
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Deducing generic causal relations between RNA transcript features and protein expression profiles from endogenous gene expression data remains a major unsolved problem in biology. The analysis of gene expression from heterologous genes contributes significantly to solving this problem, but has been heavily biased toward the study of the effect of 5′ transcript regions and to prokaryotes. Here, we employ a synthetic biology driven approach that systematically differentiates the effect of different regions of the transcript on gene expression up to 240 nucleotides into the ORF. This enabled us to discover new causal effects between features in previously unexplored regions of transcripts, and gene expression in natural regimes. We rationally designed, constructed, and analyzed 383 gene variants of the viral HRSVgp04 gene ORF, with multiple synonymous mutations at key positions along the transcript in the eukaryote S. cerevisiae. Our results show that a few silent mutations at the 5′UTR can have a dramatic effect of up to 15 fold change on protein levels, and that even synonymous mutations in positions more than 120 nucleotides downstream from the ORF 5′end can modulate protein levels up to 160%–300%. We demonstrate that the correlation between protein levels and folding energy increases with the significance of the level of selection of the latter in endogenous genes, reinforcing the notion that selection for folding strength in different parts of the ORF is related to translation regulation. Our measured protein abundance correlates notably(correlation up to r = 0.62 (p=0.0013)) with mean relative codon decoding times, based on ribosomal densities (Ribo-Seq) in endogenous genes, supporting the conjecture that translation elongation and adaptation to the tRNA pool can modify protein levels in a causal/direct manner. This report provides an improved understanding of transcript evolution, design principles of gene expression regulation, and suggests simple rules for engineering synthetic gene expression in eukaryotes.
Introduction
Arguably, the major challenge of functional genomics is to decipher how information encoded in an RNA transcript (e.g. the 5′UTR, ORF and 3′UTR) affects various aspects of its expression. Most previous studies aimed at understanding such signals are based on the analysis of endogenous gene expression.Citation1-10
However, endogenous transcripts are subject to different forms of evolutionary selection, not necessary acting on their expression level. Thus, for example, if a highly expressed gene has a certain feature it can be difficult to discern if it is related to its function or rather to the corresponding protein abundance. In addition, endogenous transcripts vary widely in their features (for example, they have different length, promoters, UTRs, amino acid content, etc.); therefore, the effect of any particular feature on expression levels is significantly masked. For example, if a gene x with certain codons has higher protein levels than gene y with different codons we cannot be sure if these differences in protein levels are not due to the fact that they have different promoters or different UTRs, etc. Eventually, it is extremely difficult to determine causality based on the correlation between endogenous features and their expression levels measurements.
Previous large scale studies with heterologous genes shed considerable light on the subject, but were all performed on E. coli and either focused on the very start of the ORF or included synthetic libraries that did not resemble the sequence properties of endogenous transcripts.Citation11-17 As a result, many questions regarding the causal relations between transcript features and protein levels in endogenous genes and specifically in eukaryotes remain poorly understood.
For example, it was suggested that in E. coli the folding strength of the mRNA near the START codon affects the translation initiation rate (and thus the protein levels), as it is related to the efficiency with which the pre-initiation complex recognizes the start codonCitation11,13; however, most of the open questions in the field remain unanswered due to the fact that almost all previous studies focused on the analysis of endogenous genes and on indirect measures of translation; and due to the fact that the answers to these questions may be condition- and organism-dependent.Citation16 The ribosome elongation speed and its association with expression regulation is also not fully understood: some studies suggested that it is constant,Citation18,19 while others suggested that different codons have different decoding times, for example due to different tRNA levels.Citation11,17,20-22 Other important questions relate to the effect of codon and nucleotide composition in different parts of the transcript on protein levels: while some studies proposed that the codon distribution in all parts of the ORF, and specifically in those related to translation elongation, can affect the protein abundance,Citation1,17,23 others claimed that only the nucleotide composition near the beginning of the ORF impacts protein levels.Citation11 In addition the exact cause and effect of codon usage bias on organismal fitness has not been identified yet: does it evolve due to neutral (or near neutral) evolution, or does it significantly affect fitness? Do highly expressed genes have stronger codon bias to improve their protein levels, or rather due to other reasons (e.g., ribosome allocation)?Citation24-29
The aim of this study is to tackle these problems in a quantitative manner using a combined computational-synthetic biology approach in S. cerevisiae. Specifically, we aimed at deciphering how protein abundance is encoded in several regions of the transcript, some of them previously unexplored, by addressing the following questions: 1) What is the magnitude of the effect of synonymous changes in different parts of the transcript on protein abundance? 2) How does folding in different parts of the transcript affect protein abundance? 3) How do ribosome codon decoding rates affect protein abundance? 4) Do transcript features that are selected for in S. cerevisiae endogenous genes determine protein abundance?
Results
Generating YFP libraries to study the effect of silent nt composition of the 5′UTR and synonymous nt composition in the coding region on protein levels
We analyzed the gene HRSVgp04 and generated 3 libraries to understand the distinct effect of the nucleotide composition in different regions of the transcript on protein levels. The structure of all 3 libraries was identical: all had the same promoter followed by the 5′UTR (14nt) of the TEF gene, and the HRSVgp04 gene fused with a YFP reporter (). The aim of the first library was to study the effect of the nucleotide composition of the 5′UTR (i.e. silent mutations in the 5′UTR) on protein levels; to this end, we randomized the 14 nt composing the 5′UTR, maintaining the nucleotide composition of the rest of the transcript (). The aim of the second library was to study how the protein levels are affected by synonymous nucleotide substitutions in the first 40 codons of the ORF. To this end, we modified only the third nt of codons 2–41 of the HRSVgp04 ORF, maintaining the encoded protein and the nucleotide composition outside this region (). Finally, the third library aimed at studying the effect of synonymous nucleotide substitutions in codons 42–81 of the ORF. To this end, we modified only the third nt of each of the codons 42–81 of the HRSVgp04 coding sequence, again maintaining the encoded protein and the nucleotide composition outside this region (). As explained in the Methods section, we designed the library variants such that their features (adaptation to the tRNA pool and mRNA folding) resemble endogenous genes. Thus, the relations reported here are expected to represent the effect of mutations on S. cerevisiae endogenous genes (see, for example, Citation16). The three libraries include a total of 207/151/25 variants respectively, and were named L5UTR, L2-41C, L42-81C (more details in the Methods section).
Figure 1. (A and B) Generating YFP libraries to study the effect of different parts of the transcript on translation efficiency and protein levels. Three libraries were generated and analyzed: in the first, L5UTR, we randomized the last 14 nt of the 5′UTR, but did not change the codons of the analyzed gene and YFP; in the second library, L2-41C, we modified only the first 40 codons of the ORF while maintaining the encoded protein; in the third, L42-81C, we modified only codons 42–80 of the ORF but maintained the encoded protein. (C) Upper part: The distribution of nucleotides in the modified positions in each of the 3 libraries. Lower part: The distribution of nucleotides in the 3rd position of each codon corresponding to the L2-41C and L42-81C libraries.
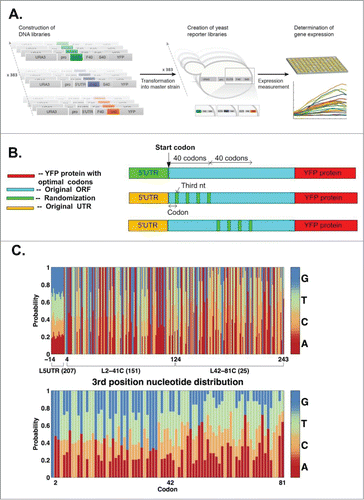
Even a small number of synonymous modifications in the transcript can significantly affect protein abundance
We measured the YFP and Optical Density (OD) of each variant over time and calculated their estimated protein levels (See and Methods section). Interestingly, though the number of randomized nucleotides is relatively very low (only a few dozen), and all the changes are strictly synonymous or silent, the changes in protein levels are between dozens to hundreds of percentages in all 3 cases. Specifically, the ratio (maximal estimated protein level)/( minimal estimated protein level) in the L5UTR, L2-41C and L42-81C libraries was 15.3, 3, and 1.6, respectively (after correcting for the different number of points in the 3 libraries it was 9.1, 2, and 1.6; see details in and its caption), while the (STD/mean) (i.e., the Coefficient of Variance) was 0.42, 0.12 and 0.12, respectively (see ). These results suggest that while synonymous or silent mutations in all parts of the ORF/UTR can significantly affect protein levels, mutations at the 5′UTR end tend to have a higher effect compared to adjacent synonymous mutations at the beginning of the ORF. In contrast, synonymous mutations in different parts of the ORF have a relatively comparable effect with an a bit higher effect at the region closer to the 5′end of the ORF. Our findings also show () that the protein level values related to the variant that achieves the highest protein level in each of the 3 libraries are relatively similar (the differences between the maximal protein levels values in the 3 libraries are less than ∼8.6%).
Figure 2. Total fluorescence levels (YFP) (A) and number of yeast cells (OD) (B) over 17 hours of a library generated by fusing the gene human respiratory syncytial virus HRSVgp04, after introducing modifications to the last 14 nucleotides of the 5′UTR (maintaining the encoded protein), to a YFP in S. cerevisiae. Each row corresponds to the measurements of one variant of the library over 17 hours; red denotes higher levels, and green denotes lower levels. (C) The resultant estimated protein levels, which are the mean YFP/OD over the period (Methods). (D–F) Include the mean estimated protein levels for the 3 libraries: UTR randomization (L5UTR) (D.), first 40 codons randomization (L2-41C) (E), second 40 codons randomization (L42-81C) (F); as can be seen, in all cases, the differences between the resultant fluorescence level per cell are between dozens to hundreds of percentages. (G) Blue: The ratio (maximal estimated protein levels)/(minimal estimated protein levels) for each library; Green: The ratio (maximal estimated protein levels)/(minimal estimated protein levels) for each library when sampling for the libraries L5UTR and L2-41C the same number of points as in the library L42-81C (average over 100 samples). (H). The CV (Coefficient of Variance) of the estimated protein levels in each library.
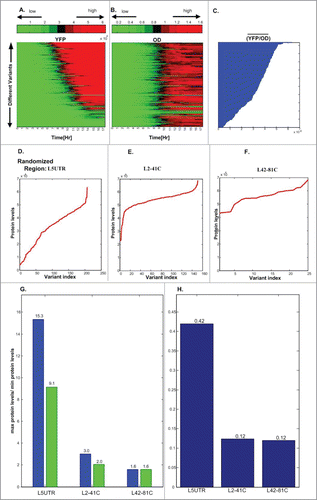
However, the lower protein level values among the libraries have more significant differences: the lowest protein level value in L2-41C was found to be 5.54 times higher than the lowest protein level in L5UTR; the lowest protein level value in L2-41C was 10.5 times higher than the lowest protein level in L5UTR. This result supports the conjecture that silent mutations (in terms of the encoded protein) near the beginning of the ORF may have strong negative effect on protein levels, probably due to their effect on translation initiation efficiency.Citation11,23,30-32 Here we provide a novel quantification of this effect in eukaryotes.
Silent mutations at the 5′UTR affect protein levels via their effect on translation initiation
To study the effect of various characteristics of the 5′UTR on protein levels, we generated 96 different features (see Table S1, and the Methods section) related to this region. Among others, the features include: the folding energy in different parts of the UTR (with respect to the beginning of the ORF); distance from the Kozak sequence (‘ACCATGG’), suggested to be the optimal sequence for START codon recognition by the pre-initiation complexCitation33; START codon context score,Citation34 which is a score aiming at estimating the optimality of the affinity of the nucleotide content surrounding the START codon to the pre-initiation complex; and similarity to binding sites of different RNA binding proteins.Citation35 Additional features are the frequency of each nucleotide in different positions of the 5′UTR; specifically, according to the Kozak ruleCitation30,31,34 and analysis of endogenous genes the nucleotide A at distance 3nt before the beginning of the ORF is the most favorable and nucleotide T is the least favorable nucleotide. It was suggested that this nucleotide interacts with the pre-initiation complex and the existence of ‘A’ in this position improves the recognition of the START codon by the pre-initiation complex and thus the initiation efficiency.
All the correlations throughout this study were based on Spearman's rank correlation coefficient. We found that the features with the top correlation with estimated protein abundance (PA) were (see Table S1 which includes the correlation and also correction for FDR): the START codon context score (r = −0.46; p = 2.3·10−12), the frequency of the nucleotide A at distance 3nt before the beginning of the ORF (r = −0.45; p = 6.6·10−12), average folding energy (FE) over all the UTR windows (r = 0.36, p = 10−7), the frequency of the nucleotide T at distance 3nt before the beginning of the ORF (r = −0.34; p = 3.3·10−7), and the FE of the window starting 7 nt before the beginning of the ORF (r = 0.34, p = 3.3·10−7). We also checked other features such as the affinity to 22 different RNA binding proteins (RBP); however, all the correlations were found to be either not significant, or (in one case) borderline significant with a very low correlation (r = 0.14, p = 0.044 which didn't not pass FDR filtering). Since the single nucleotide features mentioned above are related to the Kozak and context scores,Citation30,31,33,34 we conclude that the PA variability of the L5UTR can be significantly explained by features related to the translation initiation efficiency, the affinity to the pre-initiation complex and/or the folding of the mRNA in the region surrounding the start codon.
Correlation of protein levels with mean codon decoding times inferred based on Ribo-Seq
Ribosome profiling (Ribo-Seq) is a new approach that enables measuring ribosomal densities over the entire transcriptome at a single nucleotide resolutionCitation36,37 (see ). The method includes the following steps: cells are treated with cyclohexamide to arrest translation, ribosomes are fixed and ribosome-protected RNA fragments are recoveredCitation36,37 (see ). After processing and reverse-transcription, these are sequenced, mapped, and used to derive ribosomal density profiles. In order to estimate the typical nominal relative codon decoding times in endogenous S. cerevisiae genes under natural conditions, we implemented a novel statistical approach that filters biases and considers ribosomal traffic jamsCitation20 ( and Methods). Subsequently, we aimed at estimating the effect of codon usage bias on protein levels via the effect of codons on ribosomal elongation rates in natural conditions. To this end, we computed the correlation between the estimated Mean of the Typical codon Decoding Rates (MTDRCitation20; based on data fromCitation37, details in the Methods section) and PA in the L2-41C and L42-81C libraries. Interestingly, the correlation in the first library was relatively low but significant when controlling for the folding energy (FE) of the mRNA in this region, specifically: r (MTDR, PA) = 0.14 (p = 0.08); r (MTDR, PA | FE) = 0.24 (p = 0.0034). However, in the case of the second library, it was found to be both relatively high and significant: r (MTDR, PA) = 0.56 (p = 0.004); r (MTDR, PA | FE) = 0.6175 (p = 0.0013) (details in the Methods section).
Figure 3. Correlation between YFP levels and codon decoding times estimated based on Ribo-Seq in endogenous genes. The mean of the codon decoding rate of a variant is unit-less and is expected to be proportional to the mean decoding rate (1/time) of its codon (details in the Methods section). (A). The Ribo-Seq approach and estimating typical codon decoding times. (B). Correlation between mean typical codon decoding times and YFP levels for the first 40 codons library (the inset is related to bins of size 15 (Methods)). (C). Correlation between mean typical codon decoding times and YFP levels for the second 40 codons library (the inset is related to bins of size 3 (Methods)).
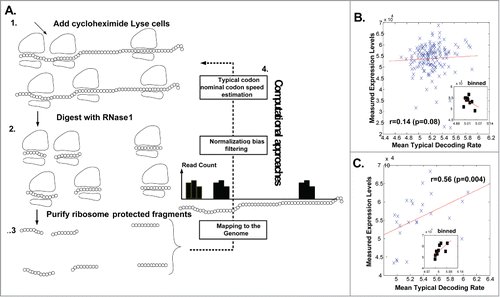
It is important to mention that results obtained in a similar analysis based on the tRNA adaptation index (tAI) (MethodsCitation38) were quantitatively similar to the ones obtained for MTDR, albeit weaker, demonstrating the advantage of the MTDR measure: in the case of L2-41C, r (tAI, PA | FE) = 0.24 (p = 0.0029); in the case of L42-81C, r (tAI, PA | FE) = 0.41 (p = 0.0466).
These findings may suggest that in S. cerevisiae the effect of codon usage at the very beginning of the ORF is strongly related to mRNA folding via its effect on initiation (as suggested in prokaryotesCitation11,13,14), but it is also significantly related to elongation rates. Moreover, the results may also suggest that the decoding times of codons is different at the beginning of the ORF than afterwards.Citation39
Furthermore, our results also suggest that after the beginning of the ORF the frequency of codons can affect protein abundance in a causal/direct way due to the fact that different codons have different decoding rates. Therefore, elongation rates can directly affect expression levels; this relation can be partially explained via the fact that codons that are recognized by tRNA species (and other translation factors) with higher abundance tend to have higher decoding rates.
Finally, the result demonstrates that it is possible to deduce codon decoding times for heterologous genes from the analysis of ribosome profiling in endogenous genes, potentially enabling new design tools for engineering gene expression in synthetic systems.
The correlation with estimated protein levels cannot be explained by changes in mRNA levels
To demonstrate that the reported signals and changes in the estimated protein levels are not due to effects on mRNA levels, but are directly related to translation, we measured the mRNA levels of some of the variants for each of the libraries (Methods). For example, in the case of the L42-81C library, we found that mRNA levels do not correlate with protein levels (r = −0.371; p = 0.497; correlation in the “wrong” direction). Fot these measurements, the correlation between MTDR and protein levels was found to be significant (r = 0.886, p = 0.033), and remained significant even after controlling for mRNA levels ( r(PA, MTDR | mRNA) = 0.89, p = 0.04). In contrast, no significant correlation was found between MTDR and mRNA levels (r = 0.371, p = 0.497). These results support the conjecture that the correlation between protein levels and MTDR is indeed related to ribosome elongation speed and not to mRNA levels. Similar conclusions were obtained for the other libraries: non-significant correlation between mRNA levels and protein levels (r = 0.19 p = 0.66 for L5UTR; r = −0.22, p = 0.4 for L2-41C ) were observed.
The direction of the effect of synonymous codons and silent mutations on protein abundance may vary along the coding sequence
At the next step, we aimed at understanding the effect of synonymous codons in different parts of the transcript on protein levels via their effect on mRNA folding. To this end, we computed the local mRNA folding energy in different windows for all the variants in all the libraries (Methods). The local mRNA folding energy is predicted for overlapping/sliding windows of length 40nt with a slide of 1nt (Methods); the folding energy of a nucleotide sequence is related to the energy needed for its unfolding after folding to its strongest structure: a more negative number is related to stronger folding.
For each nt position (which induces a window of length 40nt as described above), we computed the correlation between folding energy and protein levels. As can be seen in , the correlation between local mRNA folding energy and protein levels is significant and positive at the 5′UTR and the first ∼10–13 positions/windows inside the ORF (stronger folding at the 5′ end of the ORF tends to have a negative effect on protein levels). This result supports previous conclusions based on the experimental analysis of the prokaryote E. coliCitation11; however, this is the first time such a causal result is reported for a eukaryote. In addition, it supports previous evolutionary analyses of transcripts that suggested that in many organisms there is selection for weak local folding of the mRNA sequence at the beginning of the ORF, probably to improve initiation efficiency and the recognition of the START site by the pre-initiation complex.Citation23,32 It may also explain the difference in the ‘linearity’ of the profiles that appear in , relative to the profile in E. (that is less ‘linear’), which may be related to the central effect of mRNA folding near the 5′end of the ORF on protein levels in L2-41C. It is probable that this affect is distributed in a bi-modal manner in the database, so that it is relatively easy to randomly generate variants with either strong folding / many base-pairs / relatively low protein levels, or weak folding / few base-pairs / relatively high protein levels.
Figure 4. The effect of codons on the protein levels varies along the coding sequence. (A) Correlation with folding window (40nt) in the beginning of the UTR and the beginning of the ORF based on the L5UTR library. (B) Upper-part – Correlation with folding windows (40nt) at the beginning of the ORF based on the L2-41C library in heterologous and endogenous genes (red/green denotes significant positive/negative correlation respectively). Lower-part – the mean genomic mRNA folding energy in endogenous genes (red/green denotes selection for significant weak/strong folding respectively). (C) Correlations between mean local FE and PA in S. cerevisiae endogenous genes vs. correlations between local FE and PA in the heterologous genes (details in the main text). (D) Local FE Z-scores in S. cerevisiae endogenous genes corresponding to the FE in the real genome in comparison to randomized versions of the genome vs. correlations between the local FE and PA in the heterologous genes (details in the main text). (E) The effect of codons on the protein levels in the L2-41C library vs. the effect of codons on protein levels based on the L42-81C library.
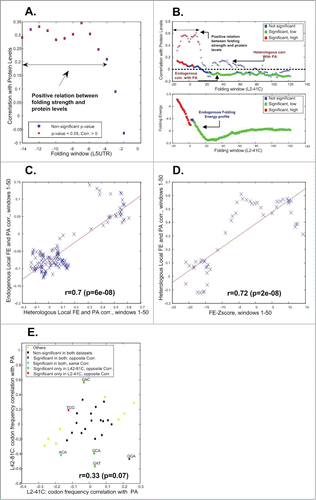
Interestingly, as can be seen in , after the first 20 positions/windows both in the endogenous genes and in the heterologous genes there is a region where the correlation between folding energy (FE) and protein levels (PA) is reversed (i.e. negative, strong folding tends to increase protein levels).
In the case of the endogenous genes, the region is relatively long (dozens of codons afterwards) and significant. In the case of the heterologous genes, there are several such short regions which are not significant (probably due to the lower number of points).
This result supports a previous study that demonstrated based on a comparison to randomized versions of the genome that there is a selection for strong mRNA folding in after the first ˜14 positions/windows of the ORFCitation40 (); this region appears downstream of region with a selection for weak mRNA folding at the very beginning of the ORF. A possible explanation for this result is that strong mRNA folding further downstream improves the fidelity of translation initiation by blocking the pre-initiation complex from scanning it, and increasing the probability that it will remain in the vicinity of the START codon and recognize it correctly.Citation34,41-43 It is also possible the strong structure downstream may help prevent strong folding at the START codon. In order to further demonstrate that the reported relation between mRNA folding and protein levels in our heterologous system can explain folding evolution in S. cerevisiae endogenous genes, we observed that the effect of mRNA folding on protein levels is similar in endogenous genes and in our heterologous libraries. To this end, we computed the correlation between 1) the vector of correlations between the mean local FE and PA in S. cerevisiae endogenous genes and 2) the vector of correlations between the local FE and PA in the first 50 windows heterologous gene; we found it to be highly significant (r = 0.7; p = 6.28·10−8; ). Similarly, we showed that the observed correlation between protein levels and folding energy in the heterologous library increases with the significance of the level of selection of the latter in endogenous genes. To this end, we computed the correlation between 1) the vector of the local FE Z-score in the first 50 windows of the wild-type S. cerevisiae endogenous genes, with respect to the corresponding randomized variants maintaining various genomic properties (Methods) and 2) the vector of correlations between the local FE and PA in the first 50 windows in the heterologous gene; we found it to be highly significant (r = 0.7; p = 2.4·10−8; ).
At the next step, we aimed at understanding how the effect of different codons on protein levels changes along the coding sequence. Each dot in correspond to one codon, and includes the correlation between its frequency and protein levels in the library L2-41C (x-axis) vs. the correlation between its frequency and protein levels in the library L42-81C (y-axis). Interestingly, while in general an agreement between the 2 libraries (r = 0.33; p = 0.07) can be observed, there are cases where the effect of codon frequency on protein levels in the first 40 codons is different from its effect in the second 40 codons, supporting the conjecture that the effect of codon frequency on protein levels is context dependent and changes along the ORF. For example, the codon GGA has a positive effect on protein levels when it appears in codons 2–42 (r = 0.24, p = 0.0035 ), but it has negative effect on protein levels when it appears in codons 42–81 (r = −0.47, p = 0.018 ) ().
Discussion
Our synthetic biology driven approach to study the effect of synonymous or silent mutations in different parts of the transcript on protein abundance, enabled us to gain an improved understanding of the relation between transcript features and their corresponding protein levels. Previous studies based on evolutionary systems biology of endogenous genes could not infer the causality of the relations between transcript features and protein levels. Our systematic study of rationally designed heterologous genes bypasses many of the pitfalls characterizing the investigation of endogenous gene expression. This approach is becoming increasingly available due to rapid advances in DNA library construction methodologies. The results reported in here emphasize the utility of applying synthetic biology for deciphering how transcripts modulate their expression and enables us to provide quantitative estimations of the relations between various features of the transcript and translation/protein levels in a eukaryote.
Our analyses support the hypothesis that in eukaryotes weak mRNA folding near the beginning/end of the ORF/5′UTR respectively, improves translation initiation and increases protein levels.Citation11,13,23,32,40-42,44,45 Specifically, the results emphasize the negative effect of strong mRNA folding at the beginning/end of the ORF/5′UTR on translation initiation and protein levels.Citation11,13,23,32,41,44,45 The analysis also quantifies the effect of the affinity of the nucleotide context surrounding the START codon to the pre-initiation complex on protein levels, demonstrating that this is one of the major determinants that can explain the effect of silent/synonymous mutations in this region on translation and protein levels. Previous studies addressing this problem either analyzed small numbers of heterologous or mutated variants,Citation30,31 or analyzed endogenous genes.Citation34,46
Our analysis provides the first comparative estimation of the possible effect of silent/synonymous mutations in different parts of the transcript on protein levels. Specifically, it suggests that the most deleterious mutations (the ones resulting in the lowest protein levels) at the initiation region (the 5′UTR) have one order of magnitude higher effect on protein levels abundance than the most deleterious mutations in the elongation region (the coding region): the lowest protein level obtained for 5′UTR (L5UTR) mutations was around 10 times lower than the lowest protein level obtained for coding region mutations (codons 42–81; L42-81C). It was suggested that the beginning of the ORF (codons 2–41; L2-41C) is related to both initiation and elongationCitation16; indeed the lowest protein level obtained for a mutation in this region was around 5 times higher than the lowest protein level obtained for 5′UTR mutations (i.e., between the 2 other regions).
Finally, we show that codon decoding rates, inferred via ribosome profiling measurements, affect protein levels in a direct and casual way. This result supports previous studies performed on endogenous genes that suggested, based on various proxies of elongation rates, that protein levels can be affected by codon distributions in the ORF, possibly due to their effect on translation elongation.Citation1,17,23,47 Our study demonstrates for the first time the strong relation between codon decoding times and protein levels in a direct causal manner. It is important to emphasize that this result does not contradict previous findings suggesting that the correlation between measures of codon frequencies and protein levels in endogenous genes is partially due to non-direct reasons such as global ribosomal allocation, protein folding, and translation fidelity, etc.Citation16,24,26,48 Thus, predictors based on ribosomal profiling data may be used for inferring protein levels of heterologous genes. Such predictors may be helpful specifically in cases of genes that do not enable reliable measurements of protein levels (e.g. very short onesCitation49), and can be used for engineering heterologous genes for tailored gene expression. Furthermore, this result demonstrates that elongation speed is not constant and that both this speed and the associated protein levels can be affected by synonymous features of the transcript (as was suggested for example inCitation17,20-22), and not only by initiation rates and/or the amino acid content encoded in the coding sequence (as was suggested inCitation18,19,50).
The current accepted model is that translation initiation is the rate limiting step of the translation process, and thus synonymous mutations near the beginning of the ORF modulate protein levels, while synonymous mutations downstream of this region do not (see, for example,Citation11,51). The analyses reported here demonstrate that elongation and codon distribution downstream of the ORF 5′ end do significantly modulate protein levels (specifically, when the region near the START codon is ‘optimal’). This does not contradict the fact that the nucleotide composition near the beginning of the ORF, as well as translation initiation, can have stronger effects on protein levels compared to codon frequencies downstream of the ORF 5′ end and translation elongation.
Our study differentiates the nature and strength of the effect of synonymous mutations in different parts of the transcript/ORF on protein levels, and may be used to guide the design of synthetic genes.Citation52 The reported results support the notion that the term ‘optimal codons’ (see, for example,Citation27,53), which describes the preferred codon for each amino acid in a certain organism, should be fine-tuned; optimal codons are context dependent and may vary among different parts of the ORF. Specifically, we analyzed a viral gene (HRSVgp04) and demonstrated that silent and synonymous mutations in different parts of its transcript can significantly affect its protein levels. Thus, these results may serve as a proof of concept for the use of accurate design of such mutations to generate rationally tailored expression of genes.
Materials and Methods
Methods for DNA library construction
Construction of all the DNA variants of the HRSV genes fused to the reporter gene was performed according to the methods described in.Citation54,55 A cloned and sequenced wild type version of the HRSV gene was constructed as a Minigene (IDT DNA). HRSV variants were generated by fully or partially randomizing specific nucleotide positions within the HRSV gene. Randomized nucleotide positions were ordered as machine mixed synthetic nucleotides (IDT DNA) within DNA Ultramers (IDT DNA), that were used to edit the wild type HRSV fragment and fuse it to the YFP gene, following the methods described inCitation54,55 with slight modifications, as follows:
The HRSV wild type Minigene was edited to generate variants by using it as a template in extension PCR reactions using different randomized Ultramers as the reverse primer. The Ultramers also contained at their 5′ a segment for homologous recombination into a promoter-less YFP in the yeast genome. The forward primer of the HRSV Minigene PCR amplification contained at its 5′ an overlap segment for its fusion with a second PCR fragment, that contained a promoter for the HRSV-YFP fusion and a URA3 selection cassette (amplified from a pre-made template).
Primer phosphorylation
300 pmol of single stranded DNA in a 50 μl reaction containing 70 mM Tris–HCl, 10 mM MgCl2, 7 mMdithiothreitol, pH 7.6 at 37 °C, 1 mM ATP and 10 units T4 Polynucleotide Kinase (NEB) was used. Reaction is incubated at 37 °C for 30 min, then at 42 °C for 10 min and inactivated at 65 °C for 20 min.
Elongation between single stranded DNA fragments
1 pmol of single stranded DNA of each progenitor in a 25 μl reaction containing 2.5 μl of Hot Start DNA Polymerase (Novagen, 71086-3) reaction according to manufacturer's guidelines was used. Three cycles of annealing were executed for each elongation to ensure full yield of elongation.
PCR of elongation reaction
All PCR reactions were performed in 96 well PCR microplates, using KOD Hot Start DNA Polymerase (Novagen, 71086-3) according to its protocol.
Digestion of phosphorylated PCR strand by Lambda exonuclease
1–5 pmol of 5′ phosphorylated DNA termini in a 30 μl reaction containing 67 mM Glycine–KOH, 2.5 mM MgCl2, 0.01% Triton X-100, 5 mM 1,4-dithiothreitol, 5.5 units Lambda exonuclease (Epicentre) and SYBR Green diluted 1:50,000. Thermal Cycler program is 37 °C for 15 min, 42 °C for 10 min, enzyme inactivation at 65 °C for 10 min.
Chemical oligonucleotide synthesis
Standard PCR primers for all experiments were ordered from IDT with standard desalting.
DNA Ultramers™ (IDT DNA) harboring modified HRSV sequences were used as PCR primers in order to insert the variable segments of the HRSV variants. Specifically, we integrated precise mixes of degenerate (N, K and others) bases at predefined positions that effectively recoded the genes according to a predefined DNA sequence specification.
DNA purification
DNA purifications required in the process of DNA construction were performed with the ZR-96 DNA Clean & Concentrator (Zymo research) kit using standard protocols.
The master strain
The master strain was created by integrating into the yeast genome a cassette containing a promoter-less YFP, followed by a NAT (Nourseothricin) resistance marker under its own promoter. The entire sequence was inserted into the his3Δ1 locus.
Transformations of the library into yeast his3Δ1 locus
All HRSV variants were transformed into the master strain using the LiAc/SS carrier DNA/PEG method following the procedures described in.Citation56 Cells were plated on solid agar SD-URA selective media and incubated at 30 °C for 3–4 days. Transformant colonies were handpicked and patched on SD-URA + NAT (Werner BioAgents) agar plates in 384 format. Correct transformation was verified for all variants by PCR amplification from the yeast's genome, gel electrophoresis and DNA Sequencing. The constructs were transformed into the master strain which contained a promoter-less YFP coding sequence at the his3Δ1 locus. Each synthetic construct contained a URA3 selection marker under its own promoter followed by a TEF promoter, the relevant HRSV gene ORF, and the beginning of the YFP ORF (for recombination purposes).
Sequencing
Colonies were picked manually from the plates of each variant, the specific integration locus was PCR amplified from each clone. Correct size amplifications were verified by gel electrophoresis. Amplicons were sequenced in house using Sanger sequencing. Colonies with the correct sequences were chosen for analysis.
Culture, fluorescence measurements and mRNA quantification
The variant strains of all genes were maintained in 384 well format on SD-URA + NAT solid medium using the Singer colony arrayer (RoToR, Singer instruments). In order to measure growth and fluorescent protein expression, the Singer colony arrayer was used to inoculate all colonies of the library from solid medium into 100 μl of SD-URA media in a 384 well growth microplate (Greiner bio-one, 781162). Following 24 h of pre-incubation, 5 μl of the yeast cultures was diluted into 80 μl of SD complete media in a 384 well microplate, to reach a starting O.D600 of ∼0.1–0.2.
A microplate reader (Neotec Infinite M200 monochromator) was then set to measure the 384 well plates following parameters in cycles of 10 min: Cell growth (as extracted from absorbance at 600 nm) and YFP expression (Ex. 500 Em. 540). Each cycle contained 4 min of orbital shaking at amplitude of 3 mm. The number of cycles was set to 100 (16h) and the temperature to 30 °C. We performed triplicates of the expression measurements.
mRNA quantification of YFP reporter
mRNA level measurements were performed using quantitative real-time PCR (qPCR). Yeast strains were grown to mid-log and RNA purification was performed using the MasterPure™ yeast RNA purification kit (Epicentre) according to manufacturer's protocol. First strand cDNA synthesis was performed using the SuperScript® III First Strand Synthesis kit (Invitrogen) according to manufacturer's protocol, and qPCRs on yeast cDNA's were performed in a LightCycler 480 Real-Time PCR system (Roche) in 384 well-format using SYBR®. Green detection mode and relative quantification analysis was performed using default parameters of the ΔΔCT method. Normalization of mRNA levels was performed according to mRNA levels of the highly expressed Actin gene, and several negative controls were performed to validate our results including (1) no reverse transcription, (2) no PCR template, (3) no YFP (Yeast strain without the HRSV fusion), as well as a positive control for RNA extraction with a known RNA extract. The estimated mRNA levels were based on the average of all replicates.
Sequences of primers for RT-PCR are as follows:
Actin Fwd: ‘CTGGGACGATATGGAAAAGAT’;
Actin Rev: ‘GTTCACTCAAGATCTTCAT’;
YFP Fusion Fwd: ‘ATTCACTTGGTGTTGTCCCAATTTTGG’;
YPF Fusion Rev: ‘GATCTGGGTATCTAGCAAAACACATC’.
Computational analysis
Designing the heterologous gene variants
As describe in the main text, we analyzed the gene HRSVgp04 and generated 3 libraries (L5UTR, L2-41C, L42-81C) to understand the distinct effect of the nucleotide compositions in different regions of the transcript on protein levels. The structure of all 3 libraries was identical: All had the same promoter, followed by the 5′UTR (14nt) of the TEF gene, and the HRSVgp04 gene fused with a YFP reporter ().Citation23,32
To make sure that the effect of the mutations of protein levels will resemble its effect in natural conditions,Citation16,57 we chose for each amino acid of the HRSVgp04 protein the codon with the highest tRNA adaptation index (tAI) in S. cerevisiae coding sequencesCitation38_ENREF_23; then we choose the first 39 nucleotides of the coding sequence such the strength of the folding there will be minimal (Folding energy close to zero)Citation23,32; in addition, the context/Kozak sequence related to the 6 last nucleotides of the 5′UTR was optimized based on the optimal sequence of S. cerevisiae endogenous genes.Citation30,34 All the variant sequences appear in Table S2.
By randomizing this basic transcript we generated the 3 libraries (L5UTR, L2-41C, L42-81C).
Normalizations and filtering of the data
Estimated protein levels were based on the mean YFP/OD over all cycles. Note that the reported results are robust to various definitions of outlier filtering.
Inferring the mean typical decoding rate of an ORF based on ribosomal profiling data
The method for estimating codon decoding times, MTDR, was published in.Citation20 For clarity we briefly describe the method here: S. cerevisiae ribosomal profiles were reconstructed using the data published in the GEO database, accession number GSE13750 (GSM346111, GSM346114).Citation57
Ribosomal profiles were normalized (to get normalized footprint counts, NFCs) as in previous studies, by dividing each profile by its mean read count; this enables to control for variation in initiation rates and mRNA levels of different genes, and analyzing/comparing all the genes/profiles in a unified manner. Next, for each codon type we generated a vector consisting of NFC values originating from all analyzed genes. These vectors were used to generate, for each codon type, a histogram reflecting the probability of observing each NFC value in the expressed genes (the number of times each NFC value occurs in the data normalized by the total number of times the codon appears in the data), that was named the ‘NFC distribution’ of the codon.
Based on the characteristics of the NFC distributions (see some explanations below and inCitation20) we suggest https://www.google.com/search?q=we+hypothesizedandspell=1andsa=Xandei=U6sEUtG6NcXmOaPVgfgMandved=0CCoQvwUoAA that their topology could result from a superposition of 2 distributions/components: the first one describes the ‘typical’ decoding time of the ribosomes, which was modeled by a normal distribution characterized by its mean and variance
with a probability density function
(for a random variable
) of Citation20:
(1)
(1) The second component describes relatively rare translational pauses and ribosomal interactions, such as traffic jams due to the codons' different translation efficiency, and was modeled by a random variable with an exponential distribution characterized by one parameter
, with a probability density function
(for a random variable
) of:
(2)
(2) The mean of the exponential distribution is 1/ λ and can reflect the average NFC of a non-typical/non-nominal phenomenon such as traffic jams, pauses, and biases.
It is known that the distribution of a random variable w(t,) which is the sum of 2 independent random variables and
(i.e. w(t) =
+
), is calculated as a convolution between the 2 distributionsCitation58:
(3)
(3) Thus, the summation of 2 independent normal and exponential random variables corresponding to the distributions mentioned above results in a distribution which is named ‘exponentially modified Gaussian’ (EMG), which is a convolution of a normal and exponential distribution. Formally, the EMG distribution function
, of a random variable
(where
)Citation59 is:
(4)
(4) where
(5)
(5) The parameters
were estimated by fitting the measured NFC distributions to the EMG distribution, under the log-likelihood criterion.
Intuitively, the model above is supported by the following points (see all the details inCitation20): 1) A simulation of ribosome profiling when there are no traffic jams, biases, or pauses (the simulation was based on the S. cerevisiae genome and ribosomal profiling measurements). In this case we found the NFC of the codon to be normal/Gaussian. Thus, we determined that the component of typical decoding time to be normal/Gaussian. 2) When traffic jams (various codon decoding rates) and extreme NFC values are added to the model/simulation the distribution is skewed and it looks log normal or like an EMG. 3) We expect that extreme pauses or traffic jams (due to slower codons for example) will increase the NFC values of a codon, resulting in a right tail. A natural and simple way to describe such a right tail is via an exponential distribution. This is the reason we added the second component of the EMG distribution, the exponential distribution. 4) We performed a simulation of ribosomal profiling where we know the translation rate/time of each codon. We run the EMG filter on the simulation and show that it accurately estimates the true translation times (r = 0.99). 5) We used Akaike information criterion (AIC) to show that the NFC distributions are better described by an EMG distribution than by either exponential or normal distributions. 6) We found that the estimated typical decoding rate correlates with measurements related to the translation decoding rate (such as tRNA levels) and the mean typical decoding rate correlates with protein levels and proteins per mRNA levels of endogenous genes.
The typical decoding time of a codon is its μ; the mean typical decoding time/rate of a gene is the geometric mean of μ or 1/ μ respectively.
All the per-codon inferred values appear in Supplemental Table 3.
Note that the normalized footprint count (Z) is dimensionless since it is obtained via normalizing/dividing the vector of read count (RC) related to each coding region by the mean RC of the coding region. This means that in our case μ, λ, and σ are dimensionless parameters of this distribution(s) (normal distribution, exponential distribution, and EMG). Therefore (by definition) the normalized read count and also μ are dimensionless values that describe the ratio between 1) the read count of the codon and 2) the mean read count of the codons.
We expect that (given a certain initiation rate and mRNA levels) the read count related to a codon be proportional to the time the ribosome spends on it (or to the probability to see the ribosome on the codon relatively to other codons), thus, the normalized read count and also μ (and λ, and σ) are dimensionless values that describe the ratio between 1) the decoding time of the codon and 2) the mean decoding time of codons.
Hence we multiply the μ for a certain codon c by a constant related to the mean decoding time of codons in a genome, we should get an estimation of the decoding time of the codon c.
mRNA folding predictions
The local pre-mRNA folding profiles were computed based on the ViennaRNA PackageCitation60 with default parameters (e.g., temperature is 37 °C). We used a 40 nt length sliding window (with 1 nt step), corresponding to the approximated ribosome size in fungi.
In the case of the correlations reported in , we considered the mean folding energy of all the windows intersecting with the variable part of the library. In the case of , we considered the folding in windows of size 40 nt (details in the main text).
Correlation between codon frequencies and protein levels
For the correlation reported in we considered only the 31 codons with non-constant frequency distribution both in the L2-41C and the L42-81C library.
Folding profiles and protein abundance of endogenous S. cerevisiae genes ()
The coding sequences and UTRs of S. cerevisiae were downloaded from.Citation61 The coding sequences sequences were randomized as follows: for each amino acid in each gene, we sampled a codon from the distribution of genomic codon frequencies/codon-bias in the S. cerevisiae (i.e., more frequent codons in the genome have a higher probability of being sampled). Thus, the randomized variants maintain both the amino acid content of each coding sequence, and the codon frequencies of the original genome. 20 randomized versions of the genome were generated in this manner; local folding vectors were computed for each gene in the randomized genome, and were used to generate the z-score profiles that appear in .
For S. cerevisiae endogenous genes we considered 4 quantitative large scale measurements of Protein Abundance (PA).Citation6,62,63 We averaged across the 4 datasets (after normalizing each data set by its mean) to reduce experimental noise (resulting with 1,448 genes with measurements in all datasets).
Statistical analyses
Statistical analyses were performed using Matlab. All the reported correlations (including partial correlations) are Spearman. In this study we computed, among others, partial correlations between 2 variables (x and y) when controlling for the third variable (z), which is denoted by r(x,y|z). Partial correlation is a standard way to measure the degree of association between 2 random variables, when the effect of a set of controlling random variables is removed. False Discovery Rate (FDR see Table S1) was performed based on Benjamini and HochbergCitation64 and StoreyCitation65 methods (we used FDR cutoff of 5%).
The tAI Index
The tAI indexCitation38 uses the adaptiveness of the codons of a gene to the tRNA pool. Denote the adaptiveness value of codon of type with
. Let
be the copy number of the
-th anti-codon that recognizes the
-th codon, and let
be the selective constraint of the codon-anti codon coupling efficiency. The
vectorCitation38,66
,
,
,
,
,
,
,
,
] was defined for eukaryotes as [0, 0, 0, 0, 0.561, 0.28, 0.9999, 0.68, 0.89]. Then, the absolute adaptiveness value of a codon is defined by
(6)
(6) Let us mark the relative adaptiveness value of codon
with
, by normalizing each
with the maximal
value among the 61
values. The tAI index for a gene is an average over the
of its codons.
The analyzed UTR features for L5UTR
In addition to local mRNA folding calculated across 40nt windows, we analyzed the following features:
ATG context score was computed as inCitation34:
Specifically, we calculate the context score (corresponding to 6 nt upstream of the START codon and 3 nt downstream of it) according to the following steps: 1. Select percentage of highly expressed/translated endogenous genes (in our case we used 2% of highly expressed genes, according to the ribosomal load). 2. Calculate a position specific scoring matrix (PSSM) based on the nucleotide context surrounding the start codon of the selected highly expressed genes. Let 3. Calculate the context score for a START codon according to the PSSM:
, where j is the variant index, i the nucleotide position,
the probability that the ith nucleotide of the j-th gene appears in the i-th position (based on the PSSM).
The Kozak score was computed as the hamming distance (i.e. number of mutations) from the Kozak consensus sequence: ‘ACCATGG’.Citation33
The similarity to binding sites of different RNA binding proteins was based on consensus sequences of 22 RBP taken fromCitation35. The score of each variant was based on the hamming distance of a window in the variant (we checked all sliding windows with a length identical to the consensus sequence) with the minimal number of mutations relative to the consensus sequence.
In addition, for each position in the UTR, and for each of the 4 nucleotides, we defined a binary variable (i.e., 14·4= 56 variables) as follows: its value is ‘1’ if the nucleotide appears in the position; otherwise the value is 0. Correlation of the different UTR features with protein levels appear in Table S1.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary_Files.zip
Download Zip (46 KB)Funding
This study was supported in part by a fellowship from the Edmond J. Safra Center for Bioinformatics at Tel-Aviv University. The study was partially supported by the Israel Cancer Research Fund (ICRF) and German-Israeli Foundation (GIF).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Lithwick G, Margalit H. Hierarchy of sequence-dependent features associated with prokaryotic translation. Genome Res (2003) 13:2665-73; PMID:14656971; http://dx.doi.org/10.1101/gr.1485203
- Zur H, Tuller T. Transcript features enable accurate prediction and understanding of gene expression in S. cerevisiae. BMC Bioinfomatics. (2013) 14:S1; http://dx.doi.org/10.1186/1471-2105-14-S15-S1
- Tuller T, Kupiec M, Ruppin E. Determinants of protein abundance and translation efficiency in S. cerevisiae. PLoS Comput Biol (2007) 3:2510-19; http://dx.doi.org/10.1371/journal.pcbi.0030248
- Vogel C. Abreu Rde S, Ko D, Le SY, Shapiro BA, Burns SC, Sandhu D, Boutz DR, Marcotte EM, Penalva LO. Sequence signatures and mRNA concentration can explain two-thirds of protein abundance variation in a human cell line. Mol Syst Biol (2010) 6:1-9; http://dx.doi.org/10.1038/msb.2010.59
- Huang T, Wan S, Xu Z, Zheng Y, Feng KY, Li HP, Kong X, Cai YD. Analysis and prediction of translation rate based on sequence and functional features of the mRNA. PLoS One (2011) 6:e16036; PMID:21253596; http://dx.doi.org/10.1371/journal.pone.0016036
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature (2003) 425:737-41; PMID:14562106; http://dx.doi.org/10.1038/nature02046
- Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol (1999) 19:1720-30; PMID:10022859
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature (2011) 473:337-42; PMID:21593866; http://dx.doi.org/10.1038/nature10098
- Lu P, Vogel C, Wang R, Yao X, Marcotte EM. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat Biotechnol (2007) 25:117-24; PMID:17187058; http://dx.doi.org/10.1038/nbt1270
- Wang M, Weiss M, Simonovic M, Haertinger G, Schrimpf SP, Hengartner MO, von Mering C. PaxDb, a Database of Protein Abundance Averages Across All Three Domains of Life. Mol Cell Proteomics (2012) 11:492-500; http://dx.doi.org/10.1074/mcp.O111.014704
- Kudla G, Murray AW, Tollervey D, Plotkin JB. Coding-sequence determinants of gene expression in Escherichia coli. Science (2009) 324:255-8; PMID:19359587; http://dx.doi.org/10.1126/science.1170160
- Welch M, Govindarajan S, Ness JE, Villalobos A, Gurney A, Minshull J, Gustafsson C. Design parameters to control synthetic gene expression in Escherichia coli. PLoS One (2009) 4:1-10; http://dx.doi.org/10.1371/journal.pone.0007002
- Goodman DB, Church GM, Kosuri S. Causes and effects of N-terminal codon bias in bacterial genes. Science. (2013) 342:475-9; http://dx.doi.org/10.1126/science.1241934
- Allert M, Cox JC, Hellinga HW. Multifactorial determinants of protein expression in prokaryotic open reading frames. J Mol Biol (2010) 402:905-918; http://dx.doi.org/10.1016/j.jmb.2010.08.010
- Bentele K, Saffert P, Rauscher R, Ignatova Z, Bluthgen N. Efficient translation initiation dictates codon usage at gene start. Mol Syst Biol (2013) 9:675., 10.1038/msb.2013.1032.; PMID:23774758; http://dx.doi.org/10.1038/msb.2013.32
- Tuller T, Zur H. Multiple Roles of the Coding Sequence 5′ End in Gene Expression Regulation. Nucleic Acids Res (2015) 43:13-28; PMID:25505165; http://dx.doi.org/10.1093/nar/gku1313
- Supek F, Smuc T. On relevance of codon usage to expression of synthetic and natural genes in Escherichia coli. Genetics (2010) 185:1129-34; PMID:20421604; http://dx.doi.org/10.1534/genetics.110.115477
- Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell (2011) 147:789-802; PMID:22056041; http://dx.doi.org/10.1016/j.cell.2011.10.002
- Qian W, Yang J-R, Pearson NM, Maclean C, Zhang J. Balanced codon usage optimizes eukaryotic translational efficiency. PLoS genetics (2012) 8:e1002603; PMID:22479199; http://dx.doi.org/10.1371/journal.pgen.1002603
- Dana A, Tuller T. The effect of tRNA levels on decoding times of mRNA codons. Nucleic Acids Res. (2014) 42:9171-81; PMID:25056313
- Chu D, Kazana E, Bellanger N, Singh T, Tuite MF, von der Haar T. Translation elongation can control translation initiation on eukaryotic mRNAs. EMBO J. (2014) 33:21-34. Epub 201382013 Dec 201385619; PMID:24357599; http://dx.doi.org/10.1002/embj.201385651
- Gardin J, Yeasmin R, Yurovsky A, Cai Y, Skiena S, Futcher B. Measurement of average decoding rates of the 61 sense codons in vivo. Elife. (2014); 3 10.7554/eLife.03735.; PMID:25347064; http://dx.doi.org/10.7554/eLife.03735
- Tuller T, Waldman YY, Kupiec M, Ruppin E. Translation efficiency is determined by both codon bias and folding energy. Proc Natl Acad Sci U S A (2010) 107:3645-50; PMID:20133581; http://dx.doi.org/10.1073/pnas.0909910107
- Plotkin JB, Kudla G. Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet (2010) 12:32-42; PMID:21102527; http://dx.doi.org/10.1038/nrg2899
- Chamary JV, Parmley JL, Hurst LD. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat Rev Genet (2006) 7:98-108; PMID:16418745; http://dx.doi.org/10.1038/nrg1770
- Sauna ZE, Kimchi-Sarfaty C. Understanding the contribution of synonymous mutations to human disease. Nat Rev Genet (2013) 12:683-91; http://dx.doi.org/10.1038/nrg3051
- Hershberg R, Petrov DA. Selection on codon bias. Annu Rev Genet (2008) 42:287-99; PMID:18983258; http://dx.doi.org/10.1146/annurev.genet.42.110807.091442
- Gingold H, Pilpel Y. Determinants of translation efficiency and accuracy. Mol Syst Biol. (2011) 7:481., 10.1038/msb.2011.1014.; PMID:21487400; http://dx.doi.org/10.1038/msb.2011.14
- Novoa EM, Ribas de Pouplana L. Speeding with control: codon usage, tRNAs, and ribosomes. Trends Genet. (2012) 28:574-81; http://dx.doi.org/10.1016/j.tig.2012.07.006
- Kozak M. Point mutations close to the AUG initiator codon affect the efficiency of translation of rat preproinsulin in vivo. Nature (1984) 308:241-6; PMID:6700727; http://dx.doi.org/10.1038/308241a0
- Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell (1986) 44:283-92; PMID:3943125; http://dx.doi.org/10.1016/0092-8674(86)90762-2
- Gu W, Zhou T, Wilke CO. A universal trend of reduced mRNA stability near the translation-initiation site in prokaryotes and eukaryotes. PLoS Comput Biol. 2010 6:1-8 (2010); http://dx.doi.org/10.1371/journal.pcbi.1000664
- Hamilton R, Watanabe CK, de Boer HA. Compilation and comparison of the sequence context around the AUG startcodons in Saccharomyces cerevisiae mRNAs. Nucleic Acids Res. (1987) 15:3581-93.; PMID:3554144
- Zur H, Tuller T. New Universal Rules of Eukaryotic Translation Initiation Fidelity. PLoS Comput Biol (2013) 9:e1003136; PMID:23874179; http://dx.doi.org/10.1371/journal.pcbi.1003136
- Li X, Quon G, Lipshitz HD, Morris Q. Predicting in vivo binding sites of RNA-binding proteins using mRNA secondary structure. RNA. (2010) 16:1096-107. Epub 2012010 Apr 2017223.; PMID:20418358
- Ingolia NT. Ribosome profiling: new views of translation, from single codons to genome scale. Nat Rev Genet. (2014) 15:205-13; PMID:24468696; http://dx.doi.org/10.1038/nrg3645
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science (2009) 324:218-23; PMID:19213877; http://dx.doi.org/10.1126/science.1168978
- dos Reis M, Savva R, Wernisch L. Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res (2004) 32:5036-44; PMID:15448185; http://dx.doi.org/10.1093/nar/gkh834
- Dana A, Tuller T. Properties and Determinants of Codon Translation Speed Distributions. BMC Genomics (2014); 15 Suppl 6:S13; PMID:25572668
- Tuller T, Veksler-Lublinsky I, Gazit N, Kupiec M, Ruppin E, Ziv-Ukelson M. Composite effects of gene determinants on the translation speed and density of ribosomes Genome Biol (2011) 12:R110; PMID:22050731
- Kozak M. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene (2005) 361:13-37; PMID:16213112; http://dx.doi.org/10.1016/j.gene.2005.06.037
- Kozak M. Downstream secondary structure facilitates recognition of initiator codons by eukaryotic ribosomes. Proc Natl Acad Sci (1990) 87:8301-05; http://dx.doi.org/10.1073/pnas.87.21.8301
- Kochetov AV, Palyanov A, Titov II, Grigorovich D, Sarai A, Kolchanov NA. AUG_hairpin: prediction of a downstream secondary structure influencing the recognition of a translation start site. BMC Bioinformatics. (2007) 8:318.; PMID:17760957; http://dx.doi.org/10.1186/1471-2105-8-318
- Robbins-Pianka A, Rice MD, Weir MP. The mRNA landscape at yeast translation initiation sites. Bioinformatics. (2010) 26:2651-2655; PMID:20861026; http://dx.doi.org/10.1093/bioinformatics/btq509
- Eyre-Walker A, Bulmer M. Reduced synonymous substitution rate at the start of enterobacterial genes. Nucl. Acids Res. (1993) 21:4599-603; http://dx.doi.org/10.1093/nar/21.19.4599
- Nakagawa S, Niimura Y, Gojobori T, Tanaka H, Miura K. Diversity of preferred nucleotide sequences around the translation initiation codon in eukaryote genomes. Nucleic Acids Res (2008) 36:861-71; http://dx.doi.org/10.1093/nar/gkm1102
- Man O, Pilpel Y. Differential translation efficiency of orthologous genes is involved in phenotypic divergence of yeast species. Nat Genet (2007) 39:415-21; PMID:17277776; http://dx.doi.org/10.1038/ng1967
- Drummond DA, Wilke CO. The evolutionary consequences of erroneous protein synthesis. Nat Rev Genet (2009) 10:715-724; PMID:19763154; http://dx.doi.org/10.1038/nrg2662
- Frith MC, Forrest AR, Nourbakhsh E, Pang KC, Kai C, Kawai J, Carninci P, Hayashizaki Y, Bailey TL, Grimmond SM. The abundance of short proteins in the mammalian proteome. PLoS Genet. (2006) 2:e52. Epub 2006 Apr 2028.; PMID:16683031; http://dx.doi.org/10.1371/journal.pgen.0020052
- Charneski CA, Hurst LD. Positively charged residues are the major determinants of ribosomal velocity. PLoS Biol (2013) 11:e1001508; PMID:23554576; http://dx.doi.org/10.1371/journal.pbio.1001508
- Jacques N, Dreyfus M. Translation initiation in Escherichia coli: old and new questions. Mol Microbiol (1990) 4:1063-7; PMID:1700254; http://dx.doi.org/10.1111/j.1365-2958.1990.tb00679.x
- Poker G, Margaliot M, Tuller T. Sensitivity of mRNA Translation. in review (2014).
- Shields DC, Sharp PM, Higgins DG, Wright F. “Silent” sites in Drosophila genes are not neutral: evidence of selection among synonymous codons. Mol Biol Evol. (1988) 5:704-16.; PMID:3146682
- Linshiz G, Yehezkel TB, Kaplan S, Gronau I, Ravid S, Adar R, Shapiro E. Recursive construction of perfect DNA molecules from imperfect oligonucleotides. Mol Syst Biol (2008) 4:191; PMID:18463615; http://dx.doi.org/10.1038/msb.2008.26
- Shabi U, Kaplan S, Linshiz G, Benyehezkel T, Buaron H, Mazor Y, Shapiro E. Processing DNA molecules as text. Syst Synth Biol (2010) 4:227-36; PMID:21189843; http://dx.doi.org/10.1007/s11693-010-9059-y
- Gietz RD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol (2002) 350:87-96.; PMID:12073338; http://dx.doi.org/10.1016/S0076-6879(02)50957-5
- Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science (2009) 324:218; PMID:19213877; http://dx.doi.org/10.1126/science.1168978
- Damelin SB, Miller Jr W. The mathematics of signal processing. (Cambridge University Press, 2011).
- Grushka E. Characterization of exponentially modified Gaussian peaks in chromatography. Anal Chem (1972) 44:1733-38; PMID:22324584; http://dx.doi.org/10.1021/ac60319a011
- Lorenz R, Bernhart SH, Höner Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. ViennaRNA Package 2.0. Algorithms Mol Biol. (2011) 6:26; PMID:22115189; http://dx.doi.org/10.1186/1748-7188-6-26
- Zur H, Tuller, T. Strong association between mRNA folding strength and protein abundance in S. cerevisiae. EMBO Rep. (2012); 13(3):272-7; PMID:22249164
- Newman JR, Ghaemmaghami S, Ihmels J, Breslow DK, Noble M, DeRisi JL, Weissman JS. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature (2006) 441:840-6; PMID:16699522; http://dx.doi.org/10.1038/nature04785
- Lee MV, Topper SE, Hubler SL, Hose J, Wenger CD, Coon JJ, Gasch AP. A dynamic model of proteome changes reveals new roles for transcript alteration in yeast. Mol Syst Biol (2011) 7:514; PMID:21772262; http://dx.doi.org/10.1038/msb.2011.48
- Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B-Methodological (1995) 57:289-300
- Storey JD. A direct approach to false discovery rates. J. R. Stat. Soc. (2002) 64:479-98; http://dx.doi.org/10.1111/1467-9868.00346
- Sabi RTT. Modeling the efficiency of codon-tRNA interactions based on codon usage bias. DNA Res (2014); 21(5):511-26; PMID:24906480