
ABSTRACT
It has been proposed that polybasic peptides cause slower movement of ribosomes through an electrostatic interaction with the highly negative ribosome exit tunnel. Ribosome profiling data—the sequencing of short ribosome-bound fragments of mRNA—is a powerful tool for the analysis of mRNA translation. Using the yeast Saccharomyces cerevisiae as a model, we showed that reduced translation efficiency associated with polybasic protein sequences could be inferred from ribosome profiling. However, an increase in ribosome density at polybasic sequences was evident only when the commonly used translational inhibitors cycloheximide and anisomycin were omitted during mRNA isolation. Since ribosome profiling performed without inhibitors agrees with experimental evidence obtained by other methods, we conclude that cycloheximide and anisomycin must be avoided in ribosome profiling experiments.
Abbreviations
| CHX | = | cycloheximide |
| GEO | = | Gene Expression Omnibus |
| K | = | lysine |
| R | = | Arginine |
| RP | = | ribosome footprint profiling |
Introduction
The translation of mRNA to proteins is an essential process of life and is subject to stringent control at several levels.Citation1 Ribosome stalling or abrupt reduction of the decoding rate is an important event that has a high energetic cost and therefore demands cellular quality-control machinery. This reduced translation can be caused by several factors such as tRNA abundance, codon decoding efficiency, mRNA structure and the physical-chemical properties of the nascent peptide on the ribosome.Citation1 It has been demonstrated that polybasic sequences cause ribosome reduced translation through the interaction of a positively charged peptide with the negatively charged ribosome tunnel.Citation2-6 Most studies investigating the influence of polybasic sequences on translation have used reporter sequences, in which stretches of sequences that code for polybasic peptides were added to or removed from a reporter protein, and the amount of protein subsequently translated was detected by western blot.Citation2,3,5 Using this approach, several authors have confirmed that the presence of poly-arginine (R), poly-lysine (K) or mixtures of both amino acids are able to reduce protein production. Polybasic sequences can also occur during the translation of aberrant mRNA lacking a termination codon (nonstop mRNA) because the translation of the polyA tail results in poly-lysines (AAA codes for lysine). The arrested products produced by polybasic peptides are able to recruit the quality-control machinery involved in the co-translational degradation of stalled peptides.Citation6-9
Another methodology used to analyze translation is ribosome footprint profiling (RP).Citation10 This methodology was created by Ingolia, Ghaemmaghami, Newman and Weissman in 2009 and is based on the deep sequencing of ribosome-protected mRNA fragments.Citation11 During translation, each ribosome encloses an approximately 30-nt portion of mRNA, protecting it against RNAase digestion. This enclosure allows protected fragments to be sequenced. Sample preparation methods differ considerably among laboratories, which makes it a challenge to interpret the results.Citation10 Most of the data generated using this technique relies on the use of the translational inhibitor cycloheximide before nuclease digestion of unprotected nucleotides. Unlike the previously described western-blot data, the effect of polybasic peptides on translation is not clear when analyzed by RP. The first 2 papers describing the effect of polybasic sequences on translation using RP concluded that these sequences were able to stall translation.Citation6,12 However, a few years later, another paper using RP data with higher sequence coverage (>6 million reads) concluded that polybasic sequences had no effect on translation speed.Citation13 Recently, it was showed by RP that ribosome stalling caused by positively charged residues could be observed in some, but not all organisms analyzed.Citation14 Our aim was to revisit this conflicting data by taking advantage of recently published RP with a greater coverage (3–66 million reads) and, more importantly, with differences in the way that the samples were prepared before sequencing with respect to the absence or presence of translational inhibitors.
We clarified the role of polybasic sequences on ribosome translation as analyzed by RP using a combination of 2 approaches. First, we mapped the genome of the yeast S. cerevisiae for polybasic sequences, meaning 6 or more R or K residues within any group of 10 amino acids. We found that approximately 19% of the yeast proteins contain at least one polybasic domain and 1.3% present 6 or more K or R residues in tandem. Second, we used the RP data generated by 5 different groups, each differing in their use or omission of the translational inhibitors cycloheximide and anisomycin. These inhibitors are usually added to stabilize the polysomes before and during cellular lysis. We observed increased ribosome density caused by 6 or more K or R residues in tandem only when the RP was performed in the absence of these drugs. When the cells were treated with cycloheximide or anisomycin before lysis, no increased ribosome density was observed. We discuss the limitations and potential applications of ribosome footprint data in the study of translation rates of proteins containing polybasic domains.
Results and discussion
The first study that systematically screened for polybasic sequences in a full genome was published in 2012 by the Weissman group.Citation6 To identify endogenous proteins containing polybasic sequences, the S. cerevisiae genome was screened for sequences with 6 or more lysines (K's) or arginines (R's) within any group of 10 amino acids. This criterion was chosen based on experimental data showing that the inclusion of 6 lysines in tandem reduces the expression of reporter genes by 45%.Citation4 The Weissman group found 75 proteins with one or more polybasic sequences and 103 total polybasic domains. This list excluded genes that presented on average less than one read per total number of nucleotides, as well as genes that contained polybasic stretches at 50 amino acids from the N or C termini. When we analyzed this list, we observed some inconsistencies in the annotation of the polybasic stretch location within the sequences, so we decided to generate a new list of yeast genes coding for polybasic domains. Therefore, we performed another search using the same criterion (6 or more lysines (K's) or arginines (R's) within any group of 10 amino acids), and we found 1187 proteins with polybasic sequences (), which comprised 19% of the yeast proteins (Table S1). As expected, we observed that the isoelectric point of these sequences was slightly higher than that of the full yeast proteome (). We then analyzed published ribosome profiling data to determine whether the polybasic sequences with 6 or more K or R residues within any group of 10 amino acids would show increased ribosomal density. We analyzed the ribosome profiling (RP) data from 5 independent groups deposited in the Gene Expression Omnibus (GEO) (). For clarity, we labeled the RP data with the name of the principal investigator, followed by information about the presence or absence of the translational inhibitors commonly used to prepare the RP sample (). Only a modest increase in ribosomal density was observed for this set of sequences (Fig. S1A, namely Brown no drug and Koller −/+). We also analyzed the 75 proteins identified in the first polybasic screen performed by the Weissman group, and observed a comparable effect on ribosome density (Fig. S1C).
Figure 1. Bioinformatic analysis of yeast polybasic sequences. (A) The full genome of the yeast S. cerevisiae was screened for polybasic sequences, meaning 6 or more R's or K's residues within any group of 10 amino acids. We found 1187 proteins containing at least one polybasic domain. These proteins were grouped according to the maximum number of consecutive K/R's residues. The inset is a magnification of the groups of proteins with 6 or more consecutive K/R's residues. (B) The isoelectric point of each protein (6198) of the full genome of S. cerevisiae was calculated and compared with the set of polybasic proteins (1182). A normalized distribution is shown.
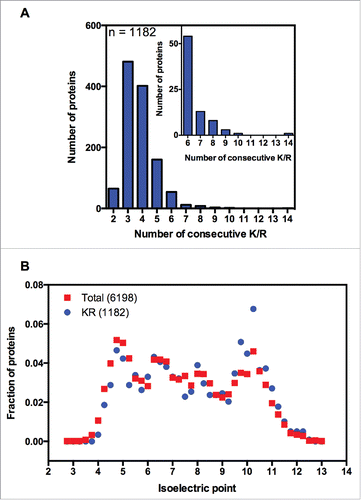
Table 1. Experimental conditions and the number of reads in each dataset mapping to the S. cerevisiae or M. musculus annotation used in the current analysis.
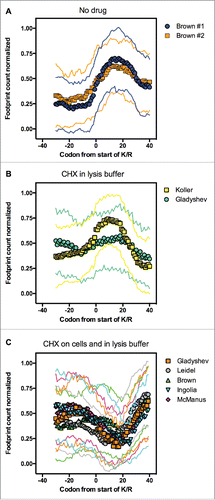
Together these data suggest that the increment of ribosome density caused by 6 or more K or R residues in a 10-amino-acid stretch is subtle. One important difference among the polybasic sequences studied with the use of reporter genes and the sequences analyzed in Fig. S1A and B is the distribution of the K/R's residues in a window of 10 amino acids. The reporter genes were built with 2, 4, 6, 8, 10 or 12 K's in tandem, and reporter expression was inversely proportional to the presence of poly-K.Citation4 Although 2 or 4 K's caused no effect on reporter expression, the presence of 6, 8, 10 or 12 K's reduced expression to 45, 9, 3 and 1%, respectively, when compared with the control. We decided to group the polybasic proteins to find polybasic sequences with 6 or more K/R's in tandem. Seventy-four percent of the sequences identified possessed 3 or 4 consecutive K/R's, whereas 81 sequences possessed 6 or more consecutive K/R's, which comprised 1.3% of the yeast proteome. This group of sequences showed a clear increase in ribosomal density compared with 1187 polybasic proteins including non-consecutive polybasic sequences (Fig. S1B). Therefore, we used the 81 consecutive polybasic sequences to perform the RP analysis on the all data sets depicted in . We observed different patterns depending on how the RP experiment was conducted (). It is important to note that all RP experiments were conducted with yeast cells grown in a rich medium (yeast peptone glucose) and harvested at the same metabolic state (fermentative); even though there are other sources of experimental variations, such as yeast strain used and details in the harvest and lysis methods, the main differences among the experiments are due to the use of the translational inhibitors cycloheximide (CHX) or anisomycin before or during cellular lysis.Citation15-19 We clearly observed increased ribosome density associated with sequences of 6 or more consecutive K/R residues when the RP was performed in the absence of any translational inhibitor (; #1 and #2 show the RP of independent replicates performed by the same authors). The reduced translation was characterized by a peak of ribosome occupancy of approximately 30 codons, starting from codon 0. The ribosomal exit tunnel accommodates approximately 30–40 amino acidsCitation20; thus, 30 codons represent 10 amino acids filling the ribosome exit channel. Another graphical representation of this data showed that the average ribosome occupancy of the polybasic sequences was twice as high as that of the regions upstream of the same sequences (Fig. S2). The coding regions near the starts are known to have a different coding biasCitation21 and it was suggested that the ribosomes decode these regions more slowly than the rest of coding sequences. To avoid potential artifacts due to differential codon usage, we excluded 100 codons downstream of start and upstream of stop codons. Even following this stringent criterion, we still observed the increased ribosome density on polybasic sequences (Fig. S3), excluding the possibility that differences in codon bias at the beginning or end of the coding sequence could be causing the increased ribosome density observed with the polybasic sequences. Another reason for ribosome pausing could be an over-representation of slow decoding arginine codon CGA.Citation22 However this is a rare codon in yeast, and only 8 sequences in the list presented this codon within the polybasic stretch.
Figure 2. Positive charges slow ribosomes when the ribosome footprint profiling is performed in the absence of translational inhibitors. Using the polybasic sequences data set containing 6 or more consecutive K/R residues (inset of ), we performed a ribosome profiling analysis on samples processed by other groups in the absence or presence of cycloheximide (CHX) (see details in ). In panel A, cell harvesting and lysis occurred in the absence of CHX (Brown #1 and #2). In panel B, the cells were lysed in a buffer with 100 μg/ml CHX (Koller and Gladyshev), whereas in panel C, the cells were pre-treated with 100 μg/ml CHX before harvesting and lysed in in a buffer with 100 μg/ml CHX (Gladyshev, Leidel, Brown, Ingolia and McManus). We show the averaged number of reads and the standard deviation (thin lines).
Our next step was to address the behavior of sequences coding for amino acids with different chemical properties that are not expected to slow down the ribosomes. We analyzed the RP of 60 randomly chosen proteins from yeast genome containing 6 or more aspartic (D) or glutamic (E) acid residues in tandem, which are negatively charged. As expected, we observed no increase in ribosomal density during translation of polyacid sequences (Fig. S4). In fact, the RP of polyacid sequences presented diminished ribosome density, indicating that this kind of sequence may have an effect on protein translation rate. This may be an interesting topic for future investigation.
The increase in ribosome density caused by polybasic sequences was strongly affected by the addition of CHX. When CHX was added only to the lysis buffer, we noticed a pattern similar to that , although it was somewhat less prominent with the Gladyshev data (). In contrast, when the cells were pre-treated with 100 μg/ml CHX (usually for 1 min) before cellular lysis (which was also performed in the presence of 100 μg/ml CHX), no reduced translation was observed (, see also Brown CHX Fig. S1A where 1189 polybasic sequences were analyzed). Interestingly, the 5 RP analyses reported in presented a very similar pattern, despite the use of different yeast strains and other experimental variations. This suggests that this methodology could be more easily reproduced when compared to the addition of CHX only in the lysis step. Therefore, adding CHX only to the lysis buffer seems to minimize the effects associated with the drug treatment, on the other hand, this protocol may introduce difficulties in data reproducibility associated with differences in the harvest or lysis methods. Another consideration is the effect of sequence coverage and the method used to normalize the occupancy of codons on the interpretation of the ribosome density analysis. Using the first RP generated by Ingolia and collaborators in 2009 (with CHX on cells and in the lysis buffer, ), Charneski and Hurst observed increased ribosome density at polybasic sites.Citation12 However, Artieri and Fraser reported that the method used by Charneski and Hurst detects stalling in any series of windows with sparse read coverage, even in the absence of polybasic sequences. Using datasets with more reads, and with CHX on cells and in the lysis buffer, Artieri and Fraser's could not observe any stalling when applying Charneski and Hurst's 2013 method.Citation13 Our data agree with Artieri and Fraser conclusion, since we did not observe reduced translation of polybasic sequences when CHX was used as translational inhibitor before lysis.
It is also worth noting that, in RP performed in the presence of cycloheximide, we observed an increase of ribosome density downstream from the expected polybasic peptides position (Fig. S5). The same pattern was observed very recently in RP analysis of rare codons.Citation23 Using different RP data sets and mathematical models the authors suggested that, rather than causing a complete halt in translation, CHX abruptly alters the codon-specific elongation rate resulting in an overall large reduction in translation during the drug treatment. Therefore, the offsets induced by CHX would be a consequence of the new codon-specific translation dynamics introduced by the drug presence.Citation23 In this model, the slow coding polybasic sites would be less sensitive to CHX treatment before lysis, so that the increase in density is shifted toward more sensitive codons downstream.
We conclude that 6 or more K/R residues in tandem are able to cause reduced translation and RP analysis is able to detect a corresponding increase in ribosome density when performed with data generated in the absence of inhibitory drugs.
In order to see whether the results presented in could be extended to another organism we repeated our analysis using previously published RP experiments performed on mammalian cells with and without translational inhibitorsCitation24, (). A BLAST search for proteins containing at least 6 K or 6 R in tandem retrieved 7 proteins from the mouse genome database (Table S1). For these few sequences, the profiles obtained in the absence of inhibitory drugs were very similar to polybasic yeast sequences (Fig. S6, compare blue circles with yellow circles). However, the interference of translation inhibitors was not so marked when compared to RP of yeast genes. These data suggested that the effect of polybasic sequences on the ribosome density is not restricted to yeast cells but the drug effect may vary between different organisms. More detailed analyses and a global quantification of all mammalian polybasic sequences are necessary to determine whether factors such as drug diffusion properties or different binding affinity can explain these differences.
The next step was to investigate the size distribution of the mRNA reads present in the data sets used for our analyses. The RP analyzed in was generated by the Brown laboratory. In a very elegant study,Citation15 the authors showed that during translation elongation, the ribosomes assumed 2 different conformations, which allowed the sequencing of 2 distinct populations of protected mRNAs: 28–30 nucleotides and 20–22 nucleotides longCitation15 (). These two populations of mRNAs were observed only when the RP was conducted in the absence of any drug.Citation15 Translational inhibitors stabilized the ribosome in specific conformations. Although CHX stabilized a conformation that protected longer mRNA fragments, anisomycin stabilized the other conformation, protecting shorter fragments of mRNA from RNase digestionCitation15 ().
Figure 3. The effect of different translation inhibitors on the ribosome profiling analysis of the polybasic sequences. The distribution of the mapped mRNA fragment lengths from untreated yeast (A), yeast treated with cycloheximide (B) or yeast treated with anisomycin (C). (D) Using the polybasic sequences containing 6 or more consecutive K/R residues (inset of ), we performed a ribosome profiling analysis using data generated by other groups using the samples described in panels A, B and C. We divided the mapped mRNA fragments for untreated yeast (panel A) into 2 groups: shorter mRNA fragments (18–25 nt, inset of panel E) and longer mRNA fragments (26–35 nt, inset of panel F). These two classes of mRNA fragments were subjected to the ribosome profiling analysis of the polybasic sequences (E and F). We show the averaged number of reads and the standard deviation (thin lines).
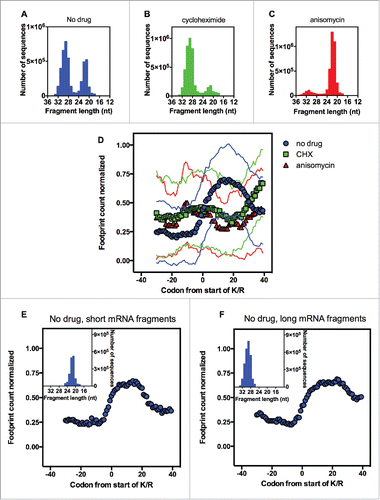
One possible explanation for the data presented in is that during the translation of polybasic sequences, the ribosome stalls in the conformation that protects shorter mRNAs, so that treatment with cycloheximide would not allow the recovery of these fragments. If this hypothesis is correct, we would expect to see ribosome reduced translation on polybasic sequences when the drug anisomycin is present (), but this was not the case (). To further confirm that the length of the mRNA analyzed is not important for the detection of increased ribosome density caused by polybasic sequences, we separated the RP data obtained in the absence of drugs into 2 categories: 26–35 nucleotides long and 18–25 nucleotides long (insets of ). In both cases, polybasic sequences caused ribosomal pausing (), which indicates that the effect of CHX on RP reduced translation on polybasic sequences is beyond the stabilization of a specific ribosome conformation. In fact, it has recently been shown that CHX distorts footprint coverage across mRNA transcripts.Citation15,16 We suggest here that CHX distorts the analysis of RP for polybasic sequences through a mechanism that is distinct from the stabilization of a specific ribosomal conformation described by the Brown group.
The next experiment was conducted to address the effect of homopolymeric-A (polyA) stretches on ribosome footprint analysis of yeast cells. Working together, the Green and Djuranovic laboratories recently described a phenomenon called ribosome sliding. Ribosome sliding occurs on mRNAs regions that are rich in homopolymeric-A stretches and leads to mRNA degradation, frameshifts and alterations in protein output.Citation25,26 Since Homopolymeric-A stretches can be associated with translation of lysine residues, consequently ribosome sliding might be related to its charge. However, it is important to note that homopolymeric-A stretches do not necessarily code only for lysines. Depending on the distribution of the A's on the mRNA, 3 lysines can be encoded by homopolymeric A's from 9 to 13 A's in length. The authors used several approaches, including the use of a reporter system and RP analysis, to show that the presence of homopolymeric-A stretches is a more important determinant than charge in ribosomal pausing.Citation26 The authors screened for homopolymeric-A stretches in several genomes and found 426 genes in S. cerevisiae with at least 8 A's in tandem (Habich, Djuranovic, Szczesny, PATACSDB - the database of polyA tracks in coding regions, in preparation). Using Green's and Djuranovic's list, we chose sequences with at least 10 homopolymeric A's, which comprised 116 proteins. Because the size of homopolymeric A's correlates positively with increased ribosome density, we expected an increased possibility to observe an effect on RP from the choice of the 116 containing at least 10 A's in tandem. Nevertheless, this was not the case: we performed the RP analysis using the data generated by other groups in the absence of any drug (Fig. S7A) or in the presence of CHX (Fig. S7B) but observed no ribosomal pausing. We conclude that RP analysis cannot detect a correlation between ribosome reduced translation and homopolymeric-A sequences in yeast cells. We also searched for homopolymeric-A sequences coding for 6 or more K/R residues in tandem and found 18 proteins (22% of total). These 18 proteins caused ribosomal pausing (data not shown), which again suggests that polybasic residues affect translation. Most biochemical data collected by the Green and Djuranovic laboratories used mammalian and insect cells as models, whereas we focused our analyses on yeast cells; this difference might explain the apparent contradiction observed here. We conclude that homopolymeric A's cause problems in translation based on the strong biochemical data presented by the Green and Djuranovic laboratories. Polybasic proteins cause similar effects, as demonstrated by our analysis and by different groups.Citation4-6,25-27
Our analysis, together with other recent studies exploring the effect of CHX on ribosome profiling,Citation23,28 showed, that the RP methodology can be more faithfully used to explore ribosomal translation if performed in the absence of translational inhibitors such as cycloheximide and anisomycin. We demonstrated that drug-free RP datasets tend to agree with the experimental evidence obtained by other methods analyzing the role of positively charged amino acids in reducing protein translation. It is important to note that polybasic sequences are only one among several different factors that can lead to reduced decoding rates or ribosome stalling. Polybasic stretches are present in only 1.3% of the yeast proteome, but these proteins are usually involved in nucleic acid or cellular membrane interaction and have important regulatory functions. Therefore, translational speed may be an important determinant for the abundance of this subset of the yeast proteome.
Materials and methods
Screening for polybasic sequences
We performed a global screening for polybasic sequences in S. cerevisiae. Coding sequences of uncharacterized and verified ORFs (5722) of S. cerevisiae were retrieved from Saccharomyces Genome Database (http://www.yeastgenome.org). We have created a binary code where codons for K or R were designated 111 and all other codons 000. After converting the sequences, our array consisted of the detection of 6 or more alkaline residues (lysine or arginine) in a sequence of 10 given residues. The analysis was performed by considering all possible sequences with 10 residues in length (e.g., aa1-aa10, aa2-aa11, aa3-aa12, etc.). Every time the same sequence was identified in a different arrangement, we grouped those sequences into one single stretch of polybasic sequence. After identifying all sequences that contained at least one polybasic domain, we searched for sequences with 6 or more consecutive K/R residues. The identification of genes coding for polyacid sequences (Fig. S4) was performed by Blast using Saccharomyces Genome Database. We searched only for sequences containing at least 6 D's or 6 E's in tandem.
For the mammalian polybasic screening we used previously identified sequences containing poly K/RCitation26 and complemented the data set by doing a Blast search against the Mouse Genome Informatics database (http://www.informatics.jax.org) (Fig. S6). We focused on sequences containing at least 6 R or 6 K in tandem.
Ribosome profiling data analyzes
Seven independent studies of ribosome profiling data sets from yeast and mammalian cells were used in our analyses (for details, see ). The data were analyzed as described by Ingolia and collaboratorsCitation10, except that the program used here was Geneious R8 (Biomatter Ltd., New Zealand) instead of CASAVA 1.8 pipeline. The data were downloaded from GEO, and the adaptors (CTGTAGGCACCATCAAT) were trimmed. The trimmed FASTA sequences were aligned to S. cerevisiae ribosomal and noncoding RNA sequences to remove rRNA reads. The unaligned reads were aligned to the S. cerevisiae S288C genome deposited in the Saccharomyces genome database. First, we removed any reads that mapped to multiple locations. Then, the reads were aligned to S. cerevisiae coding sequences database, allowing 2 mismatches per read. The first 20 codons were excluded from all analysesCitation21 and genes with <50% of positions covered were eliminated. For the analysis shown in Fig. S3, we adopted a more stringent criterion. The first and the last 100 codons were excluded and the minimum footprint coverage used was 1 read per total nucleotides in the sequence. For Fig. S5 a protocol similar to that described before for yeast was used, except that the library of transcripts was derived from Mouse Genome Informatics. We normalized the coverage within the same transcript. The average of several sequences with 30 nucleotides before and after the K/R, D/E or homopolymeric A sequences was used for all analyses.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental_Data.zip
Download Zip (1.5 MB)Acknowledgment
We thank Dr. Martha Sorenson, Dr. Debora Foguel, Dr. Cláudio A. Masuda and Dr. Maxim V. Gerashchenko for critical reading of the manuscript and helpful discussions. We thank Dr. Onn Brandman for providing us with the binary data that identified R or K residues in the full yeast genome and for helpful discussions. We thank Dr. Pawel Szczesny and Dr. Sergej Djuranovic for providing us with the data with the homopolymeric-A stretches identified in the full yeast genome. We also thank Dr. Liana Lareau and Dr. Cristina Pop for providing us with technical details regarding the ribosome footprint methodology used in their respective work. We dedicate this work to Elis Domitrovic Palhano. This work was supported by grants from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), the Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) and the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
Related Research Data
References
- Hershey JW, Sonenberg N, Mathews MB. Principles of translational control: an overview. Cold Spring Harb Perspect Biol 2012; 4; PMID:23209153; http://dx.doi.org/10.1101/cshperspect.a011528
- Lu J, Kobertz WR, Deutsch C. Mapping the electrostatic potential within the ribosomal exit tunnel. J Mol Biol 2007; 371:1378-91; PMID:17631312; http://dx.doi.org/10.1016/j.jmb.2007.06.038
- Lu J, Deutsch C. Electrostatics in the ribosomal tunnel modulate chain elongation rates. J Mol Biol. 2008; 384:73-86; PMID:18822297; http://dx.doi.org/10.1016/j.jmb.2008.08.089
- Ito-Harashima S, Kuroha K, Tatematsu T, Inada T. Translation of the poly(A) tail plays crucial roles in nonstop mRNA surveillance via translation repression and protein destabilization by proteasome in yeast. Genes Dev 2007; 21:519-24; PMID:17344413; http://dx.doi.org/10.1101/gad.1490207
- Dimitrova LN, Kuroha K, Tatematsu T, Inada T. Nascent peptide-dependent translation arrest leads to Not4p-mediated protein degradation by the proteasome. J Biol Chem 2009; 284:10343-52; PMID:19204001; http://dx.doi.org/10.1074/jbc.M808840200
- Brandman O, Stewart-Ornstein J, Wong D, Larson A, Williams CC, Li GW, Zhou S, King D, Shen PS, Weibezahn J, Dunn JG, Rouskin S, Inada T, Frost A, Weissman JS. A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell 2012; 151:1042-54; PMID:23178123; http://dx.doi.org/10.1016/j.cell.2012.10.044
- Bengtson MH, Joazeiro CA. Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature 2010; 467:470-3; PMID:20835226; http://dx.doi.org/10.1038/nature09371
- Lyumkis D, Oliveira dos Passos D, Tahara EB, Webb K, Bennett EJ, Vinterbo S, Potter CS, Carragher B, Joazeiro CA. Structural basis for translational surveillance by the large ribosomal subunit-associated protein quality control complex. Proc Natl Acad Sci USA 2014; 111:15981-6; PMID:25349383; http://dx.doi.org/10.1073/pnas.1413882111
- Shen PS, Park J, Qin Y, Li X, Parsawar K, Larson MH, Cox J, Cheng Y, Lambowitz AM, Weissman JS, Brandman O, Frost A. Protein synthesis. Rqc2p and 60S ribosomal subunits mediate mRNA-independent elongation of nascent chains. Science 2015; 347:75-8; PMID:25554787; http://dx.doi.org/10.1126/science.1259724
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc 2012; 7:1534-50; PMID:22836135; http://dx.doi.org/10.1038/nprot.2012.086
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 2009; 324:218-23; PMID:19213877; http://dx.doi.org/10.1126/science.1168978
- Charneski CA, Hurst LD. Positively charged residues are the major determinants of ribosomal velocity. PLoS Biol 2013; 11:e1001508; PMID:23554576; http://dx.doi.org/10.1371/journal.pbio.1001508
- Artieri CG, Fraser HB. Accounting for biases in riboprofiling data indicates a major role for proline in stalling translation. Genome Res 2014; 24:2011-21; PMID:25294246; http://dx.doi.org/10.1101/gr.175893.114
- Sabi R, Tuller T. A comparative genomics study on the effect of individual amino acids on ribosome stalling. BMC Genomics 2015; 16 Suppl 10:S5; PMID:26449596; http://dx.doi.org/10.1186/1471-2164-16-S10-S5
- Lareau LF, Hite DH, Hogan GJ, Brown PO. Distinct stages of the translation elongation cycle revealed by sequencing ribosome-protected mRNA fragments. eLife 2014; 3:e01257; PMID:24842990; http://dx.doi.org/10.7554/eLife.01257
- Gerashchenko MV, Gladyshev VN. Translation inhibitors cause abnormalities in ribosome profiling experiments. Nucleic Acids Res 2014; 42:e134; PMID:25056308; http://dx.doi.org/10.1093/nar/gku671
- Nedialkova DD, Leidel SA. Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell 2015; 161:1606-18; PMID:26052047; http://dx.doi.org/10.1016/j.cell.2015.05.022
- McManus CJ, May GE, Spealman P, Shteyman A. Ribosome profiling reveals post-transcriptional buffering of divergent gene expression in yeast. Genome Res 2014; 24:422-30; PMID:24318730; http://dx.doi.org/10.1101/gr.164996.113
- Pop C, Rouskin S, Ingolia NT, Han L, Phizicky EM, Weissman JS, Koller D. Causal signals between codon bias, mRNA structure, and the efficiency of translation and elongation. Mol Syst Biol 2014; 10:770; PMID:25538139; http://dx.doi.org/10.15252/msb.20145524
- Fedyukina DV, Cavagnero S. Protein Folding at the Exit Tunnel. Annu Rev Biophys 2011; 40:337-59; PMID:21370971; http://dx.doi.org/10.1146/annurev-biophys-042910-155338
- Tuller T, Carmi A, Vestsigian K, Navon S, Dorfan Y, Zaborske J, Pan T, Dahan O, Furman I, Pilpel Y. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 2010; 141:344-54.22; PMID:20403328; http://dx.doi.org/10.1016/j.cell.2010.03.031
- Letzring DP, Dean KM, Grayhack EJ. Control of translation efficiency in yeast by codon-anticodon interactions. RNA 2010; 16:2516-28; PMID:20971810; http://dx.doi.org/10.1261/rna.2411710
- Hussmann JA, Patchett S, Johnson A, Sawyer S, Press WH. Understanding biases in ribosome profiling experiments reveals signatures of translation dynamics in yeast. PLoS Genet 2015; 11:e1005732; PMID:26656907; http://dx.doi.org/10.1371/journal.pgen.1005732
- Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 2011; 147:789-802; PMID:22056041; http://dx.doi.org/10.1016/j.cell.2011.10.002
- Koutmou KS, Schuller AP, Brunelle JL, Radhakrishnan A, Djuranovic S, Green R. Ribosomes slide on lysine-encoding homopolymeric A stretches. eLife 2015; 4:e05534; PMID:25695637; http://dx.doi.org/10.7554/eLife.05534
- Arthur L, Pavlovic-Djuranovic S, Smith-Koutmou K, Green R, Szczesny P, Djuranovic S. Translational control by lysine-encoding A-rich sequences. Sci Adv 2015; 1:e1500154; PMID:26322332; http://dx.doi.org/10.1126/sciadv.1500154
- Chiabudini M, Tais A, Zhang Y, Hayashi S, Wölfle T, Fitzke E, Rospert S. Release factor eRF3 mediates premature translation termination on polylysine-stalled ribosomes in Saccharomyces cerevisiae. Mol Cell Biol 2014; 34:4062-76; PMID:25154418; http://dx.doi.org/10.1128/MCB.00799-14
- Weinberg DE, Shah P, Eichhorn SW, Hussmann JA, Plotkin JB, Bartel DP. Improved ribosome-footprint and mRNA measurements provide insights into dynamics and regulation of yeast translation. Cell Rep 2016; 14:1787-99; PMID:26876183; http://dx.doi.org/10.1016/j.celrep.2016.01.043