705; http://dx.doi.org/10.1128/MCB.25.16.6912-6920.2005
- Lei H, Day INM, Vorechovsky I. Exonization of AluYa5 in the human ACE gene requires mutations in both 3′ and 5′ splice sites and is facilitated by a conserved splicing enhancer. Nucleic Acids Res 2005; 33:3897-906; PMID:16027113; http://dx.doi.org/10.1093/nar/gki707
- Lev-Maor G, Ram O, Kim E, Sela N, Goren A, Levanon EY, Ast G. Intronic Alus influence alternative splicing. PLoS Genet 2008; 4:e1000204; PMID:18818740; http://dx.doi.org/10.1371/journal.pgen.1000204
- Schwartz S, Gal-Mark N, Kfir N, Oren R, Kim E, Ast G. Alu exonization events reveal features required for precise recognition of exons by the splicing machinery. PLoS Comput Biol 2009; 5:e1000300; PMID:19266014; http://dx.doi.org/10.1371/journal.pcbi.1000300
- Svoboda P, Di Cara A. Hairpin RNA: a secondary structure of primary importance. Cell Mol Life Sci 2006; 63:901-8; PMID:16568238; http://dx.doi.org/10.1007/s00018-005-5558-5
- Hentze MW, Caughman SW, Rouault TA, Barriocanal JG, Dancis A, Harford JB, Klausner RD. Identification of the iron-responsive element for the translational regulation of human ferritin mRNA. Science 1987; 238:1570-3; PMID:3685996; http://dx.doi.org/10.1126/science.3685996
- Piccinelli P, Samuelsson T. Evolution of the iron-responsive element. RNA 2007; 13:952-66; PMID:17513696; http://dx.doi.org/10.1261/rna.464807
- Davis RL, Homer VM, George PM, Brennan SO. A deep intronic mutation in FGB creates a consensus exonic splicing enhancer motif that results in afibrinogenemia caused by aberrant mRNA splicing, which can be corrected in vitro with antisense oligonucleotide treatment. Hum Mutat 2008; 30:221-7; PMID:18853456; http://dx.doi.org/10.1002/humu.20839
- Singh NK, Singh NN, Androphy EJ, Singh RN. Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol 2006; 26:1333-46; PMID:16449646; http://dx.doi.org/10.1128/MCB.26.4.1333-1346.2006
- Thulasi P, Pandya LK, Znosko BM. Thermodynamic characterization of RNA triloops. Biochemistry (Mosc) 2010; 49:9058-62; PMID:20843054; http://dx.doi.org/10.1021/bi101164s
- Yeakley JM, Morfin JP, Rosenfeld MG, Fu XD. A complex of nuclear proteins mediates SR protein binding to a purine-rich splicing enhancer. Proc Natl Acad Sci USA 1996; 93:7582-7; PMID:8755518
- Woese CR, Winker S, Gutell RR. Architecture of ribosomal RNA: constraints on the sequence of “tetra-loops”. Proc Natl Acad Sci USA 1990; 87:8467-71; PMID:2236056
- Zhong X-Y, Wang P, Han J, Rosenfeld MG, Fu X-D. SR proteins in vertical integration of gene expresion from transcription to RNA processing to translation. Mol Cell 2009; 35:1-10; PMID:19595711; http://dx.doi.org/10.1016/j.molcel.2009.06.016
- Tsuda K, Someya T, Kuwasako K, Takahashi M, He F, Unzai S, Inoue M, Harada T, Watanabe S, Terada T, et al. Structural basis for the dual RNA-recognition modes of human Tra2β RRM. Nucleic Acids Res 2010; 39:1538-53; PMID:20926394; http://dx.doi.org/10.1093/nar/gkq854
- Clery A, Jayne S, Benderska N, Dominguez C, Stamm S, Allain FH. Molecular basis of purine-rich RNA recognition by the human SR-like protein Tra2β1. Nat Struct Mol Biol 2011; 18:443-50; PMID:21399644; http://dx.doi.org/10.1038/nsmb.2001
- Deirdre A, Scadden J, Smith CW. Interactions between the terminal bases of mammalian introns are retained in inosine-containing pre-mRNAs. EMBO J 1995; 14:3236-46; PMID:7621835
- Donahue CP, Muratore C, Wu JY, Kosik KS, Wolfe MS. Stabilization of the tau exon 10 stem loop alters pre-mRNA splicing. J Biol Chem 2006; 281:23302-6; PMID:16782711; http://dx.doi.org/10.1074/jbc.C600143200
- Higashide S, Morikawa K, Okumura M, Kondo S, Ogata M, Murakami T, Yamashita A, Kanemoto S, Manabe T, Imaizumi K. Identification of regulatory cis-acting elements for alternative splicing of presenilin 2 exon 5 under hypoxic stress conditions. J Neurochem 2004; 91:1191-8; PMID:15569262; http://dx.doi.org/10.1111/j.1471-4159.2004.02798.x
- Warf MB, Diegel JV, von Hippel PH, Berglund JA. The protein factors MBNL1 and U2AF65 bind alternative RNA structures to regulate splicing. Proc Natl Acad Sci USA 2009; 106:9203-8; PMID:19470458; http://dx.doi.org/10.1073/pnas.0900342106
- Pedersen JS, Bejerano G, Siepel A, Rosenbloom K, Lindblad-Toh K, Lander ES, Kent J, Miller W, Haussler D. Identification and classification of conserved RNA secondary structures in the human genome. PLoS Comput Biol 2006; 2:e33; PMID:16628248; http://dx.doi.org/10.1371/journal.pcbi.0020033
- Wang J, Pantopoulos K. Regulation of cellular iron metabolism. Biochem J 2011; 434:365-81; PMID:21348856; http://dx.doi.org/10.1042/BJ20101825
- Jaffrey SR, Haile DJ, Klausner RD, Harford JB. The interaction between the iron-responsive element binding protein and its cognate RNA is highly dependent upon both RNA sequence and structure. Nucleic Acids Res 1993; 21:4627-31; PMID:8233801; http://dx.doi.org/10.1093/nar/21.19.4627
- Walden WE, Selezneva AI, Dupuy J, Volbeda A, Fontecilla-Camps JC, Theil EC, Volz K. Structure of dual function iron regulatory protein 1 complexed with ferritin IRE-RNA. Science 2006; 314:1903-8; PMID:17185597; http://dx.doi.org/10.1126/science.1133116
- Strawbridge EM, Benson G, Gelfand Y, Benham CJ. The distribution of inverted repeat sequences in the Saccharomyces cerevisiae genome. Curr Genet 2010; 56:321-40; PMID:20446088; http://dx.doi.org/10.1007/s00294-010-0302-6
- Zhao G, Chang KY, Varley K, Stormo GD. Evidence for active maintenance of inverted repeat structures identified by a comparative genomic approach. PLoS One 2007; 2:e262; PMID:17327921; http://dx.doi.org/10.1371/journal.pone.0000262
- Lynch KW, Maniatis T. Assembly of specific SR protein complexes on distinct regulatory elements of the Drosophila doublesex splicing enhancer. Genes Dev 1996; 10:2089-101; PMID:8769651; http://dx.doi.org/10.1101/gad.10.16.2089
- Sciabica KS, Hertel KJ. The splicing regulators Tra and Tra2 are unusually potent activators of pre-mRNA splicing. Nucleic Acids Res 2006; 34:6612-20; PMID:17135210; http://dx.doi.org/10.1093/nar/gkl984
- Moursy A, Allain FH, Clery A. Characterization of the RNA recognition mode of hnRNP G extends its role in SMN2 splicing regulation. Nucleic Acids Res 2014; 42:6659-72; PMID:24692659; http://dx.doi.org/10.1093/nar/gku244
- Hua Y, Vickers TA, Baker BF, Bennett CF, Krainer AR. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol 2007; 5:e73; PMID:17355180; http://dx.doi.org/10.1371/journal.pbio.0050073
- Kralovicova J, Lages A, Patel A, Dhir A, Buratti E, Searle MS, Vorechovsky I. Optimal antisense target reducing INS intron 1 retention is adjacent to a parallel G quadruplex. Nucleic Acids Res 2014; PMID:24944197; http://dx.doi.org/10.1093/nar/gku507
- Singh NN, Hollinger K, Bhattacharya D, Singh RN. An antisense microwalk reveals critical role of an intronic position linked to a unique long-distance interaction in pre-mRNA splicing. RNA 2010; 16:1167-81; PMID:20413618; http://dx.doi.org/10.1261/rna.2154310
- Pervouchine DD, Khrameeva EE, Pichugina MY, Nikolaienko OV, Gelfand MS, Rubtsov PM, Mironov AA. Evidence for widespread association of mammalian splicing and conserved long-range RNA structures. RNA 2012; 18:1-15; PMID:22128342; http://dx.doi.org/10.1261/rna.029249.111
- Butcher SE, Pyle AM. The molecular interactions that stabilize RNA tertiary structure: RNA motifs, patterns, and networks. Acc Chem Res 2011; 44:1302-11; PMID:21899297; http://dx.doi.org/10.1021/ar200098t
- Costa M, Deme E, Jacquier A, Michel F. Multiple tertiary interactions involving domain II of group II self-splicing introns. J Mol Biol 1997; 267:520-36; PMID:9126835; http://dx.doi.org/10.1006/jmbi.1996.0882
- Kralovicova J, Haixin L, Vorechovsky I. Phenotypic consequences of branchpoint substitutions. Hum Mutat 2006; 27:803-13; PMID:16835862; http://dx.doi.org/10.1002/humu.20362
- Kralovicova J, Hwang G, Asplund AC, Churbanov A, Smith CI, Vorechovsky I. Compensatory signals associated with the activation of human GC 5' splice sites. Nucleic Acids Res 2011; 39:7077-91; PMID:21609956; http://dx.doi.org/10.1093/nar/gkr306
- Kralovicova J, Vorechovsky I. Allele-dependent recognition of the 3' splice site of INS intron 1. Hum Genet 2010; 128:383-400; PMID:20628762; http://dx.doi.org/10.1007/s00439-010-0860-1
- Zhang XH, Chasin LA. Computational definition of sequence motifs governing constitutive exon splicing. Genes Dev 2004; 18:1241-50; PMID:15145827; http://dx.doi.org/10.1101/gad.1195304
- Stadler MB, Shomron N, Yeo GW, Schneider A, Xiao X, Burge CB. Inference of splicing regulatory activities by sequence neighborhood analysis. PLoS Genet 2006; 2:e191; PMID:17121466; http://dx.doi.org/10.1371/journal.pgen.0020191
- Reuter JS, Mathews DH. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics 2010; 11:129; PMID:20230624; http://dx.doi.org/10.1186/1471-2105-11-129
- Mathews DH. RNA secondary structure analysis using RNAstructure. Curr Protoc Bioinformatics 2006; 12:12-6.
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 2003; 31:3406-15; PMID:12824337; http://dx.doi.org/10.1093/nar/gkg595
- Gruber AR, Lorenz R, Bernhart SH, Neubock R, Hofacker IL. The Vienna RNA websuite. Nucleic Acids Res 2008; 36:W70-4; PMID:18424795; http://dx.doi.org/10.1093/nar/gkn188
- Raponi M, Kralovicova J, Copson E, Divina P, Eccles D, Johnson P, Baralle D, Vorechovsky I. Prediction of single-nucleotide substitutions that result in exon skipping: identification of a splicing silencer in BRCA1 exon 5. Hum Mutat 2011; 32:436-44; PMID:21309043; http://dx.doi.org/10.1002/humu.21458