
ABSTRACT
Quorum sensing is a cell density-dependent communication system of bacteria relying on autoinducer molecules. During the analysis of the post-transcriptional regulation of quorum sensing in the nitrogen fixing plant symbiont Sinorhizobium meliloti, we predicted and verified a direct interaction between the 5'-UTR of sinI mRNA encoding the autoinducer synthase and a small RNA (sRNA), which we named RcsR1. In vitro, RcsR1 prevented cleavage in the 5'-UTR of sinI by RNase E and impaired sinI translation. In line with low ribosomal occupancy and transcript destabilization upon binding of RcsR1 to sinI, overproduction of RcsR1 in S. meliloti resulted in lower level and shorter half-life of sinI mRNA, and in decreased autoinducer amount. Although RcsR1 can influence quorum sensing via sinI, its level did not vary at different cell densities, but decreased under salt stress and increased at low temperature. We found that RcsR1 and its stress-related expression pattern, but not the interaction with sinI homologs, are conserved in Sinorhizobium, Rhizobium and Agrobacterium. Consistently, overproduction of RcsR1 in S. meliloti and Agrobacterium tumefaciens inhibited growth at high salinity. We identified conserved targets of RcsR1 and showed that most conserved interactions and the effect on growth under salt stress are mediated by the first stem-loop of RcsR1, while its central part is responsible for the species-specific interaction with sinI. We conclude that RcsR1 is an ancient, stress-related riboregulator in rhizobia and propose that it links stress responses to quorum sensing in S. meliloti.
Introduction
Quorum sensing (QS) is an intercellular communication system which allows bacteria to coordinate gene expression in dependence on their population density. To achieve this, at low population density bacteria constitutively produce low amount of signal molecules called autoinducer, mostly acyl-homoserine lactones (AHLs) or autoinducer-1 in gram-negative bacteria, which can leave and re-enter the cell. At certain population density the AHL concentration reaches a threshold value leading to strong increase in the AHL production and simultaneous changes in the gene expression in the whole population.Citation1 Bacteria use quorum sensing to regulate genes related to biofilm formation, pathogenicity or symbiosis. For example in Vibrio virulence genes, and in Sinorhizobium meliloti genes for exopolysaccharide production are expressed in dependence on QS.Citation2,3
The prototype of QS relying on AHLs is the LuxR-LuxI system of Vibrio fischeri. At certain AHL concentration the AHL sensor and transcriptional regulator LuxR activates the expression of the AHL synthase gene luxI.Citation1 Similar QS systems are present in many gram-negative bacteria including our model organism S. meliloti, the nitrogen fixing symbiont of Medicago host plants.Citation4 Major players in the Sin QS system of S. meliloti are SinR and ExpR, 2 transcriptional regulators of the LuxR type, and the AHL synthase SinI. Expression of sinI depends on SinR. In addition, ExpR senses AHLs and strongly activates sinI expression at the onset of the QS response at late exponential growth. Later, at very high concentrations of AHLs, ExpR negatively regulates sinR leading to a decline of the production of AHLs.Citation5,6
In addition to the transcription regulation described above, bacterial AHL-dependent QS systems are regulated at the post-transcriptional level by mechanisms acting on luxR homologs. In Vibrio the RNA chaperone Hfq and small non-coding RNAs (sRNAs) destabilize the mRNA encoding LuxR/HapR at low cell density.Citation7 Furthermore, post-translational regulation by proteolytic degradation of the LuxR-homolog TraR was described for A. tumefaciens.Citation8 In contrast, several studies suggested that in S. meliloti the autoinducer synthase gene sinI is a direct subject of post-transcriptional regulation: The level of sinI mRNA was increased in an hfq mutant of S. meliloti and central regions of this mRNA were co-precipitated with Hfq.Citation9,10 Furthermore, it was shown that an intact RNase E gene is necessary for the 5'-degradation of the sinI transcript and for the generation of a processed 5'-end in the 5'-UTR of sinI in vivo.Citation11 Since in E. coli RNase E, Hfq and sRNAs form mRNA-destabilizing nucleoprotein complexes, the existing data suggest that sRNAs are involved in the regulation of sinI expression.Citation12
Bacterial QS systems are interconnected with other global regulatory networks like the catabolite repression and RpoS regulons, but the underlying mechanisms are mostly unknown.Citation13-15 An exception is the post-translational regulation of the autoinducer-2 synthase luxS by the sRNA CyaR in Escherichia coli. The transcription of CyaR depends on the global regulator Crp, providing a link between catabolite repression and QS.Citation16 Furthermore, bacterial QS influences stress-dependent genes and is influenced by environmental factors including stress.Citation17-19 In S. meliloti, sinR is activated under phosphate-limiting conditions, and QS of P. aeruginosa does not depend on the cell density only, but also on medium composition and oxygen availability.Citation6,19 Such interconnection of different regulatory systems is supposed to increase the versatility and adaptability of bacteria and is an important, but poorly understood aspect of the bacterial physiology.Citation18
In this work we studied the post-transcriptional regulation of sinI and identified a conserved rhizobial sRNA, which interacts with the 5'-UTR of sinI in S. meliloti impairing the autoinducer synthase translation and leading to destabilization of the sinI transcript. We found that this sRNA (formerly SmelC587) is not regulated in a cell density-dependent manner, but shows conserved changes in expression under stress in several Rhizobiaceae members.Citation20 Therefore we named it RcsR1 (rhizobial cold and salinity stress riboregulator 1). We show that RcsR1 contains a highly conserved stem-loop involved in the interaction with conserved targets, and a less conserved region responsible for its species-specific interaction with sinI. Based on our data, we propose that in S. meliloti RcsR1 links stress response to social behavior.
Results
A sRNA predicted to interact with sinI shows stress-related expression pattern
To predict sRNA(s) interacting with the 5'-UTR of sinI mRNA in S. meliloti, we performed TargetRNA analysis using the sequence of the 5'-UTR and the first 33 codons.Citation21,22 Potential, imperfect base pairing was predicted between the first 18 nt of the sinI transcript (encompassing the region from its 5'-end to the Shine-Dalgarno sequence) and a sRNA candidate previously detected in a high-throughput study and designated SmelC587.Citation20 On the chromosome the SmelC587 sequence is flanked by a putative promoter and Rho-independent terminator, suggesting that this is an orphan, trans-acting sRNA of 114 nt. Transcript of suitable length was detected by Northern blot hybridization of total RNA from S. meliloti 2011 using a SmelC587-specific probe (Fig. S1).
Assuming that SmelC587 is involved in the cell-density dependent regulation of QS, we analyzed its levels during growth along with changes in AHL levels using the strain S. meliloti 2011, which is an expR deficient mutant (Fig. S2), and in its derivative Sm2B3001 with restored expR on the chromosome ().Citation23 As expected, the AHL amount strongly increased between OD600 of 0.6 and 0.8 and declined under the limit of detection at OD600 of 2.2 in strain Sm2B3001 (), while in strain 2011 the AHL levels gradually increased during growth (Fig. S2). However, the levels of SmelC587 remained constant during growth in both strains, while the levels of the control sRNA EcpR1 (formerly Sra33; EcpR1 negatively regulates cell cycle progression) increased with increasing optical density in agreement with previous results ( and , Fig. S2).Citation24,25,26
Figure 1. The level of the sRNA RcsR1 (SmelC587) remains constant during growth. (A) Changes in the level of AHLs during growth of S. meliloti Sm2B3001, an expR+ derivative of strain 2011. Culture samples were withdrawn at the indicated OD600 between 0.4 and 2.2. AHLs were extracted from the supernatants of the samples and detected using a A. tumefaciens NTL4 reporter strain expressing β-galactosidase under the control of a QS-responsive promoter.Citation49 Shown is the result of a representative experiment. (B) Northern blot analysis of total RNA from the culture samples used for AHL extraction in A). Hybridization was performed with probes specific for RcsR1, EcpR1 and the loading control 5S rRNA. Indicated are the ODs at which the samples were analyzed and the detected RNAs. (C) Quantification of Northern blot signals. For calculation RcsR1 and EcpR1 signal intensities were normalized to 5S rRNA signal intensities. Normalized signal intensities at OD600 of 0.4 were set to 1 and fold changes at the indicated ODs were calculated. The graph shows results from 2 independent experiments with technical duplicates (means and error bars depicting the standard deviation). Representative Northern blots are shown in panel B).
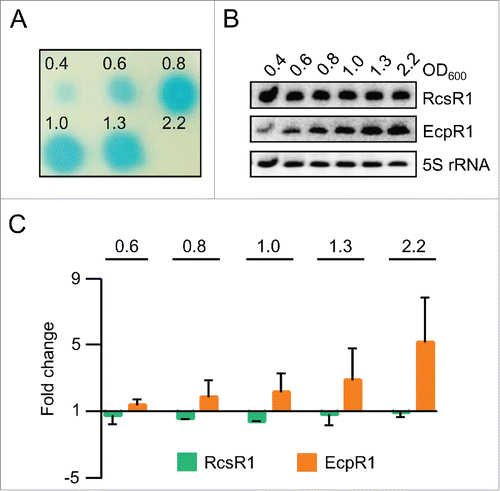
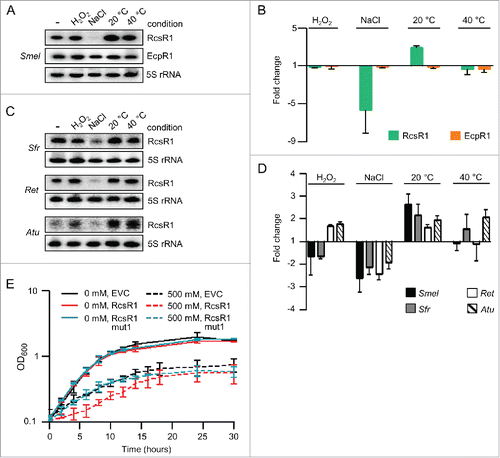
Since many sRNAs are involved in stress responses, we decided to analyze the SmelC587 levels under oxidative and salinity stress, and at different temperatures (see Methods; and ).Citation27 SmelC587 was not detectable at high salinity and its level was increased at 20°C when compared to our standard conditions (30°C). The specificity of these changes was shown by comparison to EcpR1, the level of which remained constant under the applied conditions ( and ). Since the expression pattern of SmelC587 was similar in the strains 2011 and Sm2B3001 (compare and to Fig. S2), we conclude that the expression this sRNA is not dependent on expR. Therefore all further experiments were performed in a simplified QS background using S. meliloti 2011.
Figure 2. Conserved changes in the level of RcsR1 (SmelC587) at high salinity and low temperature. (A) Northern blot analysis of total RNA isolated from S. meliloti Sm2B3001 cultures at OD600 of 1.0, which were subjected to the indicated stress conditions: 10 min exposure to 1 mM (H202), 30 min to 500 mM NaCl (NaCl), 30 min to 20°C (20°C) and 5 min to 40°C (40°C). -, RNA from non-stressed, control culture kept at 30°C. Hybridization was performed with probes specific for RcsR1, EcpR1 and the loading control 5S rRNA. (B) Quantification of Northern blot signals. RcsR1 and EcpR1 signal intensities were normalized to 5S rRNA signal intensities. Normalized signal intensities in the non-stressed culture were set to 1 and fold changes after exposure to stress were calculated. The graph shows results from 2 independent experiments with technical duplicates (means and error bars depicting the standard deviation). Representative Northern blots are shown in panel A). (C) Northern blot analysis of total RNA from S. fredii MSDJ 1536 (Sfr), R. etli CFN42 (Ret) and A. tumefaciens NTL4 (Atu) cultures exposed to stress at OD600 of 1.0. For other descriptions see panel A). (D) Quantification of Northern blot signals from 2 independent experiments with technical duplicates including the results shown in panel C) (for S. fredii, R. etli and A. tumefaciens) and A) (for S. meliloti). For other descriptions see panel B). (E) Growth curves of S. meliloti 2011 empty vector control (EVC) and strains overexpressing RcsR1 or its derivative RcsR1mut1 (see also ). NaCl concentration (in mM) in the growth medium is indicated. Shown are results from 2 independent experiments (means and error bars depicting the standard deviation).
RcsR1 (SmelC587) and its expression pattern are conserved
BLASTN analyses revealed that SmelC587 is conserved in Sinorhizobium, Rhizobium and Agrobacterium (Fig. S3). We analyzed the conservation of SmelC587 response to stress in Sinorhizobium fredii, Rhizobium etli and Agrobacterium tumefaciens and found that in all species the levels of the sRNA decrease under salt stress and increase at 20°C when compared to 30°C ( and ). Therefore we named this sRNA RcsR1 (rhizobial cold and salinity stress riboregulator 1). Under heat and oxidative stress, however, the changes in the abundance of RcsR1 were diverse in the different species ( and ).
To address the impact of RcsR1 on stress adaptation, RcsR1 was ectopically overproduced in S. meliloti 2011 under the control of an rrn promoter from pRK-SmelRcsR1 (see Methods).Citation28 At high salinity this counteracts the down-regulation of the native rcsR1 gene (Fig. S4) and leads to impaired growth in comparison to the empty vector control (EVC; ). Growth of A. tumefaciens NTL4 overproducing AtuRcsR1 was also impaired at high salinity (Fig. S4).
Stem-loop 1 of RcsR1 interacts with conserved targets
The conservation of RcsR1 and its expression pattern under salt and cold stress suggests that this sRNA plays a conserved role in rhizobia and agrobacteria. To predict conserved target mRNAs interacting with RcsR1, we used the bioinformatics tool CopraRNA.Citation29 Since RcsR1 homologs can be divided in a Sinorhizobium group and a Rhizobium/Agrobacterium group (Fig. S3), we supposed that RcsR1 may have targets conserved in the particular phylogenetic lineage, in addition to highly conserved targets present in both lineages. Therefore we performed prediction rounds with RcsR1 of S. meliloti 2011 using either sequenced Sinorhizobium, Rhizobium and Agrobacterium genomes, or only Sinorhizobium genomes.
SinI was not among the hundreds of predicted mRNA targets, the top 30 of which were inspected to select promising candidates for experimental verification. Our selection was mainly based on the free energy released upon the interaction between the potential target and RcsR1 (reflected by a low IntaRNA p-value) in addition to the conservation of the predicted interaction (reflected by a low CopraRNA p-value) and resulted in 13 candidates (Tables S1, S2 and S3).Citation29 The levels of these mRNAs in S. meliloti 2011 overexpressing rcsR1 were compared to the levels in the EVC by qRT-PCR. We detected changes for 6 mRNAs, identifying them as direct or indirect RcsR1 targets ( and Fig. S3). Three of these mRNAs were predicted to interact with RcsR1 in both the Sinorhizobium and the Rhizobium/Agrobacterium lineages (phoR, motE and sm2011_c01420 encoding the anti-σE1 factor). The remaining 3 were predicted to interact with RcsR1 only in Sinorhizobium (sm2011_c00490 encoding a GntR-type transcription regulator, flgA and trpC; ).
Figure 3. Stem-loop 1 of RcsR1 plays a role in conserved interactions. (A) Verification of predicted conserved targets by determination of fold changes in mRNA levels upon overproduction of RcsR1 and its derivatives RcsR1mut1 and RcsR1mut2 in S. meliloti 2011. For target prediction we used either genomic sequences of Sinorhizobium, Rhizobium and Agrobacterium strains, or only sequences of Sinorhizobium strains (indicated above the panel). The analyzed mRNAs are also indicated. The mRNA levels were determined by qRT-PCR. The levels in the EVC were set to 1, and fold changes in the overproducing strains were calculated. Shown are results from 3 (RcsR1) or 2 (RcsR1mut1 and RcsR1mut2) independent experiments with technical duplicates (means and error bars depicting the standard deviation). The experiments were performed with RNA from cells grown at 30°C to an OD600 of 1.0. (B) Predicted secondary structure of RcsR1 homologs. The LocARNA color annotation shows the conservation of base pairs.Citation30 Mutations in the RcsR1 derivatives RcsR1mut1 (mut1 mutation G46C in stem-loop 1) and RcsR1mut2 (mut2 mutation G69C in stem-loop2) are depicted. Target mRNAs predicted to interact with each of the stem-loops are indicated. anti-σ, sm2011_c01420 encoding the anti-σE1 factor; gntR, sm2011_c00490 encoding a GntR-type transcription regulator. The other mRNAs are indicated as annotated.
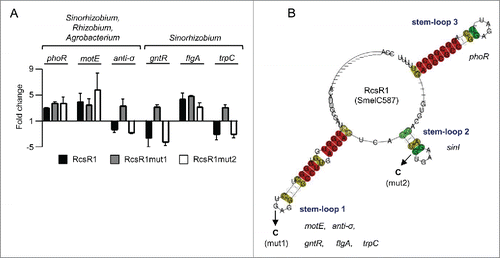
The alignment of RcsR1 and its homologs (Fig. S3) was used to predict the conserved secondary structure of this new family of riboregulators.Citation30 The secondary structure comprises 2 highly conserved stem-loops (stem-loops 1 and 3), and a less conserved stem-loop (stem-loop 2; see ). According to the bioinformatics prediction, all of the 6 likely targets with exception of phoR should interact with the conserved stem-loop 1 (; Tables S1, S2 and S3). To test the involvement of stem-loop 1 in conserved interactions, a G46C mutation was introduced into the loop resulting in RcsR1mut1. This mutation should affect the interaction with sm2011_c01420 encoding the anti-σE1 factor, sm2011_c00490 encoding a GntR-type transcription regulator, flgA and trpC but not with motE (since this G residue was not predicted to base pair with motE) and phoR (which should interact with stem-loop 3; and Fig. S5). Overproduction of RcsR1mut1 reversed the effect of the RcsR1 overproduction on the mRNAs encoding the anti-σE1 factor, GntR-type regulator and TrpC, strongly suggesting that they are direct targets: while the levels of these mRNAs were decreased in the strain overproducing the wild type sRNA, they were increased upon overproduction of RcsR1mut1 (). As expected, the levels of motE and phoR mRNAs remained increased, and in contrast to our expectations, the flgA mRNA level also remained higher in the RcsR1mut1 overproducing strain. Furthermore, the RcsR1mut1 overproducing strain was able to grow in high salinity medium similarly to the EVC ().
The bioinformatics predictions suggested that RcsR1 interacts with homologs of sm2011_c01420 encoding the anti-σE1 factor, motE and phoR in the Rhizobium/Agrobacterium lineage. Overproduction of AtuRcsR1 in A. tumefaciens NTL4 did not affect the levels of atu0419 (phoR) and atu0549 (motE) mRNAs, but the level of atu2031 mRNA (sm2011_c01420 homolog encoding an anti-σ factor) was lower (Fig. S4).
RcsR1 influences QS in an RNase E-dependent and Hfq-independent manner
Based on the predicted interaction between RcsR1 and sinI mRNA, we analyzed the effect of rcsR1 overexpression on QS in S. meliloti 2011. To this end we compared the AHL and sinI mRNA steady-state levels in the overexpressing strain to the levels in the EVC and found that they are lower ( and ). In contrast, no significant changes were observed upon rcsR1 overexpression in a S. meliloti 2011 rne mutant with a mini-Tn5 insertion in the RNase E gene ( and ; see also Fig. S6).Citation11 These results shows that RcsR1 needs RNase E for its effect on QS.
Figure 4. Overproduction of RcsR1 influences sinI mRNA and AHL levels in an RNase E-dependent manner. (A) Detection of AHLs extracted from supernatants of S. meliloti 2011 and its rne and Δhfq mutants grown to an OD600 of 1.0. Strains overproducing RcsR1 (RcsR1) were used along with the EVC. AHLs were detected using a A. tumefaciens NTL4 reporter strain .Citation49 In (B) and (C), fold changes in the level of sinI mRNA were determined by qRT-PCR. The levels in the respective EVC strains were set to 1, and fold changes in the indicated overproducing strains were calculated. Two independent experiments with technical duplicates were evaluated. Shown are means and error bars depicting the standard deviation. B) RcsR1 was overproduced in strain 2011 and its rne mutant. (C) RcsR1, strain 2011 overproducing RcsR1 ; RNase E, strain 2011 overproducing RNase E; RcsR1 + RNase E, strain 2011 simultaneously overproducing RcsR1 and RNase E. (D) Determination of the half-lives of sinI mRNA in the EVC, the RcsR1-overproducing strain and the strain overproducing both RcsR1 and RNase E. The levels of sinI mRNA before (time point 0 min) and after addition of rifampicin, which blocks transcription, were determined by qRT-PCR. The level at 0 min was set to 100 % and relative levels at defined time points after rifampicin addition were calculated. For half-life determination, 2 independent experiments with technical duplicates were evaluated. The determined half-lives in min (means +/− standard deviation) are given in parentheses. The graph shows results from a representative experiment. All experiments were performed at OD600 of 0.6.
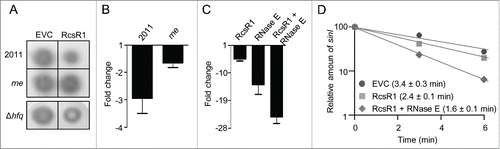
Hfq is often needed for interaction of trans-encoded sRNAs with their mRNA targets and with RNase E and therefore we analyzed the impact of Hfq using a S. meliloti Δhfq mutant.Citation12,25 Since Hfq-dependent sRNAs are destabilized in the absence of Hfq, we first compared the RcsR1 levels in the strains 2011 and 2011Δhfq and found no difference (Fig. S7).Citation25,31 This results suggests that RcsR1 acts by an Hfq-independent mechanism. Consistently, overexpression of rcsR1 in the Δhfq mutant resulted in a decreased AHL level ( and Fig. S7).
Previously it was shown that overexpression of rne reduces the half-life and the steady-state level of sinI mRNA.Citation11 This, together with our finding that RNase E is needed to achieve lower sinI mRNA steady-state levels upon rcsR1 overexpression, led to the question whether RcsR1 represents a limiting factor for sinI mRNA decay when rne is overexpressed. To address this, we used S. meliloti 2011 strains containing only pRK-SmelRcsR1 (allowing IPTG-independent overexpression of rcsR1 from an rrn promoter), only pWBrne with rne under the control of an inducible lac promoter, and both plasmids. shows that simultaneous overproduction of RcsR1 and RNase E leads to much stronger reduction of the steady-state level of sinI than overproduction of RcsR1 or RNase E alone. RNA stability measurements revealed that these changes in the steady-state levels are due to corresponding changes in the half-life of sinI mRNA ( and ref. Eleven). Thus, our results show that RcsR1 negatively influences QS by lowering the half-life of sinI mRNA. They also suggest that RcsR1 increases the negative effect of RNase E on sinI.
Stem-loop 2 of RcsR1 directly interacts with the 5'-UTR of sinI mRNA
The bioinformatics prediction suggested that stem-loop 2 of RcsR1 interacts with sinI ( and ). To test this we introduced a G69C mutation in the supposed seed region of the sRNA (labeled as mut2 in and ) resulting in RcsR1mut2 and analyzed the sinI mRNA levels in S. meliloti 2011 (). Indeed, while a 4-fold decrease in the level of sinI mRNA was measured in the strains overproducing the RcsR1 or RcsR1mut1, it was only 1.5-fold upon overproduction of RcsR1mut2. As expected, sinR mRNA, which should not interact with RcsR1, was not significantly affected (). Furthermore, consistent with a modular structure of RcsR1 in which the individual stem-loops interact with different mRNAs, the mutation in stem-loop 2 did not reverse the effect of RcsR1 overproduction on sm2011_c01420 encoding the anti-σE1 factor, sm2011_c00490 encoding a GntR-type transcription regulator and trpC ().
Figure 5. Stem-loop 2 of RcsR1 directly interacts with the 5'-UTR of sinI. (A) Predicted interaction between the sRNA and sinI and mutations used for verification of the interaction in vivo.Citation21 The sRNA numbering starts with the transcription start site (TSS).Citation20 The TSS of sinI and the RNase E-dependent 5'-end previously detected in vivo are indicated.Citation11,22 In the sinI sequence, the Shine-Dalgarno sequence is underlined and the AUG start codon is in italics. Mutation in RcsR1 (mut2) and compensatory mutation in sinI (sinI-mut) are indicated with arrows. The asterisk marks the endonucleolytic cleavage site detected in vitro in the absence of RcsR1 (see ). (B) Determination of fold change in the levels of sinI and sinR mRNAs in strain 2011 upon overproduction of RcsR1 and its mutated derivatives RcsR1mut1 and RcsR1mut2 (see ) by qRT-PCR. The levels in the EVC were set to 1, and fold changes in the overproducing strains were calculated. (C) Fluorescence of S. meliloti 2011 containing sinI'-egfp translational fusion and overexpressing RcsR1 or RcsR1mut2 was measured. A strain containing sinI-mut'-egfp and RcsR1mut2 was also included. SinI-mut'-egfp contains a compensatory mutation restoring the base pairing between RcsR1mut2 and the 5'-UTR of sinI in the reporter construct (see panel A). Six independent cultures were evaluated. Shown are means and error bars depicting the standard deviation.
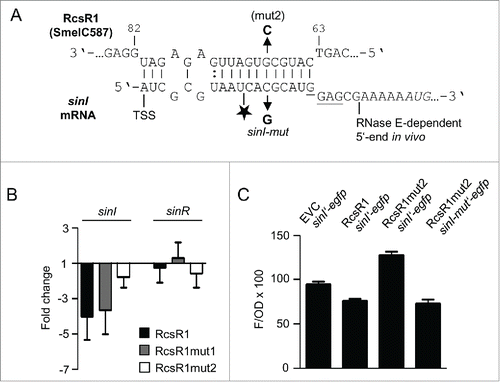
To verify a direct interaction between RcsR1 and the 5'-UTR of sinI mRNA, we implemented an in vivo reporter system based on the plasmids pLK64 and pBBR-SmelRcsR1. The plasmid pLK64 contains a sinIp-sinI′-egfp translational fusion and allows the expression of SinI′-EGFP containing the first 9 amino acid residues of SinI.Citation22 It confers tetracycline resistance like pRK-SmelRcsR1, and therefore it was necessary to use the kanamycinresistance conferring plasmid pBBR-SmelRcsR1, in which the sRNA gene is transcribed from the same rrn promoter like in pRK-SmelRcsR1. Overproduction of the sRNA in S. meliloti 2011 (pLK64, pBBR-SmelRcsR1) led to a decline in fluorescence when compared to the strain 2011 (pLK64, pBBR4352) containing the empty vector pBBR4532 (). When the mutated sRNA RcsR1mut2 was overproduced, the fluorescence was even higher than in the EVC. The compensatory mutation C to G in the 5'-UTR of sinI (see ) on pLK64 led again to a decline in fluorescence, confirming the direct interaction between RcsR1 and the 5'-UTR of sinI (). These results are consistent with a direct interaction between RcsR1 and the 5'-UTR of sinI in vivo.
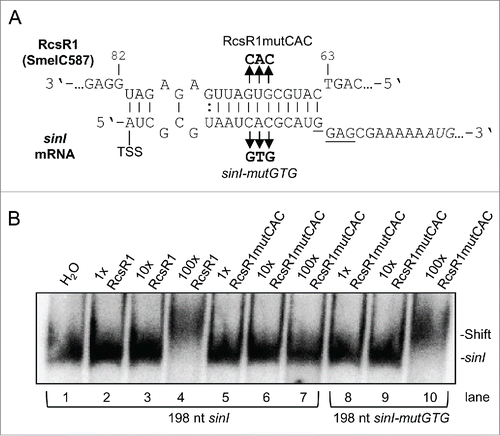
To analyze this interaction in vitro, electrophoretic mobility shift assay (EMSA) was performed using uniformly labeled transcript corresponding to the 5' 198 nt of sinI mRNA and non-labeled RcsR1, and with their mutated derivatives (). Electrophoretic mobility shift indicating interaction between wild type sinI and RcsR1 was observed in lane 4 (). Exchange of 3 nucleotides (GUG to CAC) in the supposed seed region of the sRNA abolished the shift ( and lane 7 in ), while compensatory mutations in sinI restoring the complementarity resulted in a shift ( and lane 10 in ). These results confirm the direct interaction between RcsR1 and the 5'-UTR of sinI.
Figure 6. Electrophoretic mobility shift analysis of the interaction between RcsR1 and sinI. (A) Schematic representation of the interaction between RcsR1 and sinI. The three base-mutation in the seed region of RcsR1 (RcsR1mutCAC) and the compensatory mutations in sinI (sinI-mutGTG) are depicted. (B) Internally labeled sinI transcript (150 fmol) with the length of 198 nt and containing the 5'-UTR was incubated with various concentrations of non-labeled RcsR1 as indicated above the panel and analyzed on a native 6 % polyacrylamide gel. 1x, equimolar amounts of RcsR1 and sinI were used; 10x and 100x, RcsR1 was added in 10-and 100-fold excess, respectively. The positions of bound and free sinI are shown on the right side. The use of wild type and mutant sinI and RcsR1 is indicated below and above the panel, respectively.
RcsR1 blocks cleavage in the 5'-UTR of sinI by RNase E and impairs sinI translation
The data shown in suggest that RcsR1 increases the RNase E-mediated decay of sinI. To address the mechanism by which RcsR1 directly influences sinI, we decided to purify RNase E from S. meliloti and to perform RNA degradation assays. Native RNase E and associated proteins forming an RNA degrading protein complex called degradosome were previously isolated from several bacterial species using a standard procedure (see Methods).Citation32-35 Using this procedure, we obtained an RNase E-enriched protein fraction, which in addition contains the DEAD-box RNA helicase RhlE, components of the pyruvate dehydrogenase complex, a phospholipid synthase and a putative ArsR type transcription regulator ( and Fig. S10). RNase E and a DEAD-box RNA helicase are common components of bacterial degradosomes, which in addition often contain different metabolic enzymes.Citation35
Figure 7. RNase E-enriched protein fraction of S. meliloti. RNase E of S. meliloti was enriched by a method used for purification of bacterial degradosome complexes.Citation32-35 Fractions 10 of the glycerol gradient shown on Fig. S10 was analyzed by 8 % glycine SDS PAGE (A) and 16 % tricine SDS-PAGE (B) and silver staining. Proteins identified by MALDI-TOF-MS are marked with asterisks. The corresponding genes are given in brackets. PDC, pyruvate dehydrogenase complex. To exclude RNA-mediated co-purification of proteins, 2 gradients were ran in parallel with (+) or without (-) RNase A (indicated above the panels). M, protein marker, sizes are indicated in kDa.

We used the RNase E-enriched fraction in RNA degradation assays with uniformly labeled sinI transcript of 198 nt containing the 5′-UTR, which was pre-incubated or not pre-incubated with non-labeled, 5′-monophosphorylated or 5'-triphosphorylared RcsR1.Citation36 In the absence of RcsR1, the sinI transcript was rapidly degraded, while RcsR1 strongly inhibited this degradation (). To determine the endonucleolytic cleavage site in the 5'-UTR of sinI, non-labeled 198 nt sinI transcript was incubated with the RNase E-containing fraction in the absence of RcsR1 and primer extension was performed. The detected cleavage site is located 10 nt downstream of the sinI TSS and differs from the previously determined, RNase E-dependent 5′-end in vivo ( and ).Citation11
Figure 8. RcsR1 blocks the degradation of a 5'-UTR containing sinI derivative by the RNase E-enriched protein fraction and impairs sinI translation. (A) RNA degradation assays with the RNase E-enriched protein fraction of S. meliloti. Uniformly labeled sinI transcript with the length of 198 nt and containing the 5'-UTR was incubated with the protein fraction for the indicated time (in min). Pre-incubation with 10-fold excess of 5'-monophosphorylated RcsR1 is indicated above the panel. Signals corresponding to the substrate and prominent degradation products are marked on the left side. H2O, negative control without addition of protein. (B) Primer extension analysis for determination of a processed 5'-end in the 5'-UTR of sinI. Incubation of the sinI transcript with the RNase E-enriched protein fraction is indicated above the panel. A prominent signal, which most probably corresponds to an RNase E cleavage site, is marked by an arrow in the panel. Lanes A, C, G and T each refer to the corresponding nucleotide of the DNA template (cloned sinI region) as determined by sequencing. A part of the RNA sequence with the cleavage site marked with an arrow and asterisk is indicated on the right side of the panel. (C) In vitro translation assay. The in vitro transcript containing the 5'-UTR of sinI, its first 9 codons and in frame fused egfp was subjected to in vitro translation. Equal volumes of the assay samples were loaded onto 10% SDS-PAGE and relative amounts of translated protein were analyzed by Western blot with EGFP-specific antibodies. Pre-incubation of the sinI-egfp transcript with RcsR1 is indicated above the panel.
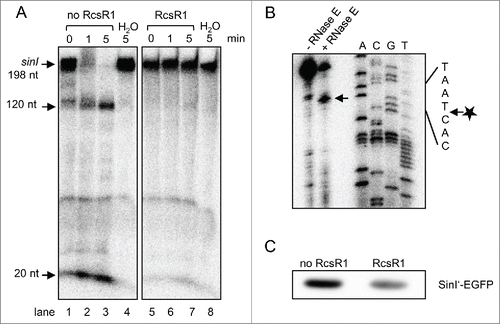
The above results strongly suggest that RcsR1 prevents degradation of the 198 nt sinI transcript by RNase E. Thus the negative influence of RcsR1 on the level and half-life of sinI in the cell shown in is indirect. It could be explained by a better accessibility of RNase E cleavage sites in the sinI coding region due to block of translation by RcsR1, which binds closely to the Shine-Dalgarno sequence ().Citation37 To analyze the influence of RcsR1 on sinI translation, we implemented in vitro translation assay using a synthetic sinI'-egfp transcript containing the 5'-UTR of sinI, its first 9 codons and translationally fused egfp. shows that pre-incubation with RcsR1 strongly decreased the amount of in vitro translated Sin'-EGFP fusion protein.
Discussion
In this work we show that RcsR1 is a conserved, trans-acting sRNA involved in response to stress and capable to directly interact with and to destabilize the autoinducer synthase encoding sinI mRNA in S. meliloti. The conservation of RcsR1 and its expression profile under salt and cold stress in the Sinorhizobium and Rhizobium/Agrobacterium lineages of Rhizobiales ( and Fig. S3) suggests a conserved role for this sRNA in the cell. This is in line with the negative influence of RcsR1 overproduction on the growth of S. meliloti and A. tumefaciens at high salinity ( and Fig. S4). It is likely that RcsR1 is involved in several independent regulatory networks, since its amount decreases at high salinity but increases at low temperature. Our finding that 3 out of the 6 verified, direct or indirect targets (the mRNAs encoding PhoR, the anti-σE1 factor and a GntR-type transcription factor ) are regulators of gene expression, also suggests that RcsR1 is a key riboregulator in rhizobia.
Three of the verified targets were predicted to be conserved in Sinorhizobium, Rhizobium and Agrobacterium: phoR encoding a sensor kinase for the response to phosphate limitation, motE encoding a chaperone for a periplasmic motility protein and sm2011_c01420 encoding the anti-σE1 factor; ).Citation38,39 Among the A. tumefaciens homologs of these targets, only atu2031 encoding the anti-σE1 factor homolog was affected upon overproduction of AtuRcsR1 (Fig. S4). This result confirms that the anti-σE1 factor encoding mRNA is a conserved target of RcsR1. However, in S. meliloti the anti-σE1 factor with its cognate σE1 factor are responsible for the induction of a limited number of genes induced under sulfite stress and thus the anti-σE1 factor encoding mRNA is probably not the RcsR1 target, which is important for adaptation to high salinity or low temperatures.Citation40 This suggests that additional conserved targets of RcsR1 remain to be discovered. Interestingly, PhoB (the response regulator in the 2 component system PhoR-PhoB) induces sinR, which is necessary for sinI expression.Citation6 This implies that RcsR1 does not only influence QS in S. meliloti by direct interaction with sinI, but also indirectly through the PhoR-PhoB signaling cascade, balancing sinI expression under phosphate limiting conditions.
RcsR1 has a modular structure containing the strongly conserved stem-loops 1 and 3 and the less conserved central part which evolved differently in the Sinorhizobium and Rhizobium/Agrobacterium lineages (Fig. S3). The mRNAs encoding the anti-σE1 factor, the GntR-type protein and TrpC seem to directly interact with the highly conserved stem-loop 1, since the G46C mutation in RcsR1mut1 reversed the effect of overproduction of the wild type RcsR1 (). The fact that the overproduction of RcsR1mut1, which presumably does not interact with these mRNAs, did not simply restore their wild-type levels but rather led to an increase, suggests the existence of an additional factor needed for the binding of RcsR1 to its targets. This factor is presumably an RNA-binding protein different from Hfq, since and Fig. S7 show that RcsR1 does not depend on Hfq. We hypothesize that this factor is occupied by the overproduced, mutated RcsR1 variant and therefore the native RcsR1 cannot exert its functions. This assumption is in line with our results demonstrating the direct interaction between stem-loop2 of RcsR1 and the 5'-UTR of sinI, since upon overproduction of RcsR1mut2 the fluorescence was not simply restored but was increased (). In contrast, the overproduction of RcsR1mut2 did not increase the level of sinI mRNA but had a weaker negative effect than the overproduction of wild type RcsR1 (), suggesting multiple interactions between sinI mRNA and RcsR1 like described for other bacterial riboregulators.Citation41 Indeed, additional base pairings between the stem-loop3 of RcsR1 and the coding region of sinI were predicted using IntaRNA (Fig. S9).
The central stem-loop 2 region of RcsR1 is conserved in the genus Sinorhizobium, although only in S. meliloti it was predicted to base-pair with the 5'-UTR of sinI mRNA. We verified the direct interaction between stem-loop 2 of S. meliloti RcsR1 and the 5'-UTR of sinI in in vivo () and in vitro (). In silico analysis of the base pairing capability between sinI (including the 30 nt upstream of the annotated ORF) and RcsR1 in S. fredii and S. medicae revealed that the most 5'-, single stranded region of RcsR1 may interact with a distinct, internal region of sinI mRNA in these species (Fig. S9). Such an interaction was not predicted for S. meliloti, suggesting that in different Sinorhizobium species RcsR1 may influence QS by different mechanisms. Modular structures of sRNAs and interactions with multiple target mRNAs are quite common among bacteria.Citation16,42
The overexpression of rcsR1 diminished the sinI level and the AHL amounts in S. meliloti, and for this intact rne gene was needed ( and ). RNase E is probably essential for growth of S. meliloti like in other bacteria, since it was not possible to obtain an rne null mutant previously.Citation11 The mini-Tn5 insertion in the rne mutant strain is in the 3'-part of the gene, which in other bacteria encodes the C-terminal, degradosome-organizing domain of RNase E, while the catalytic N-terminal domain is probably expressed.Citation11 Thus, the failure to achieve lower levels of sinI mRNA and AHLs upon overproduction of RcsR1 in the rne mutant ( and ) suggests that an RNase E-based degradosome complex is necessary for the negative influence of RcsR1 on sinI. Negative effects of the deletion of the C-terminal domain of RNase E on the action of sRNAs were previously reported for gamma-proteobacteria.Citation43,44
In this work we purified S. meliloti RNase E by a method repeatedly used by others to isolate degradosome complexes from bacteria and found that the final RNase E-enriched protein fraction contains a DEAD-box RNA helicase in addition to RNase E like many of the so far described, bacterial degradosomes.Citation35 The specificity of co-purification of components of the pyruvate dehydrogenase complex, a phospholipid synthase and a putative ArsR-type transcription regulator with RNase E () remains to be analyzed in the future. The RNA degradation assays performed with the RNase E-enriched protein fraction and the following primer extension experiments demonstrated endonucleolytic activity cleaving 10 nt downstream of the sinI TSS, at a site different from the previously in vivo determined, RNase E-dependent 5'-end ( and ). Most probably this endonucleolytic activity is due to RNase E.
Upon binding of RcsR1 to sinI, the in vitro cleavage site in the 5'-UTR should be inaccessible for RNase E, since is located in the middle of the duplex formed by the 2 RNAs () and RNase E is a single-strand specific endoribonuclease.Citation35 This is in agreement with the strong inhibition of degradation of the 198 nt sinI transcript by the RNase E-containing fraction in the presence of RcsR1 (). During this degradation, 2 major products with lengths of 120 nt and 20 nt accumulated (). These degradation products cannot be explained by simple cleavage at the position detected by primer extension, suggesting that the 198 nt sinI transcript was degraded by several enzymatic cleavages and/or activities. Obviously these additional in vitro cleavages depend on the accessibility of the RcsR1 binding site for RNase E. This result is in line with the rapid degradation of mRNA after an initial endonucleolytic cleavage as described in other bacteria.Citation45 It is tempting to speculate that the in vivo detected, RNase E-dependent 5'-end located 23 nt downstream of the sinI TSS may also result from secondary degradation events, which depend on an RNase E cleavage in the RcsR1 binding site.Citation11
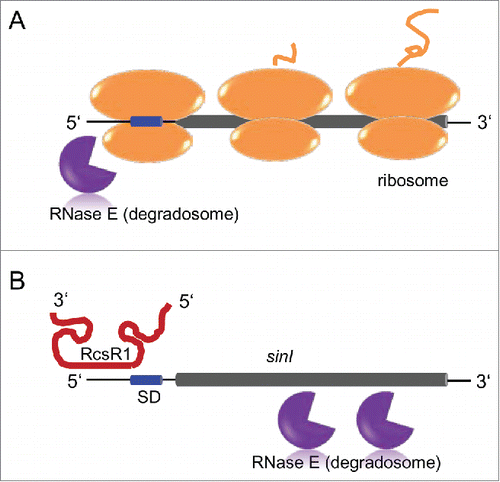
Ribosomes usually protect bacterial mRNA from degradation, because low occupancy by ribosomes allows RNase E to attack internal cleavage sites in mRNAs.Citation37 Binding of RcsR1 in the 5'-UTR of sinI is very close to the Shine-Dalgarno sequence () and impairs translation as shown in Thus the observed sinI mRNA destabilization upon overproduction of RcsR1 can be explained by RNase E cleavage(s) downstream in the sinI open reading frame. The data from this and a previous study suggest 2 RNase E-dependent pathways of degradation of sinI mRNA: (1) degradation starting in the 5'-UTR without binding of RcsR1 and (2) degradation starting in the open reading frame when translation is impaired by RcsR1 ().Citation11
Figure 9. Model of 2 RNase E-dependent pathways for sinI degradation. Thin black line, UTRs; short blue cylinder, Shine-Dalgarno sequence in the 5'-UTR; long gray cylinder, sinI open reading frame. RNase E (degradosome), ribosomes with a nascent polypeptide and the RcsR1 transcript are indicated. (A) When RcsR1 is not bound to the 5'-UTR of sinI, RNase E and ribosomes compete for interaction with the 5'-UTR. This results in a relatively stable sinI transcript due to protection of the coding region by ribosomes, and in RNase E (degradosome)-dependent transcript turnover starting in the 5'-UTR.Citation11 (B) Upon binding of RcsR1 to the 5'-UTR of sinI, the ribosome binding site is blocked. The sinI coding region is not protected by translating ribosomes and is attacked by RNase E (the degradosome), leading to transcript destabilization.
In summary, we identified the sRNA RcsR1 as a conserved riboregulator responding to environmental stress in Sinorhizobium, Rhizobium and Agrobacterium. We also identified conserved targets of this sRNA, among them 3 transcription regulators. As a species-specific, direct target of RcsR1 in S. meliloti we identified the 5'-UTR of sinI mRNA. The sinI gene encodes the autoinducer-1 synthase. Our data show that RcsR1 negatively influences the sinI translation, indirectly leading to RNase E-dependent destabilization of the transcript. This is an example of a post-transcriptional mechanism, by which environmental factors could influence the intercellular communication of bacteria.
Materials and methods
Strains and cultivation methods. Bacterial strains and their relevant characteristics are listed in Table S4. S. meliloti, S. fredii, R. etli and A.tumefaciens were cultivated on TY plates or in liquid TY cultures at 30°C with appropriate antibiotics (streptomycin 250 μg μl−1, neomycin 120 μg μl−1, gentamycin 20 μg μl−1, kanamycin 200 μg μl−1 and tetracycline 20 μg μl−1).Citation46 Escherichia coli was grown in LB broth. E. coli JM109 was used for standard cloning methods.Citation47 Plasmids were transferred from E. coli S17–1 to S. meliloti and A. tumefaciens by diparental conjugation.Citation48 For stress screening, cultures were grown to an OD600 of 1.0, before the following stresses were employed: 10 min 1 mM H2O2, 30 min 500 mM NaCl, 30 min 20 °C and 5 min 40°C. For the growth curves at high salinity conditions, NaCl was added to the freshly inoculated culture. In case of S. meliloti 500 mM NaCl were used, in case of A. tumefaciens, 250 mM NaCl were used.
Plasmid construction
The plasmids used in this work are listed in Table S4, and the primers used for cloning are listed in Table S5. For overproduction of S. meliloti RcsR1 (SmelRcsR1), the rcsR1 gene (including the terminator) was cloned between the BamHI and EcoRI restriction sites of vector pRK4352 using primers pRKSmelRcsR1fwd and pRKSmelRcsR1rev or between the BamHI and XbaI restriction sites of vector pBBR4352 using primers pBBRSmelRcsR1fwd and pBBRSmelRcsR1rev.Citation28 The resulting plasmids pRK-SmelRcsR1 and pBBR-SmelRcsR1 overexpress rcsR1 from an rrn promotor. For overproduction of A. tumefaciens RcsR1 (AtuRcsR1), the rcsR1 gene (including the terminator) was cloned between the BamHI and EcoRI restriction sites of vector pRK4352 using primers pRKAtuRcsR1fwd and pRKAtuRcsR1rev resulting in plasmid pRK-AtuRcsR1. To introduce mutations in the first and second stem-loop of RcsR1, the rcsR1 sequence was sub-cloned to vector pDrive. Inverse PCR mutagenesis was carried out with primers RcsR1mut1fwd and RcsR1mut1rev for a point mutation at position 46 and primers RcsR1mut2fwd and RcsR1mut2rev for a point mutation at position 69. The resulting mutated versions of RcsR1 were cloned to vectors pRK4352 and pBBR4352 as described above, resulting in plasmids pRK-SmelRcsR1mut1, pRK-SmelRcsR1mut2 and pBBR-SmelRcsR1mut2. For construction of plasmid pLK64mut, the sequence of the translational sinI-egfp fusion from plasmid pLK64 was subcloned to pDrive. A point mutation was introduced at position 13 to the 5′ UTR sinI by inverse PCR mutagenesis using primers sinImut2fwd and sinImut2rev.
AHL detection
The detection of AHLs is based on Agrobacterium tumefaciens NTL4 (pZLR4) expressing β-galactosidase from an AHL/TraR dependent promoter and was performed as previously described.Citation11,49 Briefly, the AHLs in supernatants from 1 ml cultures harvested at OD600 of 1.0 were extracted with 300 μl chloroform and dissolved in 20 μl acetone. Routinely 3 μl of the respective AHL solution from the trains 2011 and its rne mutant were used in the AHL detection assays. Since the AHL levels in cultures of the hfq mutant are higher, routinely 0.5 μl of the AHLs extracted from this strain were used for the assays.Citation9
RNA extraction
Cells were harvested, rapidly cooled on ice and pelleted by centrifugation (6.000 x g for 10 min at 4 °C). For Northern blot analysis, RNA was isolated with TRIzol (Ambion). To improve cell lysis, acid-washed glass beads (Sigma) were added and cells were disrupted in a tissuelyser (Retsch) for 15 min. The suspension was incubated at 65 °C for 10 min, and disruption in the tissuelyser was repeated for 15 min. Glass beads were removed by centrifugation. All further steps were performed as instructed by the manufacturer. For qRT-PCR cells were harvested by adding 1 ml of the S. meliloti culture to 1 ml of RNAprotect Bacteria Reagent (Qiagen). Pellets were resuspended in the RTL buffer provided with the RNeasy Mini Kit (Qiagen). Cells were disrupted with glass beads in the tissuelyser for 15 min. Glass beads were removed and all following steps were performed according to the RNeasy Mini Kit. To remove contaminating DNA, RNA was treated with Turbo DNA-free (Ambion).
Northern blot analysis
RNA separation and Northern blot analysis were performed as previously described.Citation50 In short, 7 μg RNA was separated in 10 % polyacrylamide-urea-gels at 300 V and blotted to Amersham HybondTM-N+ membrane (GE Healthcare) for 2 h at 100 mA. Northern blot hybridizations were carried out with labeled oligonucleotides listed in Table S5.
Quantitative RT-PCR
Conditions for qRT-PCR were as previously described.Citation11,51,52 Primers are listed in Table S6. We used a one-step RT-PCR kit (Qiagen) and added 4 ng μl−1 of total RNA into the reaction mixture. SYBR green I (Sigma) was diluted at 1:100.000 in the master mix to detect double-stranded DNA. For normalization of mRNA levels, the rpoB gene, which encodes the β subunit of RNA polymerase of S. meliloti, was used. Determination of mRNA half-lives by qRT-PCR was performed as described previously, using 16S rRNA as the reference.Citation11 Half-lives were calculated from linear-log graphs of time after rifampicin addition against relative mRNA amounts.
Bioinformatic analysis
Conservation of RcsR1 was analyzed using Basic Local Alignment Search Tool (BLAST).Citation53 To predict the conserved secondary structure of RcsR1 the conserved sequences of RcsR1 were aligned with LocARNA.Citation30 Prediction of conserved targets was done with CopraRNA.Citation29
Three rounds of CopraRNA analysis were performed, the first included species and strains from the Sinorhizobium lineage (S. meliloti 2011, NC_020528; S. meliloti 1021, NC_003047; S. meliloti GR4, NC_019845; S. meliloti Rm41, NC_018700; S. fredii USDA257, NC_018000; S. fredii NRG234, NC_012587; S. fredii HH103, NC_016812; S. medicae WSM419, NC_009636), the second included species from the Rhizobiaceae (S. meliloti 2011, NC_020528; S. fredii USDA257, NC_018000; R. etli CFN 42, NC_007761; R. tropici CIAT899, NC_020059; R. leguminosarum WSM2304, NC_011369; A. tumefaciens C58, NC_003062; A. vitis S4, NC_011989; A. radiobacter K84, NC_011985) and the third round was performed with S. meliloti 2011 (NC_020528), Rhizobium sp. IRBG74 (NC_022545) and A. radiobacter K84 (NC_011985).
Double-plasmid reporter and fluorescence assays
To verify RcsR1-sinI predicted base pairing in vivo, S. meliloti 2011 empty vector control (EVC) and strains overexpressing RcsR1 or RcsR1mut2 were co-transformed with reporter plasmids sinI′-egfp (pLK64) and its derivative sinI-mut'-egfp, containing a compensatory mutation restoring the base pairing between RcsR1mut2 and the 5'-UTR of sinI. Double transconjugants were grown in TY medium to early-stationary phase (OD600 of 1.0 to 1.2) and 100 μl aliquots cultures were transferred to a 96 well microtiter plate. OD600 and eGFP-mediated fluorescence of 6 bacterial cell cultures was measured on Tecan Infinite M200 reader (Tecan Trading AG, Switzerland) and values were normalized to the culture OD600.
Purification of RNase E
Ribonuclease E enriched protein fractions were isolated as previously described.Citation33 If not otherwise noted, all steps were performed at 4°C. All buffers were freshly prepared and contained 1 mM DTT, 1 mM PMSF, 0.8 μg/ml leupeptin, 0.8 μg/ml pepstatin A and 2 μg/ml aprotinin, and were kept at 4°C if not stated otherwise. In short, to 30 g S. meliloti cells (frozen at −80°C) 30 ml room-temperature lysozyme-EDTA-buffer (50 mM Tris-HCl pH 7.5, 100 mM NaCl, 5 % glycerol, 3 mM EDTA, 1.5 mg/ml lysozyme) was added. The suspension was incubated at 4°C for 40 min with additional stirring and short blending every 10 min. Next, 15 ml of room-temperature DNase-Triton-buffer (50 mM Tris-HCl pH 7.5, 100 mM NaCl, 5 % glycerol, 3 % Triton X-100, 30 mM magnesium acetate, 20 μg/ml DNase I) was added. Cells were lysed by 1 min of blending and incubated at 4°C for 30 min before 12 ml of 5 M NH4Cl was slowly added. The lysate was incubated for an additional 30 min at 4°C under constant stirring and then centrifuged for 1 h at 27.000 g. The clarified supernatant was subjected to ultracentrifugation for 3.5 h at 100.000 g. Proteins in the high-speed supernatant were salted out with 40 % ammonium sulfate and dissolved in 34 ml Buffer A (10 mM Tris-HCl pH 7.5, 50 mM or 300 mM or 1 M NaCl, 5 % glycerol, 0.5 % Genapol X-080, 1 mM EDTA) with 50 mM NaCl. The protein solution was loaded on a HiScreen sulfopropyl SP FF column (GE Healthcare), which was equibrilated with Buffer A (50 mM NaCl) before. The column was washed with 4 column volumes (CV) Buffer A (50 mM NaCl) and 4 CV Buffer A (300 mM NaCl). Elution was performed with 4 CV Buffer A (1 M NaCl). Peak fractions from the column were diluted 2-fold in Buffer A (50 mM NaCl) and loaded on a 10–30 % glycerol gradient in Buffer A (300 mM NaCl). For RNase A treatment, 2 μg RNase A were used. Gradient centrifugation was performed at 37.000 rpm (Beckman SW41 rotor) for 15 h. Twelve 0.5 ml fractions were collected and analyzed by SDS-PAGE.
In vitro transcription
PCR products of sinI and RcsR1 were generated using primers sinivfwd/rev and RcsR1ivfwd/rev, respectively (Table S5), resulting in a 198 bp sequence of sinI containing the 5' UTR and full-length RcsR1. In case of wild type sinI and RcsR1, chromosomal DNA was used as template. For mutated versions of sinI and RcsR1, the respective PCR products were subcloned to pDrive and inverse PCR was carried out using primers sinImutGTGfwd/rev and RcsR1mutCAC fwd/rev (Table S5), resulting in a 3 base mutation in sinI 5'-UTR from position 11 to 13 and in the second stem-loop of RcsR1 from position 69 to 71. Mutated sequences were subcloned to pDrive and used as templates for PCR as described above. PCR products were purified using the QIAquick PCR Purification Kit from Qiagen. 500 ng of each PCR product was used for in vitro transcription with the MEGAshortscript T7 kit (Ambion). The reactions were performed as instructed by the manufacturer. For monophosphorylated RcsR1, 5-fold excess of GMP over GTP was used in the reaction instead.Citation36 [α-32P]-UTP (Hartmann Analytic; FP-210) was used to label sinI transcripts internally.
Degradation assays
In vitro degradation assays were performed in 10 μl final volume as follows. 150 fmol of the 198 nt sinI in vitro transcript were incubated with 1 μl RNase E (degradosome)-enriched protein fraction in the presence of 25 mM Tris-HCl pH 8.0, 5 mM MgCl2, 60 mM KCl, 100 mM NH4Cl2, 0.1 mM DTT, 1 % glycerol and 20 U RNasin (Promega) at 30°C. 5′-mono- or triphosphorylated RcsR1 was added in 10-fold excess (1500 fmol).Citation36 No differences were observed between usage of 5'-mono- and triphosphorylated RcsR1. The sinI transcript and RcsR1 were pre-incubated for 20 min at 30°C before RNase E (degradosome) was added. Reactions were stopped by addition of 1 μl Proteinase K mix (1 mg/ml Proteinase K, 50 mM EDTA, 1 % SDS) and incubated for 15 min. Samples were mixed with formamide urea loading dye, denatured at 65°C for 10 min and separated in a 10 % polyacrylamide urea gel. Gels were dried for 90 min at 80°C and exposed to phosphoimaging screens (Bio-Rad).
Electrophoretic mobility shift assays
EMSA experiments were performed in 15 μl final volume as follows. 150 fmol internally labeled sinI in vitro transcript and cold RcsR1 in vitro transcript in different amounts (150 fmol, 1.500 fmol, 15.000 fmol) were separately denatured at 90°C for 10 min and cooled down to 4°C afterwards. They were renatured together at 30 °C for 20 min in the presence of 100 mM Tris-HCl pH 7.0, 10 mM MgCl2 and 100 mM KCl. The reaction samples were mixed with 3 μl of native loading dye (50 % glycerol, 0.5 x TBE, 0.1 % bromphenol blue) and analyzed on native 6 % polyacrylamide gels in 0.25 x TBE buffer at 200 V. Gels were dried for 90 min at 80 °C and exposed to phosphoimaging screens (Bio-Rad).
Primer extension analysis
Two μg of sinI in vitro transcript were treated with 1 μl RNase E (degradosome)-enriched protein fraction as described above, followed by phenol-chloroform extraction and ethanol precipitation. The transcript and 200.000 c.p.m. radioactively end-labeled oligonucleotide (sinIPE; Table S5) were heated for 5 min at 70°C and then incubated for 5 min at 50°C, 5 min at 37°C and 5 min at room-temperature before placed on ice. To the RNA-DNA hybrid 10 U avian myeloblastosis virus reverse transcriptase (AMV RTase; Promega), 1x AMV RTase buffer (Promega), 20 U RNasin (Promega), 25 mM dNTPs and 60 mM sodium pyrophosphate were added. Synthesis of cDNA was performed for 45 min at 42°C and additional 45 min at 48°C. For the sequencing reaction, the 5' 198 bp fragment of sinI was cloned to pDrive using primers sinIivfwd and sinIivrev. Ten μg of plasmid DNA were denatured with 2 M NaOH and precipitated with ethanol. Ten pmol of sinIPE oligonucleotide were annealed to the denatured plasmid DNA by heating for 2 min at 65 °C and slowly cooling to 4°C. The sequencing reaction was performed with the Sequenase Version 2.0 DNA Sequencing kit (Affymetrix) according to the manufacturer's instructions. Samples were analyzed in an 8 % polyacrylamide sequencing gel. Signals were visualized by phosphoimaging.
In vitro translation
A sinI'-egfp in vitro transcript was generated using primers sinIivfwd and egfpivrev and plasmid pLK64 for PCR amplification of the template and subsequent in vitro transcription with the MEGAshortscript T7 kit (Ambion) (see Tables S4 and S5). This transcript contains the 5' UTR of sinI including the Shine-Dalgarno sequence and the first 9 codons translationally fused to full-length egfp. Five μg of sinI'-egfp transcript were used for in vitro translation carried out by the PURExpress In Vitro Protein Synthesis kit (New England Biolabs) following the manufacturer's instructions. If applied, equal amounts of RcsR1 were incubated with sinI'-egfp 20 min at 30 °C prior to in vitro translation. Detection was performed by Western blot analysis using GFP-specific antibodies (Anti-GFP antibody, Clontech; Anti-Mouse IgG-Peroxidase, Sigma). Signals were visualized using a chemiluminescence imager (Fusion SL4, Vilber).
Protein identification
For mass spectrometric identification proteins band were manually cut from the gel and subsequently digested with trypsin (trypsin gold from porcine pancreas, mass spectrometry grade, Promega). Peptides were extracted from the gel with 1% aqueous trifluoroacetic acid. Matrix-assisted laser-desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS) was performed on an Ultraflex TOF/TOF mass spectrometer equipped with a nitrogen laser and a LIFT-MS/MS facility. The instrument was operated in the positive-ion reflectron mode using 2.5-dihydroxybenzoic acid and methylendiphosphonic acid as matrix. Sum spectra consisting of 200–400 single spectra were acquired. For data processing and instrument control the Compass 1.4 software package consisting of FlexControl 3.4, FlexAnalysis 3.4 and ProteinScape 3.1 was used. Proteins were identified by MASCOT peptide mass fingerprint search (Mascot 2.4.1; http://www.matrixscience.com) using a Uniprot database for bacteria (20150624 11624423 sequences; 3649040132 residues). For the search a mass tolerance of 75 ppm was allowed and carbamidomethylation of cysteine as global modification and oxidation of methionine as variable modification were used. A false positive rate of 5% was allowed.
Disclosure of Potential Conflict of Interest
No potential conflicts of interest were disclosed.
KRNB_A_1110673_supplement.zip
Download Zip (1.4 MB)Acknowledgments
We thank Gabriele Klug, Lennart Weber (University of Giessen) and Anke Becker (University of Marburg) for valuable discussions. We thank Hans-Günter Welker (Institute of Biochemistry, University Giessen) for excellent technical assistance in mass spectrometry. We thank Hendrik Melior for help in some experiments. K.B. is a member of IRTG 1384 "Enzymes and multienzyme complexes acting on nucleic acids" supported by Deutsche Forschungsgemeinschaft (DFG). K. Š. was supported by the Erasmus program of the European Union and Hlávka's foundation travel grant.
References
- Fuqua WC, Winans SC, Greenberg EP. Quorum sensing in bacteria: the LuxR-LuxI family of cell density-responsive transcriptional regulators. J Bacteriol 1994; 176:269-75; PMID:8288518.
- Zhu J, Miller MB, Vance RE, Dziejman M, Bassler BL, Mekalanos JJ. Quorum-sensing regulators control virulence gene expression in Vibrio cholerae. Proc Natl Acad Sci U S A 2002; 99:3129–34; PMID:11854465; http://dx.doi.org/10.1073/pnas.052694299
- Marketon MM, Glenn SA, Eberhard A, González JE. Quorum sensing controls exopolysaccharide production in Sinorhizobium meliloti. J Bacteriol 2003; 185:325–31; PMID:12486070; http://dx.doi.org/10.1128/JB.185.1.325-331.2003
- Marketon MM, Gronquist MR, Eberhard A, González JE. Characterization of the Sinorhizobium meliloti sinR/sinI locus and the production of novel N-acyl homoserine lactones. J Bacteriol 2002; 184:5686–95; PMID:12270827; http://dx.doi.org/10.1128/JB.184.20.5686-5695.2002
- Hoang HH, Becker A, González JE. The LuxR homolog ExpR, in combination with the Sin quorum sensing system, plays a central role in Sinorhizobium meliloti gene expression. J Bacteriol 2004; 186:5460–72; PMID:15292148; http://dx.doi.org/10.1128/JB.186.16.5460-5472.2004
- McIntosh M, Meyer S, Becker A. Novel Sinorhizobium meliloti quorum sensing positive and negative regulatory feedback mechanisms respond to phosphate availability. Mol Microbiol 2009; 74:1238–56; PMID:19889097; http://dx.doi.org/10.1111/j.1365-2958.2009.06930.x
- Lenz DH, Mok KC, Lilley BN, Kulkarni RV, Wingreen NS, Bassler BL. The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell 2004; 118:69–82; PMID:15242645; http://dx.doi.org/10.1016/j.cell.2004.06.009
- Costa ED, Chai Y, Winans SC. The quorum-sensing protein TraR of Agrobacterium tumefaciens is susceptible to intrinsic and TraM-mediated proteolytic instability. Mol Microbiol 2012; 84:807–15; PMID:22515735; http://dx.doi.org/10.1111/j.1365-2958.2012.08037.x
- Gao M, Barnett MJ, Long SR, Teplitski M. Role of the Sinorhizobium meliloti global regulator Hfq in gene regulation and symbiosis. Mol Plant Microbe Interact 2010; 23:355–65; PMID:20192823; http://dx.doi.org/10.1094/MPMI-23-4-0355
- Torres-Quesada O, Reinkensmeier J, Schlüter JP, Robledo M, Peregrina A, Giegerich R, Toro N, Becker A, Jiménez-Zurdo JI. Genome-wide profiling of Hfq-binding RNAs uncovers extensive post-transcriptional rewiring of major stress response and symbiotic regulons in Sinorhizobium meliloti. RNA Biol 2014; 11:563–79; PMID:24786641; http://dx.doi.org/10.4161/rna.28239
- Baumgardt K, Charoenpanich P, McIntosh M, Schikora A, Stein E, Thalmann S, Kogel KH, Klug G, Becker A, Evguenieva-Hackenberg E. RNase E affects the expression of the acyl-homoserine lactone synthase gene sinI in Sinorhizobium meliloti. J Bacteriol 2014; 196:1435–47; PMID:24488310; http://dx.doi.org/10.1128/JB.01471-13
- Morita T, Maki K, Aiba H. RNase E-based ribonucleoprotein complexes: mechanical basis of mRNA destabilization mediated by bacterial noncoding RNAs. Genes Dev 2005; 19:2176–86; PMID:16166379; http://dx.doi.org/10.1101/gad.1330405
- Dunlap PV, Greenberg EP. Control of Vibrio fischeri lux gene transcription by a cyclic AMP receptor protein-luxR protein regulatory circuit. J Bacteriol 1988; 170:4040–6; PMID:3410823.
- Lyell NL, Colton DM, Bose JL, Tumen-Velasquez MP, Kimbrough JH, Stabb EV. Cyclic AMP receptor protein regulates pheromone-mediated bioluminescence at multiple levels in Vibrio fischeri ES114. J Bacteriol 2013; 195:5051–63; PMID:23995643; http://dx.doi.org/10.1128/JB.00751-13
- Schuster M, Hawkins AC, Harwood CS, Greenberg EP. The Pseudomonas aeruginosa RpoS regulon and its relationship to quorum sensing. Mol Microbiol 2004; 51:973–85; PMID:14763974; http://dx.doi.org/10.1046/j.1365-2958.2003.03886.x
- De Lay N, Gottesman S. The Crp-activated small noncoding regulatory RNA CyaR (RyeE) links nutritional status to group behavior. J Bacteriol 2009; 191:461–76; PMID:18978044; http://dx.doi.org/10.1128/JB.01157-08
- Uzureau S, Lemaire J, Delaive E, Dieu M, Gaigneaux A, Raes M, De Bolle X, Letesson JJ. Global analysis of quorum sensing targets in the intracellular pathogen Brucella melitensis 16 M. J Proteome Res 2010; 9:3200–17; PMID:20387905; http://dx.doi.org/10.1021/pr100068p
- Schuster M, Greenberg EP. A network of networks: quorum-sensing gene regulation in Pseudomonas aeruginosa. Int J Med Microbiol 2006; 296:73–81; PMID:16476569; http://dx.doi.org/10.1016/j.ijmm.2006.01.036
- Wagner VE, Bushnell D, Passador L, Brooks AI, Iglewski BH. Microarray analysis of Pseudomonas aeruginosa quorum-sensing regulons: effects of growth phase and environment. J Bacteriol 2003; 185:2080–95; PMID:12644477; http://dx.doi.org/10.1128/JB.185.7.2080-2095.2003
- Schlüter JP, Reinkensmeier J, Daschkey S, Evguenieva-Hackenberg E, Janssen S, Jänicke S, Becker JD, Giegerich R, Becker A. A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing (-proteobacterium Sinorhizobium meliloti. BMC Genomics 2010; 11:245; PMID:20398411; http://dx.doi.org/10.1186/1471-2164-11-245
- Tjaden B. TargetRNA: a tool for predicting targets of small RNA action in bacteria. Nucleic Acids Res 2008; 36(Web Server issue):W109–113; PMID:18477632; http://dx.doi.org/10.1093/nar/gkn264
- McIntosh M, Krol E, Becker A. Competitive and cooperative effects in quorum-sensing-regulated galactoglucan biosynthesis in Sinorhizobium meliloti. J. Bacteriol 2008; 190:5308–17; PMID:18515420; http://dx.doi.org/10.1128/JB.00063-08
- Bahlawane C, McIntosh M, Krol E, Becker, A. Sinorhizobium meliloti regulator MucR couples exopolysaccharide synthesis and motility. Mol Plant Microbe Interact 2008; 21:1498–509; PMID:18842098; http://dx.doi.org/10.1094/MPMI-21-11-1498
- Ulvé VM, Sevin EW, Chéron A, Barloy-Hubler F. Identification of chromosomal (-proteobacterial small RNAs by comparative genome analysis and detection in Sinorhizobium meliloti strain 1021. BMC Genomics 2007; 8:467; http://dx.doi.org/10.1186/1471-2164-8-467
- Voss B, Hölscher M, Baumgarth B, Kalbfleisch A, Kaya C, Hess WR, Becker A, Evguenieva-Hackenberg E. Expression of small RNAs in Rhizobiales and protection of a small RNA and its degradation products by Hfq in Sinorhizobium meliloti. Biochem Biophys Res Commun 2009; 390:331–6; PMID:19800865; http://dx.doi.org/10.1016/j.bbrc.2009.09.125
- Robledo M, Frage B, Wright PR, Becker A. A stress-induced small RNA modulates (-rhizobial cell cycle progression. PLoS Genet. 2015; 11:e1005153; PMID:25923724; http://dx.doi.org/10.1371/journal.pgen.1005153
- Repoila F, Majdalani N, Gottesman S. Small non-coding RNAs, co-ordinators of adaptation processes in Escherichia coli: the RpoS paradigm. Mol Microbiol 2003; 48:855–61; PMID:12753181; http://dx.doi.org/10.1046/j.1365-2958.2003.03454.x
- Mank NN, Berghoff BA, Hermanns YN, Klug G. Regulation of bacterial photosynthesis genes by the small noncoding RNA PcrZ. Proc Natl Acad Sci U S A 2012; 109:16306–11; PMID:22988125; http://dx.doi.org/10.1073/pnas.1207067109
- Wright PR, Georg J, Mann M, Sorescu DA, Richter AS, Lott S, Kleinkauf R, Hess WR, Backofen R. CopraRNA and IntaRNA: predicting small RNA targets, networks and interaction domains. Nucleic Acids Res 2014; 42(Web Server issue):W119–23; PMID:24838564; http://dx.doi.org/10.1093/nar/gku359
- Smith C, Heyne S, Richter AS, Will S, Backofen R. Freiburg RNA Tools: a web server integrating INTARNA, EXPARNA and LOCARNA. Nucleic Acids Res 2010; 38(Web Server issue):W373–7; PMID:20444875; http://dx.doi.org/10.1093/nar/gkq316
- Moll I, Afonyushkin T, Vytvytska O, Kaberdin VR, Bläsi U. Coincident Hfq binding and RNase E cleavage sites on mRNA and small regulatory RNAs. RNA 2003; 9:1308–14; PMID:14561880; http://dx.doi.org/10.1261/rna.5850703
- Carpousis AJ, Van Houwe G, Ehretsmann C, Krisch HM. Copurification of E. coli RNAase E and PNPase: evidence for a specific association between two enzymes important in RNA processing and degradation. Cell 1994;76:889–900; PMID:7510217; http://dx.doi.org/10.1016/0092-8674(94)90363-8
- Jäger S, Fuhrmann O, Heck C, Hebermehl M, Schiltz E, Rauhut R, Klug G. An mRNA degrading complex in Rhodobacter capsulatus. Nucleic Acids Res. 2001; 29:4581–8; http://dx.doi.org/10.1093/nar/29.22.4581
- Zhang Y, Hong G. Post-transcriptional regulation of NifA expression by Hfq and RNase E complex in Rhizobium leguminosarum bv. viciae. Acta Biochim Biophys Sin (Shanghai). 2009;41:719–30; PMID:19727520; http://dx.doi.org/10.1093/abbs/gmp060
- Aït-Bara S, Carpousis AJ. RNA degradosomes in bacteria and chloroplasts: classification, distribution and evolution of RNase E homologs. Mol Microbiol. 2015 Jun 19; 97(6):1021–135; PMID:26096689; http://dx.doi.org/10.1111/mmi.13095
- Bandyra KJ, Said N, Pfeiffer V, Górna MW, Vogel J, Luisi BF. The seed region of a small RNA drives the controlled destruction of the target mRNA by the endoribonuclease RNase E. Mol Cell 2012;47:943–53; PMID:22902561; http://dx.doi.org/10.1016/j.molcel.2012.07.015
- Deana A, Belasco JG. Lost in translation: the influence of ribosomes on bacterial mRNA decay. Genes Dev 2005 Nov 1; 19:2526–33; http://dx.doi.org/10.1101/gad.1348805
- Eggenhofer E, Haslbeck M, Scharf B. MotE serves as a new chaperone specific for the periplasmic motility protein, MotC, in Sinorhizobium meliloti. Mol Microbiol 2004; 52:701–12; PMID:15101977; http://dx.doi.org/10.1111/j.1365-2958.2004.04022.x
- Danhorn T, Hentzer M, Givskov M, Parsek MR, Fuqua C. Phosphorus limitation enhances biofilm formation of the plant pathogen Agrobacterium tumefaciens through the PhoR-PhoB regulatory system. J Bacteriol 2004; 186:4492–501; PMID:15231781; http://dx.doi.org/10.1128/JB.186.14.4492-4501.2004
- Bastiat B, Sauviac L, Picheraux C, Rossignol M, Bruand C. Sinorhizobium meliloti sigma factors RpoE1 and RpoE4 are activated in stationary phase in response to sulfite. PLoS One 2012; 7:e50768; PMID:23226379; http://dx.doi.org/10.1371/journal.pone.0050768
- Chabelskaya S, Bordeau V, Felden B. Dual RNA regulatory control of a Staphylococcus aureus virulence factor. Nucleic Acids Res 2014; 42:4847–58; PMID:24510101; http://dx.doi.org/10.1093/nar/gku119
- Thomason MK, Fontaine F, De Lay N, Storz G. A small RNA that regulates motility and biofilm formation in response to changes in nutrient availability in Escherichia coli. Mol Microbiol 2012; 84:17–35; PMID:22289118; http://dx.doi.org/10.1111/j.1365-2958.2012.07965.x
- Viegas SC, Pfeiffer V, Sittka A, Silva IJ, Vogel J, Arraiano CM. Characterization of the role of ribonucleases in Salmonella small RNA decay. Nucleic Acids Res 2007; 35:7651–64; PMID:17982174; http://dx.doi.org/10.1093/nar/gkm916
- Prévost K, Desnoyers G, Jacques JF, Lavoie F, Massé E. Small RNA-induced mRNA degradation achieved through both translation block and activated cleavage. Genes Dev 2011; 25:385–96; http://dx.doi.org/10.1101/gad.2001711
- Hui MP, Foley PL, Belasco JG. Messenger RNA degradation in bacterial cells. Annu Rev Genet 2014;48:537–59; PMID:25292357; http://dx.doi.org/10.1146/annurev-genet-120213-092340
- Beringer JE. R factor transfer in Rhizobium leguminosarum. J Gen Microbiol 1974; 84:188–98; PMID:4612098; http://dx.doi.org/10.1099/00221287-84-1-188
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1989.
- Simon R, Priefer U, Pühler A. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram-negative bacteria. Biotechnology 1982; 1:784–91; http://dx.doi.org/10.1038/nbt1183-784
- Cha C, Gao P, Chen YC, Shaw PD, Farrand SK. Production of acyl-homoserine lactone quorum-sensing signals by gram-negative plant-associated bacteria. Mol Plant Microbe Interact 1998; 11:1119–29; PMID:9805399; http://dx.doi.org/10.1094/MPMI.1998.11.11.1119
- Madhugiri R, Evguenieva-Hackenberg E. RNase J is involved in the 5'-end maturation of 16S rRNA and 23S rRNA in Sinorhizobium meliloti. FEBS Lett 2009; 583:2339–42; PMID:19540834; http://dx.doi.org/10.1016/j.febslet.2009.06.026
- Glaeser J, Klug G. Photo-oxidative stress in Rhodobacter sphaeroides: protective role of carotenoids and expression of selected genes. Microbiology 2005; 151:1927–38; PMID:15942000; http://dx.doi.org/10.1099/mic.0.27789-0
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001; 29:e45; PMID:11328886; http://dx.doi.org/10.1093/nar/29.9.e45
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990; 215:403–10; PMID:2231712; http://dx.doi.org/10.1016/S0022-2836(05)80360-2