
ABSTRACT
Genome-wide single-cell analysis represents the ultimate frontier of genomics research. In particular, single-cell RNA-sequencing (scRNA-seq) studies have been boosted in the last few years by an explosion of new technologies enabling the study of the transcriptomic landscape of thousands of single cells in complex multicellular organisms. More sensitive and automated methods are being continuously developed and promise to deliver better data quality and higher throughput with less hands-on time. The outstanding amount of knowledge that is going to be gained from present and future studies will have a profound impact in many aspects of our society, from the introduction of truly tailored cancer treatments, to a better understanding of antibiotic resistance and host-pathogen interactions; from the discovery of the mechanisms regulating stem cell differentiation to the characterization of the early event of human embryogenesis.
Abbreviations
| aRNA | = | antisense RNA |
| CEL-seq | = | Cell Expression by Linear amplification and sequencing |
| CNV | = | Copy-Number Variant |
| CTCs | = | Circulating Tumor Cells |
| DR-seq | = | DNA and RNA sequencing |
| ES | = | End Sequence |
| FACS | = | Fluorescence Activated Cell Sorting |
| gDNA | = | genomic DNA |
| G&T-seq | = | Genome and Transcriptome sequencing |
| HT-IFC | = | High-Throughput Integrated Fluidic Circuit |
| IE | = | Inside End |
| IFC | = | Integrated Fluidic Circuit |
| InDrop | = | Indexing Droplets |
| IVT | = | In-Vitro Transcription |
| Kb | = | Kilobases |
| LCM | = | Laser Capture Microdissection |
| LNA | = | Locked Nucleic Acid |
| lncRNA, | = | long non-coding RNA |
| MACS | = | Magnetic-Activated Cell Sorting |
| MALBAC | = | Multiple Annealing and Looping Based Amplification Cycles |
| ME | = | Mosaic End |
| miRNA | = | microRNA |
| MMLV | = | Moloney Murine Leukemia Virus |
| mRNA | = | mRNA |
| MSND | = | Multi-Sample Nano-Dispenser |
| NMP | = | (deoxy)Nucleoside MonoPhosphates |
| OE | = | Outside End |
| ONT | = | Oxford Nanopore Technologies |
| piRNA | = | piwi-interacting RNA |
| PNPase | = | PolyNucleotide Phosphorylase |
| RCA | = | Rolling Circle Amplification |
| rNDP | = | RiboNucleoside Diphosphates |
| rRNA | = | rRNA |
| RT | = | Reverse Transcription |
| SBL | = | Sequencing By Ligation |
| SBS | = | Sequence-by-Synthesis |
| scBS | = | single-cell Bisulfite Sequencing |
| scRNA-seq | = | single-cell RNA-sequencing |
| SMART-seq | = | Switching Mechanism At the end of the 5´-end of the RNA Transcript sequencing |
| SMRT-seq | = | Single Molecule Real-Time Sequencing |
| SOLiD | = | Sequencing by Oligonucleotide Ligation and Detection |
| ssRRBS | = | single-cell Reduced Representation Bisulfite Sequencing |
| STAMPs | = | Single-cell Transcriptomes Attached to MicroParticles |
| STRT-seq | = | Single-cell Tagged Reverse Transcription sequencing |
| tRNA | = | tRNA |
| TS | = | Template Switching |
| TSO | = | Template Switching Oligonucleotide |
| UMI | = | Unique Molecular Identifier |
| WGA | = | Whole Genome Amplification |
Introduction
The transcriptome is a crucial constituent for the maintenance of cell identity and the survival of any organism given the multiple roles of cellular RNA as messenger, in the formation of regulatory complexes and as an essential component of housekeeping complexes.Citation1 Genome-wide transcriptome analysis is therefore the tool of choice for profiling all coding and non-coding RNA species in a cell and thus address fundamental questions that have remained unanswered for decades: how do we define a cell? What causes a normal cell to turn into a malignant one? What are the earliest differentiation events in human embryogenesis?
In the last decade several groundbreaking discoveries have revolutionized our view of the eukaryotic genome and transcriptome. It is now clear that, although protein-coding genes constitute approximately only 2% of the human genome, more than 80% of it may be transcribed.Citation2 Additional layers of complexity are given by the fact that a single genomic locus can show multiple and variable splicing patterns originating different isoforms, which in turn might also have different transcriptional start site or poly-adenylation sites.Citation3 Moreover, genes could show a random monoallelic expression, where only the maternal or paternal allele is expressed at each time.Citation4 Much of our knowledge of the transcriptome derives from bulk studies conducted on cell populations, thus averaging the expression of thousands or even million of cells. It is now well established that seemingly homogeneous cell populations in vivo or cell cultures in vitro can display considerable heterogeneity in expression patterns, due to both intrinsic stochastic processes and extrinsic factors, such as the surrounding microenvironment.Citation5 Knowing exactly the expression pattern of each individual cells is especially important if we consider that the majority of transcripts are expressed in few copies per cell, with most of long non-coding RNAs (lncRNAs) present in one or less than one copy per cell (i.e., not all cells in a particular tissue or organ express a certain transcript). Furthermore, neighboring cells sharing the same microenvironment can express a transcript at different levels due either to deterministic reasons (cell cycle effects, for example) or random factors. This randomness accounts for the so-called transcriptional noise, a random and burst-like fluctuation in expression levels that is now recognized to have transcriptome-wide effects and plays a key role in cell fate decisions.Citation6
As will be reviewed here, performing successful scRNA-seq experiments requires that the expertise from various disciplines is brought together to answer a very well defined biological question. In the next section I will first briefly go through the most common methods for isolating individual cells from the tissue or culture of interest.
The main part of the review will then be devoted to describing the techniques that currently represent the state-of-the-art, with a mention of some promising high-throughput approaches that have recently been introduced but not yet become mainstream. In the last part I will cover the future challenges that still remain and discuss the impact and practical implications scRNA-seq can have in our daily life.
The bioinformatics analysis will not be discussed here and the interested reader is referred to some excellent reviews recently published elsewhere.Citation7,8
Which method suits you?
One of the major limitations of all the single-cell sequencing methods is probably that they are, without exceptions, all DNA-based.Citation9 It is currently not yet possible to sequence RNA directly from single cells, which implies that RNA must first be converted to cDNA and amplified, either via PCR or linear amplification (In-Vitro Transcription, IVT). This harbors 2 challenges: on the one hand RNA losses have to be kept to a minimum during the conversion. On the other hand the amplification should provide enough DNA for sequencing without introducing too much quantitative bias or distorting the original picture of the cell transcriptome. Eukaryotic cells contain many different RNA species but often only some are relevant for researchers. Ideally, one would like to amplify all of them except tRNA and rRNA that would otherwise compose >90% of the reads in the final sequencing library.Citation10 To deal with this problem most (if not all) scRNA-seq methods employ an oligo-dT primer that specifically captures only polyadenylated RNA, such as mRNAs and some lncRNAs. Unfortunately, other potentially interesting but non-polyadenylated RNAs such as many lncRNAs and all the other shorter species like miRNAs and piRNAs will be lost. While many efficient protocols exist for rRNA depletion prior to library preparation, all of them require several nanograms of RNA as starting material, the equivalent of hundreds or even thousands of cells. Even if one would be able to selectively remove rRNA in single cells and use random primers to amplify all the remaining RNA species, another problem would arise: the presence of genomic DNA (gDNA), which potentially could also interfere in the downstream processes. However, it has been shown that it is possible to efficiently remove gDNA in single cells prior to library preparation and, although challenging, this should not represent an obstacle anymore (Patent No. US 2011/0136180).
Every researcher has a specific biological question that he/she wants to answer and this will dictate the method that should be chosen to best address this question.
Full-length scRNA-seq protocols allow sequencing the transcripts in their entirety and thus enabling both gene expression quantification and detection of transcript isoforms, SNPs and mutations. Citation11,12
On the other hand, tag-based methods enable the sequencing of the 5´-end or 3´-end of the transcripts, producing an estimate of transcript abundance at the cost of coverage across splice isoforms but allow early multiplexing, often have higher throughput and can be combined with molecular counting.Citation13 An additional advantage of the tag-based methods is that they retain the information from which genomic DNA strand a RNA molecule was transcribed from, an important feature considering the prevalence of pervasive antisense transcription in virtually all Eukaryotes.Citation14
How can we isolate single cells?
One of the major challenges for performing high-throughput and unbiased single cell studies probably lies in the very first step, isolating individual cells from the system of interest. Micromanipulation was the tool of choice until a few years ago and still remains the only alternative when a limited number of cells are available or when cells are too fragile and would not survive the isolation with other methods (such as FACS for example, see below). Micromanipulation has the advantage that every single cell can be directly observed under the microscope but it is unfortunately very laborious and has a relatively low-throughput, approximately in the order of 50 cells/hour and person.
Fluorescence Activated Cell Sorting (FACS) has a much higher throughput than micromanipulation, in the order of thousands or tens of thousands of cells/day. Cells can be isolated either based on their size and morphology or using complex mixtures of fluorescently labeled antibodies targeting cell surface markers, which makes possible the isolation of very rare cell types in a very efficient way. Several instrument on the market allow sorting of single cells directly into common 96- and 384-well plates, while sorting in 1536-well plates still represents an insurmountable obstacle for most research labs.
More recently, microfluidics devices such as the C1™ Single-Cell Auto Prep System (manufactured by Fluidigm) have been introduced, allowing automated capture of single cells using special Integrated Fluidic Circuits (IFC) and interesting results have already been reported by several research groups.Citation3,15 Cells immobilized in specific capture sites on the IFCs are sequentially imaged, lysed, the RNA converted to cDNA and the cDNA amplified via PCR. Notably, all reactions are performed in nanoliter-scale volumes and in a completely automated way, with beneficial effects on reaction efficiency and significant saving of reagents. Although microfluidics instruments might sound as the solution to all the problems, several obstacles remain. The fixed chip architecture introduces a bias in the cells that can be selected; the chip performs sub-optimally with primary cells, having capture rates as low as 40-50% (unpublished data); rare cell types, such as circulating tumor cells (CTCs), often present in single digits numbers among million of blood cells, require anyway an enrichment step with an alternative method (FACS or the MACS® MicroBead technology, for example) before loading them on the chip.
The first IFC commercialized by Fluidigm allowed the analysis of up to 96 cells but recently a new High-Throughput IFC (HT-IFC) capable to capture and process up to 800 cells in parallel has reached the market (https://www.fluidigm.com/press/high-throughput-single-cell-mrna-sequencing-preparation-comes-to-the-fludigm-c1-system).
As mentioned above, the need to first purchase the C1™ Single-Cell Auto Prep instrument and the IFCs often annuls the benefits of scaling down the reaction volumes, even though the HT should cut the costs to some extent. More recently, Fluidigm made available to researchers the C1™ Open App™ program that allows building and sharing new single-cell applications. It is now possible to use C1 Script Builder™ to design and develop new protocols and later share them on Script Hub™ (http://www.fluidigm.com/c1openapp#overview). The major advantage is that researchers don´t need to purchase expensive kits and can customize virtually any protocol to best suit their needs.
Other ultra-high throughput methods performing reactions on the nanoliter scale have started appearing on the market. The ICELL8 Single-Cell System (WaferGen Biosystems), for example, allows the unbiased isolation of cells of any size. Cells can be isolated in special proprietary chips composed of 5184 nanowells already pre-filled with unique combinations of barcoded adaptors. A Multi-Sample Nano-Dispenser (MSND) can dispense cells from a suspension, while the microscope coupled to the instrument is imaging all the wells in just few minutes. With the proper dilution of the cell suspension one can expect up to 37% of the wells containing a single cell according to the Poisson distribution, or approximately 1800 wells. Based on the well occupancy, the MSND is then capable to add reagents only to those containing a single cell, with significant savings in terms of money and time.
Other labor-intensive methods such as Laser Capture Microdissection (LCM) or not yet widespread methods like the use of optical tweezers are not described here but can be a valid alternative in specific cases. A more detailed description of all cell isolation methods has been published recently and the interested reader is referred to that article.Citation1
Full-length RNA-sequencing methods
Tang method
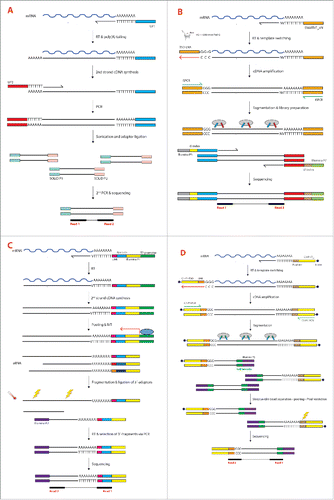
The first scRNA-seq method was developed 7 years ago by the Surani group and for researchers in the field it is now known as the “Tang method” from the first author of that landmark paper.Citation16,17 The protocol was a modified and substantially improved version of another one originally developed for single-cell microarray analysis, adapted to render it more suitable for scRNA-seq.Citation18 In that very first paper the authors used single mouse blastomers given their high RNA content (1 ng/cell). The cells were manually picked under the microscope, lysed and the polyadenylated RNA was reverse transcribed into cDNA using an oligo-dT primer carrying a specific anchor sequence (UP1) (). This generated cDNA molecules of up to 3 kb in length, a noticeable improvement from the previous method that restricted the analysis only to 0.85 kb from the 3´-end of the transcripts. After reverse transcription (RT) a poly(A) tail was added to the 3´-end of the first strand cDNA by using a terminal deoxynucleotidyl transferase. The second cDNA strand was then synthesized by using a complementary poly(T) primer carrying a second anchor sequence at its 5´-end (UP2). As a result, the double-stranded cDNA thus generated carried known sequences (UP1 and UP2) at both ends and could be efficiently amplified via PCR. After shearing and adaptor ligation the libraries were then sequenced using a SOLiD sequencer (Applied Biosystems) ().
Figure 1. (see previous page) (A)Tang method. Polyadenylated mRNA is reverse transcribed from an oligo-dT primer carrying the Universal Primer 1 (UP1) sequence at its 5´-end. After RT a poly(A) tail is added to the 3´-end of the first strand cDNA by using a terminal deoxynucleotidyl transferase. The second cDNA strand is synthesized by using a complementary poly(T) primer carrying the Universal Primer 2 (UP2) sequence at its 5´-end. Double-stranded cDNA is then amplified via PCR using complementary primers to the UP1 and UP2 sequences. After shearing and adaptor ligation the fragments undergo a second PCR that allows the introduction of SOLiD-compatible sequences (SOLiD P1 and SOLiD P2). (B) Smart-seq2. Polyadenylated mRNA is reverse transcribed from an oligo-dT primer (SMARTdT30VN). When the reverse transcriptase reaches the end of the RNA template, 2–5 cytosines are added to the newly synthesized cDNA (3 in the figure). The reaction mix also contains a TS oligonucleotide (TSO-LNA) carrying 2 riboguanosines (rG) and a LNA-modified guanosine (+G) at its 3´-end. After annealing of the 3 terminal nucleotides of the LNA-TSO with the 3 cytosines, the reverse transcriptase synthesizes a cDNA strand using the LNA-TSO as template (red arrow). The cDNA is then amplified via PCR using only one primer (ISPCR), since both the SMARTdT30VN and LNA-TSO oligonucleotides share the same sequence at their 5´-end (here in orange). The amplified cDNA is then fragmented by tagmentation using the Tn5 transposase. Simultaneously, the Tn5 ligates different 5′ and 3′ primers to the fragments (red and blue sequences). Another round of PCR introduces Illumina-compatible sequences (Illumina P5 and P7) as well as index sequences (i5 and i7 indices) to allow sample multiplexing. (C) CEL-seq. Polyadenylated mRNA is reverse transcribed from an oligo-dT primer containing the T7 promoter, the Illumina P1 adaptor and a cell barcode. In MARS-seq and CEL-seq2 the sequencing primer also accommodates a UMI downstream of the cell barcode. After RT and second-strand synthesis the cDNA from all the cells is pooled and amplified by IVT from the T7 promoter to generate aRNA. The Illumina P2 adaptor is ligated after heat fragmentation, followed by generation of double-stranded DNA and sequencing of the 3´-terminal fragments. (D) STRT/C1. Polyadenylated mRNA is reverse transcribed from a biotinylated oligo-dT primer (C1-P1-T31) containing the Illumina P1 adaptor (here in yellow) and a PvuI restriction site. The TS reaction occurs in a similar way as for Smart-seq2 but the TSO used in STRT/C1 is different. The TSO is biotinylated and contains the Illumina P1 adaptor, a UMI and 3 riboguanosines at the 3´-end. After TS the cDNA is amplified via PCR using a single primer (C1-P1-PCR) as in the Smart-seq2 method. Fragmentation and ligation of the Illumina P2 adaptor and the cell barcode are performed simultaneously utilizing in-house Tn5 transposase. After pooling all the samples, streptavidin-coated magnetic beads are used to collect only the biotinylated fragments (5´- and 3´-ends of the transcripts). The 3′-ends are then digested by the PvuI restriction enzyme and only the 5´-ends are used for sequencing.
The Tang method allowed the detection of thousands of genes and hundreds of new splice junctions more than a standard microarray experiment, something that made researchers fully appreciate the importance of analyzing single cells rather than bulk samples. Although representing a huge leap forward for the then-nascent scRNA-seq field, the method had the major limitation of still displaying a quite pronounced 3´-bias, with the majority of the reads mapping to the 3´-terminal portion of the transcripts. This constituted a severe limitation for the study of transcriptional start sites (TSS) as well as in the analysis of the different splice variants. Furthermore, inefficiencies in the enzymatic reactions resulted in decreased sensitivity with consequent loss of lowly expressed transcripts. Similarly to other full-length scRNA-seq protocol, the Tang method is not strand-specific since this information is lost during the PCR amplification step. Coupled with the short reads typical of the SOLiD instrument (but would be the same for the Illumina sequencing) this results in the impossibility of uniquely assign reads mapping on overlapping transcripts transcribed by both strands.
Smart-seq and Smart-seq2
A different approach was chosen by the Sandberg group few years later, in the attempt to markedly improve the transcriptome coverage.Citation11 This method exploits the so-called SMART reaction (Switching Mechanism At the end of the 5´-end of the RNA Transcript), developed by scientists at CLONTECH several years earlier.Citation19,20 The protocol, commercialized as SMARTer® Ultra® Low RNA kit (Patent No. JP4043516 and EP0871780) is since then known as Smart-seq and has been designed for the Illumina sequencing instruments, more common that the SOLiD instruments chosen by the Surani group. Smart-seq simultaneously exploits 2 intrinsic properties of the Moloney Murine Leukemia Virus (MMLV) reverse transcriptase: Reverse Transcription (RT) and template switching (TS). The mRNA was initially primed with an anchored oligo-dT primer, similarly to the Tang method, but additional modifications were introduced to reduce the 3´-bias (). Template switching is the ability of the MMLV reverse transcriptase to introduce a few untemplated nucleotides, predominantly 2-5 cytosines, when it reaches the 5´-end of the RNA template, corresponding to the 3´-end of the newly synthesized cDNA strand. These extra nucleotides work as a docking site for a helper oligonucleotide (“Template Switching Oligonucleotide,” or TSO) that carries 3 riboguanosines at its 3´-end. The reverse transcriptase is then able to “switch template” (from mRNA to the DNA of the TSO) and synthesize a cDNA strand using the helper oligonucleotide as template. Thus, TS makes possible the introduction of an arbitrary sequence at the end of the transcript and, along with the known sequence located at the 5´-end of the oligo-dT primer, allows the efficient amplification of all the transcripts in a cell. Because the sequence on both sides of each cDNA is the same the PCR can be carried out with just one primer, thus exploiting the PCR suppression effect.Citation19
Although Smart-seq dramatically improved the coverage of the transcriptome and had a much higher sensitivity compared to the Tang method, it bore some important limitations. A lower read coverage toward the 5´-end of the transcripts, especially long several kilobases, was still quite pronounced. Moreover, in the final sequencing library, an under-representation of transcripts with a high GC-content was observed, presumably an effect of the complex secondary structure of the RNA that the DNA polymerase could not overcome during the PCR.Citation12 Most importantly, having to buy an expensive commercial kit made library preparation prohibitive for research groups on a tight budget and planning to sequence hundreds or thousands of cells. To address all these issues and improve the existing method a fairly large selection of buffers, additives and enzymes as well as of reaction conditions were tested. Hundreds of experiments resulted in a dramatically better protocol that, perhaps without too much creativity, was named Smart-seq2.Citation12,21
From experiments on different cell lines the authors observed a substantial increase in the ability of detecting gene expression and a lower technical variation for low- and medium-abundance transcripts compared to any other full-length single-cell method. The improved sensitivity led to the detection of a couple of thousands genes more than with Smart-seq, especially those with a high GC-content.Citation12
The protocol is detailed in . Smart-seq2 also begins with the RT reaction using a MMLV-based enzyme and an oligo-dT primer with a known sequence at its 5´-end. Many researchers in the field regard the RT reaction as one of the most inefficient steps of the entire library preparation.Citation22 Unfortunately, all the retroviral-based reverse transcriptases used nowadays have intrinsically low-fidelity and processivity, which reflects the fact that they evolved to help retroviruses evade host defenses by introducing mutations and rapidly propagating the favorable ones by RNA recombination. Since no alternative solution other than using this class of enzymes was available the authors focused their efforts on increasing the processivity by using additives such as betaine that, in combination with a higher concentration of magnesium chloride, helped in maximizing the cDNA yield.Citation12 The TSO was also redesigned by replacing the terminal riboguanosine with a locked nucleic acid (LNA)-modified deoxyguanosine (). Locked nucleotides are characterized by an internal bond between the O2′ and the C4′ of the furanose ring, linked by a methylene group. The modification introduces a conformational lock in the molecule, which nonetheless still retains the physical properties of the native nucleic acid. Two interesting properties of LNAs are advantageous for this application: the enhanced thermal stability of the LNA monomers and their ability to anneal strongly to the untemplated 3′ extension of the cDNA.Citation23 However, the inclusion of one LNA-based nucleotide at the 3´-end of the TSO might be a double-edged sword, increasing the efficiency of the TS on one side but creating artifacts on the other. In fact, if the polymerized region has sequence complementarity with the 3´-end of the TSO it can invade and hybridize with the first strand cDNA that is being generated, displacing the mRNA template. The reverse transcriptase then switches template and synthesize a cDNA strand using the TSO as template, like in a normal TS reaction. The result is a cDNA molecule that is shorter than the original mRNA template and that will be spuriously regarded as an alternative splice variant with a different 5´-end when, in reality, it is just a chimera.Citation24
It is also widely accepted that a major source of bias in scRNA-seq is the PCR reaction. It is estimated that a single cell contains on average 10 picograms of total RNA, of which only 1-5% is polyadenylated mRNA. Without amplification the tiny amount of single-stranded cDNA generated after RT would be neither sufficient nor suitable for direct sequencing.
Ideally, one would like to have a DNA polymerase that is capable to amplify each transcript with the same efficiency, regardless of its length, secondary structure or GC-content. In an attempt to minimize this bias the authors tested different DNA polymerases and found out that the (then) newly released KAPA High-Fidelity DNA polymerase performed the best, allowing efficient amplification both of GC-rich and AT-rich transcripts.Citation12 Incidentally, the same enzyme had already been tested few months earlier by another group that sequenced several bacterial genomes with very different GC- and AT-content, showing that it was the one introducing the least bias when compared to unamplified samples.Citation25
While generally performing well on large cells, fine-tuning of the protocol showed to be necessary when working with small cells with an extremely low RNA content such as many primary cells and, in particular, immune cells. The accumulation of oligo-concatamers, a byproduct of secondary TS events turned out to be a problem that prevented the generation of usable libraries. This is caused by an imbalance of oligonucleotides in the reaction compared to the little amount of cell mRNA and it can easily be prevented by 5´-blocking the TSO with some iso-nucleotides, bulky blocking groups such as biotin or, like the authors did, by drastically decreasing the amount of oligonucleotides used in the experiments.Citation26-28 Blocking the 3´-end of the TSO was shown to eliminate spurious priming during RT and PCR but when tested in the Smart-seq2 protocol did not perform well and was not pursued further.Citation12,29
Probably the most important advantage of Smart-seq2 is that it entirely relies on off-the-shelf reagents, especially when performing the library preparation by using the in-house version of Tn5 transposase described below.Citation30 This makes scRNA-seq accessible and affordable to all researchers around the world who now have the possibility of analyzing hundreds of cells for a fraction of the cost of the commercial kits. However, some of the same issues already discussed for Smart-seq were not addressed and remained unsolved also in Smart-seq2. For example, the information about strand-specificity is lost in the PCR step unless the long full-length transcripts are sequenced by using the Single Molecule Real-Time (SMRT) Sequencing technology developed by Pacific Biosciences which has an average read length of > 10 kb. Such a methods is not as widespread as Illumina yet, it is still plagued by high sequencing error rates and therefore probably not a viable option at the moment.
A common disadvantage of both Smart-seq and Smart-seq2 is that the samples can be pooled just prior to sequencing, making them more labor-intensive than the tag-based sequencing methods (see below). However, this is a relatively small issue now that microfluidics devices, nano-dispensers and liquid handling robots have become common instruments in many the laboratories.
CLONTECH has recently released the new SMART-Seq® v4 Ultra® Low Input RNA Kit that implements some or all of the Smart-seq2 changes (Patent No. US 2014/052233). To the best of my knowledge no data from independent researchers has been made publicly available at the time this review was written, but CLONTECH claims that the performance of this new kit is superior to Smart-seq2´s.
Quartz-seq
A simplified and improved version of the Tang method, called Quartz-seq, was published some years later by a Japanese group.Citation31 Although based on the same principles, Quartz-seq was much more straightforward and required less hands-on time since all the steps could be completed in a single tube without any purification. The authors observed that Quartz-Seq had a higher reproducibility and sensitivity in single-cell transcriptome data compared Smart-seq.Citation31 However, the Smart-seq2 protocol published just few months later showed a much better performance than Quartz-seq.Citation12
Tag-based sequencing methods
CEL-seq, CEL-seq2 and MARS-seq
In the CEL-seq (Cell Expression by Linear amplification and Sequencing) method, Hashimshony and collaborators decided to take a different approach for amplifying the RNA and generating enough material for sequencing.Citation32 Instead of using an exponential PCR-based amplification they opted for the more linear IVT. IVT on single cells had already been carried out before but the protocol was labor-intensive, required 3 rounds of amplification and was not suitable for multiplexing.Citation33 Different attempts to perform IVT on single cells have always been challenged by the lower input of total RNA needed for a single round of amplification, which is about 400 pg, much higher that the amount of RNA present in a single cell. CEL-seq elegantly solved this problem by pooling barcoded samples to obtain sufficient material for performing IVT in an efficient way (). By pooling many samples to a single IVT, a single round of amplification is sufficient, thus reducing hands-on time for the downstream processing.
The CEL-seq method starts with a RT reaction using a composite primer carrying a T7 promoter sequence at the 5´-end followed by the 5´-Illumina sequencing adaptor sequence, a unique barcode and an anchored poly(T) at the 3´-end (). After RT and second strand cDNA synthesis, the samples are pooled and undergo IVT reaction. The antisense RNA (aRNA) is then fragmented to the appropriate size for sequencing, a 3´-Illumina adaptor is added by ligation, the RNA is reverse transcribed to DNA and, finally, the 3´-most fragments (the only ones containing both Illumina adaptors and a barcode) are selected. Paired-end sequencing is performed, where the first read identifies the barcode, and thus the cell identity, while the second recovers the mRNA transcript ().
CEL-seq has some interesting features lacking in other methods such as strand specificity (with more than 98% of exonic reads coming from the sense strand) and high barcoding efficiency (>96 %).Citation32 Moreover, because only the RNA fragments that are the closest to the poly(A) tail are selected, the estimation of expression levels is much easier than with full-length RNA-seq methods, since no normalization by gene length is necessary.
Among the disadvantages of CEL-seq are the strong 3´-bias and the low sensitivity for lowly expressed transcripts. It was calculated that if a transcript is present in 5 copies per cell, CEL-seq has only a 50% chance of identifying it.Citation32 While sequencing the 3´-terminal portion of each transcript is certainly sufficient for determining cell identities in a heterogeneous population, it is not suitable for obtaining a detailed picture of the different splice isoforms and their relative abundance.
CEL-seq was implemented and modified by another research group to become what they called automated massively parallel RNA single-cell sequencing, or MARS-seq.Citation34 The major innovation was the introduction of a Unique Molecular Identifier (UMI) in the oligo-dT primer, thus allowing random barcoding during the cDNA synthesis, an important feature that enables counting unique RNA molecules.Citation35 The method relied on sorting single cells into 384-well plates via FACS. Every step of the protocol was automated and carried out by a liquid handling robot, with the library preparation done mostly on pooled and barcoded samples, thus leading to an enormous increase in throughput and reproducibility.
One of the most interesting features of MARS-seq is that it uses 3 levels of multiplexing: molecular-, cellular- and plate-level tags to allow molecular counting and a high degree of multiplexing. To drastically cut the sequencing costs the authors decided to initially characterize the different cell subpopulations in their samples using a low-depth RNA sampling and then study the transcriptional profile at higher resolution by combining data from hundreds, potentially thousands, of cells within each unsupervised class.Citation34 This bottom-up approach is readily applicable to any tissue or organ and allows researchers to obtain a snapshot of their cell type and cell state compositions. Given the large tissue heterogeneity, thousands of cells should be sequenced to make sure even rare subpopulations are sampled. In fact, it is not uncommon to have differences of several orders of magnitude in abundance between cell types within the same tissue. Thus, at least a thousand cells are required to guarantee the necessary power to detect such rare cell populations and states.Citation36
A new version of the CEL-seq protocol, called CEL-seq2, has recently been developed by the same authors and is reported to have higher sensitivity, lower costs and require even less hands-on time.Citation37 Among the key changes are the addition of UMI and the elimination of the ligation step by inserting the Illumina adaptor directly at the RT step as a 5´-tail attached to the random hexamer primers.Citation37 CEL-seq2 has also been implemented on the Fluidigm C1 system, offering the advantage that only a single library needs to be prepared for a single chip (http://www.fluidigm.com/c1openapp/scripthub/script/2015-06/celseq-v1-1433774312876-5).
Single-cell tagged reverse transcription sequencing (STRT-seq)
In the Single-cell Tagged Reverse Transcription Sequencing (STRT-seq) protocol developed by Sten Linnarsson´s group single cells are also reverse transcribed using an anchored oligo-dT primers and a MMLV-based enzyme.Citation27,38 In a similar way to Smart-seq2, the RT mix contained a TSO for the TS reaction. However, the TSO used in STRT-seq carried a 6-base random barcode just upstream of the 3 riboguanosines of the TSO (). Two uracils residues were also included in the TSO to facilitate its degradation before PCR, thus preventing the oligonucleotide from cross-reacting as primer during the PCR. Finally, the TSO was biotinylated at the 5´-end to prevent secondary template switch reactions and an undesired accumulation of concatamers.Citation27 The resulting cDNA was then amplified by PCR, immobilized on streptavidin beads and enzymatically fragmented with dsDNA Fragmentase. Only the 5´-terminal fragments were recovered by collecting the streptavidin beads and removing the supernatant. The pulled-down cDNA underwent end-repair, A-tailing, adapter ligation and an additional round of PCR amplification before being sequenced on the Illumina platform.Citation27
The original STRT-seq protocol was later simplified, improved and adapted to the C1 Single-Cell Auto Prep system (Fluidigm) and it is now known as STRT/C1 (). The most interesting improvement is the introduction of UMIs in the TSO, in a similar way to MARS-seq.Citation39 The authors still relied on a TSO with 3 riboguanosines at the 3´-end, known to be less prone to undergo the strand invasion side-reaction, thus increasing the probability of detecting the authentic 5´-terminal end of mRNAs and TSS.Citation24 By counting the number of cDNA molecules in their experiments, they reported a 5-fold improvement in efficiency for the STRT/C1 compared to the original version, probably due to a combination of the reaction being carried out in a microfluidic chip as well as the use of additives and a newly designed TSO.Citation39 Furthermore, the protocol was significantly simplified, so that after PCR the double-stranded DNA was fragmented using in-house Tn5 transposase (see below) carrying different combinations of barcode sequences, thus uniquely and efficiently indexing each sample (). All the samples could then be pooled, the 5´- and 3´-terminal fragments recovered by using streptavidin magnetic beads (the TSO and the oligo-dT were both biotinylated), while all the internal fragments were discarded. The 3′-fragments were then cleaved by using the restriction enzyme PvuI that has its recognition sequence upstream the oligo-dT sequence used in the RT reaction ().Citation39 What was left and sequenced in the end was therefore only the 5´-terminal portion of each transcript.
As mentioned above, the introduction of UMIs in the STRT/C1 protocol offers the possibility of counting the number of unique transcripts expressed in each cell and tell them apart from PCR duplicates. Other advantages are the strand specificity and the possibility of performing an early multiplexing, thus dramatically increasing the throughput.
Among the disadvantages of STRT-seq and STRT/C1 is the impossibility of detecting SNPs or splice variants located outside the 5´-terminal portion of the transcript, a feature common to all the tag-based sequencing methods.
Array-based and emulsion-based methods: The new generation
Although the methods described above can be scaled up and automatized in order to reduce reaction volumes and costs as well as increase the efficiency, they still remain rather cumbersome and labor-intensive. Due to the many steps involved and the extensive manipulations required before getting a library ready for sequencing they are also prone to cross-contaminations and can be plagued by high failure rates. To address this issue, several different solutions have been recently developed in which the library preparation is done in picoliter wells (CytoSeq) or emulsion droplets (Drop-seq and InDrops). These new technologies allow the sequencing of thousands of cells per day for a fraction of the cost of the traditional methods from the previous generation. The main features of traditional and new generation scRNA-seq methods have been summarized in .
Table 1. Principal characteristics of the most widely used scRNA-seq methods.
CytoSeq
The first method to be published in early 2015 was called CytoSeq and enables routine digital gene expression profiling of thousands of single cells across an arbitrary but limited number of genes without using traditional liquid handling robots.Citation40 The approach exploits a recursive Poisson strategy, where a cell suspension is loaded onto custom arrays fabricated using standard lithography and containing up to 100,000 microwells, each with the diameter of approximately 30 μm and able to accommodate droplets of about 20 picoliters. The number of cells in the suspension is carefully adjusted in order to have just 1 in 10 microwells containing a cell, while the other 9 remain empty. Cells simply settle into the microwells by gravity and get trapped. Next, the array is loaded with a magnetic bead library, at a concentration that most microwells are occupied. The beads have a size of 20 μm and their concentration has been optimized to prevent or reduce the possibility that 2 beads are occupying the same well. The beads are functionalized with hundreds of millions of oligonucleotides bearing a universal PCR priming sequence, a unique 8 bp cell label (identical for all the oligonucleotides on the same bead but unique for each bead), another 8 bp molecular index (variable among oligonucleotides on the same beads) and finally an oligo-dT sequence at the 3´-end. It derives that all oligonucleotides on a bead carry the same cell label but a diversity of molecular indices. Because only about 1% of the total available cell label diversity is used, it is very unlikely that 2 single cells are tagged with the same label (the probability is in the order of 1 in 10,000). The same is true for the molecular indices on the same bead, making it extremely unlikely that 2 transcripts of the same gene and from the same cell are tagged with the same index.
Once the single cell and the bead are co-localized in the same microwell, a lysis solution is added on top of the array. The high local concentration of mRNA is efficiently driving the hybridization to the beads via pairing of the poly(A) tail with the oligo-dT present on the beads.
Once the hybridization has taken place the beads are collected and all the reactions from this point on are carried out in a single tube, dramatically simplifying the downstream procedures. All the mRNA molecules are then reverse-transcribed to cDNA, amplified and sequenced. Computational analysis groups the reads based on the cell label, collapsing the reads with the same molecular index and same gene sequence into a single entry, thus discarding the PCR duplicates and correcting for the amplification bias. This results in the possibility of counting the absolute number of transcripts expressed in each cell.
As the authors showed in a proof-of-principle study, CytoSeq is a very powerful tool for studying the expression profiles of large and heterogeneous cell populations and is able to detect rare cell types in a large background population.Citation40 Since the reactions are carried out in nanoliter volumes, the cost of the library preparation is extremely low and easily allow to scale up the methods to 10,000s or even 100,000s cells per day.
Another advantage of this method is of not being restricted to a specific cell size, thus allowing the study of complex mixtures of cells with very different cell size and shape. We can consider CytoSeq as a more flexible way to capture single cells compared to FACS or mass-spectrometry methods that often require surface markers and the availability of high-affinity antibodies.
The major limitation of CytoSeq is probably the fact that, in its current form, it is designed for analyzing only a pre-defined set of genes, mostly to reduce the costs. In fact, it was argued in the original paper that with tens or even hundreds of thousands of cells the sequencing depth required for a meaningful characterization of the cell population would result in a price unaffordable for most research labs. A solution would be to perform a shallow sequencing, as it has already been demonstrated that as few as 50,000 reads per cell are sufficient to accurately and reliably classify cell types in a population.Citation41 In fact, the majority of genes with the largest contribution to the transcriptional variance between cells can be detected already with less than 10,000 reads per cell, although finer distinctions within heterogeneous populations can be resolved only when sequencing at higher depths.Citation41
Similarly to all other tag-based sequencing approaches, CytoSeq is also able to retrieve only the 3´-most terminal portion of the transcripts.
Drop-seq & InDrop
Similarly to CytoSeq, 2 other methods carry out all the reactions in nanoliter volumes but instead of requiring a physical support in the form of an array use emulsion droplets.Citation42,43 Both methods allow the indexing of thousands of cells for RNA-seq but have some important differences and they will therefore be treated separately here.
The first platform, called InDrop (Indexing Droplets), encapsulates single cells into droplets with lysis buffer, RT reagents and barcoded hydrogel microspheres. Each microsphere carries approximately 109 covalently coupled, photo-releasable oligonucleotides encoding one of 147,456 possible unique barcodes, although more could be created if needed. By using this set, up to 3,000 cells could be barcoded with 99% unique labeling.Citation43 The authors developed a microfluidic device consisting of 4 inlets for adding cells, RT/lysis reagents, barcoded hydrogel microsphere and carrier oil and one outlet for the collection of the droplets. Due to the large cross-section of the microfluidic device (60 × 80 μm2) cell bias is not a concern. The authors optimized the protocol in a way that the cells occupy only 10% of the droplets, thus minimizing the risk of having 2 cells in the same droplet. Indeed, over 90% of the “loaded” droplets analyzed in the original paper contained exactly one cell and one microsphere.Citation43 Using these settings, the microfluidic device was able to capture 4,000–12,000 cells/hour but higher throughputs can be envisaged once the sequencing costs will drop even further.
As mentioned above, each hydrogel microsphere is covalently linked to the barcodes via a photo-releasable bond and thus, by simply exposing the solution containing the droplets to the UV light, the barcodes are released from the beads. Each barcode contains the promoter for the T7 RNA Polymerase, a 5´-Illumina sequencing primer, a cell barcode, a UMI and an oligo-dT tail. The RT reaction is then performed inside the droplets, before all the droplets are broken up and the library preparation continues in a single tube according to the CEL-seq/MARS-seq protocol.Citation32,34
InDrop enables the sequencing of large numbers of cells from heterogeneous populations in a extremely fast way, opening up the possibility of identifying even very rare cell types. However, some important limitations restrict its utility to studies aimed at the discrimination between different cell types or at the profiling of medium to highly abundant transcripts. In fact, the major technical drawback of InDrop is represented by the very low capture efficiency (only 7%), which allows to reliably detecting only transcripts present at 20-50 copies per cell.
Drop-seq shares some similarities with InDrop (and vice versa), although the 2 methods were developed independently. Drop-seq also requires a cell suspension and co-encapsulates each single cell with a barcoded bead in nanoliter-scale droplets. The oligonucleotides on all beads contain a common sequence for PCR amplification, followed by a cell barcode (shared by all the oligonucleotides on the same bead), a UMI (different between all the oligonucleotides on each bead) and an oligo-dT sequence to capture mRNA molecules. Drop-Seq does not require photoactivation of the oligonucleotides but the cells are simply lysed after being isolated in droplets, so that the poly(A) tail of the mRNAs hybridize to the oligo-dT tail on the beads forming STAMPs (Single-cell Transcriptomes Attached to MicroParticles). The droplets are broken and the RT is performed in a single tube. The single-stranded cDNA is then amplified via PCR and undergo fragmentation with the commercial Nextera XT kit. However, only the 3´-most terminal fragments can be used for sequencing, in the same way as with InDrop. The authors calculated that Drop-seq allows the preparation of 10,000 single-cell libraries per day for just 6.5 cents per cell, which is more than 100 times cheaper than the former generation methods, both in terms of time and costs. The reported capture efficiency for Drop-seq is higher than for InDrop, and lies around 12.8%, but suffers the same limitations of all the other tag-based sequencing methods.
Library preparation using Tn5 transposase
Regardless of which method is used to generate double-stranded DNA or whether a tag-based or full-length approach is chosen, the samples need to undergo an additional library preparation step to append sequencing adaptors suitable for sequencing on the Illumina instruments. I decided here to focus only on Illumina sequencing since this company is dominating the market and its instruments are present in almost all the research labs.
Library preparation represents an important cost element, in particular for the full-length methods where early indexing is not feasible and in which separate reactions need to be performed for each sample.
A few approaches can be chosen for this purpose but 2 are probably the most common: shearing with the Covaris focused-ultrasonicator or enzymatic fragmentation.
The Covaris instrument was the tool of choice until few years ago, especially when dealing with long DNA or cDNA molecules. Unfortunately, this method has the disadvantages of being labor-intensive, requires large reaction volumes and in its current version cannot be scaled up beyond the 96-well plate format. Furthermore, sonicated samples need to undergo end-repair, single adenosine addition, ligation and PCR before being ready for multiplexing and sequencing.
A much more efficient and faster approach uses a hyperactive variant of the Tn5 transposase that carries out the fragmentation of double-stranded DNA and ligates synthetic oligonucleotides at both ends in a 5-minute reaction.Citation44 In its wild-type form the Tn5 is a composite transposon with 2 near-identical Insertion Sequences (IS50L and IS50R) flanked by 3 antibiotic resistance genes.Citation45 Each IS50 sequence contains 2 inverted 19-bp End Sequences (ESs), an Outside End (OE) and an Inside End (IE) (). Wild-type ESs have a limited utility due to their relatively low activity and were therefore replaced in vitro by hyperactive Mosaic End (ME) sequences. The formation of a complex between the Tn5 transposase and the 19-bp MEs is the only necessary event for the transposition to occur, provided that the intervening DNA is long enough to bring 2 of these sequences close together to form an active Tn5 transposase homodimer.Citation46 As mentioned above, transposition is a very rare event in vivo and hyperactive mutants were derived by introducing several missense mutations in the 476 residues of the Tn5 protein (E54K, M56A, L372P), which is encoded by IS50R.Citation47 Transposition works through a “cut-and-paste” mechanism, where the Tn5 excises itself from the donor DNA and inserts into a target sequence, creating a 9-bp duplication of the target.Citation45 In the current commercial solution marketed by Illumina (Nextera and Nextera XT DNA kits), free synthetic ME adaptors are end-joined to the 5′-end of the target cDNA by the Tn5 transposase in a reaction called “tagmentation,” where the DNA is fragmented and simultaneously tagged with adaptors.Citation44 A second PCR is thus needed to append barcode adaptors for multiplexing (). Although the kit is extremely robust and versatile, the price represents a problem when thousands of single cells need to be analyzed or when the financial resources are limited.
Figure 2. (A) Structure of the transposon Tn5. In the transposon Tn5 2 near-identical Insertion Sequences (IS50L and IS50R, where “L” and “R” stand for “Left” and “Right,” respectively) bracket 3 antibiotic resistance genes [kan (kanamycin resistance), ble (bleomycin resistance) and str (streptomycin resistance)]. Each IS50 sequence contains 2 inverted 19-bp End Sequences (ESs), an Outside End (OE) and an Inside End (IE). IS50R encodes the functional transposase protein (Tnp) as well as an inhibitor of transposition (Inh). Wild-type ESs have a limited utility due their relatively low activity and were therefore replaced in vitro by hyperactive Mosaic End (ME) sequences which is, as the name indicates, contains elements from both the ES. Adapted and modified from ref. 45. (B) Tn5 transposase-mediated library preparation. Each monomer of Tn5 transposase contains one of 2 partly double-stranded oligonucleotides (here indicated in cyan and red). The double-stranded portion of each oligonucleotide is the hyperactive ME sequence necessary for transposition and is always 19 bp long. The gray-color bar is a Connecting Sequence (CS), which in the Nextera applications is 14 bp long. In the presence of magnesium chloride 2 Tn5 transposase monomers dimerize and become capable of cutting double-stranded DNA in a near-to-random fashion. The ME sequences are then appended to the DNA in a 5-minutes reaction, creating a 9 bp gap in the non-transferred strand. The gap is later filled by a DNA Polymerase. All the fragments carrying different adaptors are amplified by an “enrichment PCR” that also introduces a 8 bp index sequence (for multiplexing purposes) as well as the Illumina P5 and P7 adaptors (necessary for sequencing).
![Figure 2. (A) Structure of the transposon Tn5. In the transposon Tn5 2 near-identical Insertion Sequences (IS50L and IS50R, where “L” and “R” stand for “Left” and “Right,” respectively) bracket 3 antibiotic resistance genes [kan (kanamycin resistance), ble (bleomycin resistance) and str (streptomycin resistance)]. Each IS50 sequence contains 2 inverted 19-bp End Sequences (ESs), an Outside End (OE) and an Inside End (IE). IS50R encodes the functional transposase protein (Tnp) as well as an inhibitor of transposition (Inh). Wild-type ESs have a limited utility due their relatively low activity and were therefore replaced in vitro by hyperactive Mosaic End (ME) sequences which is, as the name indicates, contains elements from both the ES. Adapted and modified from ref. 45. (B) Tn5 transposase-mediated library preparation. Each monomer of Tn5 transposase contains one of 2 partly double-stranded oligonucleotides (here indicated in cyan and red). The double-stranded portion of each oligonucleotide is the hyperactive ME sequence necessary for transposition and is always 19 bp long. The gray-color bar is a Connecting Sequence (CS), which in the Nextera applications is 14 bp long. In the presence of magnesium chloride 2 Tn5 transposase monomers dimerize and become capable of cutting double-stranded DNA in a near-to-random fashion. The ME sequences are then appended to the DNA in a 5-minutes reaction, creating a 9 bp gap in the non-transferred strand. The gap is later filled by a DNA Polymerase. All the fragments carrying different adaptors are amplified by an “enrichment PCR” that also introduces a 8 bp index sequence (for multiplexing purposes) as well as the Illumina P5 and P7 adaptors (necessary for sequencing).](/cms/asset/b5fd7592-a0d6-4cd9-aa1f-20df503a3694/krnb_a_1201618_f0002_c.gif)
A (partial) solution to this problem could be to reduce the reaction volumes, as it has recently been shown in 3 independent studies.Citation48-50 In particular, Shapland and collaborators showed that the volume of the tagmentation reaction can be reduced by 100-fold using acoustic droplet ejection without negatively affecting the quality of the final data.Citation49
Although extremely useful this does not solve either the problem of dependence from commercial kits nor allows much flexibility in the choice of adaptors to load on the Tn5 transposase. In fact, the Nextera kit comes pre-loaded with Illumina adaptors, thus precluding the development of novel applications.
After developing Smart-seq2 we decided to tackle the cost issue by trying to set up a completely kit-independent protocol from cell lysis to sequencing-ready libraries. We devised a simple method for producing high-quality Tn5 transposase and optimized buffers and reagents in order to be able to generate sequencing libraries from nanograms and even sub-picogram amounts of double-stranded DNA.Citation30 That resulted in a in-house Tn5 transposase capable of generating libraries of comparable quality as with the commercial Nextera kit at a fraction of the cost and entirely free for the entire research community (www.addgene.com, plasmid #60240).
The combination of Smart-seq2 for generating double-stranded cDNA which gets then tagmented with in-house Tn5 transposase in a low volume results in a flexible protocol fully accessible to the research community. Depending on the sequencing needs of each project the cost of sequencing can also be amortized over a larger number of samples by designing custom-made barcode adaptors to use in the post-tagmentation PCR to allow the multiplexing of 1536 cells or more.
Spatial transcriptomics
Regardless of the method that is used for scRNA-seq, when studying solid tissues and organs the first step is always a mechanic or enzymatic dissociation into single cells. This causes a loss of positional information, rendering impossible to link transcriptional profiles with cell location throughout a tissue.
An interesting solution to the problem is to sequence the RNA directly inside the cell without prior lysis, a method known as in situ RNA-seq.Citation51 The RNA is first converted to cDNA by using either gene-specific or random primers, amplified by Rolling Circle Amplification (RCA) and sequenced by in situ Sequencing By Ligation (SBL) using a fluorescence microscope. This and similar methods are going to have a huge impact in the near future, especially for all the cases where the preservation of the spatial relationships is key for understanding the underlying biology, such as in tissue and organ development or disease progression.
For an in-depth analysis of all the emerging methods the interested reader is referred to an excellent review that has been recently published.Citation52
Nanopore sequencing
Single-molecule nanopore sequencing has recently gained attention as an ultrarapid, low-cost platform for sequencing DNA and RNA molecules. The concept of nanopore sequencing has been around for 20 years, when it was first described that a DNA strand could be translocated through a pore formed by the α-hemolysin secreted by S. aureus secreted to attack a lipid bilayer.Citation53 The transfer is facilitated by the polarization of the membrane, which is negatively charged in cis (the side where the molecule to sequence is located initially) and positively charged in trans (where the molecule ends up after going through the nanopore). It was immediately clear that only single-stranded but not double-stranded nucleic acids could be transferred.Citation53 The transfer is facilitated from base-dependent transitions in the ionic current flowing through the pore and several additional features, such as directionality of entry (5´- or 3´- end first), base modifications and length are recorded (for a review see ref. Citation54). Many studies indicated that the translocation through the pore occurs at speeds around 1-10 μs per nucleotide in a single-stranded DNA but the noise associated with such a fast movement represents a problem for correct base identification. The most significant innovation that addressed this has been ratcheting of DNA molecules in various ways, especially by molecular motors. Already in 1989 Church and collaborators found out that a DNA Polymerase molecule could be the ideal “motor” to precisely control DNA translocation through the nanopore (US Patent 5,795,782). Several modifications have been introduced since but are not the focus of this review. An alternative approach has been the so-called exonuclease-assisted nanopore sequencing which differs from the previous method because it uses a polynucleotide phosphorylase (PNPase), which processively cleaves single-stranded RNA or DNA in the 3´-to-5´ direction using inorganic phosphate (Pi) to attack the phosphodiester bond and liberates ribonucleoside diphosphates (rNDPs) or deoxynucleoside monophosphates (NMPs), respectively.Citation55 It has been also hypothesized that the additional charge of rNDP (over NMP) might result in a more efficient capture by the nanopore, and thus a more accurate sequencing, when sequencing RNA compared to DNA.
However, the focus has remained primarily on DNA, with RNA receiving less attention. Sequencing RNA molecules with this technology would overcome most, if not all, the limitations associated with all the current methods for library preparation (tailored for obtaining mostly short reads). For example, it will make possible the estimation of mRNA expression without prior amplification; or reveal splice isoforms and other post-transcriptional modifications that might play a regulatory role in the cell. It also holds the promise of investigating the entire transcriptome, without being limited to just polyadenylated RNAs, retaining strand directionality and providing information on editing event and base modifications that have not been explored at all with current technologies. One of the strengths of Nanopore sequencing is the ability of sequencing even very long molecules, as it has been recently shown with RNA strands as long as 6 kb that were successfully threaded through the nanopore.Citation56
Oxford Nanopore Technologies (ONT) is the world-leading company in nanopore sequencing and has recently released a new futuristic device called MinION that works by just plugging it in to a laptop via a USB port (http://www.nanoporetech.com/products-services/minion-mki). The MinION has the size of a cell phone and represents the first really portable sequencing instrument, being equipped with 512 pores that can be individually addressed and provide information about the transcriptome in real-time and with read lengths exceeding 50 kb. Interestingly, the analysis can be stopped when enough data has been collected. A more powerful version called PromethIon with an array of 144,000 nanopores is also available to customers (http://www.nanoporetech.com/products-services/promethion).
Combined methods
More recently, several methods have combined the analysis of the transcriptome with the genome or epigenome from the same cell. Here, I will just briefly mention them since they are not the major focus of this review.
In some cases they are simply a modification of existing protocols but in other cases they have been developed independently for that specific purpose.
Genome and Transcriptome Sequencing (G&T-seq) is a method that allows the parallel sequencing of full-length mRNA using Smart-seq2 and of the genomic DNA using various Whole Genome-Amplification (WGA) methods, according to the desired readout of the experiment.Citation57 While G&T-seq physically separates the 2 nucleic acids immediately after cell lysis, an alternative method called DR-seq (DNA and RNA sequencing), does the same but without prior nucleic acid separation, thus minimizing losses of material.Citation58 However, DR-seq uses CEL-seq for the transcriptome, therefore targeting only the 3´-end of mRNAs. Additionally, the genome is sequenced using a modification of the Multiple Annealing and Looping Based Amplification Cycles (MALBAC) method, making DR-seq a less open-platform method compared to G&T-seq.Citation57
In recent years other methods have also been developed for investigating the changes in the methylation profiles (methylome) at a single-cell level, such as single-cell Reduced Representation Bisulfite Sequencing (scRRBS) and single-cell Bisulfite Sequencing (scBS).Citation59,60 Hou and colleagues have recently combined all these methods and developed a single-cell triple omics sequencing approach that they called scTrio-seq for analyzing the relationship between the genome (copy-number variants, CNVs), DNA methylome and the transcriptome of the same mammalian cell.Citation61
Challenges ahead
The ideal scRNA-seq method would profile all the coding and non-coding cellular transcripts with high efficiency, revealing even the subtler changes in gene expression. Being able to follow transcriptional bursting in real time as well as precisely pinpoint in which cells and subcellular locations different genes are expressed is what several methods have already achieved and it is the path the field should undertake.
At present, one of the major limitations is that all the methods rely on the reverse transcription of RNA to cDNA, followed by amplification either via PCR or IVT before sequencing. Both approaches are prone to losses and biases but can´t be avoided since current second generation sequencing technologies don´t offer the possibility of directly sequencing the RNA. Illumina Sequence-by-Synthesis (SBS) technology represents today about 75% of the sequencing market but, although the recent introduction of higher-capacity instruments such as the X-Ten, have still the important limitations of being able to sequence only 2 × 300 bp (MiSeq series).
Long-read sequencing technologies have been developed, like the Real-Time Single Molecule (SMRT) commercialized by Pacific Biosciences. Although this enables RNA-seq with a median read length of up to 1.5 kb, it still requires the conversion of RNA into cDNA by using MMLV-based RT enzymes, thus suffering the same sensitivity issues and losses of rare transcripts as all the other methods.Citation62 Furthermore, the company reports a raw error rate for SMRT sequencing of 12%, much higher than the 0.1-1% with the Illumina SBS technology.
All these problems could soon be solved by the introduction of Nanopore sequencing that will also minimize the time required for sample preparation, allowing the real-time analysis of nucleic acids from any biological entity or fluid, being it a single cell or circulating DNA and RNA.
Conclusions and future perspectives
The technological development we have witnessed since the first scRNA-seq protocol was published in 2009 is simply astonishing. With sequencing costs steadily dropping and methods with significantly higher throughput being introduced every year, it will soon be possible to sequence the genome, transcriptome, epigenome and proteome of millions of cells simultaneously. This is going to increase our understanding of biology in a way that was unimaginable until just few years ago.
Being able to analyze a single cell, the most fundamental unit of life, is not just interesting per se, but is going to shed light into the complex relationships that cells entertain with each other and with the surrounding environment. All the methods described in this review and new ones that are continuously developed will yield an unprecedented amount of information in the coming years, bringing new challenges in data analysis and interpretation but surely having an enormous impact both in basic research and clinical science.
The discovery and classification of all cell types in the human body will allow, for example, the reconstruction of cell lineage trees in higher organisms; the analysis of rare circulating tumor cells will make possible early cancer detection; the dissection of the complex heterogeneity of primary tumors will allow tailored cancer treatments, with an associated vast development of the personalized medicine field.
Other exciting applications that can be envisaged are the characterization of the molecular basis determining stem cell differentiation or embryonic development in higher organisms.
I personally believe that the entire field of personalized medicine will receive a tremendous boost and it will be possible to analyze biological samples in just hours, or even minutes, providing clinicians with a very powerful tool that will make tailored treatments a reality. The time frame for this revolution is difficult to predict since many challenges still lie ahead. The major issues remain the efficient isolation of single cells from biological samples (being them a tumor, few cells from an embryo or circulating nucleic acids) and the challenges posed by the analysis of such a huge amount of sequencing data, as difficult as to find the classic needle in a haystack.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
I apologize to all scientists whose important work could not be cited in this review due to space constraints. I am grateful to Karolina Wallenborg from the Eukaryotic Single Cell Genomics Facility for valuable comments on the manuscript.
References
- Saliba AE, Westermann AJ, Gorski SA, Vogel J. Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res 2014; 42:8845-60; PMID:25053837; http://dx.doi.org/10.1093/nar/gku555
- Hangauer MJ, Vaughn IW, McManus MT. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS genetics 2013; 9:e1003569; PMID:23818866; http://dx.doi.org/10.1371/journal.pgen.1003569
- Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, Schwartz S, Yosef N, Malboeuf C, Lu D, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 2013; 498:236-40; PMID:23685454; http://dx.doi.org/10.1038/nature12172
- Deng Q, Ramskold D, Reinius B, Sandberg R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 2014; 343:193-6; PMID:24408435; http://dx.doi.org/10.1126/science.1245316
- Bengtsson M, Stahlberg A, Rorsman P, Kubista M. Gene expression profiling in single cells from the pancreatic islets of Langerhans reveals lognormal distribution of mRNA levels. Genome Res 2005; 15:1388-92; PMID:16204192; http://dx.doi.org/10.1101/gr.3820805
- Chang HH, Hemberg M, Barahona M, Ingber DE, Huang S. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature 2008; 453:544-7; PMID:18497826; http://dx.doi.org/10.1038/nature06965
- Grun D, van Oudenaarden A. Design and Analysis of Single-Cell Sequencing Experiments. Cell 2015; 163:799-810; PMID:26544934; http://dx.doi.org/10.1016/j.cell.2015.10.039
- Stegle O, Teichmann SA, Marioni JC. Computational and analytical challenges in single-cell transcriptomics. Nat Rev Genet 2015; 16:133-45; PMID:25628217; http://dx.doi.org/10.1038/nrg3833
- Shapiro E, Biezuner T, Linnarsson S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat Rev Genet 2013; 14:618-30; PMID:23897237; http://dx.doi.org/10.1038/nrg3542
- He S, Wurtzel O, Singh K, Froula JL, Yilmaz S, Tringe SG, Wang Z, Chen F, Lindquist EA, Sorek R, et al. Validation of two ribosomal RNA removal methods for microbial metatranscriptomics. Nat Methods 2010; 7:807-12; PMID:20852648; http://dx.doi.org/10.1038/nmeth.1507
- Ramskold D, Luo S, Wang YC, Li R, Deng Q, Faridani OR, Daniels GA, Khrebtukova I, Loring JF, Laurent LC, et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol 2012; 30:777-82; PMID:22820318; http://dx.doi.org/10.1038/nbt.2282
- Picelli S, Bjorklund AK, Faridani OR, Sagasser S, Winberg G, Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods 2013; 10:1096-8; PMID:24056875; http://dx.doi.org/10.1038/nmeth.2639
- Sandberg R. Entering the era of single-cell transcriptomics in biology and medicine. Nat Methods 2014; 11:22-4; PMID:24524133; http://dx.doi.org/10.1038/nmeth.2764
- Pelechano V, Steinmetz LM. Gene regulation by antisense transcription. Nat Rev Genet 2013; 14:880-93; PMID:24217315; http://dx.doi.org/10.1038/nrg3594
- Zeisel A, Munoz-Manchado AB, Codeluppi S, Lonnerberg P, La Manno G, Jureus A, Marques S, Munguba H, He L, Betsholtz C, et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015; 347:1138-42; PMID:25700174; http://dx.doi.org/10.1126/science.aaa1934
- Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods 2009; 6:377-82; PMID:19349980; http://dx.doi.org/10.1038/nmeth.1315
- Tang F, Barbacioru C, Nordman E, Li B, Xu N, Bashkirov VI, Lao K, Surani MA. RNA-Seq analysis to capture the transcriptome landscape of a single cell. Nat Protoc 2010; 5:516-35; PMID:20203668; http://dx.doi.org/10.1038/nprot.2009.236
- Kurimoto K, Yabuta Y, Ohinata Y, Ono Y, Uno KD, Yamada RG, Ueda HR, Saitou M. An improved single-cell cDNA amplification method for efficient high-density oligonucleotide microarray analysis. Nucleic Acids Res2006; 34:e42; PMID:16547197; http://dx.di.org/10.1093/nar/gkl050
- Chenchik A, Diachenko L, Moqadam F, Tarabykin V, Lukyanov S, Siebert PD. Full-length cDNA cloning and determination of mRNA 5′ and 3′ ends by amplification of adaptor-ligated cDNA. BioTechniques 1996; 21:526-34; PMID:8879595
- Zhu YY, Machleder EM, Chenchik A, Li R, Siebert PD. Reverse transcriptase template switching: a SMART approach for full-length cDNA library construction. BioTechniques 2001; 30:892-7; PMID:11314272
- Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 2014; 9:171-81; PMID:24385147; http://dx.doi.org/10.1038/nprot.2014.006
- Carninci P, Nishiyama Y, Westover A, Itoh M, Nagaoka S, Sasaki N, Okazaki Y, Muramatsu M, Hayashizaki Y. Thermostabilization and thermoactivation of thermolabile enzymes by trehalose and its application for the synthesis of full length cDNA. Proc Natl Acad Sci U S A 1998; 95:520-4; PMID:9435224; http://dx.doi.org/10.1073/pnas.95.2.520
- Petersen M, Wengel J. LNA: a versatile tool for therapeutics and genomics. Trends Biotechnol 2003; 21:74-81; PMID:12573856; http://dx.doi.org/10.1016/S0167-7799(02)00038-0
- Tang DT, Plessy C, Salimullah M, Suzuki AM, Calligaris R, Gustincich S, Carninci P. Suppression of artifacts and barcode bias in high-throughput transcriptome analyses utilizing template switching. Nucleic Acids Res 2013; 41:e44; PMID:23180801; http://dx.doi.org/10.1093/nar/gks1128
- Quail MA, Otto TD, Gu Y, Harris SR, Skelly TF, McQuillan JA, Swerdlow HP, Oyola SO. Optimal enzymes for amplifying sequencing libraries. Nat Methods 2012; 9:10-1; http://dx.doi.org/10.1038/nmeth.1814
- Kapteyn J, He R, McDowell ET, Gang DR. Incorporation of non-natural nucleotides into template-switching oligonucleotides reduces background and improves cDNA synthesis from very small RNA samples. BMC Genomics 2010; 11:413; PMID:20598146; http://dx.doi.org/10.1186/1471-2164-11-413
- Islam S, Kjallquist U, Moliner A, Zajac P, Fan JB, Lonnerberg P, Linnarsson S. Highly multiplexed and strand-specific single-cell RNA 5′ end sequencing. Nat Protoc 2012; 7:813-28; PMID:22481528; http://dx.doi.org/10.1038/nprot.2012.022
- Bjorklund AK, Forkel M, Picelli S, Konya V, Theorell J, Friberg D, Sandberg R, Mjosberg J. The heterogeneity of human CD127(+) innate lymphoid cells revealed by single-cell RNA sequencing. Nat Immunol 2016; 17:451-60; PMID:26878113; http://dx.doi.org/10.1038/ni.3368
- Pinto FL, Lindblad P. A guide for in-house design of template-switch-based 5′ rapid amplification of cDNA ends systems. Anal Biochem 2010; 397:227-32; PMID:19837043; http://dx.doi.org/10.1016/j.ab.2009.10.022
- Picelli S, Bjorklund AK, Reinius B, Sagasser S, Winberg G, Sandberg R. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res 2014; 24:2033-40; PMID:25079858; http://dx.doi.org/10.1101/gr.177881.114
- Sasagawa Y, Nikaido I, Hayashi T, Danno H, Uno KD, Imai T, Ueda HR. Quartz-Seq: a highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity. Genome Biol 2013; 14:R31; PMID:23594475; http://dx.doi.org/10.1186/gb-2013-14-4-r31
- Hashimshony T, Wagner F, Sher N, Yanai I. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell reports 2012; 2:666-73; PMID:22939981; http://dx.doi.org/10.1016/j.celrep.2012.08.003
- Eberwine J, Yeh H, Miyashiro K, Cao Y, Nair S, Finnell R, Zettel M, Coleman P. Analysis of gene expression in single live neurons. Proc Natl Acad Sci U S A 1992; 89:3010-4; PMID:1557406; http://dx.doi.org/10.1073/pnas.89.7.3010
- Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, Mildner A, Cohen N, Jung S, Tanay A, et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 2014; 343:776-9; PMID:24531970; http://dx.doi.org/10.1126/science.1247651
- Kivioja T, Vaharautio A, Karlsson K, Bonke M, Enge M, Linnarsson S, Taipale J. Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods 2012; 9:72-4; http://dx.doi.org/10.1038/nmeth.1778
- Jaitin DA, Keren-Shaul H, Elefant N, Amit I. Each cell counts: Hematopoiesis and immunity research in the era of single cell genomics. Semin Immunol 2015; 27:67-71; PMID:25727184; http://dx.doi.org/10.1016/j.smim.2015.01.002
- Hashimshony T, Senderovich N, Avital G, Klochendler A, de Leeuw Y, Anavy L, Gennert D, Li S, Livak KJ, Rozenblatt-Rosen O, et al. CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol 2016; 17:77; PMID:27121950; http://dx.doi.org/10.1186/s13059-016-0938-8
- Islam S, Kjallquist U, Moliner A, Zajac P, Fan JB, Lonnerberg P, Linnarsson S. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res 2011; 21:1160-7; PMID:21543516; http://dx.doi.org/10.1101/gr.110882.110
- Islam S, Zeisel A, Joost S, La Manno G, Zajac P, Kasper M, Lonnerberg P, Linnarsson S. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods 2014; 11:163-6; PMID:24363023; http://dx.doi.org/10.1038/nmeth.2772
- Fan HC, Fu GK, Fodor SP. Expression profiling. Combinatorial labeling of single cells for gene expression cytometry. Science 2015; 347:1258367; PMID:25657253; http://dx.doi.org/10.1126/science.1258367
- Pollen AA, Nowakowski TJ, Shuga J, Wang X, Leyrat AA, Lui JH, Li N, Szpankowski L, Fowler B, Chen P, et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat Biotechnol 2014; 32:1053-8; PMID:25086649; http://dx.doi.org/10.1038/nbt.2967
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015; 161:1202-14; PMID:26000488; http://dx.doi.org/10.1016/j.cell.2015.05.002
- Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, Kirschner MW. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015; 161:1187-201; PMID:26000487; http://dx.doi.org/10.1016/j.cell.2015.04.044
- Adey A, Morrison HG, Asan, Xun X, Kitzman JO, Turner EH, Stackhouse B, MacKenzie AP, Caruccio NC, Zhang X, et al. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol 2010; 11:R119; PMID:21143862; http://dx.doi.org/10.1186/gb-2010-11-12-r119
- Reznikoff WS. Transposon Tn5. Ann Rev Genet 2008; 42:269-86; PMID:18680433; http://dx.doi.org/10.1146/annurev.genet.42.110807.091656
- Reznikoff WS. Tn5 as a model for understanding DNA transposition. Mol Microbiol 2003; 47:1199-206; PMID:12603728; http://dx.doi.org/10.1046/j.1365-2958.2003.03382.x
- Goryshin IY, Reznikoff WS. Tn5 in vitro transposition. J Biol Chem 1998; 273:7367-74; PMID:9516433; http://dx.doi.org/10.1074/jbc.273.13.7367
- Lamble S, Batty E, Attar M, Buck D, Bowden R, Lunter G, Crook D, El-Fahmawi B, Piazza P. Improved workflows for high throughput library preparation using the transposome-based Nextera system. BMC Biotechnol 2013; 13:104; PMID:24256843; http://dx.doi.org/10.1186/1472-6750-13-104
- Shapland EB, Holmes V, Reeves CD, Sorokin E, Durot M, Platt D, Allen C, Dean J, Serber Z, Newman J, et al. Low-Cost, High-Throughput Sequencing of DNA Assemblies Using a Highly Multiplexed Nextera Process. ACS Synthetic Biol 2015; 4:860-6; PMID:25913499; http://dx.doi.org/10.1021/sb500362n
- Combs PA, Eisen MB. Low-cost, low-input RNA-seq protocols perform nearly as well as high-input protocols. PeerJ 2015; 3:e869; PMID:25834775; http://dx.doi.org/10.7717/peerj.869
- Ke R, Mignardi M, Pacureanu A, Svedlund J, Botling J, Wahlby C, Nilsson M. In situ sequencing for RNA analysis in preserved tissue and cells. Nat Methods 2013; 10:857-60; PMID:23852452; http://dx.doi.org/10.1038/nmeth.2563
- Crosetto N, Bienko M, van Oudenaarden A. Spatially resolved transcriptomics and beyond. Nat Rev Genet 2015; 16:57-66; PMID:25446315; http://dx.doi.org/10.1038/nrg3832
- Kasianowicz JJ, Brandin E, Branton D, Deamer DW. Characterization of individual polynucleotide molecules using a membrane channel. Proc Natl Acad Sci U S A 1996; 93:13770-3; PMID:8943010; http://dx.doi.org/10.1073/pnas.93.24.13770
- Bayley H. Nanopore sequencing: from imagination to reality. Clin Chem 2015; 61:25-31; PMID:25477535; http://dx.doi.org/10.1373/clinchem.2014.223016
- Ayub M, Hardwick SW, Luisi BF, Bayley H. Nanopore-based identification of individual nucleotides for direct RNA sequencing. Nano letters 2013; 13:6144-50; PMID:24171554; http://dx.doi.org/10.1021/nl403469r
- Cracknell JA, Japrung D, Bayley H. Translocating kilobase RNA through the Staphylococcal alpha-hemolysin nanopore. Nano letters 2013; 13:2500-5; PMID:23678965; http://dx.doi.org/10.1021/nl400560r
- Macaulay IC, Haerty W, Kumar P, Li YI, Hu TX, Teng MJ, Goolam M, Saurat N, Coupland P, Shirley LM, et al. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat Methods 2015; 12:519-22; PMID:25915121; http://dx.doi.org/10.1038/nmeth.3370
- Dey SS, Kester L, Spanjaard B, Bienko M, van Oudenaarden A. Integrated genome and transcriptome sequencing of the same cell. Nat Biotechnol 2015; 33:285-9; PMID:25599178; http://dx.doi.org/10.1038/nbt.3129
- Smallwood SA, Lee HJ, Angermueller C, Krueger F, Saadeh H, Peat J, Andrews SR, Stegle O, Reik W, Kelsey G. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods 2014; 11:817-20; PMID:25042786; http://dx.doi.org/10.1038/nmeth.3035
- Guo H, Zhu P, Wu X, Li X, Wen L, Tang F. Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Res 2013; 23:2126-35; PMID:24179143; http://dx.doi.org/10.1101/gr.161679.113
- Hou Y, Guo H, Cao C, Li X, Hu B, Zhu P, Wu X, Wen L, Tang F, Huang Y, et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res 2016; 26:304-19; PMID:26902283; http://dx.doi.org/10.1038/cr.2016.23
- Sharon D, Tilgner H, Grubert F, Snyder M. A single-molecule long-read survey of the human transcriptome. Nat Biotechnol 2013; 31:1009-14; PMID:24108091; http://dx.doi.org/10.1038/nbt.2705