
ABSTRACT
Our ability to map and quantify RNA modifications at a genome-wide scale have revolutionized our understanding of the pervasiveness and dynamic regulation of diverse RNA modifications. Recent efforts in the field have demonstrated the presence of modified residues in almost any type of cellular RNA. Next-generation sequencing (NGS) technologies are the primary choice for transcriptome-wide RNA modification mapping. Here we provide an overview of approaches for RNA modification detection based on their RT-signature, specific chemicals, antibody-dependent (Ab) enrichment, or combinations thereof. We further discuss sources of artifacts in genome-wide modification maps, and experimental and computational considerations to overcome them. The future in this field is tightly linked to the development of new specific chemical reagents, highly specific Ab against RNA modifications and use of single-molecule RNA sequencing techniques.
Abbreviations
| RT | = | reverse transcription |
| NGS | = | next generation sequencing |
| Ab | = | antibody |
| 1D(2D) | = | mono/bi-dimensional |
| TLC | = | thin layer chromatography |
| NMP | = | nucleotide monophosphate |
| RACE | = | Rapid Amplification of cDNA Ends |
| TTO | = | Terminal Tagging Oligo |
| HPLC | = | high-performance (pressure) liquid chromatography |
| MS | = | mass-spectrometry |
| CMCT | = | N-Cyclohexyl-N′-(2-morpholinoethyl)carbodiimide metho-p-toluenesulfonate |
| MT | = | methyltransferase |
| HITS-CLIP | = | High-throughput sequencing of RNA isolated by crosslinking immunoprecipitation |
| PAR-CLIP | = | Photoactivatable Ribonucleoside-Enhanced Crosslinking and Immunoprecipitation |
| SMRT | = | single-molecule real time |
| ZMWs | = | zero-mode waveguides |
| WC | = | Watson-Crick |
Introduction
RNA modifications have been studied for over 60 years, and yet many aspects pertaining to their function and mechanisms of action have remained elusive. All known RNA modified residues (over 150 altogether) are formed post-transcriptionally, by complex enzymatic mechanisms involving hundreds of different catalytic proteins and cofactors. RNA modifications occur with concomitantly with other RNA maturation processes, including 5′- and 3′-trimming, editing, splicing and polyadenylation.Citation1,2 These modifications affect RNA stability and folding, cellular localization, as well as interactions with various RNA and protein partners.Citation3-5 Modifications of RNA are increasingly recognized as an important layer of post-transcriptional regulation of gene expression, analogous to DNA methylation and post-translational histone modifications. Recent approaches for transcriptome-wide mapping of RNA modifications have revealed that RNA modifications occur not only on stable non-coding RNAs such as tRNA and rRNA, in which they have classically been studied, but also in coding mRNAs.Citation6-13 It is meanwhile further recognized that some mRNA modifications can be dynamically modulated, reversible, and that they can impact mRNA biology at various levels. A major challenge in the field is the precise mapping and quantification of modified RNA residues at the transcriptome level. Next-generation sequencing (NGS) technologies have revolutionized our ability to map some mRNA modifications, paving the path for understanding their physiological importance. Here we provide an overview to these technologies and their applications in recent years, along with the remaining challenges at both experimental and analytic levels.
Historical methods for detection and mapping of RNA modifications
Mono and bi-dimensional chromatography
One of the most ancient methods for detection of RNA modifications is a mono-dimensional (1D) or 2-dimensional (2D) thin layer chromatography (TLC). Modified nucleotides differ from their non-modified counterparts by net charge, polarity and hydrophobicity, thus allowing their chromatographic separation. A key advantage of this method is the analysis of 5′- or 3′-nucleotide monophosphates (NMPs), which allows to increase the sensitivity with [32P] radioactive labeling. TLC separation of NMPs can be done on different stationary phases, but microcrystalline cellulose, which provides rather efficient separation, remains a reference in the field. In addition, using this stationary phase, 2D TLC migration maps have been established for numerous modified NMPs.Citation14-16
Faint spots for modified nucleotides in RNA can be detected even by UV-shadowing, but the most sensitive approach requires pre- or post-labeling with [32P]. For in vitro assays, 1D TLC can be used, while more complex cases generally require 2D separation. At least 3 solvent systems were proposed for 1D and 2D separation, depending on the chemical properties of the target nucleotide. In 2D separation, a commonly used technique employs the same solvent system for the first dimension, while 2 alternative buffers (solvents) are used for the second dimension. Despite several attempts, the polar and charged nature of NMPs prevent the use of modern and highly efficient stationary phases based on derivatized SiO2 matrixes. Even if successful, the replacement of microcrystalline cellulose would require substantial optimization, since the reference migration maps would have to be reestablished for >100 of known modified 5′- and 3′-nucleotide monophosphates.
Recently, a TLC-based method was developed to accurately determine and quantify modification levels within individual genes, termed site-specific cleavage and radioactive-labeling followed by ligation-assisted extraction and thin-layer chromatography (SCARLET).Citation17
HPLC and MS coupling
The modification content of RNA digested to mononucleosides can be investigated by high-performance (pressure) liquid chromatography (HPLC) with simple UV detection or coupled to mass-spectrometry (MS). These methods were extensively used in the past for detection and quantification of RNA modifications, but do not provide any information on the exact localization of the modified residue. Modern approaches use MS-coupled micro or nano-chromatography systems, which reduces the amount of required material to single digit picomolar range. These methods are extensively reviewed elsewhere.Citation18-20
Direct methods of RNA modification analysis by Matrix-Assisted Laser Desorption/Ionization -Time Of Flight (MALDI-TOF) MS are still under development, and not applied in routine use. Their application is restricted by the limited sensitivity of MS detectors in negative mode and difficulties in ionization of negatively charged RNA molecules or even short oligonucleotides.Citation21,22
RT-based techniques and their link to NGS approaches
Reverse-transcription (RT)-based techniques use primer extension to detect modified RNA nucleotides.Citation23 Some modified RNA residues naturally block or pause primer extension, thus allowing detection of modified position in a given RNA. In most cases the comparison with unmodified RNA transcript (or RNA extracted from mutant strains or cells) is necessary to exclude structural RT stops. The main advantage of RT-based techniques is their sensitivity and applicability to complex mixtures of RNA, while the majority of the listed techniques require pure individual RNA molecules for analysis. Analysis of primer extension stops was traditionally done with [32P]labeled DNA primers, but radioactivity can also be replaced by fluorescent labels, albeit with some loss of sensitivity. Since primer extension is an almost indispensable part of any NGS library preparation protocol, the modern methods for analysis of RT stops employ high-throughput sequencing.
Modern next-generation sequencing (NGS) technologies
Modern systems for massive parallel DNA sequencing can be grouped into 2 major classes: physical or virtual cluster sequencing and single-molecule sequencing (). The current protocols used by sequencing machines on the market (so called second generation sequencing or NGS) include an amplification step for cluster generation (physical DNA clusters on flow-cell surface for Illumina or virtual on-bead clusters for IonTorrent/PGM). Thus, ∼1000 DNA molecules with the same sequence generate measurable sequencing signals (). Key advantages of cluster sequencing technologies include their high quality of sequencing (with an error rate typically less than 1 error per 1,000-10,000 sequenced bases) and extremely high sequencing output, compared to other technologies. However, the length of the sequencing read remains limited and does not exceed 500-600 nt in most cases.
Figure 1. Second- and third-generation sequencing technologies (NGS and NNGS). Second generation sequencing uses cluster amplification of DNA strands prior to fluorescent or potentiometric sequencing (A). Multiple molecules generate detectable signal. Third-generation sequencing technologies use single molecule sequencing with specially designed fluorescent detection systems (Zero-mode Waveguides, ZMW), or nanopore sequencing using exonuclease or DNA polymerase activities (B).
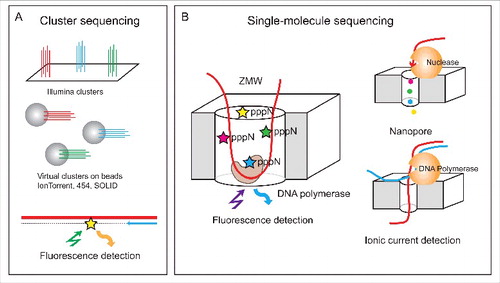
Single-molecule sequencing approaches are also called third generation sequencing or NNGS.Citation24 Two independent principles were proposed: single-molecule real time (SMRT) technology using nanowells (called zero-mode waveguides, ZMWs, Pacific BioSciences) and nanopore sequencing (MinION, Oxford Nanopore Technologies) (). An important advantage of both technologies is their very long read length >10,000 nt in average, but the overall precision of the sequencing remains low. These technologies hold a lot of promise for the future, as in principle they are more suitable for the analysis of modified RNA, as they should allow direct sequencing of a single RNA molecule without requiring its conversion into DNA which entails the loss of the modified nucleotide. However, at the moment these methodologies are mostly being applied and optimized in the context of DNA sequencing, and will require substantial optimization until they are suitable also for RNA. Nonetheless, the potential of these methodologies to identify RNA modifications was demonstrated for m6A, which can be discriminated from non-methylated A in SMRT sequencing.Citation25,26
In this review we will address only the well-established method for RNA-modification analysis employing the second generation sequencing technologies.
Library preparation protocols suitable to studies of RNA modifications
Classical protocols, which were originally used for sequencing RNA and estimation of gene expression levels, relied on randomly primed reverse transcription and randomly primed second strand synthesis, followed by ligation of adapters to double-stranded cDNA fragments. This approach has important disadvantages for identification and quantification of RNA modifications. These disadvantages include: (1) Lack of strand-specificity: this protocol cannot distinguish between transcription occurring on the Watson versus Crick strand. If a gene is transcribed on both strands, this will lead to dilution of the RT truncation and/or misincorporation signal. (2) This protocol cannot provide information pertaining to the precise position at which RT terminated (), which is critical for techniques relying on such RT-signatures. Knowledge of the precise start and end of the original RNA fragment can also improve the resolution of peak calling in methods relying on antibody-based capture of modified fragments.Citation8 (3) Finally, random priming is less efficient for short RNAs, which serve as the starting step of antibody-based capture techniques.
Figure 2. Library preparation issues. Detection of RNA modification relies on exact determination of cDNA 3′-end (A). Priming at the 3′-end of RNA depends on its size. Short RNAs (typically < 100 - 200 nt) require adapter ligation step prior to primer annealing (left), while random priming (right) can be used for long RNA species (B). Methods for treatment of cDNA 3′-end for exact determination of RT stop. Four common techniques have been developed. One - single-strand DNA adapter ligation, 2 - oligonucleotide tailing terminal transferase followed by dsDNA primer ligation, 3 - CircLigase protocol with 5′-phosphorylated cDNA followed by second strand synthesis, and 4 - templated cDNA extension with 3′-blocked NNNNNN primer. In all cases the final step includes PCR amplification and appropriate barcoding.
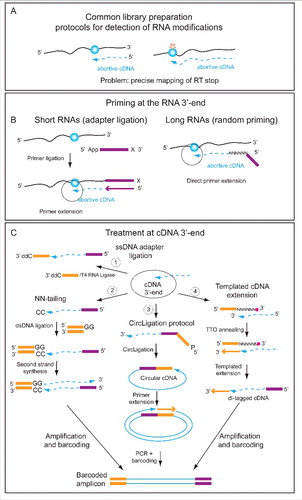
To adequately capture information pertaining to the 3′-end of the RNA, most protocols are based on techniques used to sequence small RNAs, involving ligation of an adapter to the 3′ of the RNA. If the 5′- and 3′- extremities of RNA are compatible for ligation, this step can be performed directly, otherwise m7G(m2,2,7G)-cap removal and/or dephosphorylation/phosphorylation steps are required. The 3′ ligated adapter is subsequently used to prime reverse transcription (). To adequately capture the precise position of the 3′ of the cDNA (reflecting the point at which RT dropped off, either having reached the original 5′ of the RNA or due, for example, to a bulky modified position) various alternatives have been described. One possibility is to employ direct single-stranded ligation of 3′-blocked DNA oligonucleotide to cDNA using T4 RNA ligaseCitation7,8 (). A second approach for 3′-cDNA mapping is derived from 5′-RACE (Rapid Amplification of cDNA Ends) protocol.Citation27,28 The cDNA is first extended with 2-3 homonucleotides (C or G) in a non-templated tailing reaction, and a double stranded DNA adapter with complementary (GG or CC) overhang is ligated. The second strand synthesis uses the free 3′-end of the DNA adapter. A third possibility was originally described in ribosome profiling and uses a particular DNA ligase (CircLigase) to make a circular cDNA.Citation13 This circular cDNA is then used for PCR amplification. The last alternative is the use of Terminal Tagging Oligo (TTO) which is a random primer with N6 and blocked 3′-end. In contrast to conventional random priming described above, TTO is not extended, but provides a template for cDNA 3′-end extension (see ). As a result, a di-tagged cDNA is obtained and can be used for subsequent PCR amplification and barcoding. Additional, more complex variants have been utilized for direct 5′-OH and 3′-P (or >P) ligation of fragments in RiboMethSeq protocol. This particular ligation step requires home-made preparation of 3′-cyclophosphate adapters and uses a RNA ligase mutant,Citation29 but avoids additional treatment steps which can be prone to introduce some unpredictable bias.
Principles for NGS detection of RNA modifications
The availability of high-throughput sequencing technologies in recent years has revolutionized our ability to obtain transcriptome-wide overviews on RNA modifications. Such specialized protocols have been already developed for 6-methyladenosine (m6A),Citation10,12 5-methylcytosine (m5C),Citation9,30,31 pseudouridine,Citation7,13,32,33 2′-O-methylationsCitation29,34 and 1-methyladenosine (m1A).Citation11,35 These protocols have revealed that rather than being present only on rRNA, tRNA and snRNA molecules, on which RNA modifications were classically studied, some of these modifications are also widespread on mRNAs (mRNA). In addition, in some cases these modifications are dynamically regulated, suggesting a post-transcriptional regulatory role. Importantly, currently there are no ‘general solutions’ for identifying RNA modifications using next-generation sequencing, in contrast to HPLC-MS based methodologies, which can analyze a large number of modifications in a single run. Instead, a specialized experimental protocol - relying on the unique chemistry or properties of a modified nucleotide - has to be developed for each modification of interest. In parallel, dedicated analytical approaches have to be developed to carefully analyze resulting datasets, identify putative modified sites, and filter out false positives which can arise at numerous levels.
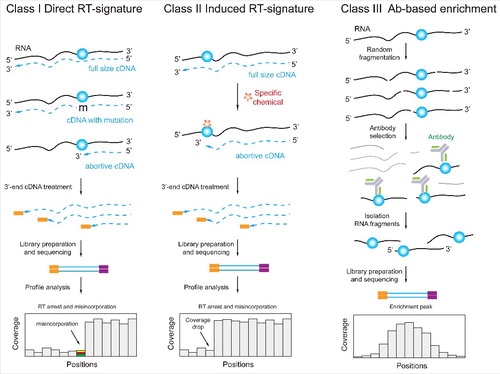
Sequencing-based methodologies for identification of RNA modifications can be broadly categorized into 3 classes (). Some modifications directly impact the ability to reverse transcribe them, and thus leave characteristic signatures either in the form of cDNA misincorporations (“mutations”) or in the form of premature termination of the reaction. Such modifications can be detected by examination of sequencing reads generated via suitable library preparation protocols (‘Class I’). Other modifications that are ‘silent’ in terms of their impact on reverse-transcription can be converted into non-silent counterparts using chemical pre-treatment of the RNA (‘Class II’). Alternatively, modified sites can be enriched using a specific reagent (e.g. an antibody) that selectively binds to the modified RNA fragments (‘Class III’). Methodologies pre-enriching methylated fragments and leading to distinct RT signatures can also be combined, thereby potentially decreasing false-detection rates. Below we discuss experimental and computational aspects, pertaining to these modification-discovery approaches.
Figure 3. Three classes of NGS-based techniques for detection and mapping of RNA modifications. Class I methods are based on “natural” RT-signature generated by the modified nucleotide. Such signature may include RT-arrest (interpreted as a coverage drop) at the modification site or/and nucleotide misincorporation at the same position (left). Class II methods are similar to Class I, but the RNA modification is RT-silent and the visible signature is induced via appropriate chemical treatment (middle). Class III techniques are based on enrichment of RNA fragments containing the RNA modification with a specific Ab. In these cases, the position of modification is typically not determined at single nucleotide resolution, but rather as an enriched region (right).
We have limited the discussion in this review only to approaches that can be applied to purified RNA. Approaches relying on steps performed in-vivo, such as cross-linking of modification readers or writersCitation36-38 will hence not be discussed.
Class I: Modification-specific RT signature
The chemical structure of the base in the RNA nucleotide determines its behavior during reverse-transcription and cDNA synthesis. Some modified nucleotides leave specific signatures in the cDNA sequences, which can include either abortive primer extension and/or misincorporation at or around the modified site (). A classic example for such a misincorporation is inosine, which is derived from A by enzymatic deamination, but behaves as G during RT reaction. Thus inosine residues can be detected by almost complete “misincorporation” of G instead of A in the sequencing profile.Citation39 In contrast, other modified nucleotides can affect the cDNA synthesis either due to their inability to base-pair with their normal partner, or by slowing down the rate of cDNA synthesis due to their bulky or highly hydrophobic structure. Modified nucleotides with additional chemical groups at the WC edge (e.g., m1A, m1G, m3U, m3C) are unable to base-pair with any partner and thus can lead to relatively efficient RT-stops and/or nucleotide misincorporation. Traces of these RNA modifications in tRNAs and other short RNAs have been found using small RNA sequencing data.Citation40 More recently we established a specific RT signature for m1A, which allows its identification through high-throughput screening.Citation28 It is noteworthy that properties of different RT-enzymes are known to be different, and that the same modification may differentially affect the RT-incorporation profile, depending on the enzyme and buffer conditions used.
Figure 4. Applications of Class (I)techniques to A-to-(I)editing and m1A. Inosine, which is derived from A by RNA editing, generates “misincorporation” into cDNA, due to its base-pairing to C. The RT-signature contains only the “misincorporated” nucleotide compared to the reference sequence (left). m1A and other modified nucleotides with altered Watson-Crick edge generate complex RT-signatures composed of both RT-arrest and misincorporation at the modification site (right).
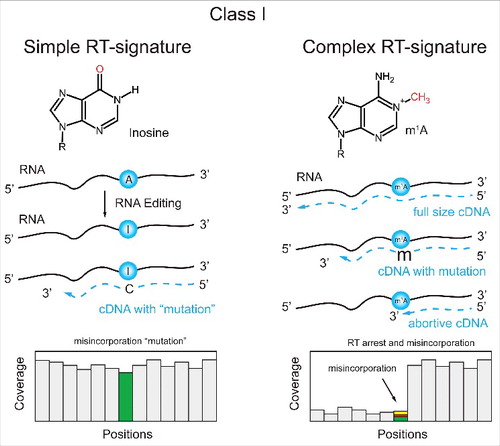
Figure 5. Applications of Class II techniques to RT-silent pseudouridine (Ψ) and m5C. Pseudouridine is reactive with water-soluble carbodiimide (CMCT) and forms stable adduct, while U-CMC adducts are removed by alkaline treatment. The resulting Ψ-CMC generates RT-arrest, detectable in the sequencing profile (left). 5-methylcytosine (m5C) is RT silent, but its detection is based on its insensitivity to bisulfite deamination. All C residues in RNA are deaminated by bisulfite, while m5C remains non-deaminated after treatment. The presence of residual C is thus detected by sequencing (right).Citation43
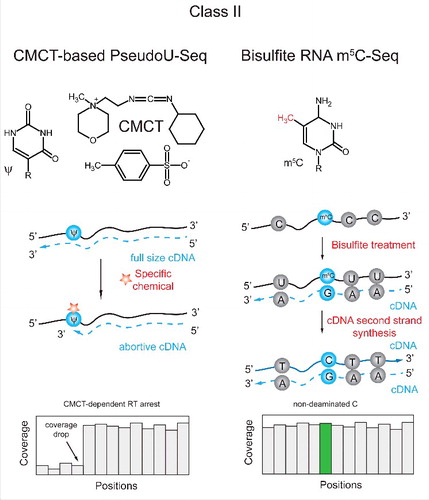
Class II: Chemical or biological induction of modification-specific RT signatures
Many modified RNA nucleotides are naturally RT-silent and do not generate any signal upon RT primer extension under normal conditions. However, the unique chemistry of the modification can be exploited to provoke misincorporation or truncation during reverse transcription. One example for such a modification is pseudouridine (Ψ). This modification is naturally silent, but can form an irreversible bulky chemical adduct with soluble carbodiimide (CMCT), that will lead to truncation of reverse transcription one nucleotide downstream of the modified site. This technique was classically coupled with primer extension assays to monitor pseudouridine presence at individual sites within tRNA and rRNA.Citation41,42 More recently, we and others employed this methodology to monitor pseudouridine levels in a transcriptome-wide manner, revealing widespread and dynamically regulated presence of pseudouridine in mRNA.Citation7,13,32,33 The hallmark of a pseudouridylated site in these approaches is a relatively high number of reads beginning precisely one nt downstream of the site with respect to the number of reads overlapping the site.
An additional modification which can be chemically induced to leave distinctive RT signature is 5-methylcytosine. Bisulfite treatment is a well known method for deoxy 5-methylcytosine (dm5C) mapping in DNA. dm5C residues are resistant to bisulfite deamination, while all unmodified dC are converted to dU (BS-Seq for DNA). A similar approach was developed also for ribo-m5C in RNA, although RNA is rather unstable under strong alkaline conditions required for efficient deamination. This approach was applied for mapping of m5C residues in tRNAs,Citation43,44 rRNAsCitation45,46 as well as in archaeal, yeast and human transcriptomes.Citation9,30
Ribose 2′-O-methylation can also yield specific RT profiles, if chemically pre-treated appropriately. Specifically, RiboMethSeq protocols for mapping 2′-O-Me rely on the ability of 2′-O-Me to protect the 3′-adjacent phosphodiester bond from alkaline cleavage ().Citation29,34 This reduced cleavage was explored to develop different versions of RiboMethSeq procedure for mapping and quantification of 2′-O-methylation in RNA, by seeking sites consistently failing to undergo cleavage. The hallmark of such sites in sequencing data are sites lacking reads beginning or ending precisely at them, i.e. gaps in 5′- (and 3′-) coverage profiles. The depth of the gap is proportional to the methylation rate, which provides the possibility for relative quantification. The method was successfully applied to map all known 2′-O-methylations in yeast and human rRNA, and also revealed several new, previously unknown modified positions.Citation29,34 Application of RiboMethSeq to other lower abundant RNAs is possible in principle, but will require substantial sequencing depth and/or pre-enrichment of RNAs of interest. Alternatively, 2′-O-Me can be detected based on pausing of reverse transcription that can be induced at 2′-O-Me sites at low dNTP concentration.Citation47 This property was extensively used for mapping of 2′-O-Me residues in rRNA and snRNAs, and has recently also been coupled with NGS approaches.Citation48,49
Figure 6. Complex cases of Class II protocols. Application for 2′-O-methylated residues, m1A and Inosine. Left: Detection of 2′-O-methylation is based on the 2′-O-Me-mediated protection of 3′-adjacent phosphodiester bond from nucleophilic cleavage. RNA is first randomly fragmented and a sequencing library is prepared from all generated fragments. Calculation of 5′-end (or cumulated 5′- and 3′-end) coverage reveals characteristic “gaps” one nucleotide downstream of the 2′-O-methylated site. The depth of the gap is proportional to methylation level of the nucleotide. Center: Conversion of m1A to m6A by Dimroth rearrangement. m1A residues present in RNA generate characteristic RT-signature, which disappears after alkaline treatment catalyzing m1A-to-m6A conversion. Comparison of 2 profiles allows identification of m1A site. Right: Specific Inosine detection by reaction with acrylonitrile (ICE-Seq). As discussed above, Inosine generates a simple RT-signature, which can be confounded with SNPs or with sequencing error. Acrylonitrile treatment converts Inosine into an RT-arresting residue, detected by characteristic coverage drop and misincorporation, thus confirming the presence of modified residue (right).
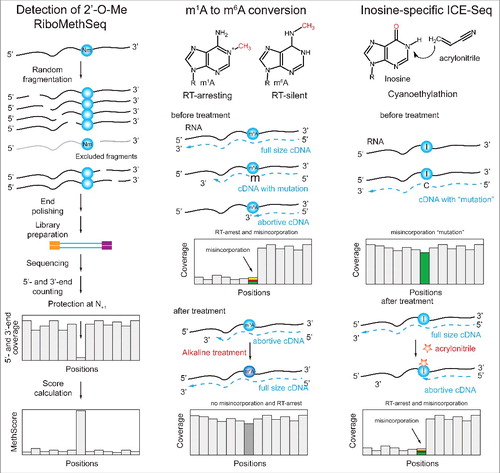
Recently, chemical approaches have also been utilized for mapping m1A. Under alkaline conditions, m1A can undergo a rearrangement to m6A (), also known as a Dimroth rearrangement.Citation11 Thus, the RT signature of m1A can be eliminated, by turning the m1A into m6A which is silent in terms of its RT signature. In this manner, specificity in the detection of m1A can be increased by identifying sites with mutation signatures exclusively under untreated conditions but not following rearrangement. An alternative approach for chemically converting m1A relied on pre-treating RNA with an m1A-demethylase, which, also, enabled identifying sites that responded to the demethylase treatment, and filtering out sites that did not as potential false positives.Citation35
A chemical pretreatment approach has also been developed for inosine. Although, as specified above, inosine can be directly detected based on it's A->G mutation profile, sequencing errors and genetic heterogeneity may affect interpretation of sequencing results. To allow definite identification of inosine sites, an approach relying on inosine cyanoethylation (), which leads to truncation of reverse transcription,Citation50 was combined with RNA-sequencing (ICE-Seq).Citation51,52
Class III: Enrichment of modified RNA fragments
An orthogonal approach for identifying modifications relies on a reagent that selectively binds to the modified RNA residues, and in this manner allows enriching for RNAs harboring the modification. In most cases utilized so far, this reagent is in the form of an antibody, raised against the modified RNA nucleoside. A classical modification which has been intensively studied by us and others over the past 5 years is 6-methyladenosine (m6A). The methodology to map m6A relies on the specificity of an anti-m6A antibody, that is applied to RNA fragmented into short (∼100 nt) pieces (). Following enrichment, cDNA libraries are generated and sequenced.Citation10,12 The hallmark of a methylated site in the resultant sequencing data is an accumulation of reads (‘peaks’) obtained in a specific region of the RNA upon enrichment using the antibody, but not in its absence. Importantly, in contrast to RT-based methodologies that can pinpoint modified sites at a single nucleotide resolution, antibody-based capture can only delineate a region that is likely to be modified, but lacks the single nucleotide resolution. Modification-specific antibodies coupled with high-throughput sequencing, using very similar protocols, have also been applied to map m5CCitation30 and m1A.Citation10,35
Figure 7. Approaches combining pulldown and specific chemistries. Left: Nucleotide-resolution detection of m1A residues by combination of MeRIP and iCLIP. m1A-containing RNA fragments are enriched after covalent cross-linking with specific anti m1A-Ab. After Ab removal by protease, the resulting covalent adducts are used for primer extension. Abortive RT products correspond to neighboring nucleotides around m1A site. Right: Enrichment of pseudouridine-containing RNA fragments using bi-functional “clickable” CMCT. RNA is first randomly fragmented and subjected to reaction with soluble carbodiimide CMCT bearing “clickable” azide (N3) group. After “click” reaction with alkyne-modified biotin, modified fragments are enriched by avidin-beads pull-out. Modified residues are detected based on their RT-arrest signature.
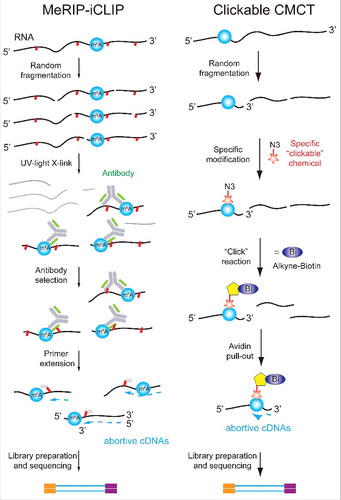
Figure 8. Sources of artifacts in Class I, II and III techniques. Left: Identification of RT-signatures in Class I methods may be affected by the presence of genomic SNPs, by possible mis-aligned nucleotides at exon-intron borders and by errors in the sequencing data set. Center: Class II approaches may generate false-positive signals due to strong non-specific cleavage sites in RNAs, due to mis-alignment of some reads to repetitive RNA sequences, due to unannotated transcription sites. Right: Class III approaches based on Ab enrichment may suffer from non-specific enrichment signals, from Ab promiscuity or mis-alignments. Important controls for these approaches include the coverage profile in the input (top trace), and the enrichment profile for KO or deleted strain (middle). Only specific peaks present in WT sample and absent in control traces should be considered as candidates. The insert shows that the position of the modified residue does not always correspond to the maximal enrichment.
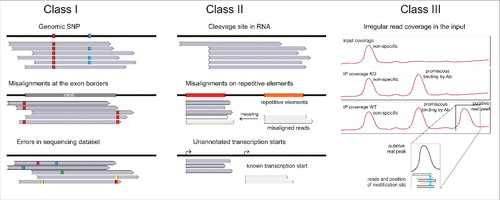
It should be pointed out that the specificity of an Ab for a given modified nucleotide is essential for successful application of these enrichment techniques. To date, highly specific Ab preparations were made only for a very limited subset of modified nucleosides. This limitation is probably related to the small size of antigen used (modified nucleoside).
Combined approaches
It is often advantageous to combine different classes of approaches, to increase specificity and/or sensitivity of detection. Several examples for such combined approaches have emerged in recent years. In particular, approaches combining pre-enrichment of modified fragments with RT mutation profiling have been very fruitful. One such example is m6A (), where UV cross-linking of an anti-m6A antibody to RNA leads to characteristic RT signature at the modified sites.Citation53,54 Use of this combined approach allows both pre-enrichment of a methylated fragment, hence reducing coverage requirements, and leads to an RT signature that provides single nucleotide resolution.
An additional example for a combined protocol is mapping pseudouridines, using a biotinylated carbodiimide (). This bi-functional reagent allows both enriching for fragments containing pseudouridine (reducing sequence coverage requirements), and to obtain single-nucleotide resolution based on the RT signature.Citation33 The recent transcriptome-wide approaches for mapping m1A have also relied on combined approaches, by combining pulldown of m1A containing fragments using an anti-m1A antibody with chemical conversion of m1A to m6A.Citation11,35
Considerations in the analysis of transcriptome-wide maps of RNA modifications
Principles of analysis of transcriptome-wide maps of RNA modifications
The different classes of experimental approaches require diverse computational approaches for their analysis. Available tools performing such analyses from start to finish are to a large extent lacking. Although existing tools may partially help in the analysis, these should be used with caution, as they were often developed for analyses of other types of experiments, which can differ in important ways in terms of underlying assumptions about the distribution of the data, sources of false positives associated with the data, and hence also appropriate background models. For example, tools have been developed to identify mutations and truncations in HITS-CLIP and PAR-CLIP data, where they are indicative of sites of cross-linking.Citation55-57 However, in these flavors of experiments aiming to identify protein binding sites, binding is typically not assumed to occur at an individual site, but across a region. Hence, these methodologies typically seek for evidence of clusters of mutations/truncations, unlike the case of RNA modifications that can often occur in isolation, and at a single nucleotide. In addition, whereas in CLIP-seq protocols there is often no ‘good’ external control for the data and hence many of the analytic pipelines do not offer the possibility of integrating such a control, its availability in type II protocols requires approaches that can make suitable use of it. Various well-used pipelines also exist for identifying mutations in DNA data. However, the complex processing of RNAs, involving splicing, cleavage and polyadenylation, renders it crucial to carefully weed out artifacts that can be caused by misaligned reads. Similarly, many approaches exist for calling peaks in ChIP-seq data. These approaches, however, are only poorly suited for peak calling in RNA-seq, because the distribution of DNA-seq and RNA-seq data are fundamentally different. Thus, for peak calling in DNA, the background ‘expected’ level of reads is typically estimated, to some extent or another, from the entire genome, based on which deviates are identified and flagged as peaks. Such a genomic background is flawed in the context of RNA, given that aggregating reads over the entire genome leads to an uninformative average of regions expressed at widely varying levels. Instead, the background needs to be estimated on a gene-by-gene, or even region by region, basis using either intrinsic or extrinsic data.Citation10,12
Nonetheless, some tools have been developed in recent years aiming to identify RNA modifications from dedicated protocols. These include tools for identifying modifications leading to RT termination,Citation58 as well as for searching for peaks in m6A-seq data.Citation59 There remains a need for additional tools, supporting more complex functionalities, such as incorporating more controls and developing models for differential analyses of modification states between conditions/perturbations.
Can quantitative information be derived from transcriptome-wide mapping?
An important aspect to consider analytically is whether a protocol for mapping an RNA modification can also be considered quantitative, i.e., whether it has the capacity to adequately provide an estimate for the proportion of transcripts modified at a given site. Class I and Class II protocols can theoretically provide such an estimate. In particular, modifications resulting in misincorporation of a single defined nucleotide, such as inosine or m5C following bisulfite treatment, allow simple counting of wild type vs. misincorporated nucleotide.Citation30,39,43 However, obtaining absolute quantifications can be made challenging through incomplete chemical conversion in Class II protocols, as well as by the dependency of RT misincorporations on sequence and structural elements.Citation28 Thus, such protocols need to be carefully calibrated to assess whether they can provide absolute quantifications of modification levels. Nonetheless, these protocols can, in principle, be suitable for relative quantifications (i.e. comparing levels of a specific modified site between samples). The same is true also for Class I and II protocols leading to RT termination. We have previously shown, for pseudouridine, that use of the ratio between reads truncating at a site and reads overlapping it provides a highly quantitative relative quantification of pseudouridine levels, as measured by synthetic spike-ins, while providing a substantial underestimate of the absolute modification levels.Citation7 Rather consistent, but still relative, quantification of 2′-O-Me residues was reported for different variants of RiboMethSeq, but this quantification is based on the assumption that 2′-O-methylation fully protects the adjacent phosphodiester bond from cleavage.Citation29,34 Class III protocols, relying on pre-enrichment, were generally considered to be even less quantitative, due to partial pulldown efficiency. However, recently a carefully calibrated pulldown procedure of m6A-containing fragments was demonstrated to provide quantitative estimates of methylation stoichiometry.Citation60
Challenges in interpretation of transcriptome-wide maps
Interpretation of transcriptome-wide maps of RNA modifications is often not trivial. A major source of challenge is due to the fact that the measurements are sought for a single nucleotide. Thus, in contrast to analyses of gene expression, for example, in which statistical robustness and power can be increased by aggregating reads from an entire region (e.g. gene), in the case of RNA modifications the measurements encompass only a single nucleotide, in particular in Class I and Class II type approaches. A second major challenge stems from substantial levels of background that is often present in such maps. Class I and Class II type measurements hinge on the ability to discriminate between misincorporation or pauses of RT that are due to a modification and between ‘background’ pauses or misincorporation, that can be due to RNA structure, RT error rate, complex processing of the RNA, genomic misalignments of sequencing reads and technical errors of the sequencing platform.Citation61-64 Similarly, antibodies can also suffer from substantial background and selectively capture unmodified fragments in a sequence dependent manner, as we demonstrated in the case of the anti-m6A antibody.Citation8 A third major challenge has to do with relatively low number of truly modified sites within the vast mRNA repertoire. Even a modification mapping approach that performs extremely well can give rise to a considerable number of false positives once it is applied to every nucleotide in the transcriptome. For instance, application of a method with a 1% false discovery rate can give rise to 10,000 false positive sites once applied to a 1 million nt transcriptome.
Accordingly, it is of crucial importance to carefully design controls for the mapping experiments. Often, intrinsic controls are available, in the form of well-documented modified sites in tRNA and in rRNA, and the sensitivity and specificity of analytic approaches can be evaluated based on such known sites. Nonetheless, it is important to keep in mind that an analytical approach that is highly sensitive and specific in detection of sites on rRNA may suffer from poorer performance when applied to mRNA. This can be due to both biological and technical reasons. At the biological level, modifications on rRNA can often be stoichiometric (i.e., be present on almost 100% of rRNA molecules), whereas within mRNA they will only be present at a fraction of the transcripts, hence decreasing signal:noise ratios. Additionally, high sensitivity and specificity at sites within rRNA are also due to the high sequencing depth that can be obtained for these highly expressed species, which can easily provide thousands of reads overlapping an individual site and hence yield robust estimates of mutations and/or truncations. In practice, such coverage is unrealistic for the majority of low abundant transcripts, and hence the RT signatures can be less robust, which can lead to difficulties both in identification and in quantification of modifications.
To further aid in the evaluation of performance, synthetic RNA oligos can be spiked into experiments. The ability to control the modification stoichiometries at these synthetic oligos and the ability to spike them in at varying concentrations offer important advantages both for establishing the signal:noise ratio of a protocol and for exploring its ability to quantify modification stoichiometry.Citation7,9,60 The disadvantages of such oligos is that they are typically spiked in following extraction of the RNA, and thus not subjected to precisely the same procedure as the endogenous sites.
Class II and III approaches offer additional important extrinsic controls, in the forms of RNA not subjected to the chemical pre-treatment or to selective pre-enrichment but otherwise identically handled. These controls can subsequently be used to define the experimental background, and help to filter out sites which are not eliminated in the control.
Additionally, performing these experiments in a number of replicates is of great importance, and can help to filter out sites that are spuriously detected in only a subset of experiments. Such replicates are of great use to evaluate the robustness and reproducibility of detection and of quantification.
In our experience, none of these controls completely eliminate the above-discussed sources of false positives. Thus, additional orthogonal controls and analyses can help further reduce the false detection rate, and allow focusing on truly modified sites. Such orthogonal controls can be genetic, in cases in which the enzymes required for RNA modification are known. Mapping the modification in mutants in which these enzymes are either eliminated or over-expressed allows identifying sites sensitive to this perturbation. An additional orthogonal confirmation can come from in-depth characterization of the modified sites. Many well-understood modifications, such as m6A or pseudouridine, occur at specific sequence and/or structural motifs.Citation7,10,12,13,32 Thus, filtering putative modified sites on the basis of such elements can help reduce the false positive rate, although if this is imposed as a hard filter this can increase the false negative rate.
Conclusion
Substantial advances have been made in recent years in expanding the view of RNA modifications from tRNA and rRNA molecules, in which they were classically studied, to other species of RNA, most notable of which mRNA. These advances have been facilitated, to a large extent, by next-generation sequencing approaches. Technologies allowing direct sequencing of RNA, such as nanopores and single molecule sequencing approaches, hold particular promise to allow direct measurement of modification states in the context of full length RNAs, although their utility remains to be established. Up to now, out of > 150 chemically different modified residues, only a small minority was detected at the transcriptome-wide level and confirmed by independent complementary approaches. We anticipate that in the future, protocols will be developed to allow mapping of additional modifications, and analytic tools will emerge enabling more precise mapping, quantification and differential detection. These should aid in addressing the major open questions, pertaining to the functions and mechanisms of action of modified RNA nucleotides.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank S. Schwartz and Y. Motorin lab members for comments on the manuscript. This work was supported by joint ANR-DFG grant HTRNAMod (ANR-13-ISV8-0001/HE 3397/8-1) to YM.
Funding
SS was supported by research grants from The Abramson Family Center for Young Scientists, the David and Fela Shapell Family Foundation INCPM Fund for Preclinical Studies, the Estate of David Turner, Berlin Family Foundation New Scientist Fund, the Abisch Frenkel Foundation and by the Israel Science Foundation (grant No. 543165).
Related Research Data
References
- Lee Y, Rio DC. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu Rev Biochem 2015; 84:291-323; PMID:25784052; http://dx.doi.org/10.1146/annurev-biochem-060614-034316
- Licht K, Jantsch MF. Rapid and dynamic transcriptome regulation by RNA editing and RNA modifications. J Cell Biol 2016; 213:15-22; PMID:27044895; http://dx.doi.org/10.1083/jcb.201511041
- Wu R, Jiang D, Wang Y, Wang X. N (6)-methyladenosine (m(6)A) methylation in mRNA with A dynamic and reversible epigenetic modification. Mol Biotechnol 2016; 58:450-9; PMID:27179969; http://dx.doi.org/10.1007/s12033-016-9947-9
- Jia G, Fu Y, He C. Reversible RNA adenosine methylation in biological regulation. Trends Genet 2013; 29:108-15; PMID:23218460; http://dx.doi.org/10.1016/j.tig.2012.11.003
- El Yacoubi B, Bailly M, de Crécy-Lagard V. Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu Rev Genet 2012; 46:69-95; PMID:22905870; http://dx.doi.org/10.1146/annurev-genet-110711-155641
- Yi C, Pan T. Cellular dynamics of RNA modification. Acc Chem Res 2011; 44:1380-8; PMID:21615108; http://dx.doi.org/10.1021/ar200057m
- Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, León-Ricardo BX, Engreitz JM, Guttman M, Satija R, Lander ES, et al. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 2014; 159:148-62; PMID:25219674; http://dx.doi.org/10.1016/j.cell.2014.08.028
- Schwartz S, Agarwala SD, Mumbach MR, Jovanovic M, Mertins P, Shishkin A, Tabach Y, Mikkelsen TS, Satija R, Ruvkun G, et al. High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell 2013; 155:1409-21; PMID:24269006; http://dx.doi.org/10.1016/j.cell.2013.10.047
- Squires JE, Patel HR, Nousch M, Sibbritt T, Humphreys DT, Parker BJ, Suter CM, Preiss T. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res 2012; 40:5023-33; PMID:22344696; http://dx.doi.org/10.1093/nar/gks144
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012; 485:201-6; PMID:22575960; http://dx.doi.org/10.1038/nature11112
- Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, Peer E, Kol N, Ben-Haim MS, Dai Q, Di Segni A, Salmon-Divon M, Clark WC, et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature [ Internet] 2016; 530:441-6; PMID:26863196; http://dx.doi.org/10.1038/nature16998
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 2012; 149:1635-46; PMID:22608085; http://dx.doi.org/10.1016/j.cell.2012.05.003
- Carlile TM, Rojas-Duran MF, Zinshteyn B, Shin H, Bartoli KM, Gilbert WV. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 2014; 515:143-6; PMID:25192136; http://dx.doi.org/10.1038/nature13802
- Grosjean H, Droogmans L, Roovers M, Keith G. Detection of enzymatic activity of transfer RNA modification enzymes using radiolabeled tRNA substrates. Methods Enzymol 2007; 425:55-101; PMID:17673079; http://dx.doi.org/10.1016/S0076-6879(07)25003-7
- Grosjean H, Keith G, Droogmans L. Detection and quantification of modified nucleotides in RNA using thin-layer chromatography. Methods Mol Biol 2004; 265:357-91; PMID:15103084; http://dx.doi.org/10.1385/1-59259-775-0:357
- Keith G. Mobilities of modified ribonucleotides on two-dimensional cellulose thin-layer chromatography. Biochimie 1995; 77:142-4; PMID:7599271; http://dx.doi.org/10.1016/0300-9084(96)88118-1
- Liu N, Parisien M, Dai Q, Zheng G, He C, Pan T. Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA 2013; 19:1848-56; PMID:24141618; http://dx.doi.org/10.1261/rna.041178.113
- Thüring K, Schmid K, Keller P, Helm M. Analysis of RNA modifications by liquid chromatography-tandem mass spectrometry. Methods [ Internet] 2016; 107:48-56; PMID:27020891; http://dx.doi.org/10.1016/j.ymeth.2016.03.019
- Kellner S, Ochel A, Thüring K, Spenkuch F, Neumann J, Sharma S, Entian K-D, Schneider D, Helm M. Absolute and relative quantification of RNA modifications via biosynthetic isotopomers. Nucleic Acids Res 2014; 42:e142; PMID:25129236; http://dx.doi.org/10.1093/nar/gku733
- Limbach PA, Paulines MJ. Going global: the new era of mapping modifications in RNA. Wiley Interdiscip Rev RNA 2016 Jun 1; PMID:27251302; http://dx.doi.org/10.1002/wrna.1367
- Beverly MB. Applications of mass spectrometry to the study of siRNA. Mass Spectrom Rev 2011; 30:979-98; PMID:20201110; http://dx.doi.org/10.1002/mas.20260
- Basiri B, Bartlett MG. LC-MS of oligonucleotides: applications in biomedical research. Bioanalysis 2014; 6:1525-42; PMID:25046052; http://dx.doi.org/10.4155/bio.14.94
- Motorin Y, Muller S, Behm‐Ansmant I, Branlant C. Identification of Modified Residues in RNAs by Reverse Transcription‐Based Methods. In: Methods in Enzymology. Academic Press; 2007. page 21-53.
- Schadt EE, Turner S, Kasarskis A. A window into third-generation sequencing. Hum Mol Genet 2010; 19:R227-40; PMID:20858600; http://dx.doi.org/10.1093/hmg/ddq416
- Vilfan ID, Tsai YC, Clark TA, Wegener J, Dai Q, Yi C, Pan T, Turner SW, Korlach J. Analysis of RNA base modification and structural rearrangement by single-molecule real-time detection of reverse transcription. J Nanobiotechnology 2013; 11:8; PMID:23552456; http://dx.doi.org/10.1186/1477-3155-11-8
- Saletore Y, Meyer K, Korlach J, Vilfan ID, Jaffrey S, Mason CE. The birth of the Epitranscriptome: deciphering the function of RNA modifications. Genome Biol 2012; 13:175; PMID:23113984; http://dx.doi.org/10.1186/gb-2012-13-10-175
- Cahová H, Winz ML, Höfer K, Nübel G, Jäschke A. NAD captureSeq indicates NAD as a bacterial cap for a subset of regulatory RNAs. Nature 2015; 519:374-7; PMID:Can't; http://dx.doi.org/10.1038/nature14020
- Hauenschild R, Tserovski L, Schmid K, Thüring K, Winz M-L, Sharma S, Entian KD, Wacheul L, Lafontaine DLJ, Anderson J, et al. The reverse transcription signature of N-1-methyladenosine in RNA-Seq is sequence dependent. Nucleic Acids Res 2015; 43:9950-64; PMID:26365242; http://dx.doi.org/10.1093/nar/gkv895
- Birkedal U, Christensen-Dalsgaard M, Krogh N, Sabarinathan R, Gorodkin J, Nielsen H. Profiling of Ribose Methylations in RNA by High-Throughput Sequencing*. Angew Chem Int Ed Engl 2015; 54:451-5; PMID:25417815; http://dx.doi.org/10.1002/anie.201408362
- Edelheit S, Schwartz S, Mumbach MR, Wurtzel O, Sorek R. Transcriptome-Wide Mapping of 5-methylcytidine RNA Modifications in Bacteria, Archaea, and Yeast Reveals m 5 C within Archaeal mRNAs. PLoS Genet 2013; 9:e1003602; PMID:23825970; http://dx.doi.org/10.1371/journal.pgen.1003602
- Hussain S, Sajini AA, Blanco S, Dietmann S, Lombard P, Sugimoto Y, Paramor M, Gleeson JG, Odom DT, Ule J, et al. NSun2-Mediated Cytosine-5 Methylation of Vault Noncoding RNA Determines Its Processing into Regulatory Small RNAs. Cell Rep 2013; 4:255-61; PMID:23871666; http://dx.doi.org/10.1016/j.celrep.2013.06.029
- Lovejoy AF, Riordan DP, Brown PO. Transcriptome-wide mapping of pseudouridines: pseudouridine synthases modify specific mRNAs in S. cerevisiae. PLoS One 2014; 9:e110799; PMID:25353621; http://dx.doi.org/10.1371/journal.pone.0110799
- Li X, Zhu P, Ma S, Song J, Bai J, Sun F, Yi C. Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat Chem Biol 2015; 11:592-7; PMID:26075521; http://dx.doi.org/10.1038/nchembio.1836
- Marchand V, Blanloeil-Oillo F, Helm M, Motorin Y. Illumina-based RiboMethSeq approach for mapping of 2′-O-Me residues in RNA. Nucleic Acids Res [ Internet] 2016; 44:e135
- Li X, Xiong X, Wang K, Wang L, Shu X, Ma S, Yi C. Transcriptome-wide mapping reveals reversible and dynamic N1-methyladenosine methylome. Nat Chem Biol [ Internet] 2016 [cited 2016 Feb 15]; 12:311-6
- Khoddami V, Cairns BR. Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nat Biotechnol 2013; 31:458-64; PMID:23604283; http://dx.doi.org/10.1038/nbt.2566
- Haag S, Warda AS, Kretschmer J, Günnigmann MA, Höbartner C, Bohnsack MT. NSUN6 is a human RNA methyltransferase that catalyzes formation of m5C72 in specific tRNAs. RNA 2015; 21:1532-43; PMID:26160102; http://dx.doi.org/10.1261/rna.051524.115
- Khoddami V, Cairns BR. Transcriptome-wide target profiling of RNA cytosine methyltransferases using the mechanism-based enrichment procedure Aza-IP. Nat Protoc 2014; 9:337-61; PMID:24434802; http://dx.doi.org/10.1038/nprot.2014.014
- Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol 2004; 22:1001-5; PMID:15258596; http://dx.doi.org/10.1038/nbt996
- Ryvkin P, Leung YY, Silverman IM, Childress M, Valladares O, Dragomir I, Gregory BD, Wang L-S. HAMR: high-throughput annotation of modified ribonucleotides. RNA 2013; 19:1684-92; PMID:24149843; http://dx.doi.org/10.1261/rna.036806.112
- Bakin AV, Ofengand J. Mapping of pseudouridine residues in RNA to nucleotide resolution. Methods Mol Biol 1998; 77:297-309; PMID:9770678
- Ofengand J, Bakin A. Mapping to nucleotide resolution of pseudouridine residues in large subunit ribosomal RNAs from representative eukaryotes, prokaryotes, archaebacteria, mitochondria and chloroplasts. J Mol Biol 1997; 266:246-68; PMID:9047361; http://dx.doi.org/10.1006/jmbi.1996.0737
- Schaefer M, Pollex T, Hanna K, Lyko F. RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res 2009; 37:e12; PMID:19059995; http://dx.doi.org/10.1093/nar/gkn954
- Tuorto F, Liebers R, Musch T, Schaefer M, Hofmann S, Kellner S, Frye M, Helm M, Stoecklin G, Lyko F. RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat Struct Mol Biol 2012; 19:900-5; PMID:22885326; http://dx.doi.org/10.1038/nsmb.2357
- Gigova A, Duggimpudi S, Pollex T, Schaefer M, Koš M. A cluster of methylations in the domain IV of 25S rRNA is required for ribosome stability. RNA 2014; 20:1632-44; PMID:25125595; http://dx.doi.org/10.1261/rna.043398.113
- Bourgeois G, Ney M, Gaspar I, Aigueperse C, Schaefer M, Kellner S, Helm M, Motorin Y. Eukaryotic rRNA modification by yeast 5-methylcytosine-methyltransferases and human proliferation-associated antigen p120. PLoS One 2015; 10:e0133321; PMID:26196125; http://dx.doi.org/10.1371/journal.pone.0133321
- Maden BE. Mapping 2′-O-methyl groups in ribosomal RNA. Methods 2001; 25:374-82; PMID:11860292; http://dx.doi.org/10.1006/meth.2001.1250
- Jorjani H, Kehr S, Jedlinski DJ, Gumienny R, Hertel J, Stadler PF, Zavolan M, Gruber AR. An updated human snoRNAome. Nucleic Acids Res 2016; 44:5068-82; PMID:27174936; http://dx.doi.org/10.1093/nar/gkw386
- Incarnato D, Anselmi F, Morandi E, Neri F, Maldotti M, Rapelli S, Parlato C, Basile G, Oliviero S. High-throughput single-base resolution mapping of RNA 2′-O-methylated residues. Nucleic Acids Res [ Internet] 2016; Available from: http://nar.oxfordjournals.org/content/early/2016/09/09/nar.gkw810.abstract; PMID:27614074
- Sakurai M, Yano T, Kawabata H, Ueda H, Suzuki T. Inosine cyanoethylation identifies A-to-I RNA editing sites in the human transcriptome. Nat Chem Biol 2010; 6:733-40; PMID:20835228; http://dx.doi.org/10.1038/nchembio.434
- Suzuki T, Ueda H, Okada S, Sakurai M. Transcriptome-wide identification of adenosine-to-inosine editing using the ICE-seq method. Nat Protoc 2015; 10:715-32; PMID:25855956; http://dx.doi.org/10.1038/nprot.2015.037
- Sakurai M, Ueda H, Yano T, Okada S, Terajima H, Mitsuyama T, Toyoda A, Fujiyama A, Kawabata H, Suzuki T. A biochemical landscape of A-to-I RNA editing in the human brain transcriptome. Genome Res 2014; 24:522-34; PMID:24407955; http://dx.doi.org/10.1101/gr.162537.113
- Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods [ Internet] 2015; 12:767-72; PMID:26121403; http://dx.doi.org/10.1038/nmeth.3453
- Chen K, Lu Z, Wang X, Fu Y, Luo GZ, Liu N, Han D, Dominissini D, Dai Q, Pan T, et al. High-resolution N(6) -methyladenosine (m(6) A) map using photo-crosslinking-assisted m(6) A sequencing. Angew Chem Int Ed Engl 2015; 54:1587-90; PMID:25491922; http://dx.doi.org/10.1002/anie.201410647
- Uren PJ, Bahrami-Samani E, Burns SC, Qiao M, Karginov FV, Hodges E, Hannon GJ, Sanford JR, Penalva LOF, Smith AD. Site identification in high-throughput RNA–protein interaction data. Bioinformatics 2012; 28:3013-20; PMID:23024010; http://dx.doi.org/10.1093/bioinformatics/bts569
- Corcoran DL, Georgiev S, Mukherjee N, Gottwein E, Skalsky RL, Keene JD, Ohler U. PARalyzer: definition of RNA binding sites from PAR-CLIP short-read sequence data. Genome Biol 2011; 12:R79; PMID:21851591; http://dx.doi.org/10.1186/gb-2011-12-8-r79
- Sievers C, Schlumpf T, Sawarkar R, Comoglio F, Paro R. Mixture models and wavelet transforms reveal high confidence RNA-protein interaction sites in MOV10 PAR-CLIP data. Nucleic Acids Res 2012; 40:e160; PMID:22844102; http://dx.doi.org/10.1093/nar/gks697
- Li B, Tambe A, Aviran S, Pachter L. Prober: A general toolkit for analyzing sequencing-based “toeprinting” assays [Internet]. 2016. Available from: http://biorxiv.org/lookup/doi/10.1101/063107
- Cui X, Meng J, Zhang S, Chen Y, Huang Y. A novel algorithm for calling mRNA m6A peaks by modeling biological variances in MeRIP-seq data. Bioinformatics 2016; 32:i378-85; PMID:27307641; http://dx.doi.org/10.1093/bioinformatics/btw281
- Molinie B, Wang J, Lim KS, Hillebrand R, Lu ZX, Van Wittenberghe N, Howard BD, Daneshvar K, Mullen AC, Dedon P, et al. m(6)A-LAIC-seq reveals the census and complexity of the m(6)A epitranscriptome. Nat Methods 2016; 13:692-8; PMID:27376769; http://dx.doi.org/10.1038/nmeth.3898
- Li M, Wang IX, Li Y, Bruzel A, Richards AL, Toung JM, Cheung VG. Widespread RNA and DNA sequence differences in the human transcriptome. Science 2011; 333:53-8; PMID:21596952; http://dx.doi.org/10.1126/science.1207018
- Pickrell JK, Gilad Y, Pritchard JK. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome.” Science 2012; 335:1302; author reply 1302; PMID:22422963; http://dx.doi.org/10.1126/science.1210484
- Kleinman CL, Majewski J. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome.” Science 2012; 335:1302; author reply 1302; PMID:22422962; http://dx.doi.org/10.1126/science.1209658
- Lin W, Piskol R, Tan MH, Li JB. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome.” Science 2012; 335:1302; author reply 1302; PMID:22422964; http://dx.doi.org/10.1126/science.1210624