
ABSTRACT
Extracellular vesicles (EVs) contain a wide range of RNA types with a reported prevalence of non-coding RNA. To date a comprehensive characterization of the protein coding transcripts in EVs is still lacking. We performed RNA-Sequencing (RNA-Seq) of 2 EV populations and identified a small fraction of transcripts that were expressed at significantly different levels in large oncosomes and exosomes, suggesting they may mediate specialized functions. However, these 2 EV populations exhibited a common mRNA signature that, in comparison to their donor cells, was significantly enriched in mRNAs encoding E2F transcriptional targets and histone proteins. These mRNAs are primarily expressed in the S-phase of the cell cycle, suggesting that they may be packaged into EVs during S-phase. In silico analysis using subcellular compartment transcriptome data from the ENCODE cell line compendium revealed that EV mRNAs originate from a cytoplasmic RNA pool. The EV signature was independently identified in plasma of patients with breast cancer by RNA-Seq. Furthermore, several transcripts differentially expressed in EVs from patients versus controls mirrored differential expression between normal and breast cancer tissues. Altogether, this largest high-throughput profiling of EV mRNA demonstrates that EVs carry tumor-specific alterations and can be interrogated as a source of cancer-derived cargo.
Introduction
Analysis of single mRNAs by PCR suggests that the presence of quantifiable tumor RNA in plasma may allow the development of non-invasive diagnostic and prognostic tools.Citation1 Extracellular RNA (exRNA) in body fluids is protected from RNases by RNA binding proteins and extracellular vesicles (EVs).Citation2-4 EVs are membrane-enclosed particles that are shed by all cell types in unicellular and multicellular organisms during normal and pathological states, including cancer.Citation5 EVs play an important role in intercellular communication.Citation6-8 Increasing in vitro and in vivo evidence indicates that they act through specific mechanisms involving the transfer of oncogenic molecules between cell and tissue compartments, including at metastatic sites.Citation7-9 It is increasingly clear that cancer cells can shed heterogeneous populations of EVs, which differ in size, molecular cargo, function, and likely in biogenesis.Citation10,11 The term “EVs” includes 30–150 nm exosomes (Exo), as well as larger particles, frequently described as microvesicles or ectosomes (up to 1 μm diameter).Citation12 A new category of atypically large EVs (1–10 μm), referred to as large oncosomes (LO), results from the shedding of non-apoptotic membrane blebs produced by highly migratory and metastatic “amoeboid” cancer cells.Citation13,14 LO are associated with advanced prostate cancer in vivo and can also be produced by cancer cells of different organ types in association with invasive behavior and other aggressive characteristics.Citation15,16 Recent studies suggest that diverse classes of EVs might contain different RNA profiles, as demonstrated by electropherogram.Citation17
Molecules carried in EVs are functional and can potentially be used as clinical biomarkers. EV RNA appears particularly promising with respect to the potential of developing minimally invasive tests with high sensitivity and specificity.Citation8 RNA types identified in EVs include miRNA (often reported as the most abundant RNA species in EVs), ribosomal RNA (rRNA), transfer RNA (tRNA), long non-coding RNA (lncRNA), piwi-interacting RNA (piRNA), small nuclear RNA and small nucleolar RNA (snoRNA).Citation18 messenger RNA (mRNA) has also been identified in EVs from cancer cells as a functional regulator of target cell behavior,Citation2,3,8 and functional transfer of RNA has been displayed in mice using a Cre recombinase system for in vivo identification of tumor cells that take up EVs.Citation19,20
Patterns of mRNA enrichment in EVs can vary significantly across cell types and populations.Citation8 For example, mRNAs involved in cell migration, angiogenesis, and cell proliferation have been identified in glioblastoma cell-derived EVs, whereas mouse mast cells (MC/9) contain mRNAs responsible for protein synthesis, RNA post-transcriptional modification and cellular development.Citation2,3 Despite some evidence suggesting the possibility of a common sequence in the 3′-UTR of mRNAs enriched in EVs,Citation21 the exact mechanism for selective packaging of mRNA in EVs remains unknown.Citation8 Previous studies on comparative miRNA profiling in EVs and donor cells suggest that miRNA secretion is an active, ATP-dependent process, and that specific types of miRNA are exported in EVs.Citation22,23 Conversely, data showing that extracellular miRNA levels mirror their intracellular abundances favor the hypothesis that miRNA export is a non-selective process.Citation8 So far, RNA profiling in EVs has been commonly performed by qRT-PCR analysis of single RNAs, rather than by large-scale profiling, an approach that relies on previously discovered RNAs.Citation3 RNA-Seq has only been employed in a few studies to characterize EV RNA.Citation10,24-26 However, because of a reported predominance of short non-coding RNA in EVs, these RNA-Seq studies have been conducted using small RNA libraries that by design exclude long mRNA.
In the present study, we employed exome capture-based RNA-Seq to profile the mRNA in 2 types of EVs, Exo and LO, and to determine whether the observed differences between EVs and originating glioblastoma cells might inform the derivation of EVs from a particular subcellular compartment. We examined differences in the mRNA splicing state, level of enrichment of mRNAs encoding a signal peptide, and the content of short half-life mRNAs between the EVs and cells. We extended the analysis to an ENCODE collection of whole-cell, cytosolic, and nuclear mRNA fractions obtained from diverse cancer cell lines in order to derive more general information on the subcellular derivation of the mRNA exported in EVs. Finally, we analyzed the whole transcriptome of EVs purified from the plasma of 10 patients with breast cancer and 5 control individuals to test the potential value of circulating EVs as carriers of tumor-derived RNA.
Results
RNA can be extracted from both large oncosomes and exosomes
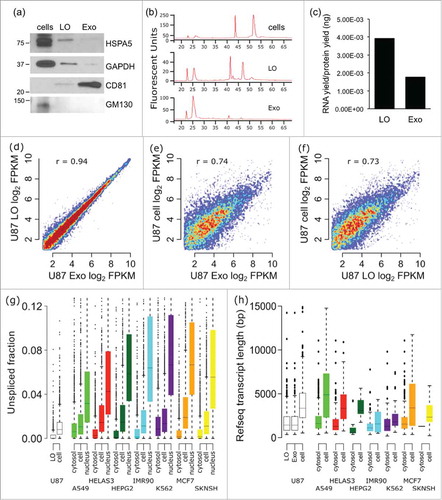
LO and Exo were isolated from the conditioned medium of glioblastoma U87 cells by a differential centrifugation-based protocol that can differentiate LO from Exo, as confirmed by flotation in iodixanol.Citation11 Western blotting with GAPDH and HSPA5, which are enriched in LO,Citation11 CD81, which is typically enriched in Exo,Citation11,27 and the Golgi protein GM130, which is typically excluded from EVs,Citation28 confirmed the purity of our preparations (). Analysis of the RNA quality by electropherogram demonstrated that 18S and 28S rRNA peaks and longer RNA species (up to 4,000 bp) were well recognizable in the LO preparations while undetectable in Exo (, Fig. S1). Along with this qualitative difference in RNA profiles, RNA yields, normalized to the number of originating cells and total protein amount, was also higher in LO (2.2-fold) than in Exo ().
Figure 1. The mRNA profile of U87 EVs and cells are significantly different. mRNA was isolated from U87 exosomes (Exo), large oncosomes (LO), and U87 cells, and was profiled by RNA-Seq. (A) The purity of LO and Exo fractions was confirmed via Western blotting for HSPA5, GAPDH, and CD81. (B) The electropherograms show the time distribution and fluorescence intensity (FU) of total RNA in LO, Exo, and cells. (C) RNA yields from U87 LO were substantially higher than RNA yields from U87 Exo. (D) The mRNA profiles of the Exo and LO EV fractions correlate very well (r = 0.94). (E-F) The mRNA profiles of both Exo and LO exhibited significant differences with the mRNA profile of U87 donor cells (r = 0.74, and 0.73 respectively). (G) The mRNA in U87 LO is less unspliced (more spliced) than the mRNA in U87 cells. The fraction of unspliced RNA-Seq reads was calculated for each gene in U87 LO and U87 cell data sets, in comparison with ENCODE RNA-Seq database on whole cell, cytosol, and nucleus from various cancer cell lines. * - p < 0.001. (H) U87 LO and Exo fractions are enriched for short mRNA transcripts in comparison with U87 cells. The annotated transcript length was found for mRNAs enriched or depleted in U87 EVs vs. cells, and in ENCODE cytosol vs. whole-cells.
Vesicular mRNA reflects the profile of the originating tumor cells
In order to determine whether the mRNA content of the EVs reflects the mRNA of the originating tumor cells, we isolated and sequenced mRNA populations from U87 cells and from derived LO and Exo. We obtained a minimum of 50 million paired end reads over 2 million uniquely mapped reads in Exo, and over 5 million uniquely mapped reads in LO (Supplementary Table S1). Applying stringent criteria on minimum transcript abundance (>5 FPKM), 6,543 transcripts were quantified in LO and 6,487 transcripts in Exo. Despite the difference in RNA yield and the relative lack of long fragments in Exo vs. LO, we were still able to identify similar numbers of transcripts in both EV fractions, and their abundance was highly correlated (r = 0.94, p ≈ 0, Pearson, ). Conversely, 4,583 genes exhibited more than 2-fold difference between donor cells and LO or Exo (r = 0.74 for Exo, r = 0.73 for LO, Pearson, ). This result suggests that the mRNA of the EVs does not represent random shedding from the source cells, and is in line with previous observations supporting active export of miRNA in EVs.Citation18,22,24 For validation, we randomly selected 13 mRNAs that showed at least 2-fold change in LO vs cells and measured their abundance using a high throughput, microfluidic system (Fluidigm). Directional trends for 7 (54%) of these transcripts was confirmed (Fig. S2, Table S2).
Vesicular mRNA is derived from cytosolic mRNA
We hypothesized that the observed differences between EV and whole-cell mRNA profiles are potentially due to the EVs being derived from specific cellular or subcellular compartments. To test this hypothesis, we queried the Cold Spring Harbor RNA-Seq data set of the ENCODE project, including various cancer cell lines and multiple subcellular compartments.Citation29 We first observed similar differences between the mRNA populations of whole cell and cytosol (r = 0.79–0.91) (Fig. S3) to those observed between the U87 EVs and cells (r = 0.73–0.74) (). Next, to assess the origin of EV mRNA from cytosolic vs. non-cytosolic cell compartments, we focused on the unspliced mRNA, i.e. intronic sequence, that is known to be comparatively less abundant in cytoplasm than in either whole-cell or nuclear compartments.Citation29 We characterized the presence of unspliced mRNA in U87 cells and LO and compared our RNA-Seq data to cancer cell line data from the ENCODE project () that included cytosolic, whole-cell, and nuclear mRNA fractions. We found that the LO fraction contains significantly less unspliced mRNA (p ≈ 0, Wilcoxon rank-sum) than the originating U87 cells. U87 Exo were not included in the analysis due to substantially fewer non-redundant reads in the data set. A similar difference was seen within the ENCODE cell lines, with the cytosolic fractions having significantly less unspliced mRNA than the corresponding whole-cell fractions, which in turn had less unspliced mRNA than the nuclear fractions. The similar pattern seen when comparing splicing between LO and whole-cell mRNA, and between cytosol and whole-cell mRNA, is suggestive of a cytosolic origin for the EV mRNA.
To investigate this further, we then compared the annotated transcript length of mRNAs enriched (>3-fold higher) or depleted (>3-fold lower) in U87 EVs compared with whole cells in our data set, and in the cytosol of ENCODE cell lines compared with whole cells (). This comparison showed that transcripts enriched in either U87 EV fraction tend to be significantly shorter than those enriched in the U87 cells (p ≈ 0 for Exo, p ≈ 0 for LO, Wilcoxon), with a similar result for the cytosol of most ENCODE cell lines, further corroborating a cytosolic origin of EVs. Lastly, as new mRNAs are not generated in the cytosol, but rather in the nucleus, it would be expected that short half-life mRNAs, which are degraded faster, would be depleted from the cytosol when compared with the whole cell. Using annotated mRNA half-life data from a published study,Citation30 we compared fold changes in mRNA abundance both between U87 EV sets and whole cells, and ENCODE data from cytosol and whole cell, grouping fold-changes by long and short mRNA half-life (Fig. S4). For both the Exo and LO data, and the majority of ENCODE cell lines, shorter half-life mRNAs showed lower fold-changes than long half-life mRNAs (p = 8.2e−5 for Exo, p = 2.7e−7 for LO, Wilcoxon), further arguing for a cytosolic origin of the mRNA cargo in EVs.
Signal peptide bearing mRNAs are depleted from EVs
The mRNA population of the cytosol is not uniformly distributed, particularly with regard to mRNAs encoding a signal peptide. Many eukaryotic genes encode an N-terminal signal peptide that is recognized by the Signal Recognition Particle and directs the mRNA to the rough endoplasmic reticulum (RER) for translation.Citation31 mRNAs encoding a signal peptide are more likely to be associated with the RER than free in the cytosol.Citation32 Based on this notion, if the RER were involved in the generation of EVs it would be expected that mRNAs encoding a signal peptide would be enriched in EVs compared with cells. Signal peptides can be accurately predicted computationally via machine learning using the SignalP utility.Citation33 We therefore used SignalP to assess all protein coding genes in the human genome for the presence of a signal peptide; we then compared the abundance of signal peptide encoding mRNAs between U87 EVs and cells, and between ENCODE cytosol and whole-cell (, Fig. S5). Surprisingly, we found that EVs were strongly depleted for signal peptide-encoded mRNAs (p ≤ 0.001, NES = −3.1, GSEA, mean log2 fold-change = −1.0.). The signal peptide depletion was not observed when we compared cytosol and whole cells in the ENCODE data set. The depletion of signal peptide-bearing mRNAs from the EVs suggests that EVs are formed to the exclusion of mRNAs co-translationally bound to the RER. Such a depletion of signal peptide encoding mRNAs also argues strongly for a cytosolic origin of the mRNA in the EVs, since signal peptide recognition is itself a cytosolic process.
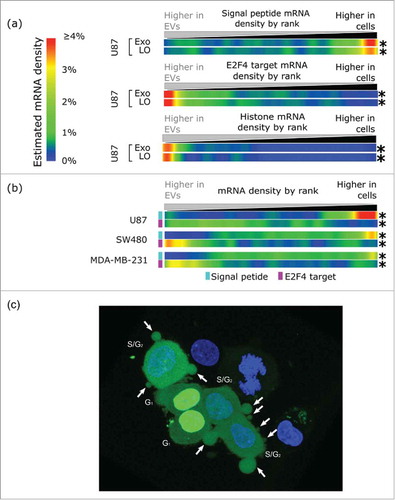
Figure 2. U87 EVs are derived from U87 cytosol and enriched for S-phase associated transcripts. (A) U87 EVs are depleted for mRNAs encoding a signal peptide, and enriched for E2F4 targets and histone mRNAs. The only phase of the cell cycle in which both E2F targets and histone mRNAs are upregulated simultaneously is the S-phase, which suggests that mRNA export in LO occurs in S-phase. The fold-changes of genes in the 3 gene sets were found between U87 EVs and cells. Density is displayed as the estimated density of genes in each gene set in 1024 ranked bins. (B) Expression microarray data comparing mRNA in EVs and cells from the indicated cell lines recapitulate the signal peptide (cyan) and the E2F4 target (light purple) patterns observed in the U87 EVs and cells. A depletion of signal peptide bearing mRNAs and an enrichment of E2F4 targets were observed in EVs from U87, SW480, and MDA-MB-231. * - p < 0.001. (C) U87 cells stably expressing DHB-YFP (green) were used for immunofluorescence imaging. Non-apoptotic blebbing, which results in LO formation, is associated with translocation of DHB-YPF from the nucleus to the cytoplasm, indicating that the cells are in the S-phase of the cell cycle when LO formation occurs. A representative confocal image is shown. Nuclei were stained with DAPI (blue). Arrows indicate the LO.
U87 EVs are enriched in S-phase specific transcripts
We found that several important cell cycle regulators were enriched in EVs, including E2F (∼40-fold) and CDK2 (∼7-fold), suggesting a possible relationship between progression through the G1-S checkpoint and mRNA export (Supporting Material File 1). New mRNA is unlikely to be generated in the EVs; rather, the mRNA content of EVs likely reflects the mRNA state of the cytosol at the time of EV biogenesis. Depletion or enrichment of mRNA that is expressed during a specific phase of the cell cycle permits inference of a phase-specific origin of EV mRNA. Notably, CDK2 is involved in the transition to the S-phase of the cell cycle via phosphorylation of the Rb family membersCitation34 and the concomitant release of E2F transcription factors.Citation35 The E2F family of transcription factors is responsible for both repressing and enhancing the cell cycle-specific expression of many genes, with targets tending to have peak expression in late G1 and S-phase.Citation36,37 Indeed, the E2F1 gene itself is known to peak during G1 and S-phases.Citation38 Comparing EV and whole cell profiles, we found that mRNAs of genes with E2F binding motifs near or in their promotersCitation39 are significantly enriched in EVs (p < 0.001, NES = 2.5) (Fig. S6). This enrichment is not seen when comparing cytosol and whole-cell RNA-Seq data in ENCODE cell lines, indicating that it is not due to the cytosolic origin of the EV mRNA. CDC2/CDK1 is a heavily E2F regulated gene; release of the repressive of E2F4, a paralog of E2F1 with a common DNA binding motif and highly overlapping target gene set,Citation40 from the CDC2 promoter region precedes the transcriptional activation of CDC241. In our RNA-Seq data, we found that CDC2/CDK1 is increased >100-fold in EVs compare with cells (Supporting Material File 1). To examine genome-wide the differences in E2F family targets between U87 EVs and cells, we defined a set of E2F regulated genes using ENCODE ChIP-seq dataCitation42 for the broadly expressed E2F4 protein.Citation43 Comparing the abundance of mRNAs from this gene set between EVs and cells we found that the EV sets are greatly enriched for these mRNAs, (p ≤ 0.001, NES = 3.6, mean log2 fold-change = 1.1) (,), suggesting that most mRNAs are exported in EVs at a time in the cell cycle when E2F4 targets are de-repressed, most likely in the G1 or S-phases. Enrichment of this E2F4 regulated gene set was not observed when comparing the cytosol to whole cell of any of the ENCODE cell lines (Fig. S7). The expression of canonical human histone genes is similarly regulated by the cell cycle, being largely confined to the S-phase, followed by their degradation in G2.Citation44 As with the E2F targets, we found that these histone mRNAs are enriched in EVs (p ≤ 0.001, NES = 3.37, mean log2 fold-change 2.5, ). The histone mRNAs were also slightly enriched in the cytosol from several ENCODE cell lines (NES = −1.2 – 2.9, Fig. S8). However, compared with the EVs, the fold-change of histone mRNAs in the cytosolic data sets was significantly lower (mean log2 fold change 0.4–1.53), suggesting that the enrichment in EVs cannot be solely explained by cytosolic localization of histone mRNAs. The enrichment of histone mRNAs in the EVs, combined with the similar strong enrichment of E2F targets, suggests that most of the mRNA is exported to EVs during the S-phase of the cell cycle, when both gene sets are highly expressed. Of course this result could also be a mere reflection of an increased expression of these transcripts in the S-phase. The implication of this result is that profiling EV mRNA could capture a snapshot of cytosolic, S-phase mRNA.
Patterns seen in U87 RNA-Seq data are recapitulated in expression microarrays
Using expression microarray data from previous studies that have characterized the mRNA content of EVs and originating cells,Citation20,45,46 we detected a robust depletion of signal peptide encoding mRNAs from the EVs of 3 diverse cancer cell lines, U87 (glioma, p < 0.001, NES = −2.7, mean fold-change = −1.3), SW480 (colon cancer, p < 0.001, NES = −2.75, mean log2 fold-change = −0.3), and MDA-MB-231 (breast cancer, p < 0.001, NES = −2.6, mean log2 fold-change = −0.1) (), in good agreement with observations from our RNA-Seq data. We additionally observed significant enrichment of E2F4 targets in the EVs of all 3 data sets, U87 (p < 0.001, NES = 1.48, mean log2 fold-change = 0.2), SW480 (p < 0.001, NES = 2.3, mean log2 fold-change = 0.3) and MDA-MB-231 (p < 0.001, NES = 2, mean log2 fold-change = 0.1), again in strong agreement with our U87 RNA-Seq data. Histone genes are not sufficiently spotted on these array platforms for similar analysis. Such concordance across different cell types and expression platforms makes it very unlikely that these patterns are either spurious or are artifacts of library preparation, and the presence of these patterns across diverse cell lines suggests that they may be universal features of EV mRNA.
Increased LO formation in S-phase
The RNA-Seq and microarray data indicate enrichment for cell cycle relevant mRNAs in EVs. To pursue this finding further, we created stable cell lines expressing the CDK2-responsive portion and nuclear export signal of the DNA helicase B (DHB) gene fused to YFP.Citation47 The DHB protein is localized in the nucleus during G0 and G1 phases, but translocates to the cytoplasm when phosphorylated by CDK2 at the G1/S transition. YFP-tagging of the CDK2 targeted region of the DHB protein can thus be used as a sensor to track the sub-cellular localization of the protein and, thus, the cell cycle phase of individual cells.Citation47 Using fluorescent microscopy, we found that LO formation was observed predominantly in the S-phase of the cell cycle (). A similar result was also observed in breast cancer cells (data not shown).
Analysis of biological pathways enriched in LO vs. Exo leads to functionally relevant VEGFA
Even though the majority of the transcripts in LO and Exo were present at similar levels in the 2 sets, 5.2% of transcripts were present at greater than 2-fold difference between LO and Exo (414 enriched and 118 depleted mRNAs in LO in vs. Exo) (). Even when we used the more stringent criteria of at least 5 FPKM for mRNA detectability, 215 mRNAs were enriched and 110 mRNAs were depleted in LO. Ten of 11 (>90%) of these mRNA, which were randomly selected among mRNAs with at least 2-fold differential abundance in LO vs Exo, were validated by a high throughput, microfluidic system (Fig. S9). The correlation between RNA-Seq and the microfluidic system data was high (r = 0.76) (, Fig. S9, Table S2). HSPA5, which was previously identified as enriched in LO vs Exo at the protein level by mass spectrometry,Citation11 appeared among the mRNAs that were enriched in LO vs Exo.
Figure 3. VEGFA appears as one component of biological pathways enriched in LO. (A) Scatter plot showing the log2 abundance in the LO set and the log2 ratio between LO and Exo for transcripts with an absolute log2 fold-change of at least 1 between the LO and Exo mRNA sets. Genes validated by qRT-PCR are highlighted, with genes found to be of higher abundance in LO labeled in red, and genes found to be of lower abundance in LO labeled in blue. (B) Validation of mRNA abundance differences between LO and Exo mRNA fractions. For 11 genes, the RNA-Seq fold-change ranking was validated via Fluidigm qRT-PCR, with a strong correlation between the 2 methods (r = 0.76). Gene ontology (GO) enrichment analysis using FunRich software indicates the cellular component (5 out of 6 GO terms show association with membrane structures) (C), molecular function (D) and biological pathways (E) for the mRNAs overrepresented in LO. 2 out of 3 and 3 out of 5 GO terms respectively associated with transporter or receptor functions. For each GO category the plots show, on the x-axis, the percentage of genes that belong to the GO term indicated in the y-axis. Only statistically significant enrichment is displayed (Bonferroni corrected; * for p < 0.05, ** for p < 0.001), and the numbers below the asterisks indicate the number of genes for each GO term. FC = fold change; (F) VEGFR mRNA levels were measured by qRT-PCR in HUVEC cells at baseline or after treatment with LO at the indicated time points (* p < 0.05).
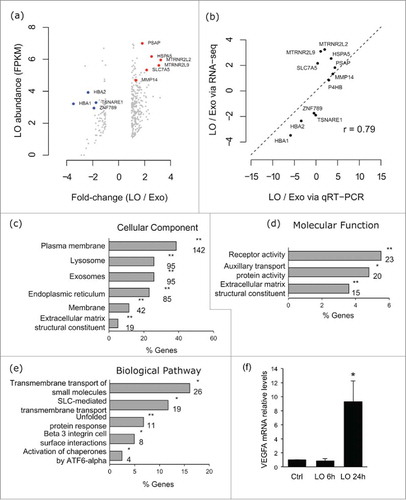
Gene ontology (GO) enrichment analysis using the FunRichCitation48 tool was performed for all LO-enriched mRNAs. The analysis demonstrated that LO contain high levels of mRNAs that encode proteins localized in membrane structures (). More specifically, the proteins encoded by the mRNAs enriched in LO belong to categories such as plasma membrane (cellular component) and transporters or receptors (molecular function and biological pathway). The biological pathway that caught our attention was the β3 integrin cell surface interactions group. Recent reports suggest that integrins are enriched in Exo, that β3 integrin signaling is important in promoting angiogenesis, and that specific integrins can direct cancer metastasis to specific organs.Citation49 This biological pathway has also been reported to play a role in glioblastoma,Citation50 and 6 out of 8 components of this pathway have been reported to be significantly enriched in glioblastoma tissue in the Oncomine database. VEGFA, which is a well-known and potent angiogenesis stimulator,Citation51 belongs to this group and has been shown to be an important mediator of angiogenesis and tumor progression in several human tumor types. Whether this mRNA can be transferred between cells via LO is unknown. Treatment of endothelial cells with LO obtained from U87 cell media for 24 hours resulted in increased VEGFA expression. This was not the case at 6 hour treatment, which is more suggestive of transcriptional induction than RNA transfer ().
Analysis of 15 plasma whole transcriptomes identifies a global EV mRNA signature and breast cancer signal in patients
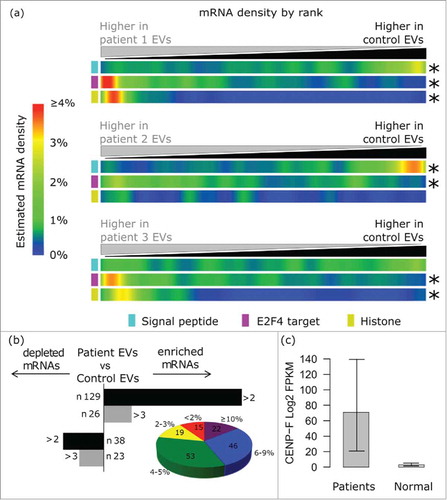
Given that our observed signature of differentially abundant mRNA sets between U87 EVs and cells (a depletion of signal peptide encoding mRNAs, and an enrichment of histones and E2F targets) was recapitulated in a range of heterologous cancer cell lines, including breast cancer MDA-MB-231 cells, we used RNA-Seq in an attempt to identify this signature in circulating EVs from the plasma of patients with advanced breast cancer. As a proof of principle, we isolated EVs from the plasma of patients with stage III breast Invasive Lobular Carcinoma (ILC) vs. healthy controls (Supplementary Table S3). On average, we achieved 26.5 million raw reads (ranging from 5 to 62 million) (Supplementary Table S4). Of these raw reads, 87–97% were uniquely mapped to annotated transcriptional loci. In EV mRNA preparations from patients with ILC, we observed a significant enrichment of E2F4 target genes, and in 2 out of these 3 patients we found enrichment of histone mRNAs and a strong depletion of signal peptide compare with healthy women (). We then performed RNA-Seq on plasma-derived EVs from an additional group of patients with different histotypes of Stage III breast cancer. 155 mRNAs were enriched and 61 were depleted in the EVs from patient plasma vs. healthy controls (, and Supplementary Material 2). 145 of the enriched mRNAs were identified in the TCGA database as upregulated (based on z-score) or amplified in patients with breast cancer at different frequency (from 1% to >10%).Citation52,53 (, Fig. S10). Additionally, we observed a significant enrichment of GATA1 target genes in plasma EVs from patients compared with healthy women (data not shown). High GATA1 mRNA levels have been identified in breast cancer tissueCitation54 Finally, CENPF was up ∼5-fold in plasma EVs from ILC patients vs. control women. Notably, a recent study reported increased expression levels of CENPF in 16% of breast cancer tissue samples from patients with ILC.Citation55 Collectively, these results suggest that an mRNA signature of the disease is visible in the circulating EV mRNA of patients.
Figure 4. Analysis of 15 plasma whole transcriptomes identifies a global EV mRNA signature and breast cancer signal in patients. (A) The EV signature (depletion of signal peptide encoding mRNAs, enrichment of E2F4 targets and enrichment of histones) was recapitulated in vivo in EVs isolated from 2 out of 3 patients with invasive lobular carcinoma (ILC) vs. control individuals. (B) The histogram on the left shows the number of transcripts that are enriched or depleted of at least 2-fold (Log 2) in EVs from all breast cancer patients in comparison with controls. The pie chart shows the distribution of the mRNAs that we found upregulated in plasma EVs in TCGA breast cancer tissues. The size of each slice is determined by the number of transcripts that are enriched in given fractions of patients. 15 mRNAs were altered in 0–1% of the cases, 19 in 2–3% of the cases, 53 in 4–5% of the cases, 46 in 6–9% of the cases, 22 in > 10% of the cases. (C) CENPF Log 2 expression (FPKM) in plasma EVs from patients (n = 15) vs. healthy controls (n = 5).
Discussion
This is the first study on global mRNA characterization of 2 types of EVs (LO and Exo) with in-depth comparison to the donor cell using RNA-Seq. It is also the first attempt to use expression data to determine what factors influence the abundance of different mRNAs in the EVs. Finally, this is the first whole transcriptome analysis of EVs circulating in the plasma of patients with breast cancer. Our results demonstrate that 1) the transcripts exported in EVs are mostly of cytosolic origin, 2) the EV transcriptome is enriched in S-phase specific transcripts and exhibits a distinct signature (high E2F targets and histones, and low signal peptide) that discriminates them from the donor cell, 3) LO and Exo exhibit an overall similar mRNA cargo, however they show differences that might be functionally relevant, and 4) the EV signature can be detected in plasma of patients with breast cancer vs. controls, along with transcripts that are upregulated in breast cancer tissues.
Our demonstration that most of the mRNAs exported in 2 different types of EVs derive from the cytosol, and that this might occur during the S-phase of the cell cycle, is in agreement with previous studies hypothesizing that EV shedding might be cell cycle-dependent.Citation3,46 The findings also highlight a common, perhaps universal feature of EV mRNA biology. In fact an mRNA pattern, similar to that we identified by RNA-Seq in our EV preparations, was found in array data from published studies on EVs across distinct systems, including breast cancer models.Citation20,21,37 The appearance of this same pattern in plasma of patients with breast cancer suggests that identification of this EV signature in vivo might be indicative of increased EV shedding in patients with cancer. This promising result could be followed up by studies aimed to test whether a panel of known E2F targets and histones, combined with signal peptide genes, could be used to screen circulating EVs in patients with cancer. The identification of breast cancer specific transcripts suggests that analysis of EV RNA could result in potentially useful biomarkers. If validated on larger cohorts, this result could help ongoing efforts to improve the current tools for early diagnosis of breast cancer using minimally invasive methods.
Our results also have important implications for future attempts to study EV mRNA cargo. As we show here, the EV content is likely derived from cytosol, rather than whole cells. While EV mRNA has typically been compared with whole-cell mRNA from the donor source,Citation20,45,46 our results suggest that comparisons between EVs and cytosolic mRNA are more appropriate to refine which mRNAs are enriched or excluded from the EVs during their formation. Importantly, this is one of the first studies of this kind, and the depth of the sequencing in our samples, both in vitro and in the circulation, was very high, allowing us to identify a larger number of genes than a previous study, in which the abundance of ribosomal and/or other RNA types precluded in-depth comparison of mRNAs expression between the Exo and the source cells.Citation25,56
Recent studies have described surprising heterogeneity in EV populations and have provided evidence that EVs originating from distinct intracellular origins (but from the same cell donor) might contain diverse cargo and play specialized functions.Citation27 In line with this, our group has demonstrated that 25% of the protein cargo of LO and Exo purified from the same cell source is significantly different.Citation11 However, this study shows a much more similar RNA profile among the 2 EV populations, with only 5% of the transcripts present at a difference greater than 2-fold in LO vs. Exo. In vivo, the EV signature was identified by whole transcriptome analysis of a mixture of EVs from patients. Whether one population of EVs contributes to this signature more than another remains unresolved. Ultimately, this study highlights the possibility that a combinatorial analysis of different EV subsets might be a more relevant source for liquid biopsy than either particle alone. Future studies to clarify whether the tumor signal can be increased by improved purification methods are warranted.
With respect to the differences between LO and Exo (5% of the transcripts are at least 2-fold different between the sets), we observed an interesting functional trend. Most of the transcripts that were enriched in LO vs. Exo encode proteins involved in important plasma membrane functions, and some of them for proteins with key functional roles in tumor progression and previously considered Exo resident molecules (e.g., VEGFA). An extracellular function for VEGFA, which is enriched in glioblastoma-derived EVs and capable to induce angiogenesis in recipient endothelial cells, has been previously demonstrated.Citation3 Our demonstration that VEGFA mRNA levels are increased in endothelial cells exposed to LO is interesting and provocative. It might be in agreement with published reports that suggest that EVs can transfer RNA to target cells.Citation2,3,20,57 However, our observation that VEGFA mRNA levels increase as a response to LO treatment for 24 hours, but not for 6 hours, argues against RNA transfer in this case, and rather suggests that LO might be inducing mRNA expression in target cells. Additional studies using tagged constructs will be necessary for a more definitive conclusion. Indeed, functionally relevant is also the observed enrichment of HSPA5 mRNA in LO. We had previously identified the protein as a potential LO marker by mass spectrometry, and here we show that it is also enriched at the mRNA level in a different type of cell line.Citation11
In summary, this study represents the most extensive use of NGS profiling of EV mRNA, including 2 distinct classes of tumor-derived EVs. Our study has shown that EVs carry tumor-specific alterations and can be interrogated as a source of cancer-derived cargo. Because sample size and the number of RNA sequencing reads directly influence the accuracy of molecular subtyping, continuing improvements in technology will allow the translation of these findings to the clinic. Our methods did not distinguish between RNA present as full-length transcripts vs. fragmented RNA. Hence, understanding whether intact transcripts can be shed in EVs is also an important question for exploration in future studies.
Materials and methods
Cell culture
U87 cells were cultured, as previously described,Citation22 in MEM medium supplemented with 10% fetal bovine serum 2 mmol/L L-glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin. Unless otherwise specified, media and supplements were from Invitrogen.
Purification of EVs
The cells were cultured in serum free media for 24 hours prior to EV purification. EVs were then purified from conditioned medium by differential centrifugation as previously described.Citation11,14,22 Briefly, cells and debris were eliminated by centrifugation at 2,800 g for 10 min. The supernatant was then centrifuged using a Beckman SW28 rotor at 10,000 g for 30 min to precipitate large EVs. The remaining supernatant was subjected to additional centrifugation at 100,000 g for 60 min for nano-sized EV isolation. Both the 10,000 g and 100,000 g pellets were collected in TRIzol reagent (Invitrogen) for RNA isolation.
EV isolation from plasma
Plasma samples from breast cancer patients (n = 10) and healthy donors (n = 5) were obtained through an Institutional Review Board approved protocol at Cedars-Sinai Medical Center in compliance with the Declaration of Helsinki. All subjects provided written informed consent for blood to be used for research purposes. EVs were isolated from 1 ml of plasma per patient by differential centrifugation. The pellets containing large and small EVs were collected in TRIzol reagent (Invitrogen) and combined for RNA isolation.
RNA isolation and profiling
Total RNA was extracted from U87 cells and derived large oncosome (LO) and nano-sized EV (Exo) fractions, as well as from plasma EVs by ethanol precipitation. RNA was quantified using NanoDrop2000 and the RNA yield was normalized over the total protein amount (ng). The quality of RNA was assessed by total RNA electropherogram profile, using an Agilent 2100 bioanalyzer (Total RNA Nano Series II).
Western blot
Protein lysates from U87 whole cells, LO and Exo were blotted with: rabbit monoclonal HSPA5 (C50B12) (Cell Signaling), GM130 (Cell Signaling), and mouse monoclonal CD81 (M38) (Abcam), at 1:1000 dilution, and HRP conjugated GAPDH (14C10) (Cell Signaling), at 1:2000 dilution.
Library preparation for Next-Generation Sequencing
Approximately 200 ng of total RNA from U87 cells, LO and Exo, and 1 to 10 ng of total RNA from the circulating EV samples were used for the preparation of paired-end libraries. These libraries were made using a pre-release version of the Illumina RNA Access kit (http://www.illumina.com/products/truseq-rna-access-kit.html). Briefly, this protocol uses random priming to create 1st and 2nd strand cDNA from total RNA. No poly-A selection or rRNA depletion is done prior to creation of cDNA. The total cDNA libraries are then enriched for protein coding regions of mRNA by hybridization and capture using a set of probes designed against 21,415 human genes. Each library was analyzed on one lane of an Illumina GAIIx instrument. RNA-Seq reads were then aligned to the human genome. See Supplementary Methods for detail of RNA-Seq mapping and transcript quantification.
RNA-Seq validation by RT-qPCR
We used high-throughput on chip quantitative RT-PCR using a 48 × 48 dynamic array (Fluidigm Corporation).Citation58 The assay plate contained specific primers for AARS, KLF2, RIN1, ADRBK1, MMP14, RRM2, ASL, MTRNR2L2, SLC7A5, DAZAP1, MTRNR2L9, TK1, E2F1, P4HB, TMEM41B, HBA1, PHF19, TPM1, HBA2, POMP, TSNARE1, HSPA5, PSAP and ZNF789 (id: 5578_FDGP_15). Primer sequences and amplicon length are shown in Supplementary Table S2. cDNA was obtained from 11 ng of input RNA from U87 cells, LO and Exo using the Fluidigm Reverse Transcription Master Mix. Additional PCR experiments included quantitative detection of VEGFA (5′-CTTGCCTTGCTGCTCTACC-3′ forward and 5′-CACACAGGATGGCTTGAAG-3′ reverse) in endothelial cells exposed to U87-derived LO.
Computational analysis and statistics
Detailed description is reported as Supplementary Methods.
Immunofluorescent (IF) imaging
The lentiviral pCSII-EF-DHB-YFP vector was kindly provided by Dr. Sabrina Spencer, University of Colorado. U87 cells stably expressing DHB-YFP were made using lentiviral pCSII-EF-DHB-YFP vector as previously described.Citation47 Stained cells were fixed in 4% paraformaldehyde (PFA), and coverslips were mounted in Vectashield mounting medium containing DAPI (5′,6′-diamido-2-phenylindole) (Vector Laboratories, Burlingame, CA). Images were taken using Leica SP5-X confocal fluorescent microscopy.
Data Availability
These sequence data have been submitted to the GenBank database under accession number SRP061372
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary_Material.zip
Download Zip (5.3 MB)Acknowledgement
We are grateful to Dr. Gary Schroth and Irina Khrebtukova (Illumina Inc., San Diego, CA, United States) for their technical assistance with the RNA-Seq experiments, and to Dr. Sabrina Spencer for generously providing the DHB-YFP construct plasmid used in . We would also like to thank our patients and their families for participation in this study.
Funding
This study was supported by grants from the National Institutes of Health (NIH UCLA SPORE in Prostate Cancer award P50 CA092131 (to D.D.V.); Avon Breast Cancer Foundation Fund 02–2013–043 (to D.D.V.); the Martz Translational Breast Cancer Research Fund (to M.R.F. and D.D.V.); Department of Defense PC150836 (to D.D.V.), the Steven Spielberg Discovery Fund in Prostate Cancer Research (to M.R.F. and D.D.V.). M.T. acknowledges support from the NIH Transformative R01 Grant R01DK085714 and NIH Extracellular RNA Communication Common Fund Grant U01 HL126499–01.
Related Research Data
References
- Garcia V, Garcia JM, Pena C, Silva J, Dominguez G, Lorenzo Y, Diaz R, Espinosa P, de Sola JG, Cantos B, et al. Free circulating mRNA in plasma from breast cancer patients and clinical outcome. Cancer Lett 2008; 263:312-20; PMID:18280643; http://dx.doi.org/10.1016/j.canlet.2008.01.008
- Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 2007; 9:654-9; PMID:17486113; http://dx.doi.org/10.1038/ncb1596
- Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT, Jr., Carter BS, Krichevsky AM, Breakefield XO. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 2008; 10:1470-6; PMID:19011622; http://dx.doi.org/10.1038/ncb1800
- Arroyo JD, Chevillet JR, Kroh EM, Ruf IK, Pritchard CC, Gibson DF, Mitchell PS, Bennett CF, Pogosova-Agadjanyan EL, Stirewalt DL, et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci U S A 2011; 108:5003-8; PMID:21383194; http://dx.doi.org/10.1073/pnas.1019055108
- Lo Cicero A, Stahl PD, Raposo G. Extracellular vesicles shuffling intercellular messages: for good or for bad. Curr Opin Cell Biol 2015; 35:69-77; PMID:26001269; http://dx.doi.org/10.1016/j.ceb.2015.04.013
- Yanez-Mo M, Siljander PR, Andreu Z, Zavec AB, Borras FE, Buzas EI, Buzas K, Casal E, Cappello F, Carvalho J, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles 2015; 4:27066; PMID:25979354; http://dx.doi.org/10.3402/jev.v4.27066
- Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, Rak J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol 2008; 10:619-24; PMID:18425114; http://dx.doi.org/10.1038/ncb1725
- Redzic JS, Balaj L, van der Vos KE, Breakefield XO. Extracellular RNA mediates and marks cancer progression. Semin Cancer Biol 2014; 28:14-23; PMID:24783980; http://dx.doi.org/10.1016/j.semcancer.2014.04.010
- Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, Garcia-Santos G, Ghajar C, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med 2012; 18:883-91; PMID:22635005; http://dx.doi.org/10.1038/nm.2753
- Ji H, Chen M, Greening DW, He W, Rai A, Zhang W, Simpson RJ. Deep sequencing of RNA from three different extracellular vesicle (EV) subtypes released from the human LIM1863 colon cancer cell line uncovers distinct miRNA-enrichment signatures. PLoS One 2014; 9:e110314; PMID:25330373; http://dx.doi.org/10.1371/journal.pone.0110314
- Minciacchi VR, You S, Spinelli C, Morley S, Zandian M, Aspuria PJ, Cavallini L, Ciardiello C, Reis Sobreiro M, Morello M, et al. Large oncosomes contain distinct protein cargo and represent a separate functional class of tumor-derived extracellular vesicles. Oncotarget 2015; 6:11327-41; PMID:25857301; http://dx.doi.org/10.18632/oncotarget.3598
- Cocucci E, Meldolesi J. Ectosomes and exosomes: shedding the confusion between extracellular vesicles. Trends Cell Biol 2015; 25:364-72; PMID:25683921; http://dx.doi.org/10.1016/j.tcb.2015.01.004
- Di Vizio D, Kim J, Hager MH, Morello M, Yang W, Lafargue CJ, True LD, Rubin MA, Adam RM, Beroukhim R, et al. Oncosome formation in prostate cancer: association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res 2009; 69:5601-9; PMID:19549916; http://dx.doi.org/10.1158/0008-5472.CAN-08-3860
- Di Vizio D, Morello M, Dudley AC, Schow PW, Adam RM, Morley S, Mulholland D, Rotinen M, Hager MH, Insabato L, et al. Large oncosomes in human prostate cancer tissues and in the circulation of mice with metastatic disease. Am J Pathol 2012; 181:1573-84; PMID:23022210; http://dx.doi.org/10.1016/j.ajpath.2012.07.030
- Minciacchi VR, Freeman MR, Di Vizio D. Extracellular vesicles in cancer: exosomes, microvesicles and the emerging role of large oncosomes. Semin Cell Dev Biol 2015; 40:41-51; PMID:25721812; http://dx.doi.org/10.1016/j.semcdb.2015.02.010
- Ciardiello C, Cavallini L, Spinelli C, Yang J, Reis-Sobreiro M, de Candia P, Minciacchi VR, Di Vizio D. Focus on Extracellular Vesicles: New Frontiers of Cell-to-Cell Communication in Cancer. Int J Mol Sci 2016; 17:175; PMID:26861306; http://dx.doi.org/10.3390/ijms17020175
- Crescitelli R, Lasser C, Szabo TG, Kittel A, Eldh M, Dianzani I, Buzas EI, Lotvall J. Distinct RNA profiles in subpopulations of extracellular vesicles: apoptotic bodies, microvesicles and exosomes. J Extracell Vesicles 2013; 2; PMID:24223256; http://dx.doi.org/10.3402/jev.v2i0.20677
- Tosar JP, Gambaro F, Sanguinetti J, Bonilla B, Witwer KW, Cayota A. Assessment of small RNA sorting into different extracellular fractions revealed by high-throughput sequencing of breast cell lines. Nucleic Acids Res 2015; 43:5601-16; PMID:25940616; http://dx.doi.org/10.1093/nar/gkv432
- Ridder K, Keller S, Dams M, Rupp AK, Schlaudraff J, Del Turco D, Starmann J, Macas J, Karpova D, Devraj K, et al. Extracellular vesicle-mediated transfer of genetic information between the hematopoietic system and the brain in response to inflammation. PLoS Biol 2014; 12:e1001874; PMID:24893313; http://dx.doi.org/10.1371/journal.pbio.1001874
- Zomer A, Maynard C, Verweij FJ, Kamermans A, Schafer R, Beerling E, Schiffelers RM, de Wit E, Berenguer J, Ellenbroek SI, et al. In Vivo imaging reveals extracellular vesicle-mediated phenocopying of metastatic behavior. Cell 2015; 161:1046-57; PMID:26000481; http://dx.doi.org/10.1016/j.cell.2015.04.042
- Bolukbasi MF, Mizrak A, Ozdener GB, Madlener S, Strobel T, Erkan EP, Fan JB, Breakefield XO, Saydam O. miR-1289 and “Zipcode”-like Sequence Enrich mRNAs in Microvesicles. Mol Ther Nucleic Acids 2012; 1:e10; PMID:23344721; http://dx.doi.org/10.1038/mtna.2011.2
- Morello M, Minciacchi VR, de Candia P, Yang J, Posadas E, Kim H, Griffiths D, Bhowmick N, Chung LW, Gandellini P, et al. Large oncosomes mediate intercellular transfer of functional microRNA. Cell Cycle 2013; 12:3526-36; PMID:24091630; http://dx.doi.org/10.4161/cc.26539
- Fujita Y, Yoshioka Y, Ochiya T. Extracellular vesicle transfer of cancer pathogenic components. Cancer Sci 2016; 107:385-90; PMID:26797692; http://dx.doi.org/10.1111/cas.12896
- Cheng L, Sharples RA, Scicluna BJ, Hill AF. Exosomes provide a protective and enriched source of miRNA for biomarker profiling compared to intracellular and cell-free blood. J Extracell Vesicles 2014; 3; PMID:24683445; http://dx.doi.org/10.3402/jev.v3.23743
- Jenjaroenpun P, Kremenska Y, Nair VM, Kremenskoy M, Joseph B, Kurochkin IV. Characterization of RNA in exosomes secreted by human breast cancer cell lines using next-generation sequencing. PeerJ 2013; 1:e201; PMID:24255815; http://dx.doi.org/10.7717/peerj.201
- Lunavat TR, Cheng L, Kim DK, Bhadury J, Jang SC, Lasser C, Sharples RA, Lopez MD, Nilsson J, Gho YS, et al. Small RNA deep sequencing discriminates subsets of extracellular vesicles released by melanoma cells - evidence of unique microRNA cargos. RNA Biol 2015; 810-23; PMID:26176991; http://dx.doi.org/10.1080/15476286.2015.1056975
- Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, Dingli F, Loew D, Tkach M, Thery C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A 2016; 113:E968-77; PMID:26858453; http://dx.doi.org/10.1073/pnas.1521230113
- Van Deun J, Mestdagh P, Sormunen R, Cocquyt V, Vermaelen K, Vandesompele J, Bracke M, De Wever O, Hendrix A. The impact of disparate isolation methods for extracellular vesicles on downstream RNA profiling. J Extracell Vesicles 2014; 3; PMID:25317274; http://dx.doi.org/10.3402/jev.v3.24858
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. Landscape of transcription in human cells. Nature 2012; 489:101-8; PMID:22955620; http://dx.doi.org/10.1038/nature11233
- Sharova LV, Sharov AA, Nedorezov T, Piao Y, Shaik N, Ko MS. Database for mRNA half-life of 19 977 genes obtained by DNA microarray analysis of pluripotent and differentiating mouse embryonic stem cells. DNA research : an international journal for rapid publication of reports on genes and genomes 2009; 16:45-58; PMID:19001483; http://dx.doi.org/10.1093/dnares/dsn030
- Martoglio B, Dobberstein B. Signal sequences: more than just greasy peptides. Trends Cell Biol 1998; 8:410-5; PMID:9789330; http://dx.doi.org/10.1016/S0962-8924(98)01360-9
- Jagannathan S, Reid DW, Cox AH, Nicchitta CV. De novo translation initiation on membrane-bound ribosomes as a mechanism for localization of cytosolic protein mRNAs to the endoplasmic reticulum. Rna 2014; 20:1489-98; PMID:25142066; http://dx.doi.org/10.1261/rna.045526.114
- Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nature methods 2011; 8:785-6; PMID:21959131; http://dx.doi.org/10.1038/nmeth.1701
- Akiyama T, Ohuchi T, Sumida S, Matsumoto K, Toyoshima K. Phosphorylation of the retinoblastoma protein by cdk2. Proc Natl Acad Sci U S A 1992; 89:7900-4; PMID:1518810; http://dx.doi.org/10.1073/pnas.89.17.7900
- Trouche D, Le Chalony C, Muchardt C, Yaniv M, Kouzarides T. RB and hbrm cooperate to repress the activation functions of E2F1. Proc Natl Acad Sci U S A 1997; 94:11268-73; PMID:9326598; http://dx.doi.org/10.1073/pnas.94.21.11268
- Johnson DG, Schwarz JK, Cress WD, Nevins JR. Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature 1993; 365:349-52; PMID:8377827; http://dx.doi.org/10.1038/365349a0
- DeGregori J, Kowalik T, Nevins JR. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol Cell Biol 1995; 15:4215-24; PMID:7623816; http://dx.doi.org/10.1128/MCB.15.8.4215
- Whitfield ML, Sherlock G, Saldanha AJ, Murray JI, Ball CA, Alexander KE, Matese JC, Perou CM, Hurt MM, Brown PO, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Molecular biology of the cell 2002; 13:1977-2000; PMID:12058064; http://dx.doi.org/10.1091/mbc.02-02-0030
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011; 27:1739-40; PMID:21546393; http://dx.doi.org/10.1093/bioinformatics/btr260
- Xu X, Bieda M, Jin VX, Rabinovich A, Oberley MJ, Green R, Farnham PJ. A comprehensive ChIP-chip analysis of E2F1, E2F4, and E2F6 in normal and tumor cells reveals interchangeable roles of E2F family members. Genome Res 2007; 17:1550-61; PMID:17908821; http://dx.doi.org/10.1101/gr.6783507
- Tommasi S, Pfeifer GP. In vivo structure of the human cdc2 promoter: release of a p130-E2F-4 complex from sequences immediately upstream of the transcription initiation site coincides with induction of cdc2 expression. Mol Cell Biol 1995; 15:6901-13; PMID:8524257; http://dx.doi.org/10.1128/MCB.15.12.6901
- Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, Wong MC, Maddren M, Fang R, Heitner SG, et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res 2013; 41:D56-63; PMID:23193274; http://dx.doi.org/10.1093/nar/gks1172
- Ferreira R, Magnaghi-Jaulin L, Robin P, Harel-Bellan A, Trouche D. The three members of the pocket proteins family share the ability to repress E2F activity through recruitment of a histone deacetylase. Proc Natl Acad Sci U S A 1998; 95:10493-8; PMID:9724731; http://dx.doi.org/10.1073/pnas.95.18.10493
- Marzluff WF, Duronio RJ. Histone mRNA expression: multiple levels of cell cycle regulation and important developmental consequences. Curr Opin Cell Biol 2002; 14:692-9; PMID:12473341; http://dx.doi.org/10.1016/S0955-0674(02)00387-3
- Kucharzewska P, Christianson HC, Welch JE, Svensson KJ, Fredlund E, Ringner M, Morgelin M, Bourseau-Guilmain E, Bengzon J, Belting M. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc Natl Acad Sci U S A 2013; 110:7312-7; PMID:23589885; http://dx.doi.org/10.1073/pnas.1220998110
- Hong BS, Cho JH, Kim H, Choi EJ, Rho S, Kim J, Kim JH, Choi DS, Kim YK, Hwang D, et al. Colorectal cancer cell-derived microvesicles are enriched in cell cycle-related mRNAs that promote proliferation of endothelial cells. BMC Genomics 2009; 10:556; PMID:19930720; http://dx.doi.org/10.1186/1471-2164-10-556
- Spencer SL, Cappell SD, Tsai FC, Overton KW, Wang CL, Meyer T. The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 2013; 155:369-83; PMID:24075009; http://dx.doi.org/10.1016/j.cell.2013.08.062
- Pathan M, Keerthikumar S, Ang CS, Gangoda L, Quek CY, Williamson NA, Mouradov D, Sieber OM, Simpson RJ, Salim A, et al. FunRich: An open access standalone functional enrichment and interaction network analysis tool. Proteomics 2015; 15:2597-601; PMID:25921073; http://dx.doi.org/10.1002/pmic.201400515
- Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder S, et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015; 527:329-35; PMID:26524530; http://dx.doi.org/10.1038/nature15756
- Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer 2010; 10:9-22; PMID:20029421; http://dx.doi.org/10.1038/nrc2748
- Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1995; 1:27-31; PMID:7584949; http://dx.doi.org/10.1038/nm0195-27
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2:401-4; PMID:22588877; http://dx.doi.org/10.1158/2159-8290.CD-12-0095
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6:pl1; PMID:23550210; http://dx.doi.org/10.1126/scisignal.2004088
- Boidot R, Vegran F, Jacob D, Chevrier S, Cadouot M, Feron O, Solary E, Lizard-Nacol S. The transcription factor GATA-1 is overexpressed in breast carcinomas and contributes to survivin upregulation via a promoter polymorphism. Oncogene 2010; 29:2577-84; PMID:20101202; http://dx.doi.org/10.1038/onc.2009.525
- Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, Zhang H, McLellan M, Yau C, Kandoth C, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015; 163:506-19; PMID:26451490; http://dx.doi.org/10.1016/j.cell.2015.09.033
- Yuan T, Huang X, Woodcock M, Du M, Dittmar R, Wang Y, Tsai S, Kohli M, Boardman L, Patel T, et al. Plasma extracellular RNA profiles in healthy and cancer patients. Sci Rep 2016; 6:19413; PMID:26786760; http://dx.doi.org/10.1038/srep19413
- Akers JC, Ramakrishnan V, Kim R, Skog J, Nakano I, Pingle S, Kalinina J, Hua W, Kesari S, Mao Y, et al. MiR-21 in the extracellular vesicles (EVs) of cerebrospinal fluid (CSF): a platform for glioblastoma biomarker development. PLoS One 2013; 8:e78115; PMID:24205116; http://dx.doi.org/10.1371/journal.pone.\0078115
- Mitra AK, Mukherjee UK, Harding T, Jang JS, Stessman H, Li Y, Abyzov A, Jen J, Kumar S, Rajkumar V, et al. Single-cell analysis of targeted transcriptome predicts drug sensitivity of single cells within human myeloma tumors. Leukemia 2015; 30(5):1094-102; PMID:26710886; http://dx.doi.org/10.1038/leu.2015.361