
ABSTRACT
Cells secrete extracellular RNA (exRNA) to their surrounding environment and exRNA has been found in many body fluids such as blood, breast milk and cerebrospinal fluid. However, there are conflicting results regarding the nature of exRNA. Here, we have separated 2 distinct exRNA profiles released by mast cells, here termed high-density (HD) and low-density (LD) exRNA. The exRNA in both fractions was characterized by microarray and next-generation sequencing. Both exRNA fractions contained mRNA and miRNA, and the mRNAs in the LD exRNA correlated closely with the cellular mRNA, whereas the HD mRNA did not. Furthermore, the HD exRNA was enriched in lincRNA, antisense RNA, vault RNA, snoRNA, and snRNA with little or no evidence of full-length 18S and 28S rRNA. The LD exRNA was enriched in mitochondrial rRNA, mitochondrial tRNA, tRNA, piRNA, Y RNA, and full-length 18S and 28S rRNA. The proteomes of the HD and LD exRNA-containing fractions were determined with LC-MS/MS and analyzed with Gene Ontology term finder, which showed that both proteomes were associated with the term extracellular vesicles and electron microscopy suggests that at least a part of the exRNA is associated with exosome-like extracellular vesicles. Additionally, the proteins in the HD fractions tended to be associated with the nucleus and ribosomes, whereas the LD fraction proteome tended to be associated with the mitochondrion.
We show that the 2 exRNA signatures released by a single cell type can be separated by floatation on a density gradient. These results show that cells can release multiple types of exRNA with substantial differences in RNA species content. This is important for any future studies determining the nature and function of exRNA released from different cells under different conditions.
Introduction
Extracellular nucleic acids (cell-free nucleic acids) were discovered in the late 1960s/early 1970s in serum/plasma and synovial fluid.Citation1-3 Since then, their potential use as biomarkers for several different pathological conditions has been investigated, including diseases such as cancer, cardiovascular disease, pre-eclampsia, and autoimmune disease.Citation4-7 Extracellular circulating DNA (cell-free DNA; cfDNA) is considered to be a result of apoptosis of lymphocytes and blood cells in healthy individuals, but it can also originate from tumors in cancer patients.Citation8 The cfDNA is usually unprotected and can be degraded quickly by circulating DNases. Extracellular RNA (exRNA) is not intrinsically resistant to endogenous RNase activity,Citation9,10 and it can be selectively released by viable cells as cargo inside extracellular vesicles (EVs) or within protective high density lipoproteins (HDLs) or protein complexes.Citation9,11,12 When protected by a lipid bilayer or a protein, the exRNA can evade degradation by circulating RNases.
EVs (including apoptotic bodies, microvesicles, and exosomes) are released by most human cells and can shuttle molecules such as proteins, RNA, and lipids from one cell to another, thereby influencing the recipient cell phenotype. This field is currently growing very rapidly, and it has vast implications for understanding cell-to-cell communication in different diseases, including cancer.Citation13-17 Also, EVs and EV cargo have the potential to be used as biomarkers in disease and as gene vectors for therapeutic nucleotide delivery.Citation18-20 EVs have been shown to contain mRNA and miRNA as well as several non-coding RNA (ncRNA) classes such as small nucleolar RNAs (snoRNA), piwi-interacting RNA (piRNA), vault RNA, Y RNA, and tRNA.Citation21 Subpopulations of EVs have been shown to contain different miRNA signaturesCitation22 and breast cancer cells release miRNA both non-selectively in large vesicles and selectively in smaller vesicles.Citation23
The non-vesicle-associated exRNA is associated with and protected by protein complexes, including argonaute 2 and nucleophosmin 1Citation9,10,24 or HDLs.Citation11,25 Until now, protein complexes and HDL have only been demonstrated to contain miRNACitation9-11,24 but not mRNACitation10,11 or other ncRNA classes. Because HDLs have an average size of 8–12 nm, and are therefore considerably smaller than EVs that are 50–5000 nm in diameter, it is not surprising that the diversity of their RNA cargo is smaller.
However, conflicting results regarding the nature of exRNA has been suggested. In the current study, we hypothesized that cells release different exRNA signatures, one consisting of full-length rRNA that can be seen as distinct peaks on an electropherogram and one that does not consist of full-length rRNA, and therefore do not show rRNA peaks. We therefore separated 2 distinct exRNA profiles from the human mast cell line HMC-1 by their buoyant density. Briefly, we first discard microvesicles and apoptotic bodies and then used centrifugation and density gradient isolation procedures to separate the 2 exRNA profiles. The exRNA was determined with both microarray (miRNA and mRNA) and next-generation sequencing (NGS; all types of coding and ncRNA classes). The exRNA-associated proteins were determined with LC-MS/MS, Western blot, and flow cytometry.
Results
Detection of 2 distinct extracellular RNA profiles
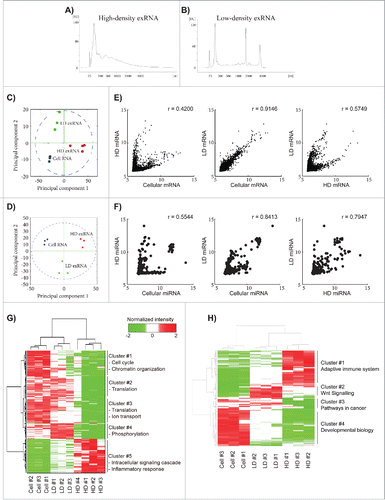
Cells, apoptotic bodies, and cellular debris were removed from the cell culture media by 300 × g and 16,500 × g centrifugations. Both filtered (0.2 µm) and non-filtered supernatants were then ultracentrifuged at 120,000 × g. Pellets collected after the 120,000 × g ultracentrifugation were further separated on a density gradient (schematics are shown in Fig. S1A). Ten fractions were collected and washed from each sucrose gradient before their RNA profile was analyzed with a Bioanalyzer. Based on the RNA profiles of the different fractions, it could be concluded that 2 distinct exRNA signatures were present in the cell supernatant, one containing peaks for full-length rRNA and the other one not containing such peaks (Fig. S1B). The first exRNA profile was collected from fractions 8–10 (density 1.24–1.31 g/cm3), and will hereafter be called high-density (HD) exRNA. The second exRNA profile was collected from fractions 2–6 (density 1.09–1.21 g/cm3) and will hereafter be called low-density (LD) exRNA. The RNAs detected in the HD fractions showed a relatively broad peak for short RNAs (25–500 nucleotides) and no prominent rRNA peaks (), whereas the RNA detected in the LD fractions had a significantly narrower peak for the short RNAs (50–150 nucleotides) and showed distinct 18S and 28S rRNA peaks (). These results demonstrate that the exRNA must be associated with other molecules/structures because free RNA has a density of 1.6–1.9 g/cm3 Citation26 and the exRNA without the 18S and 28S rRNA peaks from the HD fractions are associated with structures that have a higher density than the structures associated with the rRNA-positive exRNA in the LD fractions. Therefore, a density gradient can be used to separate and purify these different exRNA-associated structures.
Figure 1. Microarray analysis of the RNA content in the high- and low-density fractions. Isolated samples were allowed to float into a sucrose gradient (0.4–2.5 M). High-density extracellular RNA (HD exRNA) was isolated from fractions 8–10 of the filtered sample, while low-density extracellular RNA (LD exRNA) was isolated from fractions 2–6 of the un-filtered sample. (A and B) RNA was extracted from the HD and LD fractions, and the RNA profiles were analyzed with a Bioanalyzer instrument. (C and D) HD and LD exRNA was analyzed for its mRNA and miRNA content with 3D Gene Microarray (Toray). Principal component analysis (PCA) was performed on the mRNA (C) and miRNA (D) content. PCA was used to visualize the variance in RNA among samples from cells (blue), HD exRNA (red), and LD exRNA (green). (E and F) Scatter plots were constructed to determine the relationship between mRNA (E) and miRNA (F) in the HD and LD exRNA with the cellular RNA. The correlation was determined with the Pearson's test (r = Pearson's correlation). (G and H) Hierarchical clustering of the identified mRNA (G) and miRNA (H) in cells, HD fractions, and LD fractions (normalized expression intensity is in log2 scale).
Validation of these findings was performed by loading the isolated pellets on the top of the sucrose gradient, which resulted in a similar distribution of RNA profiles as the bottom-loaded gradients, showing that the exRNA-associated structures reach their equilibrium buoyant density after the performed centrifugation (Fig. S2A). The distribution of exRNA with or without full-length rRNA peaks was also confirmed in an erythropoietic cell line, TF1 (Fig. S2B), showing that these 2 distinct exRNA profiles are not exclusive for mast cells.
The exRNA in the high- and low-density fractions have different miRNA and mRNA contents as determined by microarray
Because the exRNA profiles for the HD and LD fractions were substantially different (), both the miRNA and mRNA contents of the HD and LD fractions were determined by 3D-Gene™ microarray technology (Toray Industries, Inc.) in multiple biological replicates of both fractions. The miRNA and mRNA contents were also determined in the exRNA-producing cells. Principal component analysis (PCA) showed that the biological replicates of RNA samples from the cells and the HD and LD fractions formed distinct and separate clusters for both miRNA and mRNA (). The reproducibility was further supported by a positive correlation of RNA expression among the biological replicates (Fig. S3A and B).
A total of 3311, 4985, and 9836 mRNAs were identified in all HD, all LD, and all cellular samples, respectively (Tables S1–3). The mRNAs in the HD fractions showed relatively low correlation with the cellular mRNA, while LD mRNA showed a higher correlation with the cellular RNA (; Pearson's correlation r = 0.42 and r = 0.91, respectively). The most abundant mRNAs in the cells and the LD fractions were transcripts for several 40S and 60S ribosomal proteins, glyceraldehyde-3-phosphate dehydrogenase, α-enolase, and ubiquitin, while the most abundant transcripts in the HD fractions were those for polypeptide N-acetylgalactosaminyltransferase-like protein 2 and FK506-binding protein 14 (Table S1–3). Furthermore, the relationship between the mRNA in the HD and LD fractions was relatively weak (; Pearson's correlation r = 0.57), which suggests that the loading and release of mRNAs is different for the structures found in the HD and LD fractions.
A total of 401, 369, and 490 miRNAs were reproducibly identified in all HD, all LD, and all cellular samples, respectively (Tables S4–6). In similarity with the mRNA analysis the miRNAs in the HD fractions showed relatively low correlation with the cellular miRNA, while LD miRNA showed a higher correlation with the cellular RNA (; Pearson's correlation r = 0.55 and r = 0.84, respectively). Even though the miRNAs in the HD and LD samples correlated to some degree (Pearson's correlation r = 0.79), PCA showed that component 1 separates the cells from the exRNA fractions, while component 2 separates the HD fractions from the LD fractions ().
We could observe 4 clusters (clusters 1–4) of mRNAs that were more abundant in both the cells and the LD fractions, and we found one cluster (cluster 5) that was more abundant in the HD fractions (). Cluster analysis of the miRNAs showed 4 clusters, one that contained miRNA that was abundant in both cells and LD fractions (cluster 3) and 3 clusters that were uniquely up-regulated in the HD fractions (cluster 1), in the LD fractions (cluster 2), or in cells (cluster 4; ). This microarray analysis suggests that the HD and LD fractions contain different structures that harbour fundamentally different RNA cargo.
The exRNA in the high- and low-density fractions have different non-coding RNA content as determined by NGS
In addition to the microarray, NGS was used to get a better understanding of the RNAs that are not targeted by the probes used in the arrays described above, including several ncRNA species. To obtain high coverage, both a long (Nugen Ovation V2) and a short (TruSeq small RNA by Illumina) RNA library was constructed and sequenced for 2 replicates of RNA from the HD and LD fractions as well as the RNA-producing cells. PCA showed good reproducibility among the biological replicates (Fig. S4A–B), and there were positive correlations in the RNA expression between the biological replicates (Fig. S4C–D). In the long RNA library, 0.8 × 106 HD reads and approximately 10 × 106 LD and cellular reads were mapped to the human genome after normalization. Coding reads were more abundant percentage-wise in the exRNA in the HD fraction than in the LD exRNA ( and Table S7; 75% in the HD exRNA compared to 20% in the LD exRNA). Furthermore, the mRNA in LD fractions had a higher correlation with the cellular mRNA than the HD mRNA did (; blue box, Pearson's correlation r = 0.92 and r = 0.60, respectively), while the mRNA in the HD and LD fractions had relatively low correlation in the NGS analysis (; green box, Pearson's correlation r = 0.62). A total of 13,567, 8971, and 11,353 mRNAs with a minimum of 10 reads were identified in both HD samples, both LD samples, or both cellular samples, respectively (Tables S1–3). The non-coding reads were percentage-wise more abundant in the exRNA from the LD fractions than in the cells and the HD fractions ( and Table S7; 80% compared to 55% and 25%, respectively). When the ncRNAs were analyzed in detailed, most of the ncRNA in the LD fraction and the cells consisted of mitochondrial rRNA, while long intergenic non-coding RNA (lincRNA) and antisense RNA (, and Table S8) were the most abundant in the HD exRNA. The lincRNA “nuclear paraspeckle assembly transcript 1” (NEAT1) and “metastasis associated lung adenocarcinoma transcript 1” (MALAT1) were present in large amounts in all samples, while “long intergenic non-protein coding RNA 1206” and “long intergenic non-protein coding RNA 910” were enriched in the HD fraction. The antisense RNA “ZNFX1 antisense RNA 1” was highly expressed only in the LD exRNA and cells, while “KCNQ1 opposite strand/antisense transcript 1” was highly expressed in the HD exRNA and cells.
Figure 2. Next-generation sequencing analysis of the RNA content in the HD and LD fractions – long library. RNA was extracted from 2 biological replicates for the HD and LD fractions as well as the exRNA producing cells, and the RNA was used to construct long RNA libraries (Nugen Ovation V2) that was sequenced. (A) The graph shows the percentage of reads for each RNA biotype. (B) The correlation for the detected mRNA was determined with Pearson's test. The correlation between the biological replicates is indicated with red boxes, the correlation between the LD fractions or the HD fractions with the cellular mRNA is indicted with a blue box, and the correlation between the HD and LD fractions is indicated with a green box.
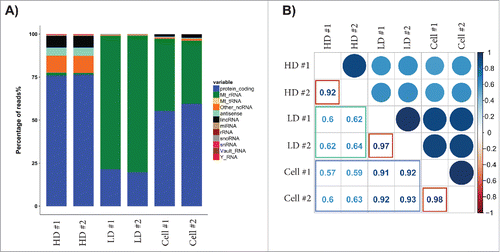
Table 1. Summary of the characteristics of the HD and LD fractions.
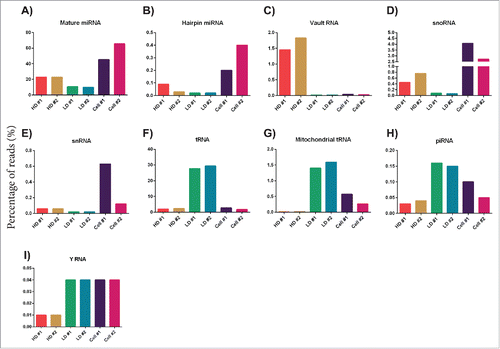
Because the long RNA sample preparation creates libraries with inserts longer than 200 nt, the short ncRNA classes would not be sequenced using this method so a separate short library was also constructed. In the short RNA library, 0.3 × 106, 0.35 × 106, and 0.55 × 106 reads were mapped to the human genome after normalization for the HD, LD, and cellular RNA, respectively. The HD fraction contained mostly mature miRNA (23%), while the LD fraction contained mainly tRNA (28%) and mature miRNA (10%). Importantly, these profiles were both different from the cells that produced these exRNAs. The cells primarily contained mature miRNA (∼55%) and snoRNA (∼3.5%) (Table 9). The exRNA in the HD fractions had more reads mapped to mature miRNA, hairpin miRNA, vault RNA, snoRNA, and small nuclear RNA (snRNA) (, Table S9, Table S10) in relation to the LD exRNA, while tRNA, mitochondrial tRNA, piRNA, and Y RNA were most abundant in the LD exRNA (, Table S9, Table S10). A total of 199, 147, and 246 miRNAs were reproducibly identified with at least 10 reads in both replicates of the HD, LD, or the cellular samples, respectively (Tables S4–6).
Figure 3. Next-generation sequencing analysis of the RNA content in the HD and LD fractions – short library. RNA was extracted from 2 biological replicates for the HD and LD fractions as well as the exRNA producing cells, and the RNA was used to construct short RNA libraries (TruSeq small RNA by Illumina) that was sequenced. The graphs show the percentage of reads for each RNA biotype in each sample – (A) mature miRNA, (B) hairpin miRNA, (C) vault RNA, (D) snoRNA, (E) snRNA, (F) tRNA, (G) mitochondrial tRNA, (H) piRNA, and (I) YRNA.
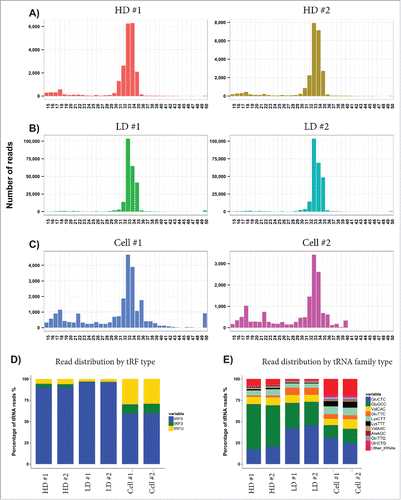
The average lengths of the tRNA reads in both the HD and LD exRNA were approximately 32 nucleotides () indicating that the tRNAs are fragmented. tRNA fragments of this size were also detected in the cells, but the cells also had tRNA fragments with average lengths of approximately 17 nucleotides (). Interestingly, 96% of the tRNA fragments present in the LD exRNA were assigned to the 5′ region (). Although cells contained fewer tRNA reads than the LD exRNA, cells had more tRNA fragments assigned to the 3′ region (). The LD fractions and cells mainly contained GluCTC tRNA, while the HD fractions primarily contained GlyGCC tRNA. The cells showed a larger variation of tRNA; while the HD and LD fractions contained 80% GluCTC, GlyGCC, and ValCAC tRNAs, these 3 only made up 50% of the tRNAs in cells ().
Figure 4. Analysis of tRNA fragments. (A–C) The graphs show the lengths of the tRNA reads identified in each sample, demonstrating an average length of ∼33 nucleotides long fragments. (D) Each tRNA fragment (tRF) was assigned to the 5′ region (blue), the 3′ region (green), or as unknown (yellow). (E) The graphs show the percentage of reads distributed in tRNA family type in all samples.
Characteristics of the structures associated with the high- and low-density exRNA
The RNA, protein, and particle concentrations were measured for the HD and LD samples to calculate the amount of RNA and protein per particle. As shown in , the RNA concentration was significantly higher per particle in the HD fraction than in the LD fraction (p < 0.01), whereas no significant difference was observed in protein concentration.
Figure 5. Characterization of the exRNA-associated structures. (A and B) The number of particles was determined with nanoparticle tracking analysis (NTA) and used to calculate the amount of RNA and protein per particle in the HD and LD fractions. Unpaired t-test; ** p-value < 0.01. ns = non-significant. (C) The proteomes of both the HD and the LD fractions were analyzed with LC-MS/MS. In total, 1436 proteins were identified in the HD fractions and 2408 proteins in the LD fractions. Of these, 388 and 417, respectively, were categorized as “RNA-binding proteins” with Gene Ontology (GO). (D) The presence of the commonly used exosomal markers TSG101, CD81, Annexin-2, and Flottilin-1 were determined with Western blot. (E) The presence of the tetraspanins CD9, CD63, and CD81 was determined by flow cytometry with anti-CD63-coated beads. (F) The total proteins identified with LC-MS/MS in the HD fractions (1436 proteins) and in the LD fractions (2408 proteins) were analyzed with GO Term Finder to identify enriched cellular components compared to the genome frequency.
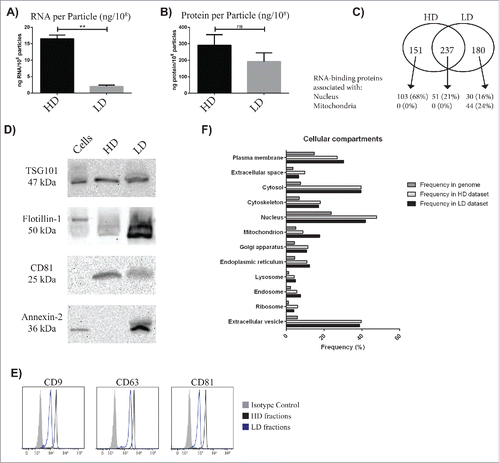
Furthermore, we analyzed the HD and LD fractions with LC-MS/MS to investigate the proteins associated with the exRNA. For the HD and LD fractions, 1436 and 2408 proteins were identified, respectively (Table 11). The proteomes of both HD and LD fractions were analyzed with Gene Ontology (GO) Term Finder to identify the associated functions that were most enriched in relation to the genome frequency. “Protein binding,” “poly(A) RNA binding,” and “RNA binding” were the top associated functions for the proteomes of both the HD and LD fractions. Because the RNA contents of the HD and LD fractions were substantially different, the RNA-binding proteins are of special interest with 27% and 17% of the total proteins being RNA-binding proteins in the HD and LD fractions, respectively. Interestingly, 24% of the uniquely identified RNA-binding proteins in the LD fractions were associated with mitochondria, while none of the RNA-binding proteins uniquely identified in the HD fractions or the RNA-binding proteins identified in both HD and LD fractions were associated with mitochondria. On the contrary, 68% of the RNA-binding proteins were associated with the nucleus in the HD fractions compared with only 16% in the LD fractions ().
Additionally, the proteomes of both fractions were analyzed to identify the associated cellular components. “Extracellular vesicle,” “cytosol,” and “extracellular region” were among the functions with the highest association for the proteomes of both the HD and LD fractions. Because exRNAs are commonly associated with EVs such as exosomes, the association with “extracellular vesicle” was particularly interesting. Thus the HD and LD fractions were analyzed with Western blot and flow cytometry for the presence of proteins that can be considered to be enriched in exosomes and are commonly termed “exosomal markers.”Citation27 Western blot showed that TSG101 and CD81 were detected in both HD and LD fractions, while Annexin 2 and Flotillin 1 were mainly found only in the LD fraction (). Particles positive for 3 transmembrane proteins – CD9, CD63 and CD81 – were also identified in both the HD and LD fractions by flow cytometry and anti-CD63 beads ().
Furthermore, 13 relevant Gene Ontology cellular component terms were specifically chosen for comparison. For most of the cellular components, the frequency was similar for the proteins in the HD and LD fractions, with the exception of “extracellular space,” “nucleus,” and “ribosome” that had a higher frequency in the proteome of the HD fractions compared to the LD proteome and “plasma membrane,” “mitochondrion,” and “endosome” that had a stronger association with the proteome of the LD fractions compared to the HD proteome ().
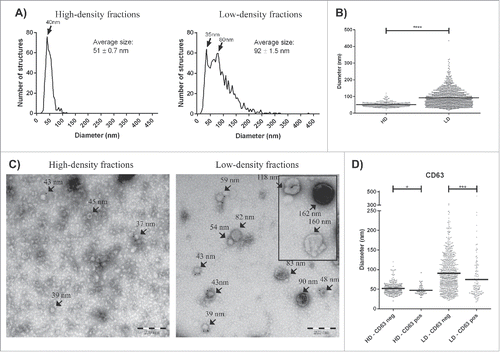
Electron microscopy was used to further analyze the structures associated with HD and LD exRNA, and this revealed the presence of vesicle-like structures in the LD fraction with an average diameter of 92 nm, whereas the vesicle-like structures in the HD fraction had an average diameter of 51 nm (, p < 0.0001). Immunogold staining could detect CD63 positivity in both the LD and HD fractions, but in both isolates the CD63+ structures were significantly smaller than the CD63− structures (; p < 0.05 and p < 0.001 for HD and LD, respectively).
Figure 6. Electron microscopy analysis of exRNA-associated structures. Samples (10 µg protein) were loaded onto grids, labeled with anti-CD63, and examined by electron microscopy. (A) All structures in approximately 100 micrographs per sample type were measured and are presented as graphs with the number of structures on the y-axis and the diameter of the structures in nm on the x-axis. In total, the diameters of 1566 structures were measured for this analysis (425 and 1141 for the HD and LD fractions, respectively). (B) The structures in the LD fractions were significantly larger (92 ± 1.5 nm) than the HD structures (51 ± 0.7 nm). (C) Representative electron micrographs are shown for each separated fraction. (D) In both the HD and LD fractions, the CD63+ structures were smaller than the CD63− structures. Unpaired t-test; * p < 0.05, *** p < 0.001, **** p < 0.0001.
Discussion
This study demonstrates that a single cell type can release several exRNA populations, as determined by both NGS and microarray techniques. These exRNA signatures are present in HD and LD extracellular fractions, which can be separated by flotation on a density gradient. Further, the RNA signatures are associated with different proteins, as determined by mass spectrometry. Some of the RNA in both fractions might be carried as cargo in EVs because electron microscopy reveals the presence of round CD63+ structures in both fractions ( summarizes the characteristics of the HD and LD exRNA).
The clear lack of correlation between both the long and short RNA in the HD and LD fractions as analyzed with both microarray and NGS (, and Fig. S4C–D) strongly argues that the exRNAs in the 2 fractions are associated with distinct pathways. This is further supported by the PCA, which showed that both mRNA and miRNA cluster differently in whole cells and in the HD vs. LD fractions ( and Fig. S4A and B). For mRNA, there is a low correlation between the cells and the HD exRNA, but a good correlation between the cells and the LD exRNA, in the microarray analysis (). This conclusion is supported by similar data acquired by NGS (). Our current data might indeed explain several observations made in the literature regarding exRNA. Several studies have demonstrated a limited relationship between cellular and extracellular mRNA when analyzing exRNA in EVsCitation20,28,29, while other studies have suggested a closer relationship between cellular and extracellular mRNA in vesicles.Citation30,31 Similarly, several studies have observed a lack of peaks for the full-length 18S and 28S rRNA subunits when analyzing exRNA in EVs.Citation20,28,32-36 In contrast, other studies have identified peaks for 18S and 28S rRNA in EV-associated exRNA.Citation30,33,37 The differences in these previous studies might be explained in part by cell or tissue-specific differences, different isolation methods, and/or the use of different RNA analysis platforms. However, our current results strongly suggest that different exRNAs are present in the extracellular space () and they may be enriched by different isolation methods, which can affect the results and can thus explain the previously contradictory findings.Citation20,28-31 It is clear that exRNA represents several entities, but it is also possible that the HD and LD fractions themselves are mixtures of multiple exRNA-containing structures that come together in the same density fractions. Special steps are clearly required to fully separate distinct exRNA profiles, which is important to understanding the differences in their biology.
The exRNA in the HD fractions consisted mainly of mRNA, miRNA, lincRNA, antisense RNA, vault RNA, snoRNA, and snRNA, while the LD exRNA consisted mainly of mRNA, miRNA, mitochondrial rRNA, mitochondrial tRNA, tRNA, piRNA, and Y RNA (). Y RNA is a family of 84–113 nt hairpin-containing ncRNAs that are involved in Ro60 inhibition and DNA replication initiation.Citation38 Y RNA has been suggested to be associated with EVs,Citation21 however there are also reports indicating that Y RNA does not circulate in vesicles but in protein complexes with a mass of 100 or 300 kDa.Citation39 One study demonstrated that Y RNA is enriched in EVs compared to the EV-producing cells.Citation40 However, we observed no enrichment in Y RNA in the HD and LD fractions compared to cellular RNA. It has also previously been shown that when dendritic cells and T cells are co-cultured the extracellular Y RNA released is mainly associated with the LD fractions and not the HD fractions,Citation21 which is in line with our results in which more Y RNA was associated with the particles in the LD fractions than the HD fractions ().
Vaults are large ribonucleoprotein particles located in the cytoplasm that consist of one major vault protein (MVP), 2 minor vault proteins (VPARP and TEP1), and 80–150 nt vault RNAs.Citation41 We report here that the vault RNA was exclusively detected in the HD fractions (). Furthermore, all 3 vault proteins (MVP, VPARP, and TEP1) were exclusively detected in the HD fractions in the mass spectrometry analysis (Table S11). Vaults are visible in electron microscopy with a characteristic form,Citation42 however we saw no evidence of whole complexes in the negative stain that we performed (). Vault RNA has previously been detected in EVsCitation21,43 and at the time was hypothesized to play a role in the sorting of RNAs into the vesicles.Citation21 It can only be speculated upon as to whether the vault RNA detected here plays any part in sorting RNA into the HD structures, but it is worth noting that the HD exRNA correlated to a lesser degree with the cellular RNA compared with the LD exRNA ( and ).
tRNAs are 76–90 nt ncRNAs that take part in translation by transporting amino acids to the protein synthesis machinery. It has recently been suggested that tRNAs can also be processed into fragments that have modulatory effects.Citation44 We observed that cellular RNA contained fragments of 2 sizes, ∼17 and ∼33 nt, while the exRNA in the HD and LD fractions only contained the 33 nt fragments (). We found that both LD and HD exRNA, but in particular LD exRNA, almost exclusively contained tRNA fragments originating from the 5′ end (). The cellular RNA also contained more 5′ end tRNA, however it also contained 3′ end tRNA fragments. This is in agreement with previous reports in which the tRNA fragments detected extracellularly in EVs from human semen and immune cells are also primarily derived from the 5′ end.Citation21,45,46 We observed that HD and LD exRNA mainly contained GlyGCC and GluCTC tRNA, while the cells had a larger variation in tRNAs (), which confirmed previous findings of extracellular tRNA in EVs.Citation45
tRNA fragments can arise through 2 main processes. The first is stressed-induced tRNA cleavageCitation47, which is angiogenin-dependent and leads to cleavage of the tRNAs at the anti-codon loop. The second is the inability of the reverse transcriptase to process cDNA of the entire tRNA due to the presence of post-transcriptional modifications (such as methylation) that hinder efficient sequencing by NGS. The tRNA fragments for both the cells and exRNA were mostly 32–34 bases long and terminated at the anticodon loop of the tRNA. Furthermore, the 2 most highly abundant tRNA fragment species, GluCTC and GlyGCC, that make up close to 70% of the tRNA fragments seen in the exRNA and ∼45% of the tRNA fragments seen in the cells have no methylation modification at the anticodon loop. This suggests that the tRNA fragments that were identified here are due to cleavage rather than incomplete sequencing due to post-transcriptional tRNA modifications.
snRNA is a class of approximately 150 nt RNA molecules, commonly called U-RNA. Together with several specific proteins, snRNA forms complexes called small nuclear ribonucleoproteins (snRNPs) that participate in the splicing of pre-mRNA. snoRNAs are a class of small RNA molecules that primarily guide chemical modifications (methylation and pseudouridylation) of other RNAs, such as rRNAs and tRNAs. Both snRNAs and snoRNAs were enriched in the exRNA in the HD fractions and the cells compared to the LD exRNA (). Both of these RNA classes are located in the nucleus, and, interestingly, the HD proteome was more strongly associated with the nucleus than the LD proteome (). On the contrary, both the RNA and the proteins in the LD fractions were highly associated with the mitochondria compared to the HD fractions (). Because some NGS studies on exRNA in EVs have focused primarily on the miRNACitation48 or only on RNA shorter than 70–100 nt,Citation21,46 they might have missed the mitochondrial RNA that we have detected here (). There are studies that have identified mitochondrial RNA in exRNA in EVs,Citation22,40 and these showed that the mitochondrial RNA was enriched in EV populations that contained rRNA peaks, but not in the EV subpopulations that did not contain rRNA peaks.Citation22 This indicates an association with the mitochondria for the rRNA-containing structures in the LD fractions. In addition to the association with the nucleus, the proteome of the HD fraction was also associated with the ribosome. This was unexpected given that the exRNA profile in the HD fraction did not show any indication of full-length 18S and 28S rRNA peaks but still seems to have a proteome associated with ribosomes. Because both the RNA and protein associated with the structures identified in the HD and LD fractions are dissimilar, it is unlikely that they have evolved from a common pathway and by chance ended up having slightly different densities and/or cargos. This suggests that the HD and LD exRNA are derived from different biogenesis pathways. Overall, based on the proteomics it seems plausible that the HD exRNA production, at least to some degree, is associated with the nucleus, whereas the LD exRNA might be more associated with the cell membrane and mitochondria.
We demonstrate by mass spectrometry and Western blot that both the HD and LD exRNA profiles are associated with structures that are positive for several commonly used exosomal markers. This is intriguing because both the RNA and protein content are substantially different in the HD and LD fractions, which suggests that the HD and LD exRNA are derived from different biogenesis pathways. Although flow cytometry demonstrates the presence of tetraspanins, which could suggestion the presence of a lipid bilayer, the experimental approach applied here cannot distinguish the exact molecular and/or vesicular structures with which the exRNA is associated. Electron microscopy suggests that at least a part of the exRNA could be associated with extracellular vesicle-like structures ().
It is possible that exRNAs and their associated extracellular vesicle-like structures, when released by other cells or under other conditions, may behave differently during the ultracentrifugation procedure and end up in different pellets compared to our study. Importantly, if a swinging bucket rotor is used all material will be pelleted at the bottom as one pellet. However, we have loaded both pellets on the same density gradient and the HD and LD exRNA was successfully separated then as well (data not shown). We therefore suggest that one density gradient can separate these 2 exRNA entities also when they have been co-isolated.
To further elucidate the biogenesis and function of subpopulations of exRNA released by a single type of cell, it is crucial to describe their molecular contents and to develop methodologies to separate them efficiently. The fact that several proteins that are associated with the HD and LD exRNA are differentially expressed suggests that the HD and LD exRNA are derived from different biogenesis pathways, which might also provide clues to developing exRNA subtype-specific markers. This study therefore describes a fundamental advance in the study of exRNA. However, our study also introduces another level of complexity to the field, and any study of exRNA must now consider the possibility that it could be studying a single subpopulation or a mixture of several subpopulations. Additionally, from a clinical perspective it is possible that RNA-based biomarkers for any disease might lie with separate subpopulations of structures, which is necessary to consider in the course of disease biomarker discovery.
Material and methods
Cells
The human mast cell line HMC-1 (Dr. Joseph Butterfield, Mayo Clinic, Rochester, MN, USA) and the erythroleukemic cell line TF-1 (ATCC: CRL-2003) were cultured in a 37°C humidified incubator with 5% CO2. HMC-1 was cultured in Iscove's Modified Dulbecco's Medium (IMDM), while TF-1 was cultured in RPMI-1640. Both media were supplemented with 10% foetal bovine serum (FBS), 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mM L-glutamine. In addition, the medium for HMC-1 also contained 1.2 mM α-thioglycerol and the medium for TF-1 was supplemented with 5 ng/mL granulocyte macrophage colony-stimulating factor (GM-CSF) (all reagents were from Sigma-Aldrich, St Louis, MO, USA).
To eliminate serum exosomes, the FBS was ultracentrifuged (Ti45 rotor, Beckman Coulter, Brea, CA, USA) for 18 hours at 120,000 × g (average) prior to use in cultures.
The cell viability was calculated using the trypan blue dye method.
Isolation of exRNA-enriched isolates
ExRNA-enriched isolates were isolated from fresh conditioned media from HMC-1 and TF-1 cell cultures. Cells, debris, and large extracellular vesicles were eliminated by centrifugation at 300 × g for 10 minutes and at 16,500 × g for 20 minutes. Only half of the supernatant was passed through 0.2 µm filters (Sarstedt, Nümbrecht-Rommelsdorf, Germany), whereas the other half was left unfiltered prior to the last ultracentrifugation step at 120,000 × g for 70 minutes (average Ti45 rotor: fixed angle rotor). In the samples isolated without the filtration step, 2 separate pellets could be identified; one pellet that was firmly attached to the tube wall (pellet 1), and a second non-adherent pellet (pellet 2) at the bottom of the tube (schematics are shown in Fig. S1A). Pellet 1 was also found in the filtered samples, whereas pellet 2 (the non-adherent) was never observed in the isolation where the 0.2 µm filtration step was applied. Because pellet 2 would normally be discarded in the supernatant removal steps, pellet 2 were collected using a pipette before the medium was poured off, while pellet 1 were collected after the medium was poured off by resuspension of the pellets in an appropriate buffer.
Separation of exRNA on sucrose gradients
Further isolation of the different subpopulations of exRNA was performed using sucrose gradients. Pellet 1 and pellet 2 were layered at the bottom of a sucrose gradient, and 10 fractions were isolated and re-pelleted for further analyses. The sucrose gradient procedure was performed as previously describedCitation49. Briefly, for the bottom-loaded gradients, isolated pellets were resuspended in 2.5 M sucrose in PBS and transferred to an ultracentrifuge tube. Fifteen consecutive 2.2 ml layers of decreasing sucrose concentrations in PBS (from 2 M to 0.4 M) were layered on top of the sample. For the top-loaded gradients, 15 consecutive 2.2 ml layers of decreasing sucrose concentrations in PBS (from 2.5 M to 0.5 M) were layered in an ultracentrifuge tube. Isolated pellets were then resuspended in 0.4 M sucrose in PBS and layered on top of the gradient. Gradients were ultracentrifuged for 16 hours, without the applications of brakes, at 4°C in a SW32 rotor (swinging bucket rotor) at 175,000 × g (maximum). Ten fractions were collected (3.9 ml each), and the density of the sucrose was measured with a refractometer. Fractions were diluted in PBS separately or pooled (fractions 2–6 or 8–10) and ultracentrifuged at 120,000 × g (Ti70 rotor).
RNA isolation
The RNA was isolated with the miRCURY™ RNA Isolation Kit - Cell and Plant (Exiqon A/S, Vedbaek, Denmark) according to the manufacturer's protocol (with 1% 2-mercaptoethanol in the lysis buffer) and analyzed on an RNA 6000 Nano chip or an RNA 6000 Pico chip using an Agilent 2100 Bioanalyzer (Agilent Technologies Inc., Palo Alto, CA, USA) according to the manufacturer's protocol.
Microarray
For the miRNA analysis, all samples were mixed with miRNA spike (Toray Industries Inc., Tokyo, Japan) and further labeled, hybridized, and washed according to the manufacture's protocol (3DGENE miRNA label Hybridization 4plex v1, Toray Industries Inc.) using the miRCURY LNA microRNA Array Power Labeling kit (Exiqon) and Human miRNA Oligo chip – 4plex (Toray Industries Inc.). The intensity of each miRNA was analyzed with the 3D-Gene Scanner 3000 (Toray Industries Inc.) with auto gain, auto focus, and auto analysis settings. Quality control was performed on all samples based on the quality-control report from the instrument.
For the mRNA analysis, the mRNA was converted into aRNA according to the manufacture's protocol (3DGENE mRNA amplification v2, Toray Industries Inc.) using the Amino Allyl MessageAmp™ II aRNA Amplification Kit (Life Technologies) and analyzed for size distribution using gel electrophoresis (Bio-Rad laboratories). The aRNA was then labeled with Cy5 Mono-Reactive Dye (GE Healthcare, Little Chalfont, UK) and purified using columns (Bio-Rad and Millipore, Billerica, MA, USA) according to the manufacture's protocol (3DGENE mRNA CyDye label v2, Toray Industries Inc.). Incorporation of Cy5 and the concentration of the labeled aRNA was determined on a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific) before being hybridized to microarray chips (Human Oligo Chip 25k ver. 2.1., Toray Industries Inc.). The hybridization and subsequent washing was performed according to the manufacture's protocol (3DGENE mRNA hybridization v2, Toray Industries Inc.). The intensity of each mRNA was analyzed with the 3D-Gene Scanner 3000 (Toray Industries Inc.) with auto gain, auto focus or focus set to zero, and auto analysis settings. Quality control was performed on all 10 samples based on the quality-control report from the instrument.
Normalization of microarrays
RNA expression in each dataset was normalized by quantile normalizationCitation50 with MATLAB (version 2010a). Fold changes (log2-ratio) were also calculated by MATLAB (version 2010a). To define HD and LD mRNA and miRNA based on the fluorescence value of microarray, we used the previous method using Gaussian mixture model.Citation30,51,52 Using false discovery of <1%, we defined mRNA and miRNA whose fluorescence values are over 5.8515 and 6.7554, as present mRNA and miRNA, respectively.
To choose the high-quality data sets to be used in further analyses, quality control was performed not only by depicting the scatter plots of relative RNA expression, but also by performing principal component analysis (PCA),Citation53 which is provided by MATLAB (version 2010a).
Next-generation sequencing
For both the whole transcriptome and small-RNA sequencing, the raw sequence image files from the Illumina HiSeq 2500 were converted to the fastq format and checked to ensure that the quality scores did not deteriorate drastically at the read ends. The fastqs were trimmed to remove the adapters for the small-RNA sequencing runs whereas for the long RNA they were left untrimmed. The fastqs were then aligned to the reference human genome (hg19, Ensembl version 75) using STAR v2.4.0 for the long RNA and Bowtie for the small RNA. STAR also aligned the reads to a splice junction database to annotate splice junctions between exons. The STAR/Bowtie-aligned .sam files were converted to .bam files and sorted by coordinate position using SAMtools v0.1.19.
For the whole-transcriptome sequencing runs, read counts for genes were generated using featureCounts with hg19, Ensembl 75 as the gene reference. The libraries for the samples were scaled to the total number of reads mapping to the reference human genome (total library normalization).
For the small-RNA sequencing runs, the samples were run through sRNAbench, which allows one to map the reads to various other RNA libraries using Bowtie. The reads mapping to rRNA (from NCBI) were removed, and the remaining reads were mapped to the miRNA database (miRBase v21). After mapping to miRNA, the remaining reads were simultaneously and competitively mapped to the other RNA libraries, namely, tRNA (from UCSC http://gtrnadb.ucsc.edu/Hsapi19/hg19-tRNAs.fa), piRNA (piRBase v1.0), and all other genes found in the Ensembl 75 gtf.
No threshold was used for the correlations and all genes were used irrespective of their expression. The detected RNA threshold was set to >10 reads for all RNA.
Protein concentration
The protein concentrations of the various samples were determined using the Pierce BCA Protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's protocol.
Nanoparticle tracking analysis (NTA)
NTA measurements were performed with a NanoSight LM10 (NanoSight, Amesbury, UK) equipped with a 0.3 ml sample chamber and a 640 nm laser. All measurements were performed at room temperature. Furthermore, the temperature was measured for each analysis and manually entered into the system. The software used for capturing and analyzing the data was NTA 2.2 Build 0381. For all samples, 0.5 µg protein was resuspended in 1 ml of 0.1 µm filtered PBS. Each sample was measured 5 × 60 seconds, and for each of these measurements a new portion of the sample was injected. The result for each sample is presented as an average of the 5 measurements. The camera settings were as follows: camera shutter = 1472 and camera gain = 509. The analysis settings were as follows: detection threshold = auto, blur = 5 × 5, minimum track length = auto, and minimum expected particle size = 30 nm.
Sample preparation for protein extraction and digestion
HD and LD fractions were initially lysed in 8 M urea, 50 mM triethylammonium bicarbonate (TEAB) buffer pH 8.5, 4% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), 0.2% sodium dodecyl sulfate (SDS), and 5 mM EDTA. The remaining pellet was further solubilised with 250 mM TEAB, 4% SDS, and 300 mM dithiothreitol (DTT) pH 7.6. The protein concentration was determined using the Pierce 660 nm Protein Assay Reagent (Thermo Fisher Scientific), and 1000 µg per sample was used for further analysis.
Each sample was incubated with tris(2-carboxyethyl)phosphine (TCEP), transferred to 3K MW cutoff filters (Pall) and diluted with 8 M urea in 0.1 M TEAB for filter-aided sample preparation (FASP)Citation54. Samples were alkylated with methyl methanethiosulfonate (MMTS) and digested with trypsin in 0.5 M TEAB at a 1:25 ratio overnight at 37°C. The peptides were eluted at 12,000 rpm for 10 minutes, and filters were rinsed with 20% acetonitrile (ACN) at 12,000 rpm for 5 minutes.
Strong cation exchange chromatography (SCX) of peptides
The concentrated peptides were acidified with 10% formic acid (FA) and diluted with SCX solvent A (25 mM ammonium formate pH 2.8, 20% ACN) and injected onto a PolySULFOETHYL A SCX column (2.1 mm i.d. × 10 cm length, 5 μm particle size, 300 Å pore size). SCX chromatography and fractionation were carried out on an ÄKTA purifier system (GE Healthcare) at a 0.25 mL/minute flow rate using the following gradient: 0% B (500 mM ammonium formate pH 2.8, 20% ACN) for 5 minutes; 0–40% B for 20 minutes; 40–100% B for 10 minutes; and 100% B for 10 minutes. UV absorbance at 254 nm and 280 nm was monitored while fractions were collected at 0.5 mL intervals. The fraction volumes were reduced in a SpeedVac. The 20 peptide-containing fractions were desalted on PepClean C18 spin columns according to the manufacturer's instructions (Thermo Fisher Scientific).
NanoLC-MS/MS analysis on an LTQ-Orbitrap Velos instrument
The desalted and dried fractions were reconstituted in 0.1% FA and analyzed on an LTQ-Orbitrap Velos (Thermo Fisher Scientific) interfaced with an in-house constructed nano-LC columnCitation55. Two-microlitre sample injections were made with an Easy-nLC autosampler (Thermo Fisher Scientific) running at 200 nL/minute. The peptides were trapped on a pre-column (45 × 0.075 mm i.d.) and separated on a reversed phase column, 200 × 0.075 mm packed with 3 μm Reprosil-Pur C18-AQ particles. The gradient was as follows: 0–70 minutes 5–25% ACN, 0.2% FA; 70–80 minutes 25–40% ACN, 0.2% FA80% ACN, 0.2% FA over 5 minutes and held for the last 10 minutes at 80% ACN, 0.1% FA.
The LTQ-Orbitrap Velos settings were a: spray voltage 1.6kV, 1 microscan for MS1 scans at 60,000 resolution (m/z 400), and a full MS mass range m/z 400–1800. The LTQ-Orbitrap Velos was operated in a data-dependent mode with one MS1 FTMS precursor ion scan followed by CID (collision-induced dissociation) MS2 scans of the 10 most abundant doubly or multiply protonated ions in each FTMS scan. Dynamic exclusion within 20 ppm for 30 seconds was used after 2 repeats of MS2 for a precursor ion. MS2 were collected with 1 microscan and a collision energy of 35%.
Database search and quantification of proteomic data
We performed peptide identification using X! Tandem incorporated in a trans-proteomic pipeline (version 4.6 OCCUPY rev 2).Citation56 Each RAW data file was first converted to mzXML using the msconvert tool incorporated in ProteoWizard (version 3.0.4205).Citation57 MS/MS scans in the converted mzXMLs were then searched in X!! Tandem against the Homo sapiens Swiss-Prot database (release 2013 08) containing 20,272 proteins. The tolerance was set to 10 ppm for precursor ions and 0.8 Da for fragment ions. Enzyme specificity for trypsin was used. Variable modification options were used for the oxidation of methionine (15.995 Da), carbamidomethylation of cysteine (57.021 Da), deamination of N-terminal glutamine (−17.027 Da), and dehydration of N-terminal glutamic acid (−18.011 Da). The number of missed cleavage sites was set to be two. We finally used TPP to statistically identify peptides from the X!! Tandem search results by PeptideProphet (PeptideProphet ≥ 0.90) and then proteins using ProteinProphet (ProteinProphet ≥ 0.90). When multiple proteins were assigned to a protein group, we selected the representative protein with the highest coverage. To measure the quantity of each protein in our density fraction proteomes, we calculated relative protein abundance using absolute protein expression (APEX)Citation58. The APEX quantification is based on spectral counting of each protein corrected by prior expectation of observing each peptide. The first 50 top-ranked proteins with high spectral counts from the ProteinProphet results were selected as a training dataset in order to generate an experiment-specific data set. With APEX normalization factor = 10,000, the protein abundance was then calculated as described in Braisted et al.Citation59.
Bioinformatic analysis
The proteins identified with LC-MS/MS were analyzed using the Gene Ontology (GO) Term Finder software (http://go.princeton.edu/cgi-bin/GOTermFinder)Citation60, the Human Protein Atlas (http://www.proteinatlas.org/), and the database for annotation, visualization and integrated discovery (DAVID; http://david.abcc.ncifcrf.gov/). DAVID was also used to analyze the identified mRNAs, while miRSystem was used to analyze the identified miRNAs. These commercially available databases and software identify cellular components, biological functions, biological processes, pathways, and networks associated or enriched in protein, mRNA, and miRNA datasets, and they also identify the cellular location of the identified protein. Venny (http://bioinfogp.cnb.csic.es/tools/venny/index.html) was used to compare lists of proteins to find common and unique moleculesCitation61.
To catalog molecules, hierarchical clustering analysisCitation62 was performed on the normalized expression intensities. The expression intensities for each molecule were normalized to have a mean of 0 and a standard deviation of 1. For hierarchical clustering analysis and normalization, the “Clustergram” function in MATLAB (version 2010a) was used with the following parameters: linkage = ward; clustering = rows and columns; and distance function = euclidean.
Data access
The complete MS proteomics data were deposited to the PRoteomics IDEntifications database (PRIDE) of the European Molecular Biology Laboratory - European Bioinformatics Institute (EMBL-EBI) under the set identifier PXD004931. The raw reads for the NGS were deposited to the Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI) under bioproject ID PRJNA343960. (http://www.ncbi.nlm.nih.gov/bioproject/343960). The microarray gene expression data were deposited in NCBI's Gene Expression Omnibus (GEO) under the GEO Series accession number GSE88755.
Western blot
Proteins were extracted from cells and HD and LD isolates by dissolving the samples in RIPA buffer and sonicating them 3 × 5 minutes with vortex mixing in-between. Thirty micrograms of proteins were loaded per well and separated on precast gels (Mini-PROTEAN TGX Gels; Bio-Rad laboratories, Hercules, CA, USA) and transferred to nitrocellulose membranes (Bio-Rad laboratories). The membranes were blocked with 5% Blotting Grade Blocker Non-Fat Dry Milk (Bio-Rad Laboratories) in TBST for one hour prior to incubation with anti-Flotillin-1 (1:1000 dilution, Santa Cruz Biotechnology, Santa Cruz, CA), anti-CD81 (1:800 dilution, Santa Cruz Biotechnology), anti-TSG101 (1:1000 dilution, Abcam, Cambridge, UK), or anti-Annexin-2 (1:1000 dilution, Abcam). All primary antibodies were diluted in 0.25% Blotting Grade Blocker Non-Fat Dry Milk in TBST and incubated with the membrane overnight at 4°C. The membranes were washed 3 times before incubation with the secondary antibody for one hour. Secondary antibodies were goat F(ab')2 anti-rabbit IgG (horseradish peroxidase conjugated) for Flotillin-1, CD81, and Annexin-2 (1:5000 dilution) (Harlan Sera-Lab, Loughborough, UK) and sheep anti-mouse Ig (horseradish peroxidase conjugated F(ab')2) for Tsg101 (1:5000 dilution; GE Healthcare, Uppsala, Sweden) diluted in 0.25% Blotting Grade Blocker Non-Fat Dry Milk in TBST. The membrane was then analyzed with the Amersham ECL Prime Western Blotting Detection System (GE Healthcare) and a VersaDoc 4000 MP (Bio-Rad Laboratories).
Flow cytometry
Isolated HD and LD samples resuspended in PBS were incubated with anti-CD63-coated beads (15 µg sample/70,000 beads for each antibody; Life Technologies, Carlsbad, CA, USA) overnight at 4°C with gentle agitation. The sample-bead complexes were washed 3 times with 1% exosome-depleted FBS in PBS, incubated with human IgG (Sigma-Aldrich) for 15 minutes at 4°C, washed 3 times, and incubated with PE-labeled anti-CD9, anti-CD63, anti-CD81, or the appropriate isotype control for 40 minutes at room temperature (all antibodies were from BD Bioscience, San Jose, CA, USA). The samples were washed 2 times before 10,000 events were acquired in a FACSAria (BD Bioscience) and analyzed using the FlowJo Software (Tri Star Inc., Ashland, OR, USA).
Electron microscopy
Formvar/carbon-coated nickel grids (Ted Pella, Inc., Redding, CA, USA) were UV-treated for 5 minutes before 10 µg of sample was loaded. The grids and samples were incubated for 10 minutes before being fixed in 2% paraformaldehyde. Samples were immunostained with anti-CD63 antibody (BD Bioscience) or an appropriate isotype control (Sigma-Aldrich). Gold-labeled secondary antibody was added before the samples were fixed in 2.5% glutaraldehyde and contrasted in 2% uranyl acetate. The preparations were examined using a LEO 912AB Omega electron microscope (Carl Zeiss NTS, Jena, Germany). Vesicles were measured with the iTEM® software (Olympus-SiS, Münster, Germany).
Statistical analysis
Where appropriate, data are expressed as mean ± SEM. Statistical analysis was performed by paired or non-paired Student's t-test in GraphPad Prism 6 (GraphPad Software Inc., La Jolla, CA, USA).
The correlations of expression were verified by Pearson'sCitation63 test calculated by MATLAB (version 2010a). If the correlation coefficient r was over 0.9, we defined the correlation as high. The existence of a correlation existence was regarded as confident if p-values were less than 0.001.
Disclosure of potential conflicts of interest
J.L. has written several patents in the field of extracellular vesicles as therapeutics, and is currently an employer of Codiak BioSciences Inc. in parallel with his position at University of Gothenburg. The other authors declare that there are no financial, personal, or professional interests that could be construed to have influenced the paper.
Author contributions
Conceived and designed the experiments: CL, JL. Performed the experiments: CL, GS, AY, SR, RC. Analyzed the data: CL, AY, DKK. Gave conceptual advice: MS, YSG, KvKJ. Wrote the paper: CL, JL, but all authors contributed to the process and all authors have read and approved the final version of the manuscript.
Supplementary_Data.zip
Download Zip (7 MB)Acknowledgment
We would like to thank Gunnar Nilsson at the Karolinska Institute, Stockholm, Sweden, for the kind gift of the human mast cell line HMC-1. We would like to acknowledge Déborah L. M. Rupert at Chalmers University of Technology, Gothenburg, Sweden, for assistance with the NTA, the staff of the Proteomics Core Facility at University of Gothenburg for their technical assistance with the proteomics, TATAA Biocenter, Gothenburg, Sweden, for their technical assistance with the transcriptomics (www.tataa.com/services), and Matthew Hogg for language editing.
Funding
This work was funded by grants from the Swedish Research Council (K2011-56X-20676-04-6), the Swedish Cancer Foundation (CAN2012/690), VBG Group Herman Krefting Foundation for Asthma and Allergy Research, and the Swedish Heart and Lung Foundation (20120528). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Related Research Data
References
- Tan EM, Schur PH, Carr RI, Kunkel HG. Deoxybonucleic acid (DNA) and antibodies to DNA in the serum of patients with systemic lupus erythematosus. J Clin Invest 1966; 45:1732-40; PMID:4959277; http://dx.doi.org/10.1172/JCI105479
- Hughes GR, Cohen SA, Lightfoot RW, Jr., Meltzer JI, Christian CL. The release of DNA into serum and synovial fluid. Arthritis Rheum 1971; 14:259-66; PMID:4927484; http://dx.doi.org/10.1002/art.1780140211
- Kamm RC, Smith AG. Nucleic acid concentrations in normal human plasma. Clin Chem 1972; 18:519-22; PMID:5026765
- Tzimagiorgis G, Michailidou EZ, Kritis A, Markopoulos AK, Kouidou S. Recovering circulating extracellular or cell-free RNA from bodily fluids. Cancer Epidemiol 2011; 35:580-9; PMID:21514265; http://dx.doi.org/10.1016/j.canep.2011.02.016
- Creemers EE, Tijsen AJ, Pinto YM. Circulating microRNAs: novel biomarkers and extracellular communicators in cardiovascular disease? Circ Res 2012; 110:483-95; PMID:22302755; http://dx.doi.org/10.1161/CIRCRESAHA.111.247452
- Hromadnikova I. Extracellular nucleic acids in maternal circulation as potential biomarkers for placental insufficiency. DNA Cell Biol 2012; 31:1221-32; PMID:22364204; http://dx.doi.org/10.1089/dna.2011.1530
- Su KY, Pisetsky DS. The role of extracellular DNA in autoimmunity in SLE. Scand J Immunol 2009; 70:175-83; PMID:19703007; http://dx.doi.org/10.1111/j.1365-3083.2009.02300.x
- Swarup V, Rajeswari MR. Circulating (cell-free) nucleic acids–a promising, non-invasive tool for early detection of several human diseases. FEBS Lett 2007; 581:795-9; PMID:17289032; http://dx.doi.org/10.1016/j.febslet.2007.01.051
- Arroyo JD, Chevillet JR, Kroh EM, Ruf IK, Pritchard CC, Gibson DF, Mitchell PS, Bennett CF, Pogosova-Agadjanyan EL, Stirewalt DL, et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci U S A 2011; 108:5003-8; PMID:21383194; http://dx.doi.org/10.1073/pnas.1019055108
- Turchinovich A, Weiz L, Langheinz A, Burwinkel B. Characterization of extracellular circulating microRNA. Nucleic Acids Res 2011; 39:7223-33; PMID:21609964; http://dx.doi.org/10.1093/nar/gkr254
- Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol 2011; 13:423-33; PMID:21423178; http://dx.doi.org/10.1038/ncb2210
- Crescitelli R, Lässer C, Szabo TG, Kittel A, Eldh M, Dianzani I, Buzás EI, Lötvall J. Distinct RNA profiles in subpopulations of extracellular vesicles: apoptotic bodies, microvesicles and exosomes. J Extracell Vesicles 2013; 2; PMID:24223256; http://dx.doi.org/10.3402/jev.v2i0.20677
- Filipazzi P, Burdek M, Villa A, Rivoltini L, Huber V. Recent advances on the role of tumor exosomes in immunosuppression and disease progression. Seminars Cancer Biol 2012; 22:342-9; PMID:22369922; http://dx.doi.org/10.1016/j.semcancer.2012.02.005
- Zhong H, Yang Y, Ma S, Xiu F, Cai Z, Zhao H, Du L. Induction of a tumour-specific CTL response by exosomes isolated from heat-treated malignant ascites of gastric cancer patients. Int J Hyperthermia 2011; 27:604-11; PMID:21846196; http://dx.doi.org/10.3109/02656736.2011.564598
- Torregrosa Paredes P, Esser J, Admyre C, Nord M, Rahman QK, Lukic A, Radmark O, Gronneberg R, Grunewald J, Eklund A, et al. Bronchoalveolar lavage fluid exosomes contribute to cytokine and leukotriene production in allergic asthma. Allergy 2012; 67:911-9; PMID:22620679; http://dx.doi.org/10.1111/j.1398-9995.2012.02835.x
- Silverman JM, Reiner NE. Exosomes and other microvesicles in infection biology: organelles with unanticipated phenotypes. Cell Microbiol 2011; 13:1-9; PMID:21040357; http://dx.doi.org/10.1111/j.1462-5822.2010.01537.x
- Vella LJ, Sharples RA, Nisbet RM, Cappai R, Hill AF. The role of exosomes in the processing of proteins associated with neurodegenerative diseases. Euro Biophys J 2008; 37:323-32; PMID:18064447; http://dx.doi.org/10.1007/s00249-007-0246-z
- Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol 2011; 29:341-5; PMID:21423189; http://dx.doi.org/10.1038/nbt.1807
- Lässer C. Exosomal RNA as biomarkers and the therapeutic potential of exosome vectors. Expert Opin Biol Ther 2012; 12 Suppl 1:S189-97; PMID:Can't; http://dx.doi.org/10.1517/14712598.2012.680018
- Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT, Jr., Carter BS, Krichevsky AM, Breakefield XO. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 2008; 10:1470-6; PMID:19011622; http://dx.doi.org/10.1038/ncb1800
- Nolte-'t Hoen EN, Buermans HP, Waasdorp M, Stoorvogel W, Wauben MH, t Hoen PA. Deep sequencing of RNA from immune cell-derived vesicles uncovers the selective incorporation of small non-coding RNA biotypes with potential regulatory functions. Nucleic Acids Res 2012; 40:9272-85; PMID:22821563; http://dx.doi.org/10.1093/nar/gks658
- Lunavat TR, Cheng L, Kim DK, Bhadury J, Jang SC, Lasser C, Sharples RA, Lopez MD, Nilsson J, Gho YS, et al. Small RNA deep sequencing discriminates subsets of extracellular vesicles released by melanoma cells - Evidence of unique microRNA cargos. RNA Biol 2015; 12:810-23; PMID:26176991; http://dx.doi.org/10.1080/15476286.2015.1056975
- Palma J, Yaddanapudi SC, Pigati L, Havens MA, Jeong S, Weiner GA, Weimer KM, Stern B, Hastings ML, Duelli DM. MicroRNAs are exported from malignant cells in customized particles. Nucleic Acids Res 2012; 40:9125-38; PMID:22772984; http://dx.doi.org/10.1093/nar/gks656
- Wang K, Zhang S, Weber J, Baxter D, Galas DJ. Export of microRNAs and microRNA-protective protein by mammalian cells. Nucleic Acids Res 2010; 38:7248-59; PMID:20615901; http://dx.doi.org/10.1093/nar/gkq601
- Tabet F, Vickers KC, Cuesta Torres LF, Wiese CB, Shoucri BM, Lambert G, Catherinet C, Prado-Lourenco L, Levin MG, Thacker S, et al. HDL-transferred microRNA-223 regulates ICAM-1 expression in endothelial cells. Nat Commun 2014; 5:3292; PMID:24576947; http://dx.doi.org/10.1038/ncomms4292
- Birnie GD, Rickwood D. Centrifugal seperations in molecular and cell biology. Butterworth & Co Ltd, 1978
- Mathivanan S, Ji H, Simpson RJ. Exosomes: extracellular organelles important in intercellular communication. J Proteomics 2010; 73:1907-20; PMID:20601276; http://dx.doi.org/10.1016/j.jprot.2010.06.006
- Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 2007; 9:654-9; PMID:17486113; http://dx.doi.org/10.1038/ncb1596
- Eldh M, Ekstrom K, Valadi H, Sjostrand M, Olsson B, Jernas M, Lotvall J. Exosomes communicate protective messages during oxidative stress; possible role of exosomal shuttle RNA. PLoS One 2010; 5:e15353; PMID:21179422; http://dx.doi.org/10.1371/journal.pone.0015353
- Hong BS, Cho JH, Kim H, Choi EJ, Rho S, Kim J, Kim JH, Choi DS, Kim YK, Hwang D, et al. Colorectal cancer cell-derived microvesicles are enriched in cell cycle-related mRNAs that promote proliferation of endothelial cells. BMC Genomics 2009; 10:556; PMID:19930720; http://dx.doi.org/10.1186/1471-2164-10-556
- Xiao D, Ohlendorf J, Chen Y, Taylor DD, Rai SN, Waigel S, Zacharias W, Hao H, McMasters KM. Identifying mRNA, microRNA and protein profiles of melanoma exosomes. PLoS One 2012; 7:e46874; PMID:23056502; http://dx.doi.org/10.1371/journal.pone.0046874
- Lässer C, Alikhani VS, Ekström K, Eldh M, Paredes PT, Bossios A, Sjöstrand M, Gabrielsson S, Lötvall J, Valadi H. Human saliva, plasma and breast milk exosomes contain RNA: uptake by macrophages. J Transl Med 2011; 9:9; PMID:Can't; http://dx.doi.org/10.1186/1479-5876-9-9
- Keller S, Ridinger J, Rupp AK, Janssen JW, Altevogt P. Body fluid derived exosomes as a novel template for clinical diagnostics. J Transl Med 2011; 9:86; PMID:21651777; http://dx.doi.org/10.1186/1479-5876-9-86
- Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem 2010; 285:17442-52; PMID:20353945; http://dx.doi.org/10.1074/jbc.M110.107821
- Palanisamy V, Sharma S, Deshpande A, Zhou H, Gimzewski J, Wong DT. Nanostructural and transcriptomic analyses of human saliva derived exosomes. PLoS One 2010; 5:e8577; PMID:20052414; http://dx.doi.org/10.1371/journal.pone.0008577
- Kogure T, Lin WL, Yan IK, Braconi C, Patel T. Intercellular nanovesicle-mediated microRNA transfer: a mechanism of environmental modulation of hepatocellular cancer cell growth. Hepatology 2011; 54:1237-48; PMID:21721029; http://dx.doi.org/10.1002/hep.24504
- Miranda KC, Bond DT, McKee M, Skog J, Paunescu TG, Da Silva N, Brown D, Russo LM. Nucleic acids within urinary exosomes/microvesicles are potential biomarkers for renal disease. Kidney Int 2010; 78:191-9; PMID:20428099; http://dx.doi.org/10.1038/ki.2010.106
- Nicolas FE, Hall AE, Csorba T, Turnbull C, Dalmay T. Biogenesis of Y RNA-derived small RNAs is independent of the microRNA pathway. FEBS Lett 2012; 586:1226-30; PMID:22575660; http://dx.doi.org/10.1016/j.febslet.2012.03.026
- Dhahbi JM, Spindler SR, Atamna H, Boffelli D, Mote P, Martin DI. 5′-YRNA fragments derived by processing of transcripts from specific YRNA genes and pseudogenes are abundant in human serum and plasma. Physiol Genomics 2013; 45:990-8; PMID:24022222; http://dx.doi.org/10.1152/physiolgenomics.00129.2013
- van Balkom BW, Eisele AS, Pegtel DM, Bervoets S, Verhaar MC. Quantitative and qualitative analysis of small RNAs in human endothelial cells and exosomes provides insights into localized RNA processing, degradation and sorting. J Extracell Vesicles 2015; 4:26760; PMID:26027894; http://dx.doi.org/10.3402/jev.v4.26760
- Stadler PF, Chen JJ, Hackermuller J, Hoffmann S, Horn F, Khaitovich P, Kretzschmar AK, Mosig A, Prohaska SJ, Qi X, et al. Evolution of vault RNAs. Mol Biol Evol 2009; 26:1975-91; PMID:19491402; http://dx.doi.org/10.1093/molbev/msp112
- Kedersha NL, Heuser JE, Chugani DC, Rome LH. Vaults. III. Vault ribonucleoprotein particles open into flower-like structures with octagonal symmetry. J Cell Biol 1991; 112:225-35; PMID:1988458; http://dx.doi.org/10.1083/jcb.112.2.225
- Li CC, Eaton SA, Young PE, Lee M, Shuttleworth R, Humphreys DT, Grau GE, Combes V, Bebawy M, Gong J, et al. Glioma microvesicles carry selectively packaged coding and non-coding RNAs which alter gene expression in recipient cells. RNA Biol 2013; 10:1333-44; PMID:23807490; http://dx.doi.org/10.4161/rna.25281
- Dhahbi JM. 5′ tRNA Halves: The Next Generation of Immune Signaling Molecules. Front Immunol 2015; 6:74; PMID:25745425; http://dx.doi.org/10.3389/fimmu.2015.00074
- Tosar JP, Gambaro F, Sanguinetti J, Bonilla B, Witwer KW, Cayota A. Assessment of small RNA sorting into different extracellular fractions revealed by high-throughput sequencing of breast cell lines. Nucleic Acids Res 2015; 43:5601-16; PMID:25940616; http://dx.doi.org/10.1093/nar/gkv432
- Vojtech L, Woo S, Hughes S, Levy C, Ballweber L, Sauteraud RP, Strobl J, Westerberg K, Gottardo R, Tewari M, et al. Exosomes in human semen carry a distinctive repertoire of small non-coding RNAs with potential regulatory functions. Nucleic Acids Res 2014; 42:7290-304; PMID:24838567; http://dx.doi.org/10.1093/nar/gku347
- Thompson DM, Parker R. Stressing out over tRNA cleavage. Cell 2009; 138:215-9; PMID:19632169; http://dx.doi.org/10.1016/j.cell.2009.07.001
- Bellingham SA, Coleman BM, Hill AF. Small RNA deep sequencing reveals a distinct miRNA signature released in exosomes from prion-infected neuronal cells. Nucleic Acids Res 2012; 40:10937-49; PMID:22965126; http://dx.doi.org/10.1093/nar/gks832
- Bobrie A, Colombo M, Krumeich S, Raposo G, Théry C. Diverse subpopulations of vesicles secreted by different intracellular mechanisms are present in exosome preparations obtained by differential ultracentrifugation. 2012; 1; PMID:24009879; http://dx.doi.org/10.3402/jev.v1i0.18397
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003; 19:185-93; PMID:12538238; http://dx.doi.org/10.1093/bioinformatics/19.2.185
- Choi DS, Yang JS, Choi EJ, Jang SC, Park S, Kim OY, Hwang D, Kim KP, Kim YK, Kim S, et al. The protein interaction network of extracellular vesicles derived from human colorectal cancer cells. J Proteome Res 2012; 11:1144-51; PMID:22149170; http://dx.doi.org/10.1021/pr200842h
- Marczyk M, Jaksik R, Polanski A, Polanska J. Adaptive filtering of microarray gene expression data based on Gaussian mixture decomposition. BMC Bioinformatics 2013; 14:101; PMID:23510016; http://dx.doi.org/10.1186/1471-2105-14-101
- Hotelling H. Analysis of a complex of statistical variables into principal components. J Educational Psychol 1933; 24:417-41; http://dx.doi.org/10.1037/h0071325
- Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods 2009; 6:359-62; PMID:19377485; http://dx.doi.org/10.1038/nmeth.1322
- Carlsohn E, Nystrom J, Karlsson H, Svennerholm AM, Nilsson CL. Characterization of the outer membrane protein profile from disease-related Helicobacter pylori isolates by subcellular fractionation and nano-LC FT-ICR MS analysis. J Proteome Res 2006; 5:3197-204; PMID:17081072; http://dx.doi.org/10.1021/pr060181p
- Keller A, Eng J, Zhang N, Li XJ, Aebersold R. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol Syst Biol 2005; 1:2005 0017; PMID:16729052; http://dx.doi.org/10.1038/msb4100024
- Kessner D, Chambers M, Burke R, Agus D, Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 2008; 24:2534-6; PMID:18606607; http://dx.doi.org/10.1093/bioinformatics/btn323
- Lu P, Vogel C, Wang R, Yao X, Marcotte EM. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat Biotechnol 2007; 25:117-24; PMID:17187058; http://dx.doi.org/10.1038/nbt1270
- Braisted JC, Kuntumalla S, Vogel C, Marcotte EM, Rodrigues AR, Wang R, Huang ST, Ferlanti ES, Saeed AI, Fleischmann RD, et al. The APEX Quantitative Proteomics Tool: generating protein quantitation estimates from LC-MS/MS proteomics results. BMC Bioinformatics 2008; 9:529; PMID:19068132; http://dx.doi.org/10.1186/1471-2105-9-529
- Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, Sherlock G. GO::TermFinder–open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 2004; 20:3710-5; PMID:15297299; http://dx.doi.org/10.1093/bioinformatics/bth456
- Oliveros JC. VENNY. An interactive tool for comparing lists with Venn Diagrams., 2007
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A 1998; 95:14863-8; PMID:9843981; http://dx.doi.org/10.1073/pnas.95.25.14863
- Rodgers JL, Nicewander WA. Thirteen ways to look at the correlation coefficient. Am Statistician 1988; 42:59-66; http://dx.doi.org/10.2307/2685263