
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Staphylococcus aureus is a versatile opportunistic pathogen that adapts readily to a variety of different growth conditions. This adaptation requires a rapid regulation of gene expression including the control of mRNA abundance. The CshA DEAD-box RNA helicase was previously shown to be required for efficient turnover of the agr quorum sensing mRNA. Here we show by transcriptome-wide RNA sequencing and microarray analyses that CshA is required for the degradation of bulk mRNA. Moreover a subset of mRNAs is significantly stabilised in absence of CshA. Deletion of the C-terminal extension affects RNA turnover similar to the full deletion of the cshA gene. In accordance with RNA decay data, the C-terminal region of CshA is required for an RNA-independent interaction with components of the RNA degradation machinery. The C-terminal truncation of CshA reduces its ATPase activity and this reduction cannot be compensated at high RNA concentrations. Finally, the deletion of the C-terminal extension does affect growth at low temperatures, but to a significantly lesser degree than the full deletion, indicating that the core of the helicase can assume a partial function and opening the possibility that CshA is involved in different cellular processes.
Introduction
Staphylococcus aureus is a Gram-positive opportunistic pathogen that colonizes 20–50% of human population.Citation1 Depending on its growth phase and bacterial density, it expresses various virulence factors, including exotoxins and haemolysins, which lead to a large spectrum of diseases, from benign skin infection to severe osteomyelitis, endocarditis, and sepsis.Citation2,3 In addition to the fact that S. aureus possesses the entire molecular arsenal to develop infections, it can also be resistant against various antibiotics, raising major public health issues.Citation4 Moreover, S. aureus is able to survive and adapt to many environmental conditions, such as biofilm formation or intracellular growth, contributing to the persistence of S. aureus infections and rendering eradication of this pathogen difficult, even with harsh treatments.Citation5 The adaptation to different growth and stress conditions requires a tight regulation of gene expression, a dynamic process that is controlled at several levels of the flow of genetic information from DNA to proteins. Part of this regulation is obtained by regulating steady-state transcript levels that result from both RNA synthesis and degradation, i.e., RNA stability.
RNA helicases of the DEAD-box family play various but important roles in RNA metabolism.Citation6 All the DEAD box RNA helicases contain 12 conserved motifs gathered together in 2 RecA-fold domains that are involved in interactions with the RNA substrate and ATP. In addition to these 2 domains, composing the core of DEAD box helicases, they can possess N- or C-terminal extensions that can drive specific interactions with RNACitation7 or regulatory proteins.Citation8,9 DEAD box RNA helicases function by coupling ATP binding and hydrolysis to changes in affinity for RNA. Many DEAD-box proteins use this basic mechanism for a mode of RNA helicase activity, separating the strands of short RNA duplexes in a process that involves no translocation. They can also function as ATP-dependent RNA clamps to provide nucleation-centers that allow binding of larger protein complexes to an RNA substrate. In prokaryotes, they are involved in ribosome biogenesis, RNA decay and translation initiation. This places the helicases as important regulators in gene expression.Citation10
In E. coli, the DEAD box helicase RhlB is associated with the degradosome, a protein complex responsible for RNA degradation.Citation11,12 The RNase E endonuclease consists of a N-terminal domain that possess the catalytic activity and a C-terminal domain that serves as scaffold for the other degradosome proteins. In addition to RhlB, a 3′->5 ′ exonuclease polynucleotide phosphorylase (PNPase) and the glycolytic enzyme enolase, whose role in the complex is not understood, are part of the core complex. Other minor components, such as RraA, RraB,Citation13 Hfq,Citation14 L4,Citation15 polyphosphate kinase (PPK), DnaK, and GroEL, were also found to be associated with the degradosome.Citation16,17 Moreover, it has been shown that the E. coli degradosome associates with translating ribosomes.Citation18 During the mRNA degradation process, RNase E cleaves 5′ monophosphorylated RNAs that are subsequently degraded by the PNPase and other 3′->5 ′ exoribonucleases that are not associated with the degradosome (for review see ref. Citation19. RNase E and the PNPase are single-strand specific enzymes and RhlB activity is required to facilitate the RNA degradation in a context where the RNAs can form double-stranded structures.Citation20,21 Moreover, it has been shown that a C-terminal region of RNase E stimulates the RNA-dependent ATPase activity of RhlB.Citation22
Bacterial two hybrid analyses have shown that the DEAD box helicase CshA of S. aureus is also part of a putative degradosome,Citation23 similar to its Bacillus subtilis counterpart.Citation24 The interaction network includes the endonuclease RNase Y, the endo and 5′->3 ′ exoribonucleases RNase J1 and RNase J2 and the 3′->5′ exonuclease PNPase. The complex also contains the RNaseP component RnpA, the glycolytic enzymes enolase and phosphofructokinase. Nevertheless, a complex containing several of these proteins has not yet been purified as such. Mutations in cshA revealed in addition to a cold sensitive phenotype a reduced biofilm formation and increased hemolysis, the latter due to a stabilization of agr mRNA.Citation25 In this context, CshA could be needed to allow the degradosome enzymes to degrade the RNA, as it was shown for RhlB. In H. pylori it has been shown that, in vitro, the DEAD box RNA helicase RhpA allows RNase J to degrade double-stranded RNA.Citation26 The ribonucleases RNase Y, RNase J1, RNase J2 and PNPase are specific for single-stranded RNA and a stable duplex on the RNA can form a protective structure to prevent degradation.Citation27 The unwinding of such structures by CshA would allow the degradosome to degrade this RNA.
In this work we show that the C-terminal domain of CshA is necessary for its function in vivo, confirming the importance of the DEAD box accessory domains. We show that CshA is implicated in the control of the stability of several mRNAs, including many that encode regulators or virulence factors. Finally we show that CshA interacts in vivo with degradosome components and that the C-terminal domain of CshA is important for these interactions.
Results
The C-terminal region of CshA plays a crucial role
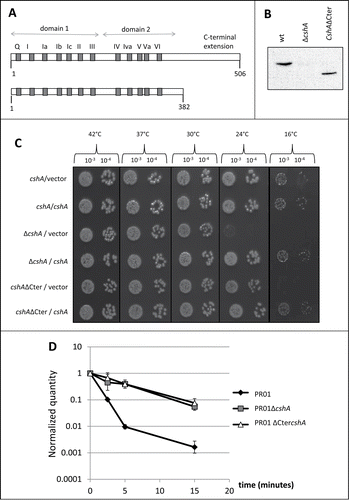
DEAD-box proteins contain 2 domains with a RecA fold, constituting together the core or motor domain involved in ATP dependent RNA binding and RNA-dependent ATP hydrolysis. Accessory N- or C-terminal extensions of the DEAD box helicases can play a regulatory or/and a specificity role. We therefore investigated whether the C-terminal extension of CshA is required for its function and deleted the region corresponding to amino acids 383 to 506, creating a protein similar in length to eIF4A, the godfather of DEAD-box proteinsCitation28 (, Fig. S1). The region after amino acid 434 appears to be unstructured by contrast to the globular catalytic core of the protein (GlobPlot 2.3,Citation29). To avoid changes in the expression level, we deleted the corresponding region of the gene directly on the chromosome using the pyrEF/5-FOA selection/antiselection systemCitation30 in a derivative of the clinical strain SA564,Citation31 resulting in strain PR01cshAΔCter. We checked by western blot that the truncated protein was still correctly expressed (). To assess the function of the truncated protein, we followed 2 readouts for CshA activity, the cold sensitive phenotype and the agr mRNA decay. The PR01cshAΔCter strain showed a cold sensitive phenotype at 16°C similar to the full deletion of the cshA gene. Interestingly, at 24°C only the PR01ΔcshA strain showed a cold sensitivity phenotype whereas the PR01cshAΔCter strain was still able to grow. Importantly, growth at 16°C can be restored in PR01cshAΔCter by expressing CshA from a plasmid. At 42°C, 37°C and 30°C both strains have a growth similar to the parental PR01 strain (). Since we have previously shown that a cshA mutant strain is affected in the decay of the agr quorum sensing mRNA, we also measured the decay of this mRNA in the PR01cshAΔCter strain by qRT-PCR. A time course after rifampicin treatment showed that the agr mRNA decay was comparable to the PR01ΔcshA strain, showing that the C-terminal extension of CshA is of great importance in vivo for the RNA turnover function of the protein ().
Figure 1. Functional analysis of CshA and CshAΔCter. (A) Schematic representation of the CshA protein. Gray boxes and the letters above represent the conserved domains characteristic of the DEAD-box family. The numbers below indicate the amino acid position. For CshAΔCter, the protein was truncated after amino acid 382. (B) Presence of CshA in the total cell extracts of PR01, PR01ΔcshA and PR01cshAΔCter. The CshA protein was detected in a western blot performed with an anti-CshA polyclonal antibody. Equal amounts of lysed cells were loaded and verified by Ponceau staining. (C) The strains PR01, PR01ΔcshA and PR01cshAΔCter, containing the plasmids pEB01 (vector) or pEB07 (containing CshA) were spotted in serial dilutions (indicated above) on rich medium containing 15 μg/ml chloramphenicol. All plates were incubated at 42°C (24 h), 37°C (24 h), 30°C (48 h), 24°C (3 days) and 16°C (6 days). Only representative 10−3 and 10−4 dilutions are shown. (D) Cultures of PR01(black diamonds), PR01ΔcshA (gray squares) and the PR01cshAΔCter (white triangles) were rifampicin treated to block de novo RNA synthesis. Samples were taken for RNA isolation at 0, 2.5, 5, 10 and 15 min after treatment, and qRT-PCR was performed using primers and probes specific for agrA, and using HU mRNA as an internal reference. The quantity of agr, relative to HU, was normalized to 1 at time zero, and plotted in the graph. Error bars represent the 99% confidence level.
The truncated version of CshA retains ATPase activity
The reduced in vivo functionality of the C-terminal truncated RNA helicase could be due to lack of interactions with partner proteins or simply be due to the absence of enzymatic activity. We therefore expressed and purified the full length and the truncated proteins from E. coli and checked for their ATPase activity in vitro in presence or absence of rRNA.
Purification of the CshA-His protein was achieved by adsorption to nickel-agarose and elution with 250 mM imidazole. The nickel-agarose preparation was highly enriched with respect to the CshA-His polypeptide, as judged by SDS-PAGE (). In parallel we purified a mutated version of CshA in which Lys52 in motif I (Walker A motif) was replaced by alanine (K52A; ). An analogous mutation in eIF4A was shown to abrogate nucleotide binding.Citation32 Recombinant wild-type CshA catalyzed the release of Pi from [gamma-32P]ATP in the presence of E. coli rRNA (rRNA) and the extent of ATP hydrolysis was proportional to enzyme concentration (). The ATPase activity of the K52A mutant was < 1% of the activity of wild-type enzyme. We therefore conclude that the observed phosphohydrolase activity is intrinsic to CshA. Moreover, the ATPase activity measured after a glycerol gradient followed exactly the position of the protein as determined by Western analysis (see below). As it was reported previously and characteristic for most DEAD-box proteins, only background ATPase activity could be detected in absence of RNA (data not shown).
Figure 2. ATPase activity of the full length and the truncated CshA proteins. (A) CshA purification. Aliquots (3 μg) of the nickel-agarose preparations of wild-type (WT) CshA, mutant K52A, deletion mutant CshA△Cter, and deletion mutant CshA△CTer-K52A were analyzed by electrophoresis through a denaturing 4–12% Bis-Tris polyacrylamide gel. The polypeptides were visualized by staining with Commassie Blue dye. The positions and sizes (in kDa) of marker proteins are indicated on the left. (B) ATPase reaction mixtures (15 μl) containing 50 mM Tris-HCl, pH 8.0, 1 mM DTT, 2 mM MgCl2, 1 mM [γ-32P]ATP, 0.2 ng/μl rRNA and wild-type (WT) CshA or mutants CshA as specified were incubated at 37°C for 15 min. The extents of ATP hydrolysis are plotted as a function of input protein. (C) ATPase reaction mixtures (15 μl) containing 50 mM Tris-HCl, pH 8.0, 1 mM DTT, 2 mM MgCl2, 1 mM [γ -32P]ATP, either 20 ng wild-type (WT) CshA or 20 ng deletion mutant CshA△Cter, and an increasing amounts of rRNA as specified were incubated at 37°C for 15 min. The extents of ATP hydrolysis are plotted as a function of input rRNA.
![Figure 2. ATPase activity of the full length and the truncated CshA proteins. (A) CshA purification. Aliquots (3 μg) of the nickel-agarose preparations of wild-type (WT) CshA, mutant K52A, deletion mutant CshA△Cter, and deletion mutant CshA△CTer-K52A were analyzed by electrophoresis through a denaturing 4–12% Bis-Tris polyacrylamide gel. The polypeptides were visualized by staining with Commassie Blue dye. The positions and sizes (in kDa) of marker proteins are indicated on the left. (B) ATPase reaction mixtures (15 μl) containing 50 mM Tris-HCl, pH 8.0, 1 mM DTT, 2 mM MgCl2, 1 mM [γ-32P]ATP, 0.2 ng/μl rRNA and wild-type (WT) CshA or mutants CshA as specified were incubated at 37°C for 15 min. The extents of ATP hydrolysis are plotted as a function of input protein. (C) ATPase reaction mixtures (15 μl) containing 50 mM Tris-HCl, pH 8.0, 1 mM DTT, 2 mM MgCl2, 1 mM [γ -32P]ATP, either 20 ng wild-type (WT) CshA or 20 ng deletion mutant CshA△Cter, and an increasing amounts of rRNA as specified were incubated at 37°C for 15 min. The extents of ATP hydrolysis are plotted as a function of input rRNA.](/cms/asset/7a218457-e09c-4391-a56e-b1b641d4cc7e/krnb_a_1035505_f0002_b.gif)
The truncated CshA△Cter and its K52A version were also expressed in E. coli and purified in parallel. The mobility of the CshA△Cter proteins during SDS-PAGE was fairly consistent with their calculated sizes of 43 kDa (). At saturating RNA concentrations, the CshA△Cter (1–382) protein retained 30% specific activity of wild type whereas the CshA△Cter-K52A was <1 % of the wild-type enzyme. We concluded that the C-terminal 124 amino acids of CshA are involved in, but not essential for, RNA dependent ATP hydrolysis ().
To test the possible effects of the C-terminal domain deletion on the rRNA interactions of CshA, we measured ATP hydrolysis as a function of the concentration of the rRNA (). Wild-type CshA and CshA△Cter displayed a typical hyperbolic dependence of ATP hydrolysis on input rRNA (). From the titration curve we estimate that the deletion of the C-terminal domain elicited about a 3-fold decrement in the activation of the CshA ATPase. From this experiment we concluded that the C-terminal domain of CshA is not the major RNA interaction domain.
Sedimentation Analysis of CshA
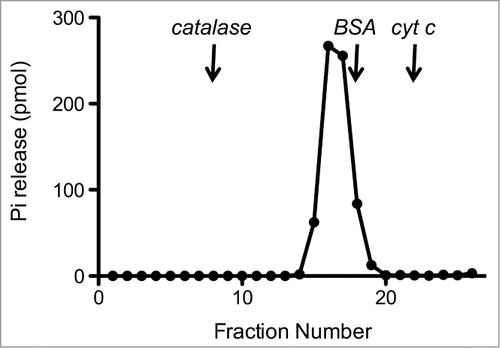
The recombinant protein was subjected to zonal velocity sedimentation in a 15–30% glycerol gradient. Marker proteins catalase (native size 248 kDa), bovine serum albumin (66 kDa), and cytochrome c (12 kDa) were included as internal standards. CshA-His6 (calculated to be a 57-kDa polypeptide) sedimented as a discrete peak (fraction 16) just slightly heavier than the BSA peak (66-kDa) (data not shown). The ATPase activity profile paralleled the abundance of the CshA-His6 polypeptide and peaked at fraction 16 (). A plot of the S values of the 3 standards versus fraction number yielded a straight line (not shown). Interpolation to the internal standard curve determined an S value of 5.6 for CshA-His by suggesting that CshA-His is a homodimer in solution.
Figure 3. Sedimentation analysis of CshA.Recombinant CshA was sedimented in a glycerol gradient as described under “Materials and Methods.” Aliquots (2 μl) of the glycerol gradient fractions were assayed for ATPase activity. The ATPase activity profile is shown. The fractions corresponding to the peaks of the internal markers catalase, BSA, and cytochrome c are indicated by vertical arrows.
CshA is required for the decay of a subset of mRNAs in S. aureus
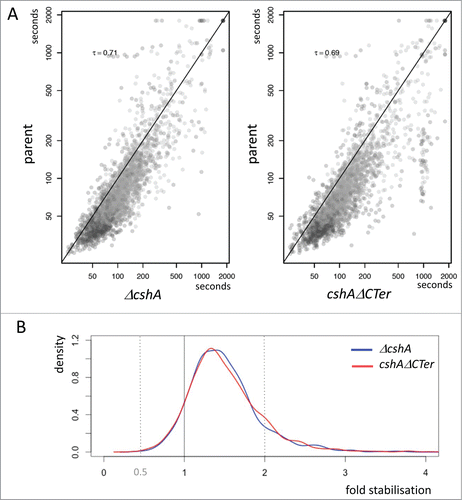
CshA was suggested to be part of the Firmicute degradosome,Citation23,24 and we have shown that CshA is involved in agr mRNA decay.Citation25 We therefore tested if the decay of other RNAs was also affected in absence of CshA or in absence of the C-terminal extension of CshA. The mutant and parental strains were grown to early exponential growth phase and transcription was stopped by the addition of rifampicin. Total bacterial RNA was purified from aliquots of cells before transcriptional arrest and at 2.5, 5, and 10 minutes post-transcriptional arrest. After depletion of rRNA, the purified RNA was then analyzed by RNA deep sequencing and the analysis of genes was fitted with the highly similar genome of N315. Genes that showed less than 100 reads on average over the 4 time points were eliminated, resulting in 1961 ORFs and tRNAs from strain SA564 out of the 2662 found in the N315 genome. The read counts were then normalized to the mRNA of HU, which was previously shown to be well expressed and relatively stable.Citation33 Similar to published studies,Citation34 these results clearly showed that most RNAs are present at background levels at >10 minutes after rifampicin treatment. Half-lives were computed by fitting a linear model on log2 transformed gene expressions. Genes whose RNA levels had the largest divergence with the fitted model are excluded to finally retain 1448 genes. Comparing the half-lives of each strain in the 2 experiments showed a good correlation of the 2 independent experiments (Fig. S2).
Interestingly, the comparison of the full deletion and the truncated cshA mutant strains revealed a large similarity in the RNA stabilization in respect to the wild type (). From the cumulative distribution of RNA stabilization in the 2 mutant strains it is clear that in both the full deletion and the truncated CshAΔCter mutant strains over 80% of the RNAs are stabilised, but only a relatively small portion is significantly (≥2 fold) stabilised in the absence of the helicase or in absence of the C-terminal extension of CshA ().
Figure 4. Stabilization of mRNAs. (A) Scatterplot showing half-life change between mutants and the parental strain. Each point represents a gene and is positioned on the x-axis according to the mean half-life estimated in both replicates of the decay experiment; similarly, the point on the y-axis is positioned according to the mean half-life estimated in both replicates of the decay experiment of the parental strain. Kendall tau correlation coefficient is given for each case. A diagonal line marks the limit between genes whose half-life is increased in the mutants (below the line), and genes whose half-life is decreased in the mutants (above the line). Only genes satisfying minimum quality criteria are reported. (B) Distribution of the half-life fold change between the mutants and the parental strain. The areas under the curves sums to 1.0, and reflects the proportion of genes affected with a given stabilization factor. The vertical line at x = 1 marks the limits between stabilization (on the right) and destabilization (on the left). The shift of the curves on the right side of the vertical line reflects the longer half-life measured in the mutants compared to the ancestor strain.
To identify genes that had RNAs stabilised at least 2 fold we compared the data of the full deletion and the CshAΔCter mutants with the wild type data. Since the 2 mutant types behaved similarly we retained genes that showed in at least 3 out of the 4 experiments (2 times the full deletion and 2 times the C-terminal truncation) a 2 fold or larger increase of the half-life. We discarded four genes in which one of the half-lives from the wt strain was longer than in one mutant strain. Using these criteria we retained 113 RNAs that were stabilised 2 fold or more (). For many RNAs, the stabilization also leads to an increase of the steady state level, as judged from the number of reads at time zero of the rifampicin treatment ().
Figure 5. Correlation of half life and steady-state levels. Scatterplot showing correlations between the changes in steady state level of RNAs a time point 0 and in the half-life from the ΔcshA and cshAΔCter mutants as compared to the parent. Each point represents a gene and is positioned on the x-axis according to log2 change in half-life (stabilization), while it is positioned on the y-axis according to log2 of the expression of the gene at time point 0 (steady-state level). Each change is computed in a conservative way making use of all replicates information in a worst case scenario strategy: to estimate the change in steady state level, the normalized gene expression of the 2 parent replicates at time point 0 are compared to the normalized gene expression of the 4 mutants at time point 0; only the combination yielding the smallest change is considered. Half-life change on the y-axis is computed with the same strategy.
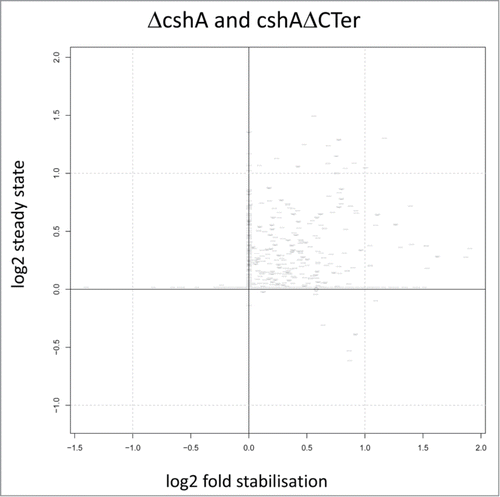
Table 1. Stabilized RNAs. The half-life of the RNAs in the 2 mutant and the parental strains in 2 independent experiments were compared. The ratio of the half-life of the mutant compared to the parental strain is indicated. Genes that showed a stabilization of the RNA at least 2-fold in 3 out of the 4 mutant strains were retained and are listed in the table
Similar results were obtained by microarray analysis for RNA preparations carried out at 0, 2.5, 5, 10, and 30 minutes after rifampicin treatment (data not shown). Among 118 identified genes in the micro array experiment, 51 are present in the list obtained by the sequencing approach. The genes that are not present in the final list of the sequencing approach were eliminated due to quality restrictions, low expression levels, or are slightly below the cut-off (≥2×) in the sequencing data set. Nevertheless the overall concordance of the 2 methods validates the sequencing approach. It should be noted that due to our RNA purification, we did not quantitatively purify small RNAs (<200 nt), and we therefore concentrate here on the analysis of coding RNAs. Interestingly, a few mRNAs seemed to be destabilised in absence of a fully functional CshA protein. At present we have no explanation for this.
Altogether these results clearly indicate that CshA is involved at least to some degree in the degradation of many RNAs. Moreover, CshA seems to play a significant role in the turnover of a subset of mRNAs. Moreover, we show that the C-terminal extension of this DEAD-box protein is important for this function within RNA decay.
Identification of CshA interaction partners
The C-terminal truncation of CshA in the CshAΔCter mutant strain showed reduced growth at 16°C but not at higher temperatures. However the analysis of the RNA-decay patterns showed virtually identical profiles for the full deletion and the C-terminal truncation. Thus it is likely that the C-terminal extension is important for interactions with the RNA-decay machinery and we set out to identify interacting proteins using a tandem affinity purification (TAP) approach. We used a PR01ΔcshA strain producing a full-length CshA protein or the C-terminal truncated CshAΔCter, containing a Strep/FLAG-tag at the C-terminus. We verified that the tagged version of full length CshA was able to restore the cold sensitive phenotype of the PR01ΔcshA strain, showing that the protein is functional (data not shown). We also checked the production of the proteins by western blot, using a polyclonal antibody raised against CshA. We noted a slight overproduction in comparison with the wild type level of CshA, probably due to the fact that the constructs are expressed from a plasmid on its native promoter (data not shown). Since some interactions could be mediated through RNA, we performed the lysis steps both in absence or presence of RNase A. The Strep/FLAG tagged versions of CshA and their potential interaction partners were purified using first a Strep-Tactin column. The corresponding elution fractions were pooled and subjected to a second round of purification on an α-FLAG resin. In order to be able to discriminate protein interactions from column background, we performed in parallel the same purifications with a lysate from a PR01 strain with the empty vector. The eluted fractions were analyzed on a SDS-PAGE and the protein partners were identified by mass spectrometry. A total of 106 proteins were identified in the elution fractions coming from the lysates containing the tagged version of CshA, with or without RNase A treatment (Table S2, Table S3 for the full data set). Among these proteins, we found proteins involved in each step of the RNA metabolism from transcription to decay, enzymes in DNA metabolism and others. Indeed, we found interactions with the β and β’ subunits of the RNA polymerase, various ribosomal proteins, translation initiation and elongation factors. Most importantly, we also identified RNase J1, RNase J2, RNase Y and enolase, components of the putative degradosome. Interestingly, the PNPase was only detected without RNase A treatment, suggesting a lose association, if any, with the degradosome. To identify the interactions mediated through the C-terminal extension, we also identified proteins that copurified with the truncated CshAΔCter. The analysis showed that out of the 106 proteins from the CshA purification, 68 proteins were absent in the CshAΔCter preparation after RNase A treatment (). Importantly, RNase J1, RNase J2, RNase Y and enolase were no longer detected, as well as many ribosomal proteins, indicating an association mediated through the C-terminal extension. We therefore also purified RNase J1 to see whether the reciprocal co-purification could be observed. Interestingly, relatively few proteins copurified with RNase J1, with the exception of ribosomal proteins and RNase J2 (Table S2). Importantly, purification of RNase J1 allowed the copurification of CshA, further confirming an interaction of CshA and RNase J1. To check for specificity of CshA, we also purified CshB by the same procedure. In contrast to CshA, relatively few proteins copurified with CshB. However, both helicases purified with a considerable number of ribosomal proteins (Table S2), which may reflect an association with the ribosome or a role in ribosome biogenesis.Citation35
Table 2. Summary of proteins identified for CshA. Proteins identified in the purifications of CshA, CshAΔCter, RNase J1, or CshB. Numbers represent the peptides identified in presence and absence (within brackets) of RNase A during the purification. The table was established by selecting, for each purification, all proteins that were represented by 2 or more peptides. Proteins were retained for the table if they were present in the CshA purification and after RNase A treatment, but not in the purification of the truncated CshAΔΧτερ. The full data set is given in Supplementary Table 3 Proteins identified in the mock sample (Elongation factor Tu, A5IQA2 (14 peptides) and Pyruvate dehydrogenase E1 P0A0A1 (5 peptides)) are not indicated
Intriguingly we were so far unable to demonstrate an in vitro interaction of individually prepared recombinant N-terminal Flag-Streptavidin tagged RNase J1 with CshA. This could be explained either by cotranslational folding or by interaction through a third partner. However, if both proteins were co-expressed together in E. coli, purification of Flag-Streptavidin tagged RNase J1 in presence of RNase A, allowed co-purification of low amounts of CshA, visible on Commassie blue stained gels and confirmed by western blot analysis (data not shown). Interestingly the H. pylori RNase J and RNA helicase interaction was also observed only by co-expression.Citation26
Discussion
We have previously shown that the S. aureus CshA DEAD-box RNA helicase is required for efficient turnover of the agr mRNA and that inactivation of cshA results in decreased biofilm formation and increased hemolysis. Here we showed that CshA is more generally involved in RNA turnover and that the C-terminal extension is required for this activity, as well as for the interaction with components of a Gram-positive degradosome. Interestingly, the cold sensitive phenotype at 24°C was almost absent with the truncated protein, but very pronounced with the full deletion. We also showed that in absence of the C-terminal extension the RNA-dependent ATPase activity was reduced.
Using the strains PR01ΔcshA and PR01cshAΔCter, we were able to show that the bulk of the mRNAs is slightly stabilised in absence of a fully functional CshA protein and that a defined subset of the mRNAs is significantly stabilised in absence of a wild-type helicase. As expected from our previous observations, the agr quorum sensing system is among the identified operons. In addition, 2 other 2-component systems, BraS/BraR and Kpd/KpdD) (KpdD was eliminated because of quality limitations) and 6 putative or established transcription factors (purR, SA1337, Rex, tcaR, SA2174, SA2495), as well as genes encoding proteins involved in intracellular signaling, such as the cyclic-di-AMP phosphodiesterase GdpP,Citation36,37 were among the genes with stabilised mRNAs. Interestingly, many genes encoding membrane-associated proteins (36) were also in the list of significantly stabilised mRNAs. At present we do not know if this observation has a biological significance, or if this is due to a potential localization of the degradosome to the membrane through the RNase Y membrane anchor. The stabilization of RNA could be due to a direct effect of a deficient degradosome, or due to an indirect effect. As an example, studies from the Dunman laboratory have shown that certain mRNAs require SarA for stabilization, which was suggested to be due its RNA binding activity.Citation34,38 Several RNAs that were identified in the study byCitation38 are also present in our list of 2 fold or more stabilised RNAs (radA, ctaA, rpmF, glnP fmtC, agrA, kdpA, lacA, moaC, pyruvate oxidase, opuCD, cidB, betA). The sarA mRNA is slightly stabilised in the ΔcshA strain (˜1.6-fold) and the cshAΔCter strain (˜2.2-fold). Moreover, DEAD-box proteins were shown to be able to remove RNA-binding proteins from their substrate,Citation39 and therefore SarA stabilised mRNAs might be stabilised in absence of CshA either by increased SarA or due to the absence of a putative RNPase activity of the RNA helicase. In a similar vein, certain genes, affected in absence of a fully functional CshA RNA helicase, comprise also genes like the one encoding α hemolysin (SA1007), which is regulated by RNA III. In this case, the stabilization of the mRNA may be due to increased translation upon up-regulation of RNA III as a consequence of the agr operon mRNA stabilization (this study andCitation25). In accordance with the analysis of CshA in B. subtilis,Citation35 the mRNA encoding the holin-like protein CidB is also largely stabilized in our experimental conditions. Altogether, this suggests that the helicase plays an important role in adaptation to changing environments and thereby influence the outcome of infections by this feared pathogen.
From the studies with the E. coli degradosome, it was strongly suggested that the helicase is required for RNAs that contain secondary structures. It is reasonable to expect that mRNAs, coding for proteins that are required only under certain growth conditions or that are involved in adaptation to stress conditions, need to be rapidly turned over when no longer necessary. Therefore one possibility could be that the CshA-dependent decay of RNAs is regulated by the RNA helicase. In this scenario the CshA protein plays a regulatory role, eventually assisted by other proteins. How the helicase itself would be regulated is not known, but in E. coli, regulatory proteins that inhibit the RhlB helicase have been described.Citation8,9 Another more direct explanation for the requirement of a DEAD-box RNA helicase in turnover would be the presence of secondary structures that perform a biological function, such as regulating the accessibility of the ribosome-binding site. For the degradation of such RNAs, the degradosome needs to be assisted by a helicase since it was shown previously that both the 5′-3′ RNase J and the 3′-5′ PNPase are sensitive to secondary structures.Citation40,41 However, it remains to be shown, whether CshA is a permanent member of the decay machinery, or if it is only recruited on demand. A detailed analysis of some of the target RNAs requiring CshA will certainly help to further distinguish between these possibilities.
RNA turnover in S. aureus has been analyzed previously in exponential phase, heat shock, cold shock, stringent and SOS response conditions.Citation42 Under heat shock, cold shock and stringent response conditions, the stability of most mRNAs increased, but so far the components required for the turnover have not been identified. Using tandem affinity purifications we were able to determine the interaction partners of CshA, CshAΔCter, RNase J1 and CshB. Importantly, CshA copurifies with the predicted degradosome proteins RNase J1, RNase J2, RNase Y and enolase. This is consistent with bacterial 2-hybrid analysesCitation23 and the finding that the minimal H. pylori degradosome is formed by RNase J and RhpA.Citation26 In addition we noted that CshA interacts with 28 ribosomal proteins and various other proteins. Most of these interactions are lost when the purification is performed with CshAΔCter, indicating that the C-terminal region drives these interactions. However, it should ne noted that many of these interactions might be indirect. Nevertheless it was shown that both the H. pylori and the E. coli degradosomes associate with poly-ribosomesCitation18,26 and that RNase J1 and RNase J2 from B. subtilis are tightly ribosome associated. Moreover, most ribosomal proteins that copurified with CshA were also found in the purification of RNase J1. Finally, CshA was reported to be involved in ribosome biogenesis in the same bacterial species.Citation35 To distinguish between an interaction with RNase J or the ribosome, we intended to co-purify recombinant RNase J1 and CshA. However, so far we were not able to demonstrate co-purification by mixing the purified proteins, but found a weak interaction if co-expressed together in E. coli (data not shown). Further experiments will be required to confirm or reject such a putative interaction between CshA and RNase J1, or an indirect interaction through the ribosome. We observed that CshA copurifies with a higher number of proteins than RNase J1 and CshB and that many more proteins unrelated to RNA metabolism co-purify with CshA. It is not clear to what extent CshA could be associated with translating ribosomes or could be anchored to the membrane through its interaction with RNase YCitation24,43 and thereby indirectly co-purify with a large number of proteins.
It has been shown for several DEAD-box proteins that N- and C-terminal extensions are important for interactions with either their substrate or partner proteins, and thereby confer specificity to these RNA-dependent ATPases. Here we showed that the deletion of the C-terminal 124 amino acids results in reduced growth at low temperature (16°C) and stabilization of the agr mRNA similar to the full deletion. The slightly better growth at 24°C indicates that the truncated helicase, despite reduced enzymatic activity (see below) and the absence of many interacting proteins, retains some functionality which may be provided by interactions through the core domain, as it is observed for the minimal RNA helicases eIF4A or eIF4AIII, interacting with eIF4G and the exon-junction complex components, respectively.Citation44,45 It may also support the hypothesis that CshA is involved in another process, such as ribosome biogenesis. Indeed, in E. coli the deletion of RhlB does not confer a cold sensitive phenotype, whereas the deletion of CsdA/DeaD or SrmB results in cold-sensitivity, probably due to erroneous 23S rRNA•5S rRNA hybridization, as elegantly shown with a srmB suppressor analysis.Citation46 Such a ribosome biogenesis function was also suggested for the B. subtilis CshACitation35 and would be in accordance with the co-purification of a large number of ribosomal proteins.
Altogether, our results show that CshA is required for efficient turnover of the bulk of mRNAs and that a selected subset of RNAs is significantly stabilised in absence of the RNA helicase. For efficient degradation, the RNA helicase interacts through its C-terminal extension with the degradosome components. The molecular function of the RNA helicase could be the destabilisation of secondary structures or the removal of hindering RNA binding proteins. Further analysis of individual RNAs and different mutants in cshA or the other components of the degradosome will help to further elucidate this exciting and important aspect of gene expression.
Materials and Methods
Bacterial strains
Bacteria () were grown under standard laboratory conditions. The deletion mutants were constructed using the pyrFE/FOA counter selection system.Citation30 Standard molecular biology methods for plasmid and strain constructions were employed according to either the manufacturer's instructions or Sambrook and Russell.Citation47 Plasmids used in this study are listed in .
Table 3. Strains
Table 4. Plasmids
RNA isolation, GeneChip analysis and RNA-sequencing
Overnight cultures were diluted in fresh Mueller-Hinton media to a final OD600 of 0.05. Cells were grown until mid-exponential phase (OD 0.3 – 0.4). A 1 ml sample was harvested and rifampicin was immediately added to arrest the transcription (final concentration 200 μg/ml). 1 ml samples were harvested at 0, 2.5, 5, and 10 minutes after rifampicin treatment. After centrifugation, the supernatant was removed and 0.5 ml of ice-cold acetone-ethanol (1:1) was added and the samples were stored at −80°C until the lysis step. For the lysis, the pellet was resuspended in 200 μl of TE containing 0.05 μg/ml lysostaphin and was incubated 10 minutes at 37°C. The RNA isolation was done immediately after, using the QIAshredder and RNeasy mini column, according to the manufacturer's recommendations (Qiagen). 1 ug of total RNA were ribo-depleted using the ribo-zero magnetic kit for Bacteria from epicenter. Libraries were then prepared using the Illumina TruSeq stranded mRNA kit according to manufacturer's recommendations. Libraries were validated on the Bioanalyzer 2100 (Agilent) and the Qubit fluorimeter (Invitrogen). Samples were multiplexed by 8 and loaded at 8 pM on one lane of a Illumina HiSeq 2500 according to a single read – 50 cycles protocol. Alternatively, RNA was then reverse transcribed, and cDNA was fragmented, 3′-biotinylated, mixed with exogenous labeled “spike-in” transcripts, and hybridized to S. aureus GeneChips by following the manufacturer's recommendations for antisense prokaryotic arrays (Affymetrix, Santa Clara, CA).
Assembly of the Staphylococcus aureus SA564 genome
Total DNA was sequenced in a HiSeq 2500 machine using Illumina technology to obtain paired 100 nt reads, which were assembled into 13 contigs with the aid of the complete genome sequence from the closely related S. aureus N315.Citation48 One contig was a circular plasmid (pSA564), and the relative order of the remaining 12 contigs was inferred from the S. aureus N315 chromosome, whereupon unique primers were designed to PCR-amplify across the gaps. All 12 gap-spanning PCR products could be obtained, thus confirming the contig-order, and the PCR products were sequenced individually by primer-walking with Sanger-sequencing to completely fill the gaps and generate a closed circular assembly. Finally, the original Illumina reads were re-mapped onto the SA564 assembly, and regions of uncertainty were PCR-amplified and confirmed/corrected with Sanger-sequencing. The final assembly was submitted to the NCBI database with accession number CP010890 and CP010891.
Bioinformatic analysis
The stranded mRNA-sequencing reads were mapped to the genome of S. aureus N315 using the software BWA.Citation49 Reads that did align to multiple positions on the genome are dropped. The number of reads overlapping a gene was counted taking into consideration the agreement between the mapped read strand and the gene strand. This was done under the R programming language environment (www.R-project.org), making use of Bioconductor packages (www.bioconductor.org). Obtained counts were normalized according to the count obtained for gene HU, and scaled to reflect the average expression level of HU in the 2 WT samples.
To include an RNA, we defined a threshold of 100 reads on average across the 4 time points to avoid too much noise in correctly estimating the half-life of a gene. The threshold of 100 was chosen arbitrary and roughly corresponds to a 5% error on the half-life estimate for a difference of 1 in the count. As reference we used the hu mRNA which was shown to be highly expressed and stable, and has been used previously.Citation33 A qRT-PCR analysis of 16S rRNA and hu mRNA showed very similar stability over a time range of 10 minutes.
Using log2 transformed expression levels, measured at the 4 time points (0, 2.5′, 5′, 10′), a linear model was fitted to estimate the half-life. Indeed, the level of expression et at time t, for a gene characterized by a half-life h, and an expression level e0 at time 0 is expected to be . The Log2 transformation of this formula results in a linear expression in t:
; so that the half-life is given by:
where a is the linear coefficient of the model fitted on the log2 transformed expressions. For simplicity, half-life estimates > 30 minutes, or negative half-lives (as stable as HU or more stable in our experimental conditions) were set to 30 minutes.
Model fitting was effectively performed by minimizing a weighted sum of square errors. Since most of the RNAs have a very short half live resulting in background levels at time point 10 minutes or even earlier, weights were set accordingly (256, 64, 16, and 1 fold, for the 0, 2.5, 5, 10 minutes time points). We empirically determined that fitted models resulting in a weighted error sum higher than 20 are not satisfying and this score was therefore used as cut-off for quality control.
Tandem affinity purification
CshA, CshAΔCter, CshB and RNase J1 were cloned in the pCG vector, resulting in the expression of proteins containing a Strep/Flag tag at their C-terminal extremity. PR01ΔcshA, PR01ΔcshB and PR01ΔJ1 strains replicating respectively the pCGCshA or the pCGCshAΔCter vector, the pCGCshB, and the pCGJ1 were grown until OD = 1. One liter of bacteria was harvested and resuspended in lysis buffer (Tris 50 mM pH 7.5; CaCl2 1 mM; Triton 0.1%; DNaseI; lysozyme) and subjected to 3 freeze/thaw cycles. The lysate was cleared by centrifugation and loaded on a Streptactin column (IBA). The elution fractions were then loaded on a α-Flag column (Sigma). The purifications were done according to the manufacturer recommendations and the yield varied between 55 and 158 μg per preparation. Proteins were identified by LC/MS starting with 10 μg of material. Proteins were considered as being present in the sample if at least 2 different exclusive peptides were detected.
Bacterial expression vectors for recombinant CshA and CshAΔCter
The pET22b-CshA plasmids were transformed into E. coli Rosetta (DE3). Cultures (500 ml) derived from single transformants were grown at 37°C in LB medium containing 50 μg/ml ampicillin and 30 μg/ml chloramphenicol until the OD600 reached 0.6. The cultures were adjusted to 0.2 mM IPTG and 2% (v/v) ethanol and incubation was continued for 20 h at 17°C. Cells were harvested by centrifugation and stored at −80°C. All subsequent procedures were performed at 4°C. Thawed bacteria were resuspended in 25 ml of buffer A (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 10% glycerol, 1 mM MgCl2) and supplemented with one tablet of protease inhibitor cocktail (Roche). The suspension was adjusted to 0.1 mg/ml lysozyme and incubated on ice for 30 min. Imidazole was added to a final concentration of 5 mM and the lysate was sonicated to reduce viscosity. Insoluble material was removed by centrifugation. The soluble extracts were mixed for 30 min with 1.6 ml of Ni2+-NTA-agarose (Qiagen) that had been equilibrated with buffer A containing 5 mM imidazole. The resins were recovered by centrifugation, resuspended in buffer A with 5 mM imidazole, and poured into columns. The columns were washed with 8 ml aliquots of 10 and 20 mM imidazole in buffer A and then eluted step-wise with 2.5 ml aliquots of buffer A containing 50, 100, 250, and 500 mM imidazole. The elution profiles were monitored by SDS-PAGE. The 250 mM imidazole eluates containing the CshA polypeptides were stored at −80°C. The protein concentrations were determined using the Bio-Rad dye reagent with BSA as the standard.
Glycerol gradient sedimentation
An aliquot (50 μg) of the nickel-agarose preparation of CshA was mixed with catalase (50 μg), bovine serum albumin (50 μg), and cytochrome c (50 μg). The mixture was applied to a 4.8-ml 15–30% glycerol gradient containing 50 mM Tris-HCl (pH 8.0), 0.25 M NaCl, 1 mM EDTA, and 2 mM DTT. The gradient was centrifuged for 20 h at 4°C in a Beckman SW50 rotor at 48,000 rpm. Fractions (0.17 ml) were collected from the bottom of the tube.
Triphosphatase assay
Reaction mixtures (15 μl) containing 50 mM Tris-HCl, pH 7.5, 1 mM DTT, 2 mM MgCl2, 1 mM [γ-32P]ATP, 400 ng/μl rRNA and CshA as specified were incubated for 15 min at 37°C. The reactions were quenched by adding 3.8 μl of 5 M formic acid. An aliquot (2 μl) of the mixture was applied to a polyethyleneimine cellulose TLC plate, which was developed using 0.5 M LiCl, 1 M formic acid. The radiolabeled material was visualized by autoradiography, and32Pi formation was quantified by scanning the TLC plate with laser Scanner Typhoon FLA 7000 (General Electric).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authors' Contributions
CG and PL developed the project. SH performed biochemical analysis of CshA. SL and JP performed the bioinformatics analysis. PR and JP performed the genome assembly and annotation. CG, SH, and PL analyzed the data. CG and PL wrote the paper.
Supplementary_files.zip
Download Zip (2.5 MB)Acknowledgments
We are grateful to Vanessa Khemici for continuous discussion and to Eckhard Jankowsky and Patrick Viollier for comments on the manuscript. RNA-sequencing experiments were performed at the iGE3 genomics platform of the University of Geneva (http://www.ige3.unige.ch/genomics-platform.php). MS/MS protein identifications were performed at the proteomics core facility of the Faculty of Medicine (http://www.unige.ch/medecine/proteomique/protocols.html), and we are very grateful for their help and advices.
Funding
This work was supported by grants from the Swiss National Science Foundation (PL), the Novartis Jubiläumsstiftung (CG), the Boninchi Foundation (PL) and by the Canton of Geneva.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Wertheim HF, Vos MC, Ott A, van Belkum A, Voss A, Kluytmans JA, van Keulen PH, Vandenbroucke-Grauls CM, Meester MH, Verbrugh HA. Risk and outcome of nosocomial Staphylococcus aureus bacteraemia in nasal carriers versus non-carriers. Lancet 2004; 364:703-5; PMID:15325835; http://dx.doi.org/10.1016/S0140-6736(04)16897-9
- Lowy FD. Staphylococcus aureus infections. N Engl J Med 1998; 339:520-32; PMID:9709046; http://dx.doi.org/10.1056/NEJM199808203390806
- Novick RP, Ross HF, Projan SJ, Kornblum J, Kreiswirth B, Moghazeh S. Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. Embo J 1993; 12:3967-75; PMID:7691599
- Chambers HF, Deleo FR. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Rev Microbiol 2009; 7:629-41; PMID:19680247; http://dx.doi.org/10.1038/nrmicro2200
- Archer NK, Mazaitis MJ, Costerton JW, Leid JG, Powers ME, Shirtliff ME. Staphylococcus aureus biofilms: properties, regulation, and roles in human disease. Virulence 2011; 2:445-59; PMID:21921685; http://dx.doi.org/10.4161/viru.2.5.17724
- Linder P, Jankowsky E. From unwinding to clamping – the DEAD box RNA helicase family. Nat Rev Mol Cell Biol 2011; 12:505-16; PMID:21779027; http://dx.doi.org/10.1038/nrm3154
- Kossen K, Karginov FV, Uhlenbeck OC. The carboxy-terminal domain of the DExDH protein YxiN is sufficient to confer specificity for 23S rRNA. J Mol Biol 2002; 324:625-36; PMID:12460566; http://dx.doi.org/10.1016/S0022-2836(02)01140-3
- Gorna MW, Pietras Z, Tsai YC, Callaghan AJ, Hernandez H, Robinson CV, Luisi BF. The regulatory protein RraA modulates RNA-binding and helicase activities of the E. coli RNA degradosome. RNA 2010; 16:553-62; PMID:20106955; http://dx.doi.org/10.1261/rna.1858010
- Pietras Z, Hardwick SW, Swiezewski S, Luisi BF. Potential Regulatory Interactions of Escherichia coli RraA Protein with DEAD-box Helicases. J Biol Chem 2013; 288:31919-29; PMID:24045937; http://dx.doi.org/10.1074/jbc.M113.502146
- Iost I, Bizebard T, Dreyfus M. Functions of DEAD-box proteins in bacteria: current knowledge and pending questions. Biochim Biophys Acta 2013; 1829:866-77; PMID:23415794; http://dx.doi.org/10.1016/j.bbagrm.2013.01.012
- Carpousis AJ. The RNA degradosome of Escherichia coli: an mRNA-degrading machine assembled on RNase E. Annu Rev Microbiol 2007; 61:71-87; PMID:17447862; http://dx.doi.org/10.1146/annurev.micro.61.080706.093440
- Kaberdin VR, Blasi U. Bacterial helicases in post-transcriptional control. Biochim Biophys Acta 2013; 1829:878-83; PMID:23291566[AQ3]
- Gao J, Lee K, Zhao M, Qiu J, Zhan X, Saxena A, Moore CJ, Cohen SN, Georgiou G. Differential modulation of E. coli mRNA abundance by inhibitory proteins that alter the composition of the degradosome. Mol Microbiol 2006; 61:394-406; PMID:16771842; http://dx.doi.org/10.1111/j.1365-2958.2006.05246.x
- Ikeda Y, Yagi M, Morita T, Aiba H. Hfq binding at RhlB-recognition region of RNase E is crucial for the rapid degradation of target mRNAs mediated by sRNAs in Escherichia coli. Mol Microbiol 2011; 79:419-32; PMID:21219461; http://dx.doi.org/10.1111/j.1365-2958.2010.07454.x
- Singh D, Chang SJ, Lin PH, Averina OV, Kaberdin VR, Lin-Chao S. Regulation of ribonuclease E activity by the L4 ribosomal protein of Escherichia coli. Proc Natl Acad Sci U S A 2009; 106:864-9; PMID:19144914; http://dx.doi.org/10.1073/pnas.0810205106
- Carpousis AJ. The Escherichia coli RNA degradosome: structure, function and relationship in other ribonucleolytic multienzyme complexes. Biochem Soc Trans 2002; 30:150-55; PMID:12035760; http://dx.doi.org/10.1042/BST0300150
- Regonesi ME, Del Favero M, Basilico F, Briani F, Benazzi L, Tortora P, Mauri P, Deho G. Analysis of the Escherichia coli RNA degradosome composition by a proteomic approach. Biochimie 2006; 88:151-61; PMID:16139413; http://dx.doi.org/10.1016/j.biochi.2005.07.012
- Tsai YC, Du D, Dominguez-Malfavon L, Dimastrogiovanni D, Cross J, Callaghan AJ, Garcia-Mena J, Luisi BF. Recognition of the 70S ribosome and polysome by the RNA degradosome in Escherichia coli. Nucleic Acids Res 2012; 40:10417-31; PMID:22923520; http://dx.doi.org/10.1093/nar/gks739
- Rochat T, Bouloc P, Repoila F. Gene expression control by selective RNA processing and stabilization in bacteria. FEMS Microbiol Lett 2013; 344:104-13; PMID:23617839; http://dx.doi.org/10.1111/1574-6968.12162
- Khemici V, Poljak L, Toesca I, Carpousis AJ. Evidence in vivo that the DEAD-box RNA helicase RhlB facilitates the degradation of ribosome-free mRNA by RNase E. Proc Natl Acad Sci U S A 2005; 102:6913-8; PMID:15867149; http://dx.doi.org/10.1073/pnas.0501129102
- Py B, Higgins CF, Krisch HM, Carpousis AJ. A DEAD-box RNA helicase in the Escherichia coli RNA degradosome. Nature 1996; 381:169-72; PMID:8610017; http://dx.doi.org/10.1038/381169a0
- Vanzo NF, Li, YS, Py B, Blum E, Higgins CF, Raynal LC, Krisch HM, Carpousis AJ. Ribonuclease E organizes the protein interactions in the Escherichia coli RNA degradosome. Genes Dev 1998; 12:2770-81; PMID:9732274; http://dx.doi.org/10.1101/gad.12.17.2770
- Roux CM, Demuth JP, Dunman PM. Characterization of components of the Staphylococcus aureus messenger RNA degradosome holoenzyme-like complex. J Bacteriol 2011; 193:5520-6; PMID:21764917
- Lehnik-Habrink M, Pfortner H, Rempeters L, Pietack N, Herzberg C, Stulke J. The RNA degradosome in Bacillus subtilis: identification of CshA as the major RNA helicase in the multiprotein complex. Mol Microbiol 2010; 77:958-71
- Oun S, Redder P, Didier JP, Francois P, Corvaglia AR, Buttazzoni E, Giraud C, Girard M, Schrenzel J, Linder P. The CshA DEAD-box RNA helicase is important for quorum sensing control in Staphylococcus aureus. RNA Biol 2013; 10:157-65; PMID:23229022; http://dx.doi.org/10.4161/rna.22899
- Redko Y, Aubert S, Stachowicz A, Lenormand P, Namane A, Darfeuille F, Thibonnier M, De Reuse H. A minimal bacterial RNase J-based degradosome is associated with translating ribosomes. Nucleic Acids Res 2013; 41:288-301; PMID:23093592; http://dx.doi.org/10.1093/nar/gks945
- Deikus G, Condon C, Bechhofer DH. Role of Bacillus subtilis RNase J1 endonuclease and 5′-exonuclease activities in trp leader RNA turnover. J Biol Chem 2008; 283:17158-67; PMID:18445592; http://dx.doi.org/10.1074/jbc.M801461200
- Rogers GW, Komar AA, Merrick WC. eIF4A: The godfather of the DEAD-box helicases. Progr Nucl Acids Res 2002; 72:307-31; http://dx.doi.org/10.1016/S0079-6603(02)72073-4
- Linding R, Russell RB, Neduva V, Gibson TJ. GlobPlot: exploring protein sequences for globularity and disorder. Nucleic Acids Res 2003; 31:3701-8; PMID:12824398; http://dx.doi.org/10.1093/nar/gkg519
- Redder P, Linder P. New range of vectors with a stringent 5-fluoroorotic acid-based counterselection system for generating mutants by allelic replacement in staphylococcus aureus. Appl Environ Microbiol 2012b; 78:3846-54; http://dx.doi.org/10.1128/AEM.00202-12
- Somerville GA, Beres SB, Fitzgerald JR, DeLeo FR, Cole RL, Hoff JS, Musser JM. In vitro serial passage of Staphylococcus aureus: changes in physiology, virulence factor production, and agr nucleotide sequence. J Bacteriol 2002; 184:1430-7; PMID:11844774; http://dx.doi.org/10.1128/JB.184.5.1430-1437.2002
- Rozen F, Pelletier J, Trachsel H, Sonenberg N. A lysine substitution in the ATP-binding site of eucaryotic initiation factor 4A abrogates nucleotide-binding activity. Mol Cell Biol 1989; 9:4061-3; PMID:2506440
- Redder P, Linder P. DEAD-box RNA helicases in Gram-positive RNA-decay. In Methods In Enzymology, E. Jankowsky, ed., 2012a:pp. 369-83
- Roberts C, Anderson KL, Murphy E, Projan SJ, Mounts W, Hurlburt B, Smeltzer M, Overbeek R, Disz T, Dunman PM. Characterizing the effect of the Staphylococcus aureus virulence factor regulator, SarA, on log-phase mRNA half-lives. J Bacteriol 2006; 188:2593-603; PMID:16547047; http://dx.doi.org/10.1128/JB.188.7.2593-2603.2006
- Lehnik-Habrink M, Rempeters L, Kovacs AT, Wrede C, Baierlein C, Krebber H, Kuipers OP, Stulke J. DEAD-Box RNA helicases in Bacillus subtilis have multiple functions and act independently from each other. J Bacteriol 2013; 195:534-44; PMID:23175651; http://dx.doi.org/10.1128/JB.01475-12
- Corrigan RM, Abbott JC, Burhenne H, Kaever V, Grundling A. c-di-AMP is a new second messenger in Staphylococcus aureus with a role in controlling cell size and envelope stress. PLoS Pathog 2011; 7, e1002217; PMID:21909268; http://dx.doi.org/10.1371/journal.ppat.1002217
- Griffiths JM, O'Neill AJ. Loss of function of the gdpP protein leads to joint beta-lactam/glycopeptide tolerance in Staphylococcus aureus. Antimicrob Agents Chemother 2012; 56:579-81; PMID:21986827; http://dx.doi.org/10.1128/AAC.05148-11
- Morrison JM, Anderson KL, Beenken KE, Smeltzer MS, Dunman PM. The staphylococcal accessory regulator, SarA, is an RNA-binding protein that modulates the mRNA turnover properties of late-exponential and stationary phase Staphylococcus aureus cells. Front Cell Infect Microbiol 2012; 2:26; PMID:22919618; http://dx.doi.org/10.3389/fcimb.2012.00026
- Fairman M, Maroney PA, Wang W, Bowers H, Gollnick P, Nilsen TW, Jankowsky E. Protein displacement by DExH/D RNA helicases without duplex unwinding. Science 2004; 304:730-4; PMID:15118161; http://dx.doi.org/10.1126/science.1095596
- Causton H, Py B, McLaren RS, Higgins CF. mRNA degradation in Escherichia coli: a novel factor which impedes the exoribonucleolytic activity of PNPase at stem-loop structures. Mol Microbiol 1994; 14:731-41; PMID:7534370; http://dx.doi.org/10.1111/j.1365-2958.1994.tb01310.x
- Laalami S, Zig L, Putzer H. Initiation of mRNA decay in bacteria. Cell Mol Life Sci 2014; 71:1799-828; PMID:24064983; http://dx.doi.org/10.1007/s00018-013-1472-4
- Anderson KL, Roberts C, Disz T, Vonstein V, Hwang K, Overbeek R, Olson PD, Projan SJ, Dunman PM. Characterization of the Staphylococcus aureus heat shock, cold shock, stringent, and SOS responses and their effects on log-phase mRNA turnover. J Bacteriol 2006; 188:6739-56; PMID:16980476; http://dx.doi.org/10.1128/JB.00609-06
- Shahbabian K, Jamalli A, Zig L, Putzer H. RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J 2009; 28:3523-33; PMID:19779461; http://dx.doi.org/10.1038/emboj.2009.283
- Andersen CB, Ballut L, Johansen JS, Chamieh H, Nielsen KH, Oliveira CL, Pedersen JS, Seraphin B, Le Hir H, Andersen GR. Structure of the exon junction core complex with a trapped DEAD-box ATPase bound to RNA. Science 2006; 313:1968-72; PMID:16931718; http://dx.doi.org/10.1126/science.1131981
- Korneeva NL, First EA, Benoit CA, Rhoads RE. Interaction between the NH2-terminal domain of eIF4A and the central domain of eIF4G modulates RNA-stimulated ATPase activity. J Biol Chem 2005; 280:1872-81; PMID:15528191; http://dx.doi.org/10.1074/jbc.M406168200
- Proux F, Dreyfus M, Iost I. Identification of the sites of action of SrmB, a DEAD-box RNA helicase involved in Escherichia coli ribosome assembly. Mol Microbiol 2011; 82:300-11; PMID:21859437; http://dx.doi.org/10.1111/j.1365-2958.2011.07779.x
- Sambrook J, Russell DR. Molecular Cloning: A Laboratory Manual, 3rd edn, Cold Spring Harbor, N. Y.: Taylor & Francis, 2001
- Kuroda M, Ohta T, Uchiyama I, Baba T, Yuzawa H, Kobayashi I, Cui L, Oguchi A, Aoki K, Nagai Y, et al. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 2001; 357:1225-40; PMID:11418146
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25:1754-60; PMID:19451168; http://dx.doi.org/10.1093/bioinformatics/btp324