
ABSTRACT
Adenosine deaminase acting on RNA (ADAR) 1 binds and edits double-stranded (ds) RNA secondary structures found mainly within untranslated regions of many transcripts. In the current research, our aim was to study the role of ADAR1 in liver homeostasis. As previous studies show a conserved immunoregulatory function for ADAR1 in mammalians, we focused on its role in preventing chronic hepatic inflammation and the associated activation of hepatic stellate cells to produce extracellular matrix and promote fibrosis. We show that hepatocytes specific ADAR1 knock out (KO) mice display massive liver damage with multifocal inflammation and fibrogenesis. The bioinformatics analysis of the microarray gene-expression datasets of ADAR1 KO livers reveled a type-I interferons signature and an enrichment for immune response genes compared to control littermate livers. Furthermore, we found that in vitro silencing of ADAR1 expression in HepG2 cells leads to enhanced transcription of NFκB target genes, foremost of the pro-inflammatory cytokines IL6 and IL8. We also discovered immune cell-independent paracrine signaling among ADAR1-depleted HepG2 cells and hepatic stellate cells, leading to the activation of the latter cell type to adopt a profibrogenic phenotype. This paracrine communication dependent mainly on the production and secretion of the cytokine IL6 induced by ADAR1 silencing in hepatocytes. Thus, our findings shed a new light on the vital regulatory role of ADAR1 in hepatic immune homeostasis, chiefly its inhibitory function on the crosstalk between the NFκB and type-I interferons signaling cascades, restraining the development of liver inflammation and fibrosis.
Introduction
In recent years, it has become evident that RNA molecules undergo extensive post-transcriptional modifications, such as RNA-editing and Methylation, shaping the epitranscriptome.Citation1-4 Adenosine deaminase acting on RNA (ADAR) 1 is a double strand (ds) RNA-binding and editing protein. It has 2 major isoforms: a shorter ADAR1p110 and a longer interferon-inducible ADAR1p150.Citation5 The promoter of the longer ADAR1p150 isoform is located in first exon and contains an interferon-stimulated response element and its translation start site (TSS) is also within exon 1A, whereas the ADAR1p110 has a different promoter and its translation start from exon 2.Citation6 The ADAR1p110 isoform is present predominantly in the nucleus whereas the interferon-inducible isoform localizes to the cytoplasm implying they have different functions.Citation6,7 The most prevalent type of RNA editing in mammalians is the conversion of adenosine to inosine by deamination (A-to-I editing), largely mediated by ADAR1 and to a lesser extent by ADAR2. RNA editing is basically divided into 2 categories: infrequent site-specific and promiscuous non-site specific “hyper” editing of repetitive elements.Citation6,8 Development of bioinformatics methodologies and tools to map A-to-I editing sites revealed that RNA editing is highly abundant in the mammalian transcriptome. The majority of these A-to-I editing sites in mammalians are in dsRNA secondary structures within UTRs that are composed of inverted repetitive sequences: i.e., the primate-specific Alu retrotransposons in the human and orthologous short interspersed repetitive elements (SINE) in the mouse. Moreover, a large difference in the number of RNA editing sites exists between the human and the mouse (∼1 million editing sites compared to low tens of thousands, respectively) and this difference is determined by the abundance of inverted primate-specific Alu sequences in human mRNAs.Citation2,4,9-11
Deletion of Adar, the gene encoding mouse ADAR1, is embryonic lethal at midgestation likely due to defective hematopoiesis and liver disintegration.Citation12 This embryonic lethality in ADAR1 null mice is linked to an excessive type-I interferons response.Citation12,13 Similarly, biallelic ADAR1 missense mutations that disrupt the catalytic function of the ADAR1 protein cause the generally fatal Aicardi-Goutières syndrome (AGS). This severe auto inflammatory disorder is characterized by systemic interferon signature, severe encephalopathy, hepatosplenomegaly and intermittent fevers.Citation14,15 These findings imply that a key physiological role of RNA editing by ADAR1 is to modify endogenous RNA duplexes in mRNAs, composed of inverted transposons, preventing aberrant activation of dsRNA sensing pathways and a toxic type-I interferons response.Citation16
Although, ADAR1 has been extensively studied for its RNA editing activity, studies from the Nishikura laboratory demonstrated, in addition, RNA editing-independent regulatory functions in global miRNA biogenesis and RNA interference.Citation7,17 More recently it was shown that ADAR1, independent of its regulatory role in innate immunity to long dsRNAs, plays a vital regulatory role in the normal development of the intestine, kidneys, and peripheral lymphoid organs.Citation18
Liver disintegration is a major feature of ADAR1 null embryos, but the biological processes underlining this pathology have not been fully elucidated.Citation12 Disruption of liver homeostasis from a wide variety of etiologies, including autoimmunity, produces the necessary signals for the induction of liver inflammation and fibrosis.Citation19,20 The functional integrity of liver hepatocytes is known to be critical for liver homeostasis and thus biological processes that lead to hepatocytes toxicity and apoptosis are a known stimuli for stellate cells activation and acquisition of a myofibroblast-like morphology with excessive production of smooth muscle α-actin (αSMA) and of extracellular matrix components leading firogenesis.Citation21,22 In this context, the NFκB pathway plays a pivotal role in normal liver physiology and pathophysiology, acting as a target and a source for inflammatory signals Citation23
In this study, we clarified the functional role of ADAR-1 in hepatocytes and liver tissue homeostasis. Thus, we generated a mouse model with Alb-Cre-dependent excision of floxed Adar in hepatocytes. Here we show that hepatocyte-specific depletion of ADAR1 leads to significant liver damage, chronic inflammation and the development of liver fibrosis within the first weeks of life. Moreover, we identified a novel NFκB-dependent RNA editing-independent molecular mechanism that regulates the paracrine activation of stellate cells by ADAR1 deficient hepatocytes.
Results
Hepatocytes specific Adar gene KO causes severe postnatal morbidity and mortality
To investigate the role of ADAR1 in adult liver physiology and its role in liver disorders, we generated mice with liver specific disruption of the Adar gene (see Materials and Methods section). Briefly, Adarf/f mice Citation24 were intercrossed with Alb-Cre+ Citation25 deleter mice, and the heterozygous offspring (Adarf/+;Alb-Cre+) were then crossed with Adarf/f to produce Adarf/f;Alb-Cre+ mutant mice (termed Adar Hep-KO). Genotyping of the mice was done by a multiplex PCR that simultaneously detects Adar wt(+), floxed(f), and excised (Δ7-9) alleles using PCR primers, and by another PCR reaction that specifically detects the Alb-Cre transgene (Fig. S1). Mice homozygous for the Adar floxed allele and encoding the Alb-Cre transgene (Adarf/f;Alb-Cre+) and appropriate littermate controls were identified and further studied.
In this model, ADAR1 is deleted specifically from hepatocytes during embryonic day E14.5.Citation25 We found that Adar Hep-KO mice were viable at birth but had profound morbidity and mortality during early weeks of life. Thus, even though the cross breeding of Adarf/+;Alb-Cre+ with Adar f/f mice is expected to produce a 1 to 4 ratio of offspring with the Adar Hep-KO genotype, only ∼10% of these mutants survived to the weaning age of 3 weeks. To exclude embryonic lethality of Adar Hep-KO embryos in uterus, we analyzed embryos from embryonic day 14.5 until birth, and found that the fraction of Adar Hep-KO mutants among fetuses surviving to birth was a quarter as expected (data not shown). These findings demonstrate the vital physiological role of ADAR1 in the maintenance of hepatocyte and normal liver homeostasis during the first weeks of life.
By western blotting (WB), we examined ADAR1 protein expression, both the p110 and p150 isoforms, in the total liver cells lysate of mice euthanized at 2 and 4 weeks of life. As expected, we found a significantly reduced level of the constitutively expressed Adar p110 isoform in Adar Hep-KO, reflecting its effective depletion from hepatocytes that are the main liver cell type. In contrast, we observed an increased expression level of the inducible p150 isoform in Adar Hep-KO livers compared to littermate control livers (). Next, we analyzed in parallel the cell lysates from purified hepatic non-parenchymal cells (NPCs) and from the total liver cells by WB for p150 and p110 levels (Fig. S2A). As predicted, the densitometric analysis of ADAR1 isoforms confirmed that the p150 to p110 ratio was significantly higher in cell lysates from total liver and NPCs of Adar Hep-KO compared to littermate controls (Fig. S2B). Importantly, the p150 isoform accounted for >35% of the total ADAR1 in the cell lysates of NPCs from Adar Hep-KO, whereas in the total liver extract from littermates it accounted only for ∼2% of total ADAR1 (Fig. S2A). Taken as a whole, these data imply that the basal levels of the p150 isoform in normal liver tissue are low, whereas hepatocyte-specific ADAR1 deletion induces the increase of the p150 isoform chiefly in NPCs.
Figure 1. Growth retardation in Hepatocytes specific Adar depleted mice. (A) Representative WB analysis of ADAR1 p110 and p150 isoforms in total cell lysates from the livers of 2- and 4-week old Adar Hep-KO mice and their littermate controls. Tubulin levels were used as loading controls. (B) Representative images of 4-week old Adar Hep-KO mice and livers compared to littermate controls. (C) Mean body weight of 4-week old Adar Hep-KO males (n = 17) and females (n = 10) compared to WT littermate controls. (D-E) Mean body weight and Liver to body mass ratio, repectively, of 2-week (n = 4) and 4-week old (n = 7) Adar Hep-KO mice compare to littermate controls.
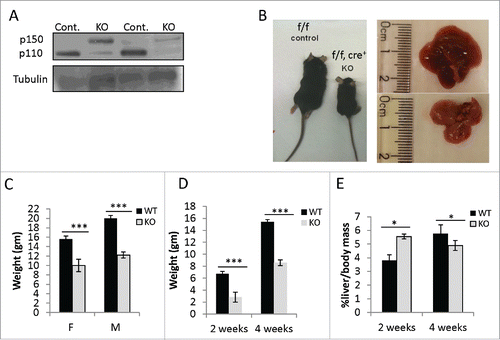
Moreover, on inspection Adar Hep-KO mice had a reduced body size with a smaller pale to yellow livers compared to their littermate controls (). This growth retardation was gender independent, so that at the age of 4 weeks both female and male Adar Hep-KO mice had significantly lower mean body weight relatively to their littermate controls (). Additionally, Adar Hep-KO mice had a significantly lower mean body weight, both at the 2 and 4 week time points (), while their liver to body mass ratio was initially higher but significantly poorer at the 4 week time point compared to littermate controls ().
Adar Hep-KO mice display extensive liver damage from early weeks of life
To evaluate liver damage in Adar Hep-KO mice, we tested levels of liver enzyme released into the serum of surviving young Adar Hep-KO mice and their littermate controls. We found that the levels of lactate dehydrogenase, alkaline phosphatase, alanine aminotransferase, aspartate aminotransferase were elevated several-fold in Adar Hep-KO compared to littermate controls (). Moreover, the serum albumin level, reflecting hepatic synthetic capacity, was significantly reduced in mutant mice. Histopathology analysis by Hematoxilin and Eosin (H&E) staining of liver sections from Adar Hep-KO mice and their littermate control, at the age of 2 and 4 weeks, showed extensive liver damage with changes in hepatic architecture (). In two- week old Adar Hep-KO mice, we observed excessive extra medullar hematopoiesis, abnormally enlarged hepatocytes with more than one nucleus, liver inflammation with extensive infiltration by immune cells in periportal areas, and zonal hepatic lipidosis with accumulation of mixed (microvesicular and macrovesicular) lipid vacuoles in hepatocyte cytoplasm. In four-week old mice, the H&E staining showed a more extensive liver damage that included more profound apoptosis with areas of coagulated necrosis surrounded by massive inflammation, bile duct hyperplasia, increased mitosis rate of hepatocytes (compensatory regeneration), hepatocytes ballooning, and more pronounced macrovasicular lipidosis ().
Figure 2. Adar Hep-KO mice show extensive liver damage with fibrogenesis. (A) Impaired level of liver enzymes detected in serum of 4-week old Adar Hep-KO mice: decreased levels of albumin, and increased levels of alkaline phosphatase (ALP), alanine aminotransferase (ALT), aspartate aminotransferase (AST) and lactate dehydrogenase (LDH) were detected in the Adar Hep-KO mice indicative of hepatocellular damage (n = 4). (B) H&E staining of representative liver sections from 2 and 4-week old Adar Hep-KO (KO) and WT control (WT) mice (n = 5). While the WT section exhibit normal characteristics the livers from the Adar Hep-KO mice show massive damages to the tissue (x40 magnification). (C) Analysis of representative liver section from 4-week old Adar Hep-KO and WT mice using specific staining to detect apoptosis (TUNEL assay, x10 magnification), compensatory proliferation (Ki67, x20 magnification), changes in liver architecture (Reticulin, x40 magnification) and hepatic fibrosis (Serius Red, x40 magnification).
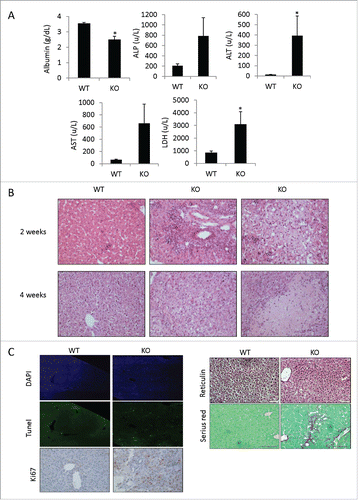
Next, we further analyzed the level of apoptosis and proliferation in liver sections from 4-week old mice. By the TUNEL assay, we detect significant increase in the number of apoptotic cells, while the Ki67 staining showed, as predicted, enhanced proliferation of surviving hepatocytes in Adar Hep-KO compared to littermate control liver sections (). These results imply that ADAR1 deficiency induces apoptotic cell death of hepatocyte and compensatory proliferation of the remaining hepatocytes that escaped Alb-Cre-dependent biallelic excision of the floxed Adar gene segment. The Reticulin stain showed loss of hepatocytes with subsequent collapse of the residual reticulin network specifically in Adar Hep-KO livers (). Moreover, Adar Hep-KO livers showed thicker hepatic plates indicating ongoing liver regeneration. Sirius red staining for the detection of collagen fibrils was positive only in liver sections of Adar Hep-KO mice, as a marker of ongoing liver fibrosis (). Thus, the liver histopathology showed increased liver injury over time with the morphological diagnosis at 4 weeks of hepatocyte apoptosis with foci of coagulated necrosis coupled with chronic multifocal inflammation, hepatocyte regeneration, and regions of fibrosis.
Extensive infiltration of adar Hep-KO livers by cytotoxic CD8+ T cells and granulocytes
Since liver Histopathology showed chronic hepatic inflammation and fibrosis, we next analyzed by flow cytometry the cellular composition of the immune cells isolated from perfused livers. We found that the immune cell infiltrate isolated from the liver tissue of 4-week old Adar Hep-KO was highly enriched for CD3+ T cells (), chiefly of the CD8+ cytotoxic T cell phenotype (). We also show enrichment of Ly6G+CD11b+granulocytes but not for liver Ly6G−F4/80+ macrophages (). The percentage of B220+CD19+ B cells and helper CD3+CD4+ T cells was conversely reduced relative to littermate controls (). Moreover, the total number of immune cells purified from the livers of Adar Hep-KO, regardless of their lineage specification, was significantly higher relative to littermate controls (data not shown).
Figure 3. Adar Hep-KO livers show increased CD8+ cytotoxic T cells and granulocyte infiltration and a type-I interferon signature. (A-C) Flow cytometry analysis of non-parenchymal hepatic cells from 4-week old Adar Hep-KO and heterozygous littermate controls. After purifications of the cells erythrocytes were depleted and cells were immunostained for CD45R/B220, CD3, CD4, CD8, CD11b, Ly6G, and F4/80: (A) Analysis for the percentage of B cells (B220+CD3− and CD19+ gating), and for CD3+B220- T cells. (B) Depicting the CD3+ gate and further analysis for the percentages of CD4+ helper T cells and CD8+ cytotoxic T cells. (C) Analysis for the percentage of Ly6G+CD11b+ granulocytes (left panel) and F4/80+Ly6G- hepatic macrophages (right panel). Shown is a representative experiment out of n = 3 performed. (D) GSEA for biological functions and disease categories of the differentially expressed genes by Qiagen's IPA tool of 2 Adar Hep-KO mice versus 2 littermate controls (using 2FC cutoff). (E) Microarray mRNA expression levels of type-I interferons stimulated genes (ISGs) in 2 and 4-week old Adar Hep-KO vs. littermate control mice.
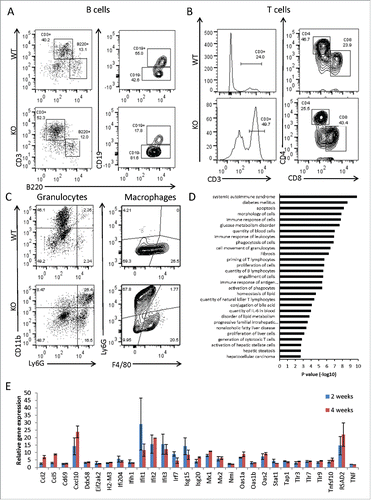
Bioinformatics analysis of Hep-Ko livers showed enrichment for the biological processes of autoimmunity and type-I interferons signaling
Homozygous disruption of Adar gene in the germline is embryonic lethal in mice, partly due to the induction of a global and toxic “autoimmune” type-I interferons response.Citation13 To determine, in unbiased manner, the gene networks and signaling pathways linked to the pathogenesis of the liver damage in Adar Hep-KO, we analyzed global gene expression in livers of 2 and 4-week old mutant and control mice by the Affymetrix GeneChip platform (see Materials and Methods for details). The gene set enrichment analysis (GSEA) of the differentially expressed genes in the livers of Adar Hep-KO, using the Qiagen's Ingenuity Pathway Analysis (IPA) software, revealed significant enrichment for biological processes highly relevant to the pathogenesis of the hepatic damage, inflammation and fibrosis. These included the following functional categories: immune response of cells, systemic autoimmune disorder, apoptosis, fibrosis, priming of T lymphocytes, quantity of IL6, generation of cytotoxic T cells, activation of hepatic stellate cells, and hepatic steatosis (). Further analysis of the up regulated (> 2-fold) immune response genes, for type-I interferons signature, revealed that a large number of interferon stimulated genes (ISGs) were specifically induced in Adar Hep-KO mice (). Next, by qPCR, we validated the up regulation of typical ISGs (Oas2, Irf7, Ifit1, Rsad2, Eif2ak2/PKR, Ccl5, TNF) in Adar Hep-KO compared to control livers at both time points (Fig. S3).
ADAR1 silencing in HepG2 cells induces the paracrine activation of hepatic stellate cells
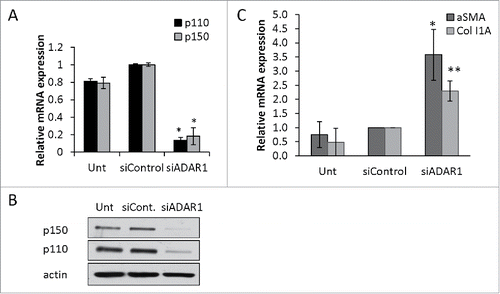
Immune cell mediated tissue injury during inflammation is a known cause of liver fibrosis.Citation20 Nevertheless, a direct paracrine interaction between ADAR1 deficient hepatocytes and stellate cells is still a potential pro-fibrotic mechanism.Citation21 To test this hypothesis, we first silenced ADAR1 expression, by targeting its mRNA with a specific siRNA, in the hepatocellular carcinoma cell line HepG2. This resulted in significant reduction of ADAR1 p110 and p150 expression both at the mRNA and protein level, at 48 hours post transfection (). Next, we collected the supernatants from 48 hours cultures of HepG2 cells transfected with siADAR1 or siControl, and incubated freshly isolated mouse hepatic stellate cells in these conditioned media. Twenty-four hours later, we purified total RNA from stellate cells treated with conditioned medium and untreated control cells. By qPCR we found > 2-fold increase in the relative expression of the fibrosis markers α smooth muscle actin (αSMA) and Collagen type IA (Col1A) in the stellate cells treated with HepG2-siADAR1 conditioned medium compared to counterpart control cells (). This result suggested that an immune-cells-independent mechanism, mediated by a direct effect of ADAR1 deficient hepatocytes on stellate cells, is involved in the induction of fibrosis.
Figure 4. ADAR1 depleted HepG2 cells induce paracrine activation of primary hepatic stellate cells. HepG2 cells were either untreated (Unt) or transfected with siControl or siADAR1. (A) 48-hours post transfection the relative mRNA expression of ADAR1 p110 and p150 in HepG2 cells was measured by qPCR, normalized to HPRT. (B) WB analysis of total cell lysates using anti-ADAR1 and anti-actin (as loading control) specific Abs. (C) Primary hepatic stellate cells were incubated with conditioned medium collected from ADAR1 HepG2-KD and control cells, and 24 hours later the relative mRNA expression levels of αSMA and Col1A were measured using qPCR, normalized to HPRT. Bar graphs show mean ± SEM of n = 3 experiments.
ADAR1 silencing in HepG2 cells upregulates the NF-kB transcriptional activity
To elucidate the biological processes induced by ADAR1 deficiency in hepatocytes that regulate the paracrine signaling to neighboring stellate cells, we examined global gene expression in HepG2 transfected with a plasmid vector containing shADAR1 or shControl. The gene array analysis showed that a large number of genes (>500) were differentially expressed (employing a 2-FC cut-off filter) subsequent to ADAR1 silencing (∼70% reduction) in HepG2 cells (see Table S1). Gene expression is usually induced by transcription factors (TFs) binding. Thus, we analyzed our upregulated gene list for over-represented transcription factor binding site motifs in the promotor region (-200 to +50 from annotated transcription start site), using the Pscan tool, as previously described.Citation26 We found a significant enrichment for NFκB complex binding motifs in the promoter regions, particularly for the p65 (RelA) subunit (p < 0.0001, by Z-test). Using the Qiagen's IPA platform, we then identified NFκB as a potential upstream regulator of the gene expression pattern observed in ADAR1 HepG2-KD data set (complete gene list, scatter plot and heat map analysis are presented Table S1 and Figs. S4A and B, respectively). Importantly, the 2 transcripts that ranked first and second in the NFκB target gene list (both >20-fold increase) encoded the cytokines IL6 and IL8 (2 outliers in scatter plot Fig. S4A). To validate this finding we next transfected HepG2 cells with either siADAR1 or siControl and measured relative IL6 and IL8 transcript levels by qPCR. We found that IL6 was indeed upregulated (), and furthermore in repeated independent experiments it was a highly consistent phenomenon though the magnitude of IL6 upregulation varied among measures [2.5 to 28-fold change (n = 10), *p < 0.01]. Since the cytokine IL6 plays an important role in liver fibrosis,Citation27 we next determined the secretion of IL6 from HepG2 cells using commercial ELISA kits. We found that ADAR1 silencing specifically induced (at 48 hours) a significant increase (>10-fold) in the secretion of IL6 from HepG2 ().
Figure 5. NFκB-dependent upregulation of IL6 and IL8 in ADAR1 HepG2-KD cells. (A) Relative mRNA expression of IL6 and IL8 in untreated, siControl and siADAR1 transfected HepG2 cells as determined by qPCR (normalized to HPRT). (B) Levels of IL6 in conditioned medium of siControl and siADAR1 transfected HepG2 cells as measured by ELISA kits. Results are representative of n = 3 experiments. (C) Untransfected, siControl and siADAR1 transfected HepG2 cells were co-transfected with pGL2-KB-Luciferase to assess the levels of NFkB activation. Luminescence values are shown relative to control Renila plasmid. (D) Relative expression of IL6 and IL8 mRNA in HepG2 cells following co-depletion of ADAR1 and p65. shP65 or shGFP control were co-transfected with siADAR1 or siControl. IL6 and IL8 expression levels were determined using qPCR. (E) The effect of IFNα treatment on mRNA expression levels of ADAR1 p110, ADAR1 p150, IL6 and IL8 in siADAR1 and siControl HepG2 transfected cells by qPCR, normalized to HPRT. Bar graphs show mean ± SEM of n = 3 experiments.
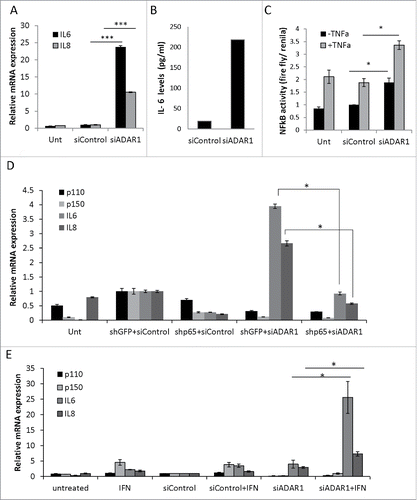
To monitor directly the relative differences in the transcriptional activity of NFκB -complex, we transfected HepG2 cells with an NFκB-responsive luciferase construct, encoding the firefly- luciferase-reporter gene under the control of a canonical NFκB transcriptional response element (TRE). We found that subsequent siRNA mediated ADAR1 silencing in these HepG2 transfectants induced a ∼2-fold increase in NFκB activity that was comparable to the up-regulation by the classic NFκB activator, TNF (). Moreover, TNF treatment further augmented the effect of ADAR1 silencing on NFκB transcriptional activity. To test specifically the contribution of p65/RelA to the enhanced transcriptional activity of NFκB in ADAR1 KD HepG2 cells, we co-silenced both ADAR1 and p65 in HepG2 cells, as validated by WB (Fig. S5). We found that this co-silencing significantly reduced the relative quantity of both IL6 and IL8 transcripts (), implying that the molecular events following ADAR1 silencing are, at least, partly mediated by p65 NFκB (RelA) transcriptional activity.
Next, to evaluate the crosstalk between type-I interferons signaling and ADAR1-dependent NFκB pathway activation, we treated ADAR1 HepG2-KD and control cells with Interferon (IFN)α. Interestingly, our results show that co-treatment of HepG2 cells with siARAR1 and IFNα induced a strong synergistic upregulation of IL6 expression and additive upregulation of IL8 expression as compared to siControl transfected counterpart cultures ().
Regulatory editing-independent interaction between ADAR1 and the NFκB cascade
We next asked whether the global effect of ADAR1 on the NFκB pathway was RNA editing-dependent or -independent. Therefore, we cloned the cDNAs of wild type (wt) ADAR1 p110 and p150 isoforms as well as their counterpart mutated (mut) editase-deficient cDNAs into plasmid vectors engineered to express FLAG-tagged proteins and further mutated to be siADAR1 resistant, as previously described.Citation28 An empty plasmid expressing only the FLAG-tag served as a negative control. Then, we co-transfected HepG2 cells with either one of these expression vectors together with siADAR1 or siControl. We found that co-transfection of HepG2 cells with plasmids encoding a catalytically inactive or normal ADAR1 induced a partial, but statistically significant, correction of the effect of ADAR1 silencing on IL6 upregulation (). These results imply that suppression of NFκB transcriptional activity by ADAR1 is, nonetheless, in part RNA-editing independent.
Figure 6. ADAR1 regulatory effect on NFκ(B)cascade is editing independent. (A) Relative IL6 mRNA expression levels normalized to HPRT as measured by qPCR, following co- transfection of HepG2 cells with siADAR1 and either one of the following 5 plasmids: p110 WT, p110 mutated in the editase domain (mut) p150WT, p150 mut and empty Flag plasmid. Transfection with siControl and empty Flag plasmid served as the negative control. Bar graphs show mean ± SEM of n = 3 experiments. (B) ADAR1-p110- GFP and ADAR1-p150-GFP plasmids were transfected into HepG2 cells. Co-IP of cell extracts using GFP-trap beads. The levels of IKKβ were detected using WB analysis with specific Abs. Expression of empty GFP vector served as negative control. (C) Co-IP of cell extracts using anti-IKKβ specific Abs and protein A-coated beads. The protein levels of ADAR1 p110 and P150 were detected using WB analysis with anti-ADAR1 specific Abs. Rabbit IgG served as negative control. Total cell lysate (input), and IP (bound) fractions served as positive control. (D) Representative WB analysis of ADAR1 p110 and p150 isoforms and IκB protein level in untreated, siControl and siADAR1 transfected HepG2 cells. TNF treatment served as positive control for NFκB pathway activation.
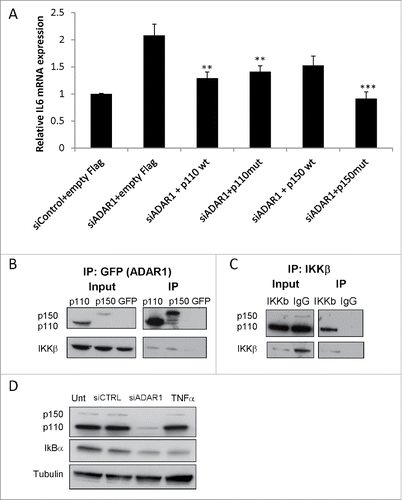
The major regulatory step of the NFκB pathway is activation of the IκB kinase (IKK) complex to induce the phosphorylation of IκB and its ensuing proteasomal degradation, enabling the active NFκB subunits to translocate into the nucleus and induce target gene transcription.Citation29 Thus, our next set of experiments were designed to answer whether ADAR1 interacts with the regulatory steps that lead to the activation of the canonical NFκB complex. First, we tested whether ADAR1 interacts with the canonical IKK complex that is composed of IKKα and IKKβ subunits. Thus, HepG2 cells were transfected with plasmid vectors expressing the p110 or p150 isoforms of ADAR1 fused to GFP. Cell lysates were analyzed by the GFP-Trap beads method, optimal for one-step IP of GFP-fusion proteins and their interacting proteins. The co-IP of IKKβ was then analyzed by immunoblotting with anti-IKKβ specific antibodies (Abs). We found that both ADAR1 isoforms indeed interacted with IKKβ (). Reciprocal experiment using untreated HepG2 cell lysates for co-IP with anti-IKKβ Abs followed by immunoblotting for ADAR1, further validated the co-IP of the ADAR1 p110 isoform (). As basal endogenous p150 protein level are very low, it is not surprising that only the interaction with the ‘housekeeping’ p110 isoform could be detected. Moreover, by WB of cell lysates from ADAR1 KD HepG2 and control cells, we validated that the silencing of ADAR1 was linked to reduced IκB levels (), the common molecular event downstream of IKK activation. These experiments imply an interaction between ADAR1 and IKKβ that down modulates its capacity to induce IκB-phosphorylation and subsequent degradation.
IL6 secreted from ADAR1 KD HepG2 cells induce hepatic stellate cells activation
To elucidate the role of NFκB-dependent IL6 secretion on the paracrine effect of ADAR1 deficient hepatocytes on stellate cells that mediate the fibrotic process, we co-silenced ADAR1 and IL6 in HepG2 cells. Thus, HepG2 cells were co-transfected with siIL6 and siADAR or with either siControl, siADAR1, or siIL6 alone. Conditioned media were collected at 48 hours post-transfection and added to primary stellate cells freshly isolated from mice livers. To validate efficient gene silencing, expression levels of ADAR1p110/p150 and IL6 in transfected HepG2 cells were measured by qPCR (see Fig. S7). The expression levels of the fibrosis markers Col1A and αSMA in stellate cells were also measured by qPCR at 24 hours post incubation with the indicated conditioned medium. As shown in , Col1A and αSMA were significantly upregulated during the incubation in ADAR1 KD conditioned medium (2.73- and 2.57-fol, respectively). In contrast, no upregulation of these fibrosis markers was observed in stellate cells incubated with conditioned medium from HepG2 co-transfected with siADAR1 and siIL6 or with siIL6 alone.
Figure 7. IL6 secreted from ADAR1 HepG2-KD cells mediates paracrine stellate cells activation. (A) Primary hepatic stellate cells were incubated with conditioned medium collected from siControl, siADAR, siIL6 or siADAR1and siIL6 co-transfected HepG2 cells. Relative mRNA expression levels of αSMA and Col1A in the HSCs were determined by qPCR analysis normalized to HPRT. Bar graphs show mean ± SEM of n = 3 experiments. (B) Representative microscopic images of the phenotypic changes of hepatic stellate cells following incubation with conditioned medium of siControl, siADAR1, siIL6, siADAR1+siIL6 transfected HepG2 cells (inverted light microscopy, x10 magnification).
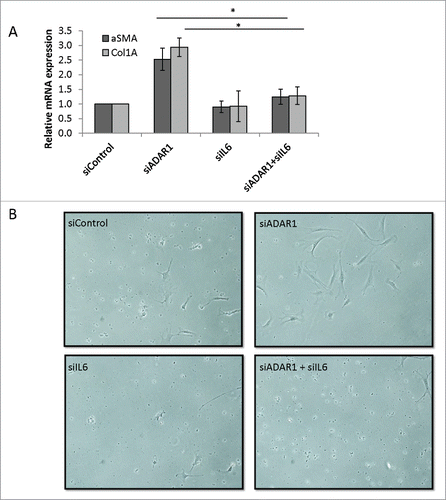
Previous studies show that IL6 induced stellate cells activation, from quiescent vitamin A storing cells to myoblast-like cells, is accompanied by visible phenotypic alteration. As can be seen in , whereas stellate cells incubated in conditioned medium from ADAR1 KD HepG2 cells display phenotypical changes characteristic of activated cells, the counterpart stellate cells treated with either siControl or siIL6 alone or co-transfected with siADAR1 and siIL6 displayed a phenotype typical of resting cells. These findings imply that IL6 secreted from “stressed” ADAR1 KD HepG2 cells is a major factor in the induction of stellate cells' activation, and importantly suggesting that in vivo a normal level of ADAR1 in hepatocytes is important for the maintenance of global liver homeostasis, including the prevention of stellate cell activation and fibrosis.
Discussion
In this study, we provide new insights into the physiological importance of ADAR1 in hepatocytes and the adult liver homeostasis. We show that somatic disruption of ADAR1 in hepatocytes induces substantial hepatocyte apoptosis, liver damage and chronic inflammation with fibrogenesis, coupled with growth retardation and increased mortality of the mice during early weeks of life. The chronic liver inflammation in ADAR1 depleted hepatocytes was associated with an interferon signature, recapitulating the interferonopathy observed in Adar−/− mice and AGS patients.Citation12,14 Using an in vitro model of silencing ADAR1 in HepG2 cells, we show for the first time that ADAR1 KD also induced an RNA-editing independent upregulation of NFκB transcriptional activity, with IL6 ranking first among the list of NFκB target genes in HepG2 KD cells. Importantly we determined that IL6 was a major player in the paracrine activation of hepatic stellate cells that is a central event in hepatic fibrosis.Citation21,30
The histopathology of liver tissue and the specific histochemical staining from 2 and 4-week old Adar Hep-KO showed significant apoptosis at the early time point that proceeded to coagulated necrosis as the liver damage progressed over time. This hepatocytes injury was accompanied by a prominent chronic inflammatory response with a mixed immune cell infiltrate, dominated by cytotoxic CD8+ T cells that are known to mediate tissue damage both in viral and in autoimmune hepatitis.Citation31,32 The ongoing hepatocyte injury in Adar Hep-KO and the ensuing chronic inflammation, as expected, induced collapse and condensation of the reticulin network, compensatory hepatic regeneration, and importantly early fibrotic changes with deposition of type I collagen fibrils.
The bioinformatics analysis of the GeneChip® array datasets from livers of Adar Hep-KO vs. littermate controls with the Qiagen's IPA platform identified interferon signaling as a potential upstream regulator of the expression pattern observed in the Adar Hep-KO data set. For instance, a large number of typical ISGs, especially Oas2, Oas1a, Irf7, Ifit1/2/3, Rsad2, Cxcl10, Mx1 and Isg15 were among the genes most significantly upregulated. This observation was in line with the robust interferon signature observed in Adar null mice and AGS patients.Citation12,13,15 Furthermore, while as expected the constitutive p110 ADAR1 protein was almost undetectable, the level of the interferon inducible p150 ADAR1 isoform was significantly higher in liver extracts from Adar Hep-KO. This observation implied a type-I interferons paracrine signaling among ADAR1-deficient hepatocytes and other tissue cells, including infiltrating immune cells that leads to increased expression of the p150 ADAR1 protein. Similarly, AdarE861A/E861A mice expressing a mutated catalytically inactive ADAR1 and die at E13.5, display an interferon signature and upregulation of the p150 isoform.Citation33
Regarding the perturbed interferon response in Adar Hep-KO livers, our group pioneered the concept that non-site-specific A-to-I editing events are abundant in the transcriptome of mammalians, occurring chiefly in inverted repetitive elements that form long dsRNA structures. Subsequent studies determined that dsRNA editing of such repetitive elements mainly within 3′UTRs of mRNA transcripts reduce their potential to serve as ligands for the cytosolic sensor of long dsRNAs, MDA5. Signaling downstream of MDA5 leads to phosphorylation of TBK1 and IKKϵ and the induction of interferons and other pro-inflammatory cytokines. This scheme has been recently confirmed by the observation that genetic ablation of MDA5 or its downstream adaptor MAVS can rescue Adar−/− mice to birth.Citation13,33
The GSEA using IPA also revealed that the biological pathways and processes enriched in the Adar KO livers dataset included immune response of cells, quantity of IL6, and activation of hepatic stellate cells. This led us to hypothesize that the crosstalk among these functional pathways contributed to the induction of liver fibrosis. Using an in vitro experimental system of primary mouse hepatic stellate cells cultured with conditioned medium from ADAR1 KD and control HepG2 cells we discovered that the medium of ADAR1 KD cells had the capacity to activate stellate cell. Importantly, the GSEA of the GeneChip® array data sets from the HepG2-KD cells revealed a significant enrichment for NFκB stimulated gene with IL6, as predicted, ranking first. We next validated the notion that ADAR1 silencing induced the enhanced transcriptional activity of NFκB both by a customary luciferase reporter assay and by specific silencing of NFκB p65 (RelA) expression.
Subsequently, we determined that the levels of the IL6 cytokine were significantly higher (∼10-fold) in the conditioned medium from siADAR1 transfected HepG2 cells. By the observation that IL6 silencing prevented this paracrine activation of stellate cells, we further proved that this process was indeed dependent on NFκB-induced IL6 upregulation. Thus, we identified an interaction among ADAR1 KD-dependent NFκB activation in hepatocytes that signals across cell boundaries, mainly via IL6, to stellate cells to adapt a pro-fibrogenic phenotype. We hypothesize that this previously unknown pro-inflammatory paracrine communication, functions in vivo together with the infiltrating immune cells to mediate liver inflammation and ensuing fibrosis.
Our analysis of the molecular mechanisms underlying the effect of ADAR1 deficiency and activation of the NFκB cascade, revealed co-imuunoprecipitation of ADAR1 and IKKβ. Importantly, the latter kinase is a major component of the IKK complex that initiates the signaling cascade resulting in IκB degradation and enhanced NFκB p65 transcriptional activity.Citation29,34 Moreover, we could partly prevent IL6 overproduction in ADAR1 KD cells, by expressing editase deficient p110 and p150 ADAR1 isoforms in these cells. Evidently, overexpression of both p110 and p1150 ADAR1 under the control of the human cytomegalovirus (CMV) promoter in these latter experiments (Fig. S6) may induce supra-physiological accumulation of both isoform in the cytosol, and thus the data should be interpreted with caution. This finding implies previously unknown, RNA-editing independent, regulatory interactions between ADAR1 and the NFκB pathway, linked to the protein-protein interaction with IKKβ. We also identified a synergistic interaction between type-I interferons and NFκB cascade signaling, as IL6 secretion was augmented several-fold by IFNα treatment of ADAR1 HepG2-KD cells.
During the preparation of this manuscript, Wang et al.Citation35 reported their findings in a similar model of hepatocyte specific ADAR1 KO. In agreement with our observations, they also show that Adar deletion in hepatocyte caused significant liver injury and alterations in synthetic liver functions coupled with increased mortality and severe growth defects of mutant mice. Their histological evaluation of the livers of 7-week-old Adar Hep-KO mice was in general similar to our assessment, revealing increased inflammation and liver infiltration by CD45+ immune cells and particularly CD68+ macrophages. Regarding this latter observation, our detailed flow cytometric analysis of the isolated NPCs from 4-week old Adar Hep-KO mice, showed mainly enrichment for CD3+ CD8+ cytotoxic T cells and Ly6G+CD11b+ granulocytes but not macrophages. They also reported, as we also describe, that the inflammation induced a higher level of hepatocyte death, fatty change, and compensatory regeneration; this coinciding with increased IL6 levels and fibrosis as detected by Sirius Red staining. However, our work provides more insight regarding the critical biological functions of ADAR1 in hepatocytes. We show in a highly detailed manner by gene chip arrays and extensive bioinformatics analysis that in vivo ADAR1 deletion induces numerous ISGs that are linked to the activation of dsRNA sensing pathways plus upregulation of numerous pro-inflammatory immune response genes. Taken together the latter study and our novel findings imply a vital regulatory role for ADAR1 in liver homeostasis as depicted schematically in .
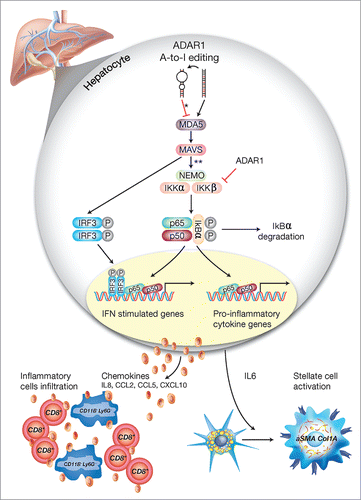
Figure 8. A schematic model of the regulatory role of ADAR1 in liver homeostasis. Under homeostatic conditions, ADAR1 catalyzes the promiscuous A-to-I editing of duplex-forming repetitive elements within mRNAs to reduce their potential to serve as ligands for the cytosolic dsRNA sensor, MDA5 (* refer to Citation33). In addition, as we now show, ADAR1 interacts with IKKβ to regulate the activity of the canonical NFκB pathway. Thus, lack of ADAR1 in hepatocyte induces aberrant signaling downstream of MDA5 leading to MAVS-dependent activation of IRF3 and NFκB transcriptional activity (** refer to. Citation13,16,18). Low ADAR1 levels are strongly associated with enhanced degradation of IκBα and the nuclear translocation of p65/p50 dimers to induce IL6 transcription. Moreover, the reciprocal activation/nuclear translocation of IRF3 promotes transcription of type-I interferons and multiple ISGs. This aberrant synergistic activation of the NFκB and type-I interferons pathways in hepatocytes, upon ADAR1 depletion, results in IL6-dependent paracrine activation of stellate cells to display a fibrogenic phenotype (Col1A+/aSMA+) alongside the recruitment of CD8+ cytotoxic lymphocytes and CD11B+ Ly6G+ granulocytes, further mediating multifocal liver damage and fibrosis.
In conclusion, we show that ADAR1 prevents dysregulation of the innate immune responses of hepatocytes. ADAR1 depletion in hepatocytes leads to activation of type I interferons and NFκB signaling cascades and more importantly to the generation of a positive feedback loop among these signal transduction pathways that results in paracrine activation of stellate cells and recruitment of cytotoxic T cells and other pro-inflammatory cells. We also determine that the regulation of the NFκB cascade and the transcription of IL6 by ADAR1 is mostly RNA editing independent.
Material and methods
Tissue culture
Human Hepatocelular carcinoma (HepG2) cells were cultured in Dulbecco's modified Eagle's medium (DMEM, cat#010551A(, supplemented with 10% FBS (cat#041211), 2 mM glutamine (cat#030201A), 100 mg ml−1 streptomycin and 100 units ml−1 penicillin (cat#030311B) (all reagents are from Biological Industries, Israel) at 37°C in a humidified incubator with 5% CO2.
Mouse model and genotyping
Mice with a loxP-flanked Adar1f7-9 (Adarf) allele in the germ line were previously described by Hartner et al.Citation24 Stuart H. Orkin (Pediatric Oncology, Dana-Farber Cancer Institute, Boston, MA) and Peter H. Seeburg (Max Planck Institute for Medical Research, Heidelberg, Germany) provided the Adarf/f mice backcrossed into the C57BL/6 background. Breeding of Adarf/f mice with Alb-Cre deleter mice Citation25 produced heterozygous ADAR1f/+;Alb-cre+ mice that were further crossed with ADAR1f/f mice to generate ADAR1f/f;Alb-Cre+ mutants and littermate controls. Genotyping PCR was performed on DNA extracted from mouse tail tips by D-Tail kit (Syntezza, cat#DE-11). The following primers were used for typing of ADAR1f/f, ADAR1f/wt, Cre+/− and Cre−/− alleles: ADAR1-1: 5′-CTGTTGGGCCAGTGG-TTCTAGTCC-3′ ADAR1-2: 5′-GCCACTTCTCCCTGACTCG-3′, ADAR1-3: 5′-GCGTACGTCGACGGTATC- 3′,CRE-1: 5′-CAGGGTGTTATAAGCAATCCC-3′, CRE-2: 5′- CCTGGAAAATGCTTCTGT CCG-3′. The mice were housed under pathogen-free conditions, and the Institutional Animal Care and Use Committee of the Weizmann Institute of Science (Rehovot, Israel) approved all animal experiments.
Blood tests
Blood was collected from 4-week old ADAR1 Hep-KO and littermate control mice and sera were analyzed for liver enzymatic profile (PathoVet Veterinary Pathology Services).
Liver histology and immunohistochemistry
For histological examination, ADAR1 Hep-KO and littermate control livers were fixed in 4% paraformaldehyde (BIO LAB, cat #06450323), embedded in paraffin, sectioned (5µm thick), and stained with hematoxylin and eosin as recommended by the manufacturer instructions (Sigma-Aldrich, cat #H9627, 230251). Selected samples were stained with Reticulin and Serius red for collagen fibrils. For immunohistochemical staining of Ki67 positive cells, deparaffinized liver sections were pre-incubated with 5% bovine serum albumin for 10 min, and then incubated with rabbit anti-mouse Ki67 antibody (1:100 dilution, Abcam, cat#ab15580) for 1 hour at room temperature in a wet chamber. After washing with phosphate buffer saline (PBS, Biological Industries, cat#020231A), the sections were incubated with an anti-rabbit secondary antibody (Vector, cat#MP-7401) for 20 min at room temperature, and detected with DAB (Vector, cat#SK-4105) and hematoxylin as the counter stain. Immunofluorescent detection of apoptotic cells was done with terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) Dead End Kit (Promega, #G3250) according to the manufacturer's instructions. Nuclei were stained with 1 ug/ml DAPI (Sigma-Aldrich, cat#D9542).
Hepatic non-parenchymal cells (NPCs) isolation
NPCs of ADAR1 Hep-KO and littermate controls were isolated according to published procedures.Citation36 Perfused livers of 4-week old mice were cut into small fragments and incubated (37°C, 250 rpm for 40 min) with 5 ml digestion buffer (5% FBS, 0.5 mg/ml collagenase VIII (Sigma-Aldrich, cat#C2139) and 0.1 mg/ml DNase I (Sigma-Aldrich, cat#D4263) in PBS+/+). This was followed by 3 cycles of washings with PBS−/− at 400 rpm from which the supernatant was taken, omitting the parenchymal cell pellet. The supernatant cell pellet was then lysed for erythrocytes by 2-min incubation with ACK buffer (Sigma-Aldrich, cat#R7757).
Flow cytometry
Single-cell suspensions of hepatic NPCs were immunostained with flourochrome-conjugated monoclonal antibodies (mAbs) against mouse B220 (cat#17045283), CD19 (cat#48019382), CD3 (cat#100206), CD4 (cat# 17004181), CD8 (cat#11008181), CD11b (cat#12011281), Ly6G (cat# 17593181), F4/80 (cat#454801) (all mAbs were from eBioscience). The cells were acquired and analyzed using a FACSAria instrument (BD Biosciences, San Jose, CA) and the FlowJo software (Tree Star Inc., Ashland, OR), respectively.
Hepatic stellate cells isolation
Livers were harvested from ICR mice (Envigo Israel cat#2icr07) anesthetized with Isoflorane (Piramal, cat# 66794-020-10). The livers were washed 3 times in GBSS buffer (Sigma-Aldrich, cat# G9779) and then dissected in 100 ml of the GBSS buffer supplemented with 6000 Units Collagenase (Worthington Biochemical Corporation, cat# LS004194), 450 Units Pronase (Roche, cat# 10 165 921 001), and 1 mM CaCl2 (Sigma-Aldrich, cat# 449709). Liver extracts were incubated for 15 min. in 37°C and then treated with 50 ml of GBSS buffer supplemented with 3000 units of collagenase, 225 units of Pronase, 1000 Units of DNAse (Sigma-Aldrich, cat# D-4263-5), and 0.5 mM CaCl2. After thirty min., the livers extracts were filtered using a mesh and centrifuged at 2000 RPM at room temperature for 7 min., and cell pellets were re-suspended in 365 mM HistodenZ (Sigma-Aldrich, cat# D-2158) in GBSS buffer and separated using Optiprep gradient. Finally, the purified stellate cells were washed twice with a DMEM 10% FCS medium.
Transfections
For single and co-transfections experiments, HepG2 cells were plated 24 hours before transfection at 2.5 × 105 cells density per well in a 6-well plates. The transfections were done with Lipofectamine 2000 reagent (Invitrogen, cat#11668027) according to the manufacture instruction. For global gene expression, cells were stabely transfected with pSuper vector containing shADAR1 or scrambled control shRNA (shControl). In all other experiments ADAR1 KD was achieved using Ambion Silencer Validated Custom ADAR1 (Ambion, cat#AM51333 ID:119581) and non-silencer control siRNAs (Ambion, cat#AM4635) alone or together with siIL6 (Santa Cruz Biotechnology, cat# sc-39627) or shP65 (and shGFP as control). ADAR1 p110 and p150 isoforms cDNA with either the normal or editing inactive mutant formCitation28 were cloned into pFLAG-CMV2 vector. All the ADAR1 expression vectors were further mutated to prevent targeting by the siADAR1 oligomers. The effectiveness of the mutagenesis was validated by WB analysis showing that treatment with siADAR1 does not reduce the plasmid driven expression of ADAR1 as compared to siControl treatment.
IFNα treatment
Twenty 4 hours post transfection with siADAR or siControl oligos, HepG2 cells were treated with IFNα (CytoLab, cat#3302A) 1000IU/ml for 24 hours and were harvested for further analyses.
Lucifrase assay
HepG2 cells were co-transfected with 120 nmol of ADAR1 or control siRNAs (and 1 ug DNA of luciferase reporter vectors: 0.9 ug/well of the NFkB luciferase reporter vector and 0.1 ug/well of Renilla luciferase reporter vector as internal control. The total DNA concentration was equalized in each well with pcDNA3.1 plasmid. Cells were harvested 48 hours after transfection, washed twice with cold PBS and lysed with passive lysis buffer (Promega, Madison, WI, USA). Luciferase levels were measured using a luminometer using Dual Luciferase Reportetr Assay System kit (Promega, cat#E1910). The luciferase activity was normalized with the internal control Renilla luciferase, and a mean value together with a standard error of triplicate samples was used to determine the reporter activity. pGL2-KB-Luciferase Citation37 and pRL-TK Renilla luciferase plasmid were used. As positive control for NFkB activation, cells were treated with 10ng/ml TNFa (Sigma-Aldrich, cat#T0157) for 1 hour.
Protein extraction and protein gel blotting
For total protein extraction, cells were washed with cold PBS and lysed with RIPA buffer (Sigma-Aldrich, cat#R0278) in the presence of protease inhibitor cocktail (complete; Roche, cat#11836145001) and phosphatase inhibitor cocktail 2 and 3 (Sigma-Aldrich, cat#P5726, P0044). Proteins were separated on a 10% SDS-PAGE gel, transferred to nitrocellulose membranes. The following primary Rabbit polyclonal antibodies were used: ADAR1 1:1,000 (Atlas Antibodies, cat#HPA003890), IkB 1:200 (Sigma-Aldrich, cat#sc-203), p65 1:1000(Santa-Cruz, cat#sc-372), GFP 1:1000 (Santa-Cruz, cat# sc-8334), Tubulin 1:50,000 (Santa-Cruz, cat#sc-5546), β Actin 1:20,000 (Santa-Cruz, cat#sc-130656) and Rabbit monoclonal anti IKKβ 1:1000 (Cell Signaling, cat#2678). Specific reactive bands were detected using Peroxidase conjugated goat anti rabbit (Jackson Immunoresearch Laboratories, cat#111035144) at a dilution of 1:10,000. Immunoreactive bands were visualized by the western blot chemiluminescence Reagent plus (Pharmacia Biotech).
RNA purification, reverse transcription and real-time quantitative PCR (qPCR)
Total RNA was extracted from HepG2 and hepatic stellate cells using TRI (Sigma-Aldrich, cat#T9424), followed by treatment with 1 U of RNase-free DNase (Roche). Reverse transcription reactions were performed on 2 ug total RNA using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Thermo Fisher Scientific Inc.), according to the manufacturer's instructions. qPCR for p150 and p110 ADAR1 isoforms, IL6, IL8, aSMA, Col1A and HPRT (as a reference control) were performed with the SYBR Green Real-Time PCR Kit (Applied Biosystems, cat#4309155) on an ABI Prism 7900 SDS Instrument (Applied Biosystems) as recommended by the manufacturer in reactions containing 50-100 ng DNA. All reactions were done in triplicates and relative gene expression values were determined using the 2−ΔΔCt method. The sequences of forward and reverse primers used in this study are specified in Table S2.
Microarray expression analysis
Gene expression was determined using the GeneChip® Gene 1.0 ST Array System for Human and Mouse (Affymetrix Inc., Santa Clara, CA), according to the manufacturer's instructions: http://www.affymetrix.com/support/technical/datasheets/gene_1_0_st_datasheet.pdf The 2100 Bioanalyzer (Agilent Technologies) was used to determine RNA quality, and biotinylated target DNA was prepared from each suitable RNA sample according to the manufacturer's instructions: https://www.affymetrix.com/support/downloads/manuals/wt_sensetarget_label_manual.pdf
Gene level RMA sketch algoritm (Affymetrix Expression Console and Partek Genomics Suite 6.2) was used for crude data generation. Genes were analyzed using unsupervised hierarchical cluster analysis (Spotfire DecisionSite for Functional Genomics; Somerville, MA) to get a first assessment of the data, and filtered according to fold change calculations. For further bioinformatics analysis, to discover interesting genes, molecular processes, upstream regulators or biological pathways enriched in the experimental dataset we used QIAGEN'S Ingenuity Pathway Analysis (IPA) software (http://www.ingenuity.com/products/ipa.)
To discover which transcription factor (TF) binding motifs were significantly enriched in promoter sequences (-200 to +50 from annotated transcription start site) of the upregulated co-expressed genes in HepG2 cells after ADAR1 silencing, we used the Pscan web tool (http://159.149.160.88/pscan/). As previously described,Citation26 the software identifies over- or under-represented motifs, describing the binding specificity of known TFs, which may be responsible for the observed expression pattern in the experimental data set.
Immunoprecipitation experiments
Co-Immunoprecipitation (co-IP) of ADAR1 p110/p150 isoforms with endogenously expressed IKKb was performed in HepG2 cells transfected with p110/p150-GFP. 1 × 107 Cells were seeded in 10 cm culture dishes and harvested in 1%NP-40 lysis buffer. Total cell lysates were incubated overnight with GFP-Trap beads (Chromotek) and the resulted complexes were washed, denatured and eluted according to the manufacturer's instruction. The co-IP of p110 and p150 ADAR1 with IKKβ was detected by protein gel blot analysis with anti GFP and IKKb specific antibodies. Reciprocal Co-IP was conducted to determine the interaction of IKKβ with ADAR1 isoforms: cells were harvested as described before, protein extracts were incubated overnight with 2.5 µg of anti-IKKb antibody in the presence of Protein A beads (Millipore), and resulting complexes were washed, denatured and eluted according to the manufacturer's instruction.
Elisa
IL6 concentration in HepG2 conditioned medium were determined 48 hours post transfection with siADAR1 and siControl, according to the manufacturer's instructions using Human IL6 ELISA kits (Invitrogen).
Statistical analysis
Statistical significance was determined by unpaired 2-tailed Student's t test. P values less than 0.05 were considered significant. Asterisks indicate the magnitude of the statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001. Error bars indicate standard error of the mean (SEM).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental_Data.pdf
Download PDF (985.5 KB)Acknowledgments
This work was supported in part by The Varda and Boaz Dotan Research Center in Hemato-oncology affiliated with the CBRC of Tel Aviv University (I.G. and G.R.). The Israeli Centers for Research Excellence I-CORE Program (ISF grants no. 41/11 and no. 1796/12 to G.R.). The Flight Attendant Medical Research Institute (G.R.). Israel Science Foundation (M.S. and G.R.). Israel Cancer Association (M.S., G.R., and Z.B.) and Israel association for the study of the liver (M.S., Z.B.).
The authors thank Ella Fudim and Yaniv Lerenthal for their technical assistance.
This work was performed in partial fulfillment of the requirements for the Ph.D. degree of S.O. and M.Sc. degree of P.K. from the Sackler School of Medicine, Tel Aviv University.
G.R. is the incumbent of the Djerassi Chair in Oncology at Tel Aviv University.
Related Research Data
References
- Li S, Mason CE. The pivotal regulatory landscape of RNA modifications. Ann Rev Genomics Hum Genet 2014; 15:127-50; PMID:24898039; http://dx.doi.org/10.1146/annurev-genom-090413-025405
- Bazak L, Haviv A, Barak M, Jacob-Hirsch J, Deng P, Zhang R, Isaacs FJ, Rechavi G, Li JB, Eisenberg E, et al. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res 2014; 24:365-76; PMID:24347612; http://dx.doi.org/10.1101/gr.164749.113
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012; 485:201-6; PMID:22575960; http://dx.doi.org/10.1038/nature11112
- Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol 2004; 22:1001-5; PMID:15258596; http://dx.doi.org/10.1038/nbt996
- Patterson JB, Samuel CE. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol Cell Biol 1995; 15:5376-88; PMID:7565688; http://dx.doi.org/10.1128/MCB.15.10.5376
- Mannion N, Arieti F, Gallo A, Keegan LP, O'Connell MA. New insights into the biological role of mammalian ADARs; the RNA editing proteins. Biomolecules 2015; 5:2338-62; PMID:26437436; http://dx.doi.org/10.3390/biom5042338
- Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem 2009; 79:321-49; PMID:NOT_FOUND; http://dx.doi.org/10.1146/annurev-biochem-060208-105251
- Amariglio N, Rechavi G. A-to-I RNA editing: a new regulatory mechanism of global gene expression. Blood Cells Mol Dis 2007; 39:151-5; PMID:17555993; http://dx.doi.org/10.1016/j.bcmd.2007.04.003
- Barak M, Levanon EY, Eisenberg E, Paz N, Rechavi G, Church GM, Mehr R. Evidence for large diversity in the human transcriptome created by Alu RNA editing. Nucleic Acids Res 2009; 37:6905-15; PMID:19740767; http://dx.doi.org/10.1093/nar/gkp729
- Eisenberg E, Adamsky K, Cohen L, Amariglio N, Hirshberg A, Rechavi G, Levanon EY. Identification of RNA editing sites in the SNP database. Nucleic Acids Res 2005; 33:4612-7; PMID:16100382; http://dx.doi.org/10.1093/nar/gki771
- Neeman Y, Levanon EY, Jantsch MF, Eisenberg E. RNA editing level in the mouse is determined by the genomic repeat repertoire. RNA 2006; 12:1802-9; PMID:16940548; http://dx.doi.org/10.1261/rna.165106
- Hartner JC, Walkley CR, Lu J, Orkin SH. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol 2009; 10:109-15; PMID:19060901; http://dx.doi.org/10.1038/ni.1680
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellaker C, Vesely C, Ponting CP, McLaughlin PJ, et al. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 2014; 9:1482-94; PMID:25456137; http://dx.doi.org/10.1016/j.celrep.2014.10.041
- Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet 2012; 44:1243-8; PMID:23001123; http://dx.doi.org/10.1038/ng.2414
- Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, Artuch R, Montalto SA, Bacino CA, Barroso B, et al. Clinical and molecular phenotype of aicardi-goutières syndrome. Am J Hum Genet 2007; 81:713-25; PMID:17846997; http://dx.doi.org/10.1086/521373
- O'Connell MA, Mannion NM, Keegan LP. The epitranscriptome and innate immunity. PLoS Genet 2015; 11:e1005687; PMID:26658668; http://dx.doi.org/10.1371/journal.pgen.1005687
- Ota H, Sakurai M, Gupta R, Valente L, Wulff BE, Ariyoshi K, Iizasa H, Davuluri RV, Nishikura K. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell 2013; 153:575-89; PMID:23622242; http://dx.doi.org/10.1016/j.cell.2013.03.024
- Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity 2015; 43:933-44; PMID:26588779; http://dx.doi.org/10.1016/j.immuni.2015.11.001
- Friedman SL. Mechanisms of Hepatic Fibrogenesis. Gastroenterology 2008; 134:1655-69; PMID:18471545; http://dx.doi.org/10.1053/j.gastro.2008.03.003
- Friedman SL. Molecular Regulation of Hepatic Fibrosis, an Integrated Cellular Response to Tissue Injury. J Biol Chem 2000; 275:2247-50; PMID:10644669; http://dx.doi.org/10.1074/jbc.275.4.2247
- Friedman SL. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 2008; 88:125-72; PMID:18195085; http://dx.doi.org/10.1152/physrev.00013.2007
- Benyon RC, Arthur MJP. Extracellular matrix degradation and the role of hepatic stellate cells. Semin Liver Dis 2001; 21:373-84; PMID:11586466; http://dx.doi.org/10.1055/s-2001-17552
- Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol 2011; 12:715-23; PMID:21772280; http://dx.doi.org/10.1038/ni.2060
- Hartner JC, Schmittwolf C, Kispert A, Müller AM, Higuchi M, Seeburg PH. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J Biol Chem 2004; 279:4894-902; PMID:14615479; http://dx.doi.org/10.1074/jbc.M311347200
- Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic β cell-specific gene knock-outs using Cre recombinase. J Biol Chem 1999; 274:305-15; PMID:9867845; http://dx.doi.org/10.1074/jbc.274.1.305
- Zambelli F, Pesole G, Pavesi G. Pscan: finding over-represented transcription factor binding site motifs in sequences from co-regulated or co-expressed genes. Nucleic Acids Res 2009; 37:W247-W52; PMID:19487240; http://dx.doi.org/10.1093/nar/gkp464
- Choi I, Kang HS, Yang Y, Pyun KH. IL-6 induces hepatic inflammation and collagen synthesis in vivo. Clin Exp Immunol 1994; 95:530-5; PMID:8137551; http://dx.doi.org/10.1111/j.1365-2249.1994.tb07031.x
- Nemlich Y, Greenberg E, Ortenberg R, Besser MJ, Barshack I, Jacob-Hirsch J, Jacoby E, Eyal E, Rivkin L, Prieto VG, et al. MicroRNA-mediated loss of ADAR1 in metastatic melanoma promotes tumor growth. J Clin Invest 2013; 123:2703-18; PMID:23728176; http://dx.doi.org/10.1172/JCI62980
- Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol 2004; 25:280-8; PMID:15145317; http://dx.doi.org/10.1016/j.it.2004.03.008
- Nieto N. Oxidative-stress and IL-6 mediate the fibrogenic effects of rodent Kupffer cells on stellate cells. Hepatology 2006; 44:1487-501; PMID:17133487; http://dx.doi.org/10.1002/hep.21427
- Thimme R, Wieland S, Steiger C, Ghrayeb J, Reimann KA, Purcell RH, Chisari FV. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J Virol 2003; 77:68-76; PMID:12477811; http://dx.doi.org/10.1128/JVI.77.1.68-76.2003
- Ichiki Y, Aoki CA, Bowlus CL, Shimoda S, Ishibashi H, Gershwin ME. T cell immunity in autoimmune hepatitis. Autoimmu Rev 2005; 4:315-21; PMID:15990080; http://dx.doi.org/10.1016/j.autrev.2005.01.005
- Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 2015; 349:1115-20; PMID:26275108; http://dx.doi.org/10.1126/science.aac7049
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol 2000; 18:621-63; PMID:10837071; http://dx.doi.org/10.1146/annurev.immunol.18.1.621
- Wang G, Wang H, Singh S, Zhou P, Yang S, Wang Y, Zhu Z, Zhang J, Chen A, Billiar T, et al. ADAR1 prevents liver injury from inflammation and suppresses interferon production in hepatocytes. Am J Pathol 2015; 185:3224-37; PMID:26453800; http://dx.doi.org/10.1016/j.ajpath.2015.08.002
- Zigmond E, Samia-Grinberg S, Pasmanik-Chor M, Brazowski E, Shibolet O, Halpern Z, Varol C. Infiltrating monocyte-derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J Immunol 2014; 193:344-53; PMID:24890723; http://dx.doi.org/10.4049/jimmunol.1400574
- DiDonato JA, Mercurio F, Karin M. Phosphorylation of I kappa B alpha precedes but is not sufficient for its dissociation from NF-kappa B. Mol Cell Biol 1995; 15:1302-11; PMID:7862124; http://dx.doi.org/10.1128/MCB.15.3.1302