
ABSTRACT
Alternative polyadenylation has been implicated as an important regulator of gene expression. In some cases, alternative polyadenylation is known to couple with alternative splicing to influence last intron removal. However, it is unknown whether alternative polyadenylation events influence alternative splicing decisions at upstream exons. Knockdown of the polyadenylation factors CFIm25 or CstF64 in HeLa cells was used as an approach in identifying alternative polyadenylation and alternative splicing events on a genome-wide scale. Although hundreds of alternative splicing events were found to be differentially spliced in the knockdown of CstF64, genes associated with alternative polyadenylation did not exhibit an increased incidence of alternative splicing. These results demonstrate that the coupling between alternative polyadenylation and alternative splicing is usually limited to defining the last exon. The striking influence of CstF64 knockdown on alternative splicing can be explained through its effects on UTR selection of known splicing regulators such as hnRNP A2/B1, thereby indirectly influencing splice site selection. We conclude that changes in the expression of the polyadenylation factor CstF64 influences alternative splicing through indirect effects.
Introduction
Eukaryotic gene expression and proteomic diversity is dependent on the appropriate removal of introns from pre-mRNAs, a process orchestrated by the spliceosome. Multiple mRNA isoforms are generated through alternative splicing (AS), a highly regulated process that has been identified in ∼95% of human genes.Citation1,2 AS requires the use of different combinations of splice sites resulting in several classes of splicing patterns, such as: alternative 5′ splice site selection (Alt5), alternative 3′ splice site selection (Alt3), the skipping of complete exons (SE), or the retention of introns (RI).
Many splicing decisions are believed to occur co-transcriptionally,Citation3-Citation7 both through the use of alternative promotersCitation8-Citation12 as well as in defining the firstCitation13 and last exons. First and last exons only have one flanking splice site and are, therefore, recognized differently than internal exons. Terminal exon definition has been shown to be aided by the polyadenylation machineryCitation14,15 through interactions between U2AF and the polyadenylation polymerase (PAP)Citation16 or cleavage factor 1m (CF1m),Citation17 or through interactions between splicing factor 3b (SF3b), a component of U2 snRNP, and the cleavage and polyadenylation specificity factor (CPSF).Citation18 Furthermore, the U1 snRNP component U1A has been shown to stimulate polyadenylation through interaction with CPSF160,Citation19 however, U1 snRNP also plays a role in preventing premature cleavage and polyadenylation through binding of U1 snRNA to cryptic poly(A) sitesCitation20 as well as through direct interactions between U1-70K and the PAP.Citation21 More recent findings have demonstrated that a terminal splice acceptor siteCitation22 and poly(A) tail may be necessary for terminal intron removal with splicing promoted through the coordinated efforts of PAP and nuclear poly(A) binding protein, PABPN1.Citation23 Thus, the interactions of splicing and polyadenylation factors at terminal exons play important roles in the regulation and enhancement of both splicing and polyadenylation.Citation24
Alternative polyadenylation (APA) has gained renewed consideration as an important regulator of gene expression. Similar to AS, the use of alternative poly(A) sites also allows for multiple mRNA isoforms to be generated from a single transcript. Recent analyses has identified APA events in about 70% of human genes.Citation25 The ability to choose a particular poly(A) site depends on the strength of the poly(A) signal, as well as surrounding cis-elements.Citation26,27 Genes/transcripts may have alternative poly(A) sites located in internal introns or exons leading to the formation of different protein isoforms. Alternatively, APA events that occur in the 3′ untranslated regions (UTR) will result in transcripts with different 3′UTR lengths, but will code for the same protein. Changing the length of the 3′UTR is believed to affect the stability, localization, transport, and even translation of the mRNA through altered interactions with microRNAs or other regulatory RNA binding proteins.Citation28-Citation32 Several polyadenylation factors have been implicated in APA including CF1mCitation33,34 and CstF.Citation35,36 For example, knockdown of CF1m25 (CF1m25 KD) or CstF64 (CstF64 KD) has been shown to result in the activation of alternative poly(A) sites.Citation34,36 Thus, APA events are mediated through factors that are also engaged in interactions with spliceosomal components.
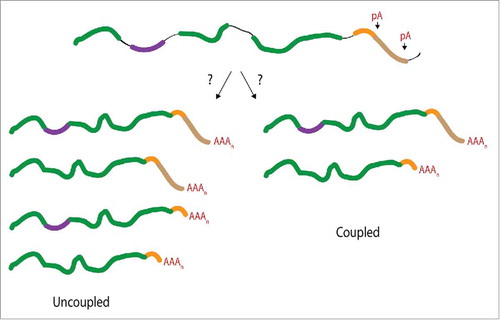
The mechanism of interaction and the extent of coupling between APA and AS are still relatively unexplored. While previous work demonstrated the coupling between splicing and polyadenylation for terminal exons, it is unknown whether there is a mechanistic connection between APA and AS events upstream of the terminal exon. In other words, does an upstream AS event dictate a preference for an APA event or vice versa? Two scenarios can be envisioned. One scenario describes AS and APA events as mechanistically uncoupled. Each process is executed independently from one another, resulting in spliced mRNA isoforms, each of which has a similar distribution of variable 3′UTR lengths as generated from APA (). However, if AS and APA events are mechanistically coupled, the resulting mRNA isoforms would be characterized by a selective preference for one type of APA with a particular AS event (). As a consequence of this coupled scenario, specific mRNA isoforms could be selectively stabilized or destabilized over other isoforms. Thus, the mechanistic coupling between AS and APA could significantly influence the expression of mRNA isoforms. Using genome-wide approaches, we tested the hypothesis that APA and upstream AS events are functionally linked.
Figure 1. Consequences of a mechanistic connection between APA and AS events upstream of the terminal exon. It is unknown whether APA and AS events are functionally coupled beyond the definition of the terminal exon. If the RNA processing events are uncoupled (left arm) mRNA isoforms generated through AS would not display a selective preference for a particular APA event. A coupled event (right arm) would preferentially associate one form of APA with a specific AS event. The alternatively spliced exon is depicted in purple, introns shown as black lines, distal poly(A) (pA) site shown in brown, and the proximal pA site is depicted in orange.
Results
Changes in APA correlate with changes in AS at the terminal intron in CF1m25 KD cells
Previous studies have elucidated several protein interactions between the splicing and polyadenylation machineries, suggesting a coupled process in vivo. To test the hypothesis that altering polyadenylation site selection induces AS changes, we induced APA through RNAi mediated knockdown of CF1m25 in HeLa cells, which was shown to induce proximal poly(A) site selection in TIMP-2, SDC2, ERCC6 and DHFR.Citation34 At efficient CF1m25 KD conditions (60-80%) (), reduced CF1m25 levels resulted in alternative TIMP-2 poly(A) site selection as demonstrated by 3′ RACE analysis (). TIMP-2, an inhibitor of matrix metalloproteinases, contains 2 functional poly(A) sites, producing mRNAs of 1.2 kb and 3.8 kb. In agreement with previous results,Citation34 PCR-mediated 3′ end amplifications demonstrate that CF1m25 KD significantly induced proximal poly(A) site selection ().
Figure 2. CF1m25 KD induces APA leading to increased last intron retention in TIMP-2. (A) Western blot illustrating the efficiency of CF1m25 KD in HeLa cells. (B) Diagram of primers used for 3′ RACE amplification of TIMP-2. The distal primer binds to the 3′ UTR sequence of the 3.8 kb mRNA isoform. The proximal primer binds to the coding sequence of TIMP-2 but amplifies only from the poly(A) tail of the 1.2 kb isoform. (C) 3′ RACE cDNA amplification of TIMP-2 from cells transfected with CF1m25 shRNA or a negative control vector. The identities of the amplified PCR products are indicated on the side of the representative gel. The results of independent experiments are displayed in the graph below as the ratio of proximal:distal poly(A) site usage. (D) Diagram of PCR primers flanking the terminal intron retention event in TIMP-2. Asterisk (*) denotes non-specific binding. (E) RT-PCR analysis of TIMP-2 intron retention in cells treated with CF1m25 shRNA or control shRNA. The unspliced and spliced products amplified are indicated on the left of the representative gel. The results of independent experiments are displayed in the graph as the ratio of unspliced:spliced.
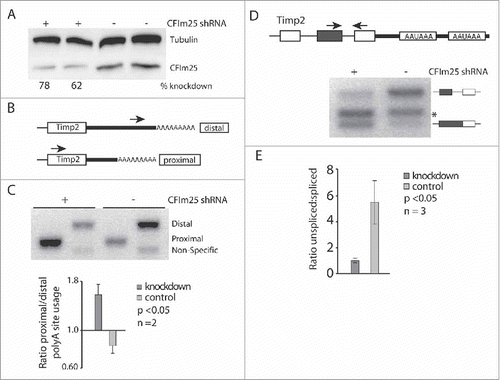
To determine if changes in APA correlate with changes in AS, RT-PCR analysis was performed along the TIMP-2 gene using cDNA from CF1m25 KD and control cells. At the terminal exon, depletion of CF1m25 resulted in a 7-fold decrease of an intron retention event (). Thus, proximal poly(A) selection increases terminal splicing efficiency in TIMP-2. These results demonstrate that proximal poly(A) site selection influences the efficiency of terminal intron removal. To address the question of whether splicing events upstream of the terminal exon were affected by CF1m25 KD, the inclusion of known alternative exons within genes that harbor conserved tandem APA eventsCitation37 were evaluated using RT-PCR approaches (TMEM135, DSTN, TSC22D2, SLC38A2, and ERCC6). None of the AS events tested displayed significant differences upon knockdown of CF1m25 (Fig. S1). These initial results suggest that changes in poly(A) site usage can influence AS near the 3′ UTR. However, there does not appear to be a correlation between upstream AS alterations and APA, supporting the notion that mechanistic ties between upstream AS events and APA are limited to terminal intron removal.
Genome-wide approach to correlate APA and upstream AS events
The major limitation of the approach described above is the small number of confirmed APA cases. To increase the power of the correlative analysis, we took advantage of a HeLa CstF64 KD cell line and used genome-wide approaches to identify APA and AS events.Citation38 After verifying >80% knockdown efficiency in CstF64 protein levels () APA events were identified using poly(A) site-sequencing (PAS-Seq)Citation39 and standard mRNA-Seq on both CstF64 KD and wild-type HeLa cells (). Subsets of these APA events were verified using RT-qPCR analysis (, Fig. S2).
Figure 3. Genome-wide approach for identification of a larger sample pool for correlation studies. (A) Western blot demonstrating CstF64 KD in HeLa cells. (B) Workflow for genome-wide analysis of CstF64 KD. To identify examples of APA genes, PAS-Seq and RNA deep sequencing was performed on both CstF64 KD and wild-type HeLa cells. APA and AS genes were then identified and the genes from each group were compared to each other to identify any overlap. (C) Fold change of the ratio of distal to proximal poly(A) sites between CstF64 KD and wild-type (WT) HeLa cells depicted for 10 genes identified by PAS-Seq, as determined by RT-qPCR, n = 3, *p < 0.05. Purple bars depict APA shifts from distal to proximal and green bars show proximal to distal APA changes. (D) Fold change of the ratio of distal to proximal poly(A) sites between CstF64 KD and WT HeLa cells depicted for 8 genes identified by MISO, as determined by qPCR, n = 3, *p<0.05. (E) Alternative splicing analysis identifies CstF64 as a potential regulator of AS. Alternative splicing analysis of RNA-Seq data identified a large number of significant AS changes upon CstF64 KD in HeLa cells (1,060 genes), indicating CstF64 as potential regulator of AS. (F) Overlap between AS and APA genes. Out of the 1,060 AS genes that were identified and 251 APA genes, only 13 genes were contained in the list of APA genes. (G) Number of CstF64 KD induced genes with intron retention located at the terminal intron (maroon) or upstream of the terminal intron (blue).
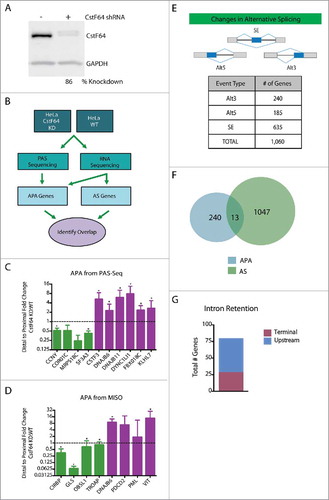
PAS-Seq and its applied bioinformatics pipeline identified 19 genes displaying statistically significant changes in APA. Differential splicing analysis of RNA-Seq datasets (by MISOCitation40) derived from CstF64 KD and wild-type HeLa cells identified 234 additional statistically significant alternative last exons (ALE), which also represent APA events. Furthermore, MISO analysis identified 1,060 genes that display statistically significant Alt3, Alt5 and SE alternative splicing events (). These results suggest that in addition to mediating APA, CstF64 also exerts a regulatory role on AS.
To determine the frequency of ‘splicing and polyadenylation linked’ events, the overlap between APA and AS events was determined. APA events detected from PAS-Seq (19 genes) and RNA-Seq (234 genes), a total of 253 genes (Table S1), were then correlated with all types of AS events. Interestingly, of the total 1,060 AS genes that were identified, only 13 genes were contained in the list of APA genes (, Table S2). This minimal overlap does not change when the statistical stringency is relaxed (see Materials and Methods). Given the small overlap between genes that display significant changes in APA and AS, we conclude that there is no general mechanistic coupling between APA and upstream AS in the conditions tested here. While this analysis does not exclude the possibility that a mechanistic link between the processing events can exist, for the majority of the alternative pre-mRNA processing events evaluated, APA and upstream AS are carried out independently.
The results evaluating the impact of induced APA in Timp2 () suggested that terminal intron removal may be influenced by the knockdown of polyadenylation factors. To test whether the knockdown of CstF64 induced intron retention events at the terminal end, we determined the extent of terminally located RI. MISO identified 77 genes displaying significant differences in the efficiency of intron removal. Interestingly, 35% of these RI events were terminal in nature (, Fig. S2C). These observations suggest that CstF64 KD preferentially impacts the removal efficiency of terminal introns, presumably through modulating interactions with the splicing and polyadenylation machinery at the 3′ end of the pre-mRNA.
Alternative splicing analysis identifies CstF64 as a potential regulator of AS
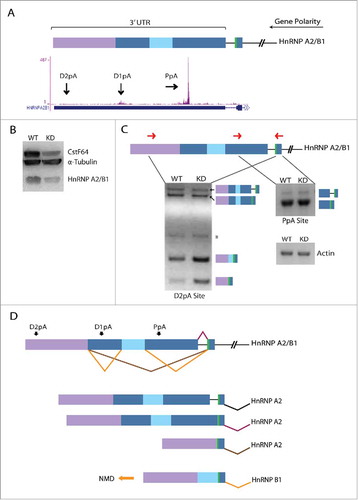
Interestingly, the large number of AS events that are detected upon CstF64 KD suggests that this polyadenylation factor may be a potential regulator of AS, either directly or through indirect effects. To test the hypothesis that CstF64 binding to the pre-mRNA directly influences AS, we mapped CstF64 iCLIP-Seq readsCitation40 to the human genome and evaluated whether unique binding signatures could be identified around alternatively spliced exons induced by CstF64 KD. Compared to control groups no significant differences in CstF64 binding densities were observed in upstream intronic binding surrounding Alt3 () or downstream intronic binding of Alt5 (). However, the upstream intronic region around alternatively included exons displayed increased CstF64 binding coverage, as demonstrated by the horizontal peak shift (). No measurable differences were detected for introns downstream of alternatively included exons (). Further analysis of the increase in CstF64 binding coverage around upstream introns of alternatively included exons identified the gene hnRNP A2/B1, which displayed strong CstF64 binding signals at APA sites (). CstF64 KD leads to a decrease in hnRNP A2/B1 protein levels (), which correlates with splicing changes within its 3′UTR (). There are 3 potential APA sites within the 3′UTR of hnRNP A2/B1 and selection of the proximal APA site leads to the formation of hnRNP A2 with no discernable difference between WT and CstF64 KD RNA levels. However, selection of the most distal APA site generates multiple splicing isoforms, one of which is the hnRNP B1 mRNA isoform, which is targeted by NMD.Citation41 Thus, CstF64 KD leads to an increase in the less stable hnRNP B1 mRNA isoform, providing a molecular basis for the reduced protein levels observed (). These observations suggest that AS changes within the 3′UTR of hnRNP A2/B1 lead to the activation of different APA sites, resulting in the generation of different terminal exons (). As hnRNP A2/B1 has also been implicated in the regulation of AS,Citation42 it is highly possible that many of the observed AS changes activated upon CstF64 KD were caused by the altered expression of this or other splicing regulatory proteins. Indeed, our analysis of CstF64 KD cells also identified hnRNP AB, hnRNP C, hnRNP H3, SRSF5 and SRSF6 as AS target genes (GEO submission GSE79157).
Figure 4. Density distribution of CstF64 binding in the vicinity of AS exons. To distinguish the location of intronic binding of CstF64, iCLIP-Seq coverage was analyzed. The density of the distribution of normalized iCLIP-Seq coverage within introns surrounding alternative exons was determined. (A) Intronic CstF64 binding distribution upstream of an alternatively spliced 3′ splice site (Alt3). (B) Intronic CstF64 binding distribution downstream of an alternatively spliced 5′ splice site (Alt5). (C) Intronic CstF64 binding distribution upstream of an alternatively skipped exon. (D) Intronic CstF64 binding distribution downstream of an alternatively skipped exon. “not differentially alternatively spliced” and “identically alternatively spliced” are control categories. The first control group includes exons that are not in the group of “differentially alternatively spliced” exons. The second control group (“identically alternatively spliced”) includes exons specifically filtered for no change in their splicing behavior. The plots show the fraction of events in those exon sets/groups that overlap with iCLIP-Seq clusters.
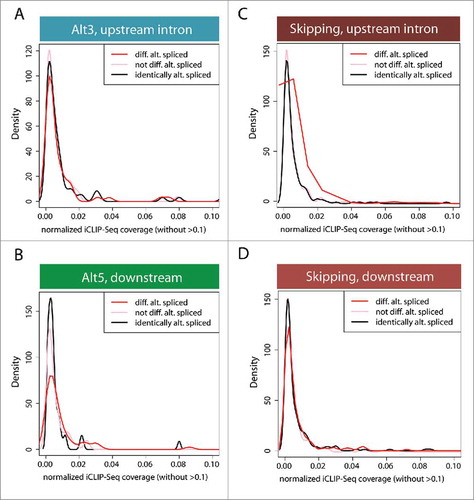
Figure 5. CstF64 binding within the 3′UTR of hnRNP A2/B1 gene triggers AS. (A) Cartoon depicts alternatively spliced regions (blue, light blue, purple boxes) located in the 3′UTR of the hnRNP A2/B1 gene. The polarity of the gene is from right (5′ end) to left (3′ end) as depicted by the long black arrow. The green bar depicts a stop codon. Snapshot from the UCSC genome browser depicts iCLIP-Seq track information for the binding of CstF64 and black arrows show the location of the proximal poly(A) site (PpA) and 2 distal poly(A) sites (D1pA and D2pA). (B) Western blot analysis demonstrating loss in hnRNP A2/B1 protein levels due to CstF64 KD. (C) RT-PCR analysis of the hnRNP A2/B1 3′UTR. Primer locations are depicted as forward and reverse red arrows. The identities of the PCR products are indicated on the side of the representative gels. Asterisk (*) denotes a non-specific amplicon. (D) Alternative splicing pattern in the 3′UTR of hnRNP A2/B1 depicting the generation of hnRNP B1 and hnRNP A2 mRNA isoforms. The orange arrow indicates the isoform (hnRNP B1) that undergoes nonsense-mediated decay (NMD).Citation41
To test the hypothesis that the reduced expression of hnRNP A2/B1 at CstF64 KD conditions accounts for many of the observed AS changes, we determined the overlap between CstF64 knockdown-mediated AS differences and hnRNP A2/B1 knockdown-mediated AS differences.Citation43 Interestingly, about one third of all genes that are affected by CstF64 KD are also affected by hnRNP A2/B1 KD (Fig. S3), thus supporting the proposal that CstF64's influence on the processing of hnRNP A2/B1 indirectly mediates changes in AS. In summary, our analysis demonstrates that CstF64 pre-mRNA binding does not correlate with induced AS. Rather, the selection of alternative poly(A) sites or the alternative splicing of several splicing regulators is altered upon CstF64 KD, suggesting that most AS differences observed are likely to be indirect effects of CstF64 KD.
Discussion
We tested the hypothesis that APA and upstream AS events are mechanistically coupled. APA was induced by the knockdown of the polyadenylation factors CF1m25 or CstF64. We then tested whether genes with APA also undergo AS upstream of the terminal exon. Using global approaches to increase the number of cases investigated, it was demonstrated that upstream AS and APA events occur independently from each other and that the major mechanistic coupling between APA and pre-mRNA splicing was limited to terminal exon processing. Direct interactions between the splicing and polyadenylation machineries have been documented previously,Citation44 as demonstrated by U1 snRNP preventing premature cleavage and polyadenylationCitation20 or the inhibition of terminal exon splicing and polyadenylation through mutations of the AAUAAA consensus poly(A) signal or the 3′ splice site.Citation14,45 In combination, these observations strongly suggest that the mechanistic coupling between APA and pre-mRNA splicing is generally limited to terminal exon definition.
The lack of a 5′ splice site at the 3′ end of the pre-mRNA dictates the need for alternative mechanisms to aid in the definition of the terminal exon. However, in the case of AS events upstream of the terminal exon, polyadenylation does not appear to exert a major influence on decision-making processes. This may be because each internal exon is efficiently recognized through the combinatorial contributions of 3′ and 5′ splice site interactions with the splicing machinery. Furthermore, kinetic barriers could limit interactions between the splicing and polyadenylation machinery to terminal exons as upstream exons are synthesized prior to polyadenylation sites. Thus, the “first come, first served” conceptCitation46-Citation48 may have a significant influence on allowing productive interactions between splicing factors recruited to upstream exons and the polyadenylation machinery assembled at the 3′ end. Moreover, it should also be noted that although transcription occurs in the 5′ to 3′ direction, not all splicing occurs in that order as demonstrated by the adenine phosphoribosyltransferase (APRT) gene where the first of 4 introns is the last to be removed.Citation49
Given the significance of AS and APA in contributing to genome diversity, it is of considerable interest to determine whether these pre-mRNA processing events are functionally linked beyond the previously demonstrated definition of terminal exons. It is known that splicing defects play a role in many human diseases,Citation50,51 including numerous types of cancer.Citation52-Citation55 Similarly, APA has a demonstrated regulatory role in human disease.Citation30,56-59 Thus, coordinated APA and upstream AS could generate alternatively spliced mRNA isoforms with unique 3′UTRs that dictate mRNA half-lives and protein product functions (). Our genome-wide analysis demonstrates that the mechanistic coupling between APA and pre-mRNA splicing is limited to terminal exon definition (). Thus, the regulation of mRNA isoform stability and translatability is mainly driven by the generation of different 3′UTRs.
Our results also identified an intriguing role for CstF64 in mediating AS. As shown, reduced levels of CstF64, led to a considerable change in AS events. However, neither enrichment nor depletion of CstF64 binding to alternatively spliced exons was observed when compared to control groups, arguing against a direct role. Instead, hnRNP A2/B1's was identified to be one of several splicing regulatory proteins that alter APA or AS patterns upon CstF64 KD. Given hnRNP A2/B1's important role in regulating ASCitation42 and mRNA stability,Citation41,60 these observations strongly suggest that CstF64 influences AS decisions through indirect mechanisms. Investigating the mechanisms that lead to 3′UTR diversification and alternative terminal exon definition will therefore be an important future research endeavor.
Materials and methods
Cell culture and transfections
CF1m25 KD was performed using shRNA constructs as previously described.Citation34 The siRNA-408 sequence (5′-GCAAUCGUCAAUGACCCAGUCUUGC-3′) was used as a target sequence in the design of an shRNA insert oligo and was cloned into the pSUPERIOR.puro vector (Oligoengine, VEC-IND-0006). To create the shRNA insert, the sense oligonucleotide 5′-GATCCCCGCAAGACTGGGTCATTGACGATTGCTTCAAGAGAGCAATCGTCAATGACCCAGTCTTGCTTTTTA-3′ was annealed with the antisense oligonucleotide 5′-AGCTTAAAAAGCAAGACTGGGTCATTGACGATTGCTCTCTTGAAGCAATCGTCAATGACCCAGTCTTGCGGG-3′ and ligated to linearized vector using T4 DNA ligase (Promega). HeLa cells were cultured in MEM and transfected with 1 µg of either CF1m25 KD plasmid or a control plasmid using Lipofectamine-2000 (Invitrogen). Media was replaced with MEM supplemented with 10% FBS and 3 mg/mL puromycin post transfection. RNA extraction was performed via Trizol and proteins were isolated with RIPA buffer.
CstF64-RNAi HeLa cell lines were obtainedCitation38 and grown in high glucose DMEM media plus 10% FBS, 1% Na-Pyruvate and 1.5 µg/mL of puromycin for selection of stably transfected cells. RNA extraction was performed via Trizol and proteins were isolated with RIPA buffer.
Western blot analysis
CF1m25 and CstF64 proteins were separated on 10% SDS-PAGE and subjected to standard western blotting procedures. Primary antibodies used were as follows: anti-rabbit NUDT21 (Proteintech Group, 10322-1-AP), anti-rabbit CstF64 (Bethyl Labs, A301-092A), anti-mouse hnRNP A2/B1 (AbCam, DP3B3) and anti-mouse α-Tubulin (Calbiochem, DM1A). Typically, 75 ug of total protein were loaded per lane.
3′ RACE and RT-PCR
3′ RACE was performed to analyze 3′ ends, as previously described.Citation61 Total RNA was reverse transcribed using the primer 5′-CCAGTGAGCAGAGTGACGAGGACTCGAGCTCAAGCTTTTTTTTTTTTTTTTT-3′. TIMP-2 gene specific primers used were: forward primer 1 5′-CGCAACAGGCGTTTTGCAAT-3′ for proximal poly(A) tail product amplification, TIMP-2 forward primer 2 5′-CTGTTCGCTTCCTGTATGGT-3′ for distal poly(A) tail product amplification,Citation34 and reverse primer 5′-CCAGTGAGCAGAGTGACG-3′.
For RT-PCR analysis, total RNA was treated with DNase I and reverse transcribed using iScript (BioRad). For the TIMP-2 gene, terminal intron retention was evaluated using forward primer 5-AGGGAAGCACACCTGCAGTA-3′ with reverse primer 5-GTGCCCGTTGATGTTCTTCT-3′. In addition to TIMP-2, 5 additional genes with documented cases of AS events were analyzed using RT-PCR. Three AS events were evaluated for each of the following genes: DSTN, TSC22D2, TMEM135, SLC38A2, and 7 AS events for ERCC6. Primer sequences are available upon request. PCR reactions were performed using Taq polymerase under standard conditions and resolved on a 0.8% agarose gel stained with ethidium bromide.
PAS-Seq APA events were verified by RT-qPCR as described previously.Citation38 RNA-Seq MISO designated AS events in the 3′UTR for hnRNP A2/B1 were evaluated in addition to APA events for eight genes: CIRBP, DNAJB6, GLS, OBSL1, PDCD2, PML, TROAP, VIT. Primer sequences are available upon request. Total RNA from CstF64 KD cells were treated with DNaseI and reverse transcribed using Oligo(dT) primers with SuperScript II (Invitrogen). RT-PCR analysis was performed with Taq polymerase under standard conditions and resolved on a 1.5–2% agarose gel stained with ethidium bromide. Quantitative real-time PCR (qPCR) was performed with the same primer pairs and iTaq Universal SYBR Green supermix (BioRad) using a 3 step qPCR protocol with an annealing temp (Tm) = 55°C and 1 minute extension at 72°C.
RNA deep sequencing
Cell lines stably transfected with shRNA plasmids targeting CstF64 were obtained from the Shi lab.Citation38 Cell lines were maintained by culturing in the presence of 1 mg/ml puromycin. Total RNA was isolated from CstF64 KD cells and wild-type control cells using Trizol according to manufacturer's protocols. CstF64 protein levels were measured via Western Blot analysis using rabbit anti-CstF64 antibody (Bethyl, A301-092A). Total RNA isolated from 2 biological replicates was used to generate independent libraries with the Illumina TruSeq RNA sample preparation kit according to manufacturer's protocols. Each library was diluted to approximately 10 pM prior to loading and sequenced using the Illumina GA IIx instrument generating 50 bp single-end reads. This produced ∼50 million reads for each wild-type and CstF64 KD biological replicate.
Computational analysis
Identification of AS and APA events
RNA-Seq results were trimmed by 10 bases at the 5′ end. Trimmed 40 bp long reads were then mapped to the human genome (GRCh37/hg19) using TopHat (version 2.0.4)Citation62 with default parameters. To identify AS events, the data sets were compared using MISO.Citation40 To do so, replicate samples were combined and only uniquely mapped reads were kept. For every known AS event expressed in both cell lines (SE, Alt3, Alt5, RI, ALE/APA) only events showing an absolute change in inclusion level or splice site usage ≥15% with a bayes factor of ≥10 were filtered for. Applying these filters resulted in the identification of 733 SE (636 genes), 267 Alt3 (240 genes), 200 Alt5 (185 genes), 109 RI (77 genes) and 328 APA (234 genes) events. To identify additional APA events, a PAS-Seq dataset was utilized comparing polyadenylation between CstF64 KD and wild-type HeLa cells.Citation38 PAS-Seq and its applied bioinformatics pipelineCitation38 identified 19 genes displaying statistically significant changes in APA, using a cutoff of ≥2 reads with FDR ≤0.05 and a change in APA ≥15% (a change in the ratio of proximal to distal shift in polyadenylation between CstF64 KD and wild-type HeLa cells).
Correlation between APA and AS events
To determine the frequency of ‘splicing and polyadenylation linked’ events, the overlap between APA and AS events was determined. APA events detected from PAS-Seq (19 genes) and RNA-Seq (234 genes), a total of 253 genes, were correlated with all types of AS events. Overlapping APA and AS genes were identified by determining those that only had AS events occurring upstream of both proximal and distal poly(A) sites. Of the 19 PAS-Seq derived list of APA genes, only one gene was located upstream of both proximal and distal poly(A) sites. Of the RNA-Seq derived list of APA genes, only 12 genes with AS events upstream of both poly(A) sites were identified.
Correlation between AS events and CstF64 binding
AS events were identified using the RNA-Seq data sets described above and MISOCitation40 filters of ≥20% change in the absolute level or splice site usage (for Alt3 or Alt5) with a bayes factor of ≥10 filtered for. Of these, events supporting only one of the 2 possible isoforms with coverage of ≥10 reads were kept for subsequent analysis. These remaining exons were considered to be “differentially alternatively spliced.” A first list included cassette exons, which either demonstrated an increase or decrease in inclusion of the knockdown sample. A second and third list consisted of exons with an Alt3 or Alt5, respectively. For each of these 3 lists, 2 different control groups were created. The first control group for each set (denoted “not differentially alternatively spliced”) included exons that did not pass the filter, but showed the same type of AS. The second control group (denoted “identically alternatively spliced”) was filtered in an opposite fashion than the test sets: events showing a change in inclusion level or splice site usage of <0.05 and a bayes factor of <0.1, which meant a change in splicing behavior was 10 times less likely than having no change.
Data from triplicate CstF64-iCLIP-Seq in HeLa cells was obtained from the Gene Expression Omnibus database (accession no. GSE40859)Citation38 and was used to identify whether AS is correlated with CstF64 binding. The frequency of CstF64 binding sites was examined within the set of differentially alternatively spliced exons and their surrounding introns, as well as within the exons (and their neighboring introns) of the 2 control groups (not differentially alternatively spliced as well as identically alternatively spliced exons).
To separate real CstF64 binding sites from background peaks, clusters of iCLIP-Seq reads in close proximity to the alternatively spliced exon were searched for. A cluster was defined as a window of at least 10 nucleotides displaying at least 2 iCLIP-Seq tags after averaging all 3 iCLIP-Seq data sets. The clusters of CstF64 binding sites were overlapped with alternatively spliced exons and the neighboring introns in both the test and control sets to compute their total iCLIP-Seq coverage. Those values were then normalized by the length of the introns. The density of the distribution of the normalized iCLIP-Seq coverage in the different sets was plotted (excluding introns with a coverage of 0). All the density functions are only plotted for normalized coverage of ≤0.1.
Correlation between CstF64 KD and hnRNP A2/B1 KD
Data from duplicate hnRNP A2/B1 KD and 5 control HeLa RNA-Seq samples were obtained from the Gene Expression Omnibus database (accession number GSE34992).Citation43 To identify AS events, the datasets were compared using MISO.Citation40 Replicate control and hnRNP A2/B1 KD samples were combined and only uniquely mapped reads were kept. For every known AS event expressed in both cell types (SE, Alt3, Alt5, RI, ALE/APA) only events showing an absolute change in inclusion level or splice site usage ≥15% with a bayes factor of ≥10 were filtered for. Overlap between CstF64 KD and hnRNP A2/B1 KD was determined for all types of AS events (SE, Alt3, Alt5, RI, ALE/APA) by comparing which exact AS events were contained in both groups.
Data accession code
All sequencing and MISO data has been submitted to the National Center for Biotechnology Information Gene Expression Omnibus database (accession no. GSE79157).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary_Data.zip
Download Zip (4.7 MB)Acknowledgments
We are grateful to the members of our laboratory, especially Angela Garibaldi for helpful discussions and comments on this manuscript as well as funding support from the NIH (R01 GM62287 and R21 CA149548 to K.J.H.)
Related Research Data
References
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 2008; 40:1413-5; PMID:18978789; http://dx.doi.org/10.1038/ng.259
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature 2008; 456:470-6; PMID:18978772; http://dx.doi.org/10.1038/nature07509
- Listerman I, Sapra AK, Neugebauer KM. Cotranscriptional coupling of splicing factor recruitment and precursor messenger RNA splicing in mammalian cells. Nat Struct Mol Biol 2006; 13:815-22; PMID:16921380; http://dx.doi.org/10.1038/nsmb1135
- Pandya-Jones A, Black DL. Co-transcriptional splicing of constitutive and alternative exons. RNA 2009; 15:1896-908; PMID:19656867; http://dx.doi.org/10.1261/rna.1714509
- Moore MJ, Proudfoot NJ. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell 2009; 136:688-700; PMID:19239889; http://dx.doi.org/10.1016/j.cell.2009.02.001
- Kornblihtt AR. Chromatin, transcript elongation and alternative splicing. Nat Struct Mol Biol 2006; 13:5-7; PMID:16395314; http://dx.doi.org/10.1038/nsmb0106-5
- Brugiolo M, Herzel L, Neugebauer KM. Counting on co-transcriptional splicing. F1000Prime Rep 2013; 5:9; PMID:23638305; http://dx.doi.org/10.12703/P5-9
- Landry J-R, Mager DL, Wilhelm BT. Complex controls: the role of alternative promoters in mammalian genomes. Trends Genet 2003; 19:640-8; PMID:14585616; http://dx.doi.org/10.1016/j.tig.2003.09.014
- Logette E, Wotawa A, Solier S, Desoche L, Solary E, Corcos L. The human caspase-2 gene: alternative promoters, pre-mRNA splicing and AUG usage direct isoform-specific expression. Oncogene 2003; 22:935-46; PMID:12584573; http://dx.doi.org/10.1038/sj.onc.1206172
- Pecci A, Viegas LR, Baranao JL, Beato M. Promoter choice influences alternative splicing and determines the balance of isoforms expressed from the mouse bcl-X gene. J Biol Chem 2001; 276:21062-9; PMID:11274164; http://dx.doi.org/10.1074/jbc.M008665200
- Wang Y, Newton DC, Robb GB, Kau C-L, Miller TL, Cheung AH, Hall AV, VanDamme S, Wilcox JN, Marsden PA. RNA diversity has profound effects on the translation of neuronal nitric oxide synthase. PNAS 1999; 96:12150-5; PMID:10518591; http://dx.doi.org/10.1073/pnas.96.21.12150
- Xin D, Hu L, Kong X. Alternative promoters influence alternative splicing at the genomic level. PLoS ONE 2008; 3:e2377; PMID:18560582; http://dx.doi.org/10.1371/journal.pone.0002377
- David CJ, Boyne AR, Millhouse SR, Manley JL. The RNA polymerase II C-terminal domain promotes splicing activation through recruitment of a U2AF65-Prp19 complex. Genes Dev 2011; 25:972-83; PMID:21536736; http://dx.doi.org/10.1101/gad.2038011
- Cooke C, Hans H, Alwine JC. Utilization of splicing elements and polyadenylation signal elements in the coupling of polyadenylation and last-intron removal. Mol Cell Biol 1999; 19:4971-9; PMID:10373547; http://dx.doi.org/10.1128/MCB.19.7.4971
- Niwa M, Rose SD, Berget SM. In vitro polyadenylation is stimulated by the presence of an upstream intron. Genes Dev 1990; 4:1552-9; PMID:1701407; http://dx.doi.org/10.1101/gad.4.9.1552
- Vagner S, Vagner C, Mattaj IW. The carboxyl terminus of vertebrate poly(A) polymerase interacts with U2AF 65 to couple 3′-end processing and splicing. Genes Dev 2000; 14:403-13; PMID:10691733; http://dx.doi.org/10.1101/gad.14.4.403
- Millevoi S, Loulergue C, Dettwiler S, Karaa SZ, Keller W, Antoniou M, Vagner S. An interaction between U2AF 65 and CF I(m) links the splicing and 3′ end processing machineries. EMBO J 2006; 25:4854-64; PMID:17024186; http://dx.doi.org/10.1038/sj.emboj.7601331
- Kyburz A, Friedlein A, Langen H, Keller W. Direct interactions between subunits of CPSF and the U2 snRNP contribute to the coupling of pre-mRNA 3′ end processing and splicing. Mol Cell 2006; 23:195-205; PMID:16857586; http://dx.doi.org/10.1016/j.molcel.2006.05.037
- Lutz CS, Murthy KG, Schek N, O'Connor JP, Manley JL, Alwine JC. Interaction between the U1 snRNP-A protein and the 160-kD subunit of cleavage-polyadenylation specificity factor increases polyadenylation efficiency in vitro. Genes Dev 1996; 10:325-37; PMID:8595883; http://dx.doi.org/10.1101/gad.10.3.325
- Kaida D, Berg MG, Younis I, Kasim M, Singh LN, Wan L, Dreyfuss G. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature 2010; 468:664-8; PMID:20881964; http://dx.doi.org/10.1038/nature09479
- Gunderson SI, Polycarpou-Schwarz M, Mattaj IW. U1 snRNP inhibits pre-mRNA polyadenylation through a direct interaction between U1 70 K and poly(A) polymerase. Mol Cell 1998; 1:255-64; PMID:9659922; http://dx.doi.org/10.1016/S1097-2765(00)80026-X
- Davidson L, West S. Splicing-coupled 3′ end formation requires a terminal splice acceptor site, but not intron excision. Nucl Acids Res 2013:gkt446; 41:7101-14
- Muniz L, Davidson L, West S. Poly(A) Polymerase and the Nuclear Poly(A) Binding Protein, PABPN1, Coordinate the Splicing and Degradation of a Subset of Human Pre-mRNAs. Mol Cell Biol 2015; 35:2218-30; PMID:25896913; http://dx.doi.org/10.1128/MCB.00123-15
- Rigo F, Martinson HG. Functional coupling of last-intron splicing and 3′-end processing to transcription in vitro: the poly(A) signal couples to splicing before committing to cleavage. Mol Cell Biol 2008; 28:849-62; PMID:17967872; http://dx.doi.org/10.1128/MCB.01410-07
- Derti A, Garrett-Engele P, Macisaac KD, Stevens RC, Sriram S, Chen R, Rohl CA, Johnson JM, Babak T. A quantitative atlas of polyadenylation in five mammals. Genome Res 2012; 22:1173-83; PMID:22454233; http://dx.doi.org/10.1101/gr.132563.111
- Fu X-D, Ares M. Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet 2014; 15:689-701; PMID:25112293; http://dx.doi.org/10.1038/nrg3778
- Millevoi S, Vagner S. Molecular mechanisms of eukaryotic pre-mRNA 3′ end processing regulation. Nucleic Acids Res 2010; 38:2757-74; PMID:20044349; http://dx.doi.org/10.1093/nar/gkp1176
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009; 136:215-33; PMID:19167326; http://dx.doi.org/10.1016/j.cell.2009.01.002
- Leung AKL. The Whereabouts of microRNA Actions: Cytoplasm and Beyond. Trend Cell Biol 2015; 25:601-10 PMID:26410406; http://dx.doi.org/10.1016/j.tcb.2015.07.005
- Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 2009; 138:673-84; PMID:19703394; http://dx.doi.org/10.1016/j.cell.2009.06.016
- Reczko M, Maragkakis M, Alexiou P, Grosse I, Hatzigeorgiou AG. Functional microRNA targets in protein coding sequences. Bioinformatics 2012; 28:771-6; PMID:22285563; http://dx.doi.org/10.1093/bioinformatics/bts043
- Szostak E, Gebauer F. Translational control by 3′-UTR-binding proteins. Brief Funct Genomics 2013; 12:58-65; PMID:23196851; http://dx.doi.org/10.1093/bfgp/els056
- Brown KM, Gilmartin GM. A mechanism for the regulation of pre-mRNA 3′ processing by human cleavage factor Im. Mol Cell 2003; 12:1467-76; PMID:14690600; http://dx.doi.org/10.1016/S1097-2765(03)00453-2
- Kubo T, Wada T, Yamaguchi Y, Shimizu A, Handa H. Knock-down of 25 kDa subunit of cleavage factor Im in Hela cells alters alternative polyadenylation within 3′-UTRs. Nucleic Acids Res 2006; 34:6264-71; PMID:17098938; http://dx.doi.org/10.1093/nar/gkl794
- Takagaki Y, Seipelt RL, Peterson ML, Manley JL. The polyadenylation factor CstF-64 regulates alternative processing of IgM heavy chain pre-mRNA during B cell differentiation. Cell 1996; 87:941-52; PMID:8945520; http://dx.doi.org/10.1016/S0092-8674(00)82000-0
- Yao C, Choi E-A, Weng L, Xie X, Wan J, Xing Y, Moresco JJ, Tu PG, Yates JR, Shi Y. Overlapping and distinct functions of CstF64 and CstF64τ in mammalian mRNA 3′ processing. RNA 2013; 19:1781-90; PMID:24149845; http://dx.doi.org/10.1261/rna.042317.113
- Ara T, Lopez F, Ritchie W, Benech P, Gautheret D. Conservation of alternative polyadenylation patterns in mammalian genes. BMC Genomics 2006; 7:189; PMID:16872498; http://dx.doi.org/10.1186/1471-2164-7-189
- Yao C, Biesinger J, Wan J, Weng L, Xing Y, Xie X, Shi Y. Transcriptome-wide analyses of CstF64-RNA interactions in global regulation of mRNA alternative polyadenylation. Proc Natl Acad Sci USA 2012; 109:18773-8; PMID:23112178; http://dx.doi.org/10.1073/pnas.1211101109
- Shepard PJ, Choi E-A, Lu J, Flanagan LA, Hertel KJ, Shi Y. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA 2011; 17:761-72; PMID:21343387; http://dx.doi.org/10.1261/rna.2581711
- Katz Y, Wang ET, Airoldi EM, Burge CB. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods 2010; 7:1009-15; PMID:21057496; http://dx.doi.org/10.1038/nmeth.1528
- McGlincy NJ, Tan L-Y, Paul N, Zavolan M, Lilley KS, Smith CW. Expression proteomics of UPF1 knockdown in HeLa cells reveals autoregulation of hnRNP A2/B1 mediated by alternative splicing resulting in nonsense-mediated mRNA decay. BMC Genomics 2010; 11:565; PMID:20946641; http://dx.doi.org/10.1186/1471-2164-11-565
- Martinez-Contreras R, Fisette J-F, Nasim FH, Madden R, Cordeau M, Chabot B. Intronic binding sites for hnRNP A/B and hnRNP F/H proteins stimulate pre-mRNA splicing. PLoS Biol 2006; 4:e21; PMID:16396608; http://dx.doi.org/10.1371/journal.pbio.0040021
- Huelga SC, Vu AQ, Arnold JD, Liang TY, Yan BY, Donohue JP, Shiue L, Hoon S, Brenner S, Ares M, et al. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep 2012; 1:167-78; PMID:22574288; http://dx.doi.org/10.1016/j.celrep.2012.02.001
- Shi Y, Di Giammartino DC, Taylor D, Sarkeshik A, Rice WJ, Yates JR, Frank J, Manley JL. Molecular architecture of the human pre-mRNA 3′ processing complex. Mol Cell 2009; 33:365-76; PMID:19217410; http://dx.doi.org/10.1016/j.molcel.2008.12.028
- Niwa M, Berget SM. Mutation of the AAUAAA polyadenylation signal depresses in vitro splicing of proximal but not distal introns. Genes Dev 1991; 5:2086-95; PMID:1657710; http://dx.doi.org/10.1101/gad.5.11.2086
- de la Mata M, Alonso CR, Kadener S, Fededa JP, Blaustein M, Pelisch F, Cramer P, Bentley D, Kornblihtt AR. A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell 2003; 12:525-32; PMID:14536091; http://dx.doi.org/10.1016/j.molcel.2003.08.001
- de la Mata M, Lafaille C, Kornblihtt AR. First come, first served revisited: Factors affecting the same alternative splicing event have different effects on the relative rates of intron removal. RNA 2010; 16:904-12; PMID:20357345; http://dx.doi.org/10.1261/rna.1993510
- de la Mata M, Muñoz MJ, Alló M, Fededa JP, Schor IE, Kornblihtt AR. RNA Polymerase II Elongation at the Crossroads of Transcription and Alternative Splicing. Genet Res Int 2011; 2011:309865
- Kessler O, Jiang Y, Chasin LA. Order of intron removal during splicing of endogenous adenine phosphoribosyltransferase and dihydrofolate reductase pre-mRNA. Mol Cell Biol 1993; 13:6211-22; PMID:8413221; http://dx.doi.org/10.1128/MCB.13.10.6211
- Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 2002; 3:285-98; PMID:11967553; http://dx.doi.org/10.1038/nrg775
- Faustino NA, Cooper TA. Pre-mRNA splicing and human disease. Genes Dev 2003; 17:419-37; PMID:12600935; http://dx.doi.org/10.1101/gad.1048803
- Brinkman BMN. Splice variants as cancer biomarkers. Clin Biochem 2004; 37:584-94; PMID:15234240; http://dx.doi.org/10.1016/j.clinbiochem.2004.05.015
- Carstens RP, Eaton JV, Krigman HR, Walther PJ, Garcia-Blanco MA. Alternative splicing of fibroblast growth factor receptor 2 (FGF-R2) in human prostate cancer. Oncogene 1997; 15:3059-65; PMID:9444954; http://dx.doi.org/10.1038/sj.onc.1201498
- Wang Z, Lo HS, Yang H, Gere S, Hu Y, Buetow KH, Lee MP. Computational analysis and experimental validation of tumor-associated alternative RNA splicing in human cancer. Cancer Res 2003; 63:655-7; PMID:12566310
- Xu Q, Lee C. Discovery of novel splice forms and functional analysis of cancer-specific alternative splicing in human expressed sequences. Nucleic Acids Res 2003; 31:5635-43; PMID:14500827; http://dx.doi.org/10.1093/nar/gkg786
- Chen J-M, Férec C, Cooper DN. A systematic analysis of disease-associated variants in the 3′ regulatory regions of human protein-coding genes II: the importance of mRNA secondary structure in assessing the functionality of 3′ UTR variants. Hum Genet 2006; 120:301-33; PMID:16807757; http://dx.doi.org/10.1007/s00439-006-0218-x
- Conne B, Stutz A, Vassalli J-D. The 3′ untranslated region of messenger RNA: A molecular “hotspot” for pathology? Nat Med 2000; 6:637-41; PMID:10835679; http://dx.doi.org/10.1038/76211
- Di Giammartino DC, Nishida K, Manley JL. Mechanisms and Consequences of Alternative Polyadenylation. Molecular Cell 2011; 43:853-66; PMID:21925375; http://dx.doi.org/10.1016/j.molcel.2011.08.017
- Lackford B, Yao C, Charles GM, Weng L, Zheng X, Choi E-A, Xie X, Wan J, Xing Y, Freudenberg JM, et al. Fip1 regulates mRNA alternative polyadenylation to promote stem cell self-renewal. EMBO J 2014; 33:878-89; PMID:24596251; http://dx.doi.org/10.1002/embj.201386537
- Fähling M, Mrowka R, Steege A, Martinka P, Persson PB, Thiele BJ. Heterogeneous nuclear ribonucleoprotein-A2/B1 modulate collagen prolyl 4-hydroxylase, alpha (I) mRNA stability. J Biol Chem 2006; 281:9279-86; PMID:16464861; http://dx.doi.org/10.1074/jbc.M510925200
- Scotto-Lavino E, Du G, Frohman MA. 3′ end cDNA amplification using classic RACE. Nat Protoc 2006; 1:2742-5; PMID:17406530; http://dx.doi.org/10.1038/nprot.2006.481
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology 2013; 14:R36; PMID:23618408; http://dx.doi.org/10.1186/gb-2013-14-4-r36