
ABSTRACT
A new screening system for artificial small RNAs (sRNAs) that inhibit the growth of Escherichia coli was constructed. In this system, we used a plasmid library to express RNAs of ∼120 nucleotides, each with a random 30-nucleotide sequence that can recognize its target mRNA(s). After approximately 60,000 independent colonies were screened, several plasmids that inhibited bacterial growth were isolated. To understand the inhibitory mechanism, we focused on one sRNA, S-20, that exerted a strong inhibitory effect. A time-course analysis of the proteome of S-20-expressing E. coli and a bioinformatic analysis were used to identify potential S-20 target mRNAs, and suggested that S-20 binds the translation initiation sites of several mRNAs encoding enzymes such as peroxiredoxin (osmC), glycyl-tRNA synthetase α subunit (glyQ), uncharacterized protein ygiM, and tryptophan synthase β chain (trpB). An in vitro translation analysis of chimeric luciferase-encoding mRNAs, each containing a potential S-20 target sequence, indicated that the translation of these mRNAs was inhibited in the presence of S-20. A gel shift analysis combined with the analysis of a series of S-20 mutants suggested that S-20 targets multiple mRNAs that are responsible for inhibiting E. coli growth. These data also suggest that S-20 acts like an endogenous sRNA and that E. coli can utilize artificial sRNAs.
Abbreviations
| IPTG | = | isopropyl-β-d-thiogalactopyranoside |
| nanoLC/ESI-MS/MS | = | nano-liquid chromatography/electrospray ionization-tandem mass spectrometry |
| nt | = | nucleotide |
| OD | = | optical density |
| Rluc | = | Renilla luciferase |
| qRT-PCR | = | quantitative reverse transcription-polymerase chain reaction |
| sRNA | = | small RNA |
| Vect. | = | vector |
Introduction
Recent advances in genome biology and functional genomics have shown that noncoding RNAs are key factors in a variety of gene regulatory systems, in both prokaryotes and eukaryotes.Citation1-3 These RNAs are not translated into proteins but act as functional RNA molecules. Because most noncoding RNAs in prokaryotes are 50-300 nucleotides (nt) long, they are called “small RNAs” (sRNAs),Citation4,5 RNA-seq analyses have shown that some sRNAs are expressed from the intergenic regions of the bacterial genome and from the antisense strands of previously annotated protein-coding genes.Citation6,7 Our group has also demonstrated that many as-yet-undiscovered sRNAs are potentially encoded in the E. coli genome.Citation8 Many of these sRNAs are reportedly expressed under stress conditions, Citation9-12 such as during temperature shock, oxidative stress, and sugar stress, or are expressed in specific growth phases.Citation13,14 These sRNAs principally inhibit the translation and/or induce the degradation of their target mRNA(s) by directly base-pairing with the target RNA molecules.Citation4,5 It has also been suggested that most sRNAs are only conserved among closely related bacterial strains,815 and therefore, that the genes encoding sRNAs evolve more rapidly than protein-coding genes.Citation16 Recent studies have shown that riboswitches are also functional sRNAs. A riboswitch is defined as an RNA sequence mainly located in the 5′-untranslated region of an mRNA that affects the expression of the downstream gene by interacting specifically with a target ligand.Citation17,18 Most of these downstream genes encode proteins that are involved in the cellular biosynthetic and transport pathways of metabolites. Therefore, riboswitches are thought to provide feedback regulation that allows cells to respond to metabolic supply and demand.Citation17,18 For example, a T-box riboswitch (tRNA-binding riboswitch) exists upstream from the glyS gene to regulate the transcription of glycyl-tRNA synthetase in Staphylococcus aureus.Citation19 The ribB flavin mononucleotide riboswitch of E. coli regulates riboflavin biosynthesis.Citation20
From a gene technology perspective, it is anticipated that functional RNAs will provide unique and effective tools for gene regulation.Citation21-24 Historically, trials of gene silencing induced by artificial antisense RNAs were first reported in the 1980s,Citation25,26 and artificial sRNAs that specifically silenced the expression of selected genes were reported in 2011.Citation27 More recently, one of the most striking technologies in the field has been genome editing with the clustered regularly interspaced short palindromic repeats (CRISPR) system.Citation28 This technology is based on the sRNA-guided defense system in prokaryotes.Citation29 In this context, we previously developed a screening system for artificial sRNAs, which inhibit the growth of E. coli.Citation30 We used a plasmid library to express artificial sRNAs (approximately 200 nt long) containing a 60-nt random sequence, and identified a set of artificial sRNAs that displayed a range of inhibitory effects on bacterial growth. We proposed a new screening strategy for artificial functional RNAs in E. coli, but it was difficult to identify their targets because the expressed artificial RNAs contained a 60-nt random sequence and ∼140 nt derived from the vector. These artificial RNA structures were quite different from endogenous sRNAs.
In the present study, we constructed a new expression system for artificial sRNAs that are more likely to mimic endogenous sRNAs than were the previously synthesized sRNAs, because (i) the vector-derived sequences are dramatically reduced, and (ii) a basic sRNA structure is introduced in which 30 nt of the target-mRNA-recognition sequence is set in the 5′ half of the sRNA sequence, followed by an Hfq-binding region. Hfq is an RNA chaperone required for efficient recognition of the target RNA.Citation4 Using this system, we cloned and focused on one highly inhibitory sRNA, S-20. With a series of experiments, including a proteomic analysis, a reporter assay, and an RNA gel shift analysis, we inferred several target mRNAs and found that S-20 acts in a similar way to endogenous sRNAs.
Results and discussion
Screening artificial sRNAs that inhibit E. coli growth
To generate an artificial sRNA expression platform, we first constructed an sRNA expression vector, pET-28DEL (), which was used as a control vector throughout the experiments. Oligonucleotides of approximately 70 bp, each including a 30-nt random sequence, were then sub-cloned into pET-28DEL (). Therefore, under the control of the T7 promoter, this plasmid expresses an ∼120-nt artificial sRNA that mimics the structure of endogenous sRNAs,Citation4 in which each random region is expected to bind a target mRNA. The constructed plasmid expressing the artificial sRNAs was designated pASRII.
Figure 1. Structure of the region inserted into the artificial sRNA expression plasmid. (A) Nucleotide sequence of the region inserted into the plasmid vector, pET-28DEL. The T7 promoter sequence is boxed in black and the transcription start site is indicated with a curved arrow. Secondary structure of the T7 terminator sequence is shown in pink. Numbers indicate the nucleotide positions from the transcription start site. Cloning site with two restriction sites is indicated in light blue. (B) Nucleotide sequence of the region inserted into the artificial sRNA expression plasmid, pASRII. ‘N’ indicates an unspecified nucleotide, and 30 Ns form a random nucleotide sequence (boxed in orange). Secondary structure of the putative Hfq-RNA-chaperone-binding region (Hfq-BR) is shown in purple. The positions of the SRA1 and SRA3 probes used for the RNA gel blotting analysis are indicated with blue lines (also see Table S1).
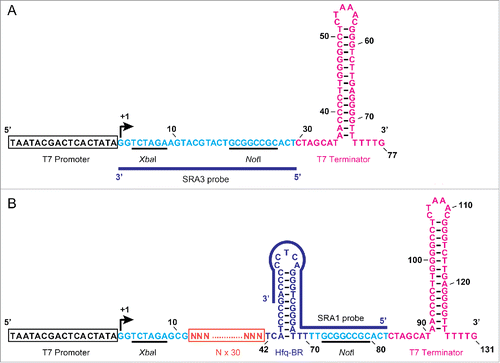
To screen for artificial sRNAs that inhibit the growth of E. coli, E. coli strain HMS174(DE3) was transformed with the pASRII library. The principal strategy of this screening technique is the same as the strategy reported previously.Citation30 In brief, 2 successive screening steps were used: (i) colony size selection on plates, and (ii) the growth of E. coli was monitored in a 96-well plate. In the first screening step, we manually selected smaller colonies (Fig. S1) under constant sRNA induction in the presence of 40 μM isopropyl-β-d-thiogalactopyranoside (IPTG). This step made it easier to handle the huge numbers of transformants. Typically, the smaller colonies appeared on the plates with a frequency of approximately 0.3%. As previously reported,Citation30 40 μM IPTG is the minimal concentration for the adequate induction of artificial sRNA expression in our system (Fig. S2). In the second step, the growth of the selected transformants was monitored continuously in a 96-well plate. As shown in , of the original 60,200 clones, 179 clones were isolated in the first step, and of these, 2 clones that exerted highly inhibitory effects on E. coli growth were selected in the second step (designated S-10 and S-20). We isolated no clones that accelerated E. coli growth with this screening strategy.
Table 1. Screening for artificial sRNAs that inhibit the growth of E. coli.
The effects of 6 sRNAs, including the 2 highly inhibitory sRNAs (S-10 and S-20), and the vector pET-28DEL on E. coli growth are shown in . S-61, S-22, S-7, and S-8 sRNAs were selected to represent a variety of growth inhibition patterns. The nucleotide sequences of the 30-nt random sequence regions in the sRNAs are shown in . The length of the random sequence region of S-10 was 28 nt, shorter than the expected size of 30 nt, probably arising from the loss of nucleotides during the PCR and/or ligation steps. The growth data were obtained with 2 independent experiments (, ). When these 2 columns in are compared, it is clear that the same sRNA reproducibly caused the same growth inhibition (see +IPTG condition). However, the growth inhibitory effects varied widely among different sRNAs. These results indicate that the growth inhibition of E. coli in this sRNA system is basically sRNA-sequence specific. For example, the effect of S-20 sRNA was very strong, from immediately after IPTG induction. However, with S-61 sRNA, E. coli grew similarly, with and without IPTG induction, until it reached an optical density at 600 nm (OD600) of 0.5, after which its growth was only inhibited in cells induced with IPTG. These observations prompted the speculation that each sRNA has a unique regulatory mechanism by which it inhibits the growth of E. coli. To evaluate whether the inhibitory effect on E. coli growth exerted by each sRNA was dependent on its nucleotide sequence rather than on its level of transcription, the sRNA expression levels were examined with northern blotting (). Major transcripts of approximately 100 nt (detected with the probe SRA1) and 50 nt (detected with the probe SRA3) were expressed in the presence of IPTG from each sRNA clone and the vector plasmid, respectively. Although a very small amount of leaky sRNA expression was observed in the absence of IPTG, the expression of the control 5S rRNA was not influenced by the induction of sRNA expression. This result confirms that there was no significant correlation between the inhibition of E. coli growth and the level of sRNA expression. Because the expression of S-10 was higher than that of the other sRNAs, we analyzed S-20 to understand the function and regulation of artificial sRNAs. We confirmed that the S-20 sequence lacks an apparent open reading frame and has no AUG initiation codon for protein synthesis, although it is possible to use the GUG or UUG initiation codon for this purpose in E. coli.Citation31 We demonstrated that purified S-20 RNA bound directly to and repressed the translation of at least 2 possible target mRNAs (see below), so S-20 probably functions as a non-coding RNA.
Figure 2. Expression of artificial sRNAs that inhibit E. coli growth. (A) Six examples of changes in E. coli growth induced by artificial sRNAs (S-10, −20, −61, −22, −7, and −8) or the empty vector pET-28DEL (Vect.). Single colonies of E. coli containing each plasmid were used to inoculate 200 μl aliquots of LB medium in 96-well plates, which were incubated at 37°C without IPTG (−) or with 40 μM IPTG (+). Cell growth was monitored by scanning the optical density at 600 nm (OD600). Data were obtained from 2 separate experiments, and the means and standard deviations of 3 cultures were calculated. (B) Northern blotting analysis of artificial sRNA expression in E. coli. E. coli containing each plasmid was grown at 37°C. After overnight incubation, the culture was diluted to OD600 = 0.3 with fresh LB medium containing 30 μg/ml kanamycin and then cultured at 37°C for 1 h. The expression of the artificial sRNAs was induced with 40 μM IPTG at 37°C. E. coli was also cultured at 37°C for 1 h without IPTG (−), as the control. The SRA1 probe was used to detect artificial sRNA and the SRA3 probe was used to detect the vector-derived sRNA (see and Table S1). The arrowheads indicate the positions of the major transcripts. 5S rRNA was used as the loading control.
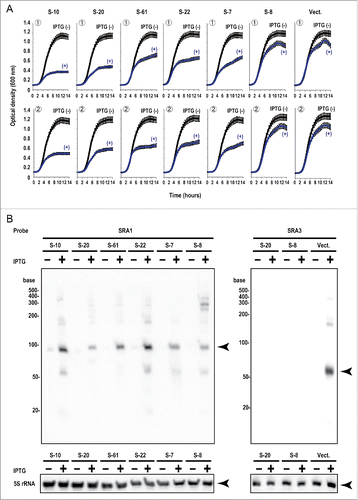
Table 2. Nucleotide sequences of cloned artificial sRNAs.
Characterization of the effects of artificial sRNA S-20 on E. coli
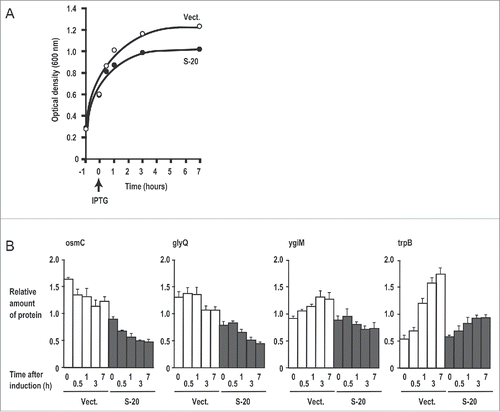
To characterize the effects of S-20 on the E. coli proteome profile, a comprehensive proteomic analysis of bacteria that express the S-20 sRNA was conducted (Table S2). For the proteomic analysis, the whole proteins were extracted at the indicated times (0, 0.5, 1, 3, and 7 h) after IPTG induction (). As a control, E. coli cells carrying the empty vector (Vect.) were analyzed in the same way. Under the culture conditions in which S-20 was induced at OD600 = 0.6, the expression of S-20 again inhibited E. coli growth: OD600 = 0.99 after incubation for 3 h in the presence of IPTG, whereas E. coli cells carrying the empty vector grew to OD600 = 1.2 after incubation for 3 h in the presence of IPTG. A nano-liquid chromatography/electrospray ionization-tandem mass spectrometry (nanoLC/ESI-MS/MS) analysis identified 1,734 E. coli proteins, from which we generated a list of proteins whose levels changed were (A) downregulated (top 35) or (B) upregulated (top 35) in response to S-20 expression. The list was sorted based on the data collected 3 h after induction. Enzymes involved in nucleic acid metabolism and energy metabolism constituted a large proportion of the proteins listed (A). The genes involved in nucleic acid metabolism were especially well represented in the list: glyQ and glyS (encoding glycyl-tRNA synthetase [GlyRS], α and β subunits, respectively), cca (encoding tRNA CCA-adding enzyme), xseA (encoding exodeoxyribonuclease 7 large subunit), ssb (encoding single-stranded-DNA-binding protein), and rnb (encoding RNase II). Examples of genes involved in energy metabolism included allB (encoding allantoinase), osmC (encoding peroxiredoxin), and trpB (encoding tryptophan synthase β chain). A computational analysis using TargetRNA232 predicted that 6 of the mRNAs encoding these 35 downregulated proteins were targets of S-20: osmC, glyQ, trpB, ygiM (encoding uncharacterized protein YgiM), gabD (encoding succinate-semialdehyde dehydrogenase), and pflB (encoding formate acetyltransferase 1). Operon information for E. coli, registered in the BioCyc Database Collection (http://biocyc.org/), showed that the gly operon includes the glyQ and glyS mRNAs. We also noted the ygiM-cca operon in the database, so these mRNAs were good candidate targets of S-20. Time-course analyses of 4 downregulated proteins are shown in . The expression of osmC and glyQ was repressed throughout the period of S-20 induction. The expression of ygiM and trpB increased with time in E. coli containing the empty vector, but S-20 suppressed this upregulation. In contrast, among the top 35 proteins upregulated were proteins involved in membrane or transporter functions, energy metabolism, and transcription. Only 2 of the mRNAs, gspG (encoding putative type II secretion system protein G) and lsrR (encoding transcriptional regulator lsrR), were predicted to be S-20 targets with TargetRNA2. However, as shown in Fig. S3, 3 of 4 examples of upregulated proteins, UPF0225 protein (ychJ), gspG, and lsrR, showed no apparent upregulation throughout the induction of S-20. Because these proteins were identified at the 3 h time point, and large differences in the levels of proteins between E. coli expressing S-20 and that expressing the empty vector were only detected at this time point, the upregulation of gspG and lsrR by S-20 was less marked throughout the induction period.
Figure 3. Four examples of proteins downregulated in response to S-20 induction. (A) Time point of S-20 induction and changes in E. coli growth. (B) Four examples of proteins downregulated in response to S-20 induction. Relative amounts of proteins (osmC, glyQ, ygiM, and trpB) at each induction time point were determined with a nanoLC-MS/MS analysis (see Materials and Methods). Mean (n = 3) and standard deviation for each value are shown. Vect.: vector.
To determine whether the changes in the amounts of these proteins correlated with the changes in their mRNA levels, we analyzed the mRNAs of 11 of the proteins in Table S2 with quantitative reverse transcription (qRT)-PCR (Fig. S4). These mRNAs were selected for 2 reasons: (i) the amounts of the proteins they encode changed when S-20 was expressed; and (ii) some of these mRNAs are predicted targets of S-20. For the control, we also selected 2 genes, glmM (encoding phosphoglucosamine mutase) and glnS (encoding glutamine tRNA ligase [GlnRS]), each protein amount of which was not influenced by the induction of S-20 (Fig. S3C) and whose mRNAs are not predicted targets of S-20 (see below). After the expression of S-20 was induced for 3 h, the expression of all of the mRNAs corresponding to the downregulated proteins (Fig. S4A) was reduced to approximately 52-90%, whereas the expression of all of the mRNAs (except sufA) corresponding to upregulated proteins (Fig. S4B) was increased to approximately 115-192%. There were no significant changes in the amounts of the 2 control mRNAs, glmM and glnS. After the expression of S-20 was induced for 7 h, the expression of all of the mRNAs was upregulated. However, the degree of upregulation differed between the mRNAs corresponding to the downregulated proteins (188% on average) and the mRNAs corresponding to the upregulated proteins (344% on average). Even the expression of the control mRNAs was upregulated (233% for glmM and 135% glnS). Therefore, the expression of the downregulated proteins listed in Table S2A did not correlate with their mRNA levels. Based on these observations, we concluded that S-20 caused the downregulation of some proteins (mentioned in ), mainly at the translational level. Therefore, we investigated whether S-20 binds to the mRNAs of these downregulated proteins and regulates their expression at the translational level, as do the endogenous sRNAs of E. coli.
Figure 4. Prediction of S-20 target mRNAs. mRNAs (operons) targeted by S-20 were predicted computationally (see Materials and Methods), and 4 examples (A-D) of the hybridization patterns between S-20 and its target mRNAs are shown. The transcription start site of each operon is indicated with a curved arrow. Numbers indicate the nucleotide positions from the translation start site of each mRNA close to the S-20 target region: (A) osmC, (B) glyQ, (C) ygiM, and (D) trpB.

Verification of the mRNAs targeted by S-20
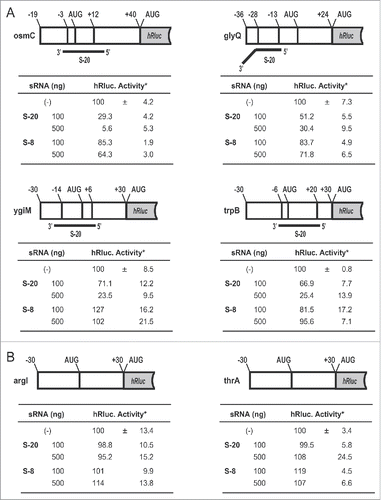
shows 4 examples of the predicted patterns of hybridization between S-20 and each candidate target mRNA (operon). S-20 hybridized to the regions that included the AUG codons in the osmC (), ygiM mRNAs () and trpB () mRNAs. S-20 also hybridized to the 5′-untranslated region, which occurs approximately 10-30 nt upstream from the initiation AUG codon, in the glyQ mRNA (). The binding sequence within the 30 nt of S-20 changed, depending on the target mRNA, and either the 5′ or 3′ half or almost the whole sequence of S-20 was involved in binding each mRNA. To verify experimentally that the translation of the predicted target mRNAs was actually controlled by S-20, as is the case with endogenous sRNAs, an in vitro translation analysis was performed using a reporter Renilla luciferase (hRluc) mRNA fused to each S-20-binding site (). For this analysis, 6 chimeric RNAs were constructed: osmC-hRluc, glyQ-hRluc, ygiM-hRluc and trpB-hRluc as S-20 target RNAs (), and arg1 (encoding ornithine carbamoyltransferase chain 1)-hRluc and thrA (encoding bifunctional aspartokinase/homoserine dehydrogenase 1)-hRluc as the control non-target RNAs (). We measured the relative amounts of hRluc activity to observe the effects of S-20. To evaluate the specificity of the sRNAs, another artificial sRNA, S-8, was also examined. The hRluc activity associated with all 4 possible target mRNAs was reduced to 5.6%-30.4% in an S-20-dose-dependent manner (). These activities were negligibly affected by the presence of S-8. However, the hRluc activity associated with the 2 control mRNAs remained the same, with or without the addition of S-20 or S-8. These results demonstrate that S-20 affects these 4 mRNAs and represses their translation, reducing their protein concentrations in E. coli.
Figure 5. Effects of artificial sRNAs on the translation of sRNA target-synthetic Renilla luciferase (hRluc) chimeric reporter RNAs. (A) Renilla luciferase activities of the S-20-targeted mRNA-hRluc chimeras. (B) Renilla luciferase activities of the negative control chimeras (no sRNA-binding region). An in vitro translation assay of the chimeric RNA was performed in the presence of the indicated amounts of sRNAs. These chimeric RNAs contained additional ribonucleotide sequences (approximately 30 nt) derived from the vector sequence at each 5′ terminal. Tables show the mean values and standard deviations of the relative luciferase activities from 3 experiments. *Normalized luciferase activity in the absence of sRNAs was set to 100%.
We then examined the RNA-RNA interactions between S-20 and its target mRNAs with an RNA gel shift assay (). For the target mRNAs, we used in vitro-transcribed 111-nt oligoribonucleotides containing each S-20-binding site (60 nt) fused to part of the hRluc mRNA, as shown in . The sequence of each target mRNA was exactly the same as the sequence of the first region of the corresponding reporter mRNAs. The results showed that S-20 directly bound to the 2 target chimeric oligoribonucleotides containing the translation initiation site sequence of either osmC or trpB. The control S-8 bound to neither of the sequences under the same conditions. However, no detectable interaction was observed between S-20 and 2 other target oligoribonucleotides from glyQ or ygiM, even after adjustment of the reaction conditions. Because S-20 repressed the translation of the reporter RNAs associated with osmC, glyQ, ygiM, and trpB, and we used an E. coli lysate for the in vitro translation experiment shown in , an additional factor, such as the RNA chaperone, Hfq protein, may be required for efficient RNA-RNA interactions in the cases of glyQ and ygiM. We also examined 2 S-20 mutants, S-20 Ma and S-20 Mf (also see ). Both mutants abolished the binding activity to trpB but not to osmC. These data support the fact that the binding sequence within the 30 nt of S-20 were altered, depending on the target mRNA (see below for the effects of these mutations on E. coli growth inhibition). In conclusion, S-20 repressed some genes at the translational level and at least 2 target mRNAs, osmC, and trpB, were directly regulated by this artificial sRNA.
Figure 6. RNA gel shift analysis of S-20 target mRNAs. RNA-RNA interactions were examined with an RNA gel shift analysis. A FITC-labeled oligoribonucleotide, either S-20 (6 pmol) or S-8 (6 pmol), was incubated with 0-20 pmol of each target oligoribonucleotide (osmC, glyQ, ygiM or trpB) at 70°C for 7 min, and then at room temperature for 1 h. RNA-RNA interactions were analyzed by electrophoresis on a non-denaturing 4% (w/v) polyacrylamide gel. Two FITC-labeled mutant oligoribonucleotides, S-20 Ma and S-20 Mf, were examined in a similar manner (also see ). Similar results were obtained in at least 2 independent experiments.
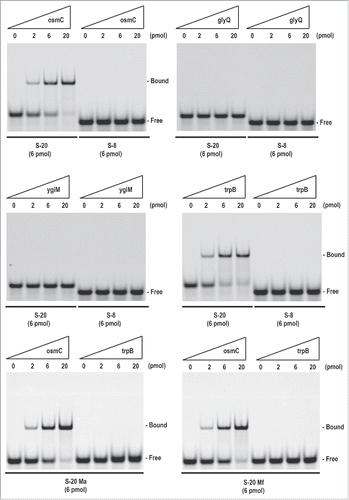
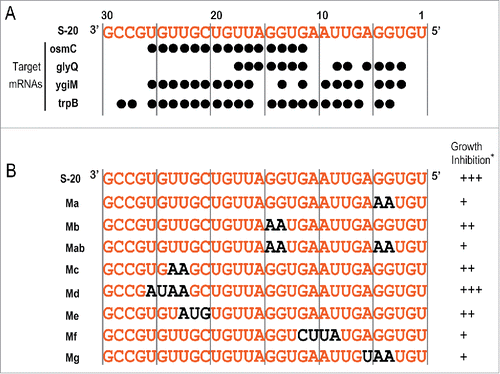
Figure 7. Analysis of S-20 mutants. (A) Nucleotide sequences (shown in orange) of the regions inserted into the artificial S-20 sRNA expression plasmid pASRIIS-20 and the ribonucleotides responsible for its hybridization with each target mRNA (indicated by black circles). See also . (B) S-20 nucleotide sequence (insert) and its mutants. S-20 nucleotides are indicated in orange and the substituted nucleotides in the S-20 sequence are indicated in black. (*) Inhibition of E. coli growth is shown as the following 3 categories: +++, maximum OD600 < 0.6; ++, 0.6 ≤ maximum OD600 < 0.8; +, 0.8 ≤ maximum OD600. See also Fig. S5.
Possible mechanism of S-20 action and further insight into artificial sRNAs
In this paper, we have shown that at least 4 mRNAs are regulated by S-20 sRNA and that S-20 reduced the amount of each protein at the translational level. The next question is: which mRNA corresponds to the inhibition of E. coli growth? To address this question, S-20 mutants were constructed and their effects on E. coli growth were investigated ( and Fig. S5A). The S-20 nucleotides required for the hybridization of S-20 to each of the 4 target mRNAs are summarized in , based on the hybridization patterns shown in . The positions of the nucleotides substituted in each mutant sRNA were mainly selected to interrupt each site of hybridization, but many of the nucleotides in S-20 that recognized the target mRNA sequences overlapped. A RNA gel blotting analysis showed that many of the S-20 mutants caused a 2-3-fold increase in the amount of each RNA (Fig. S5B), except mutants Me and Mf and the S-8 sRNA. Therefore, we carefully analyzed the strong inhibitory effect on E. coli growth when the amount of a certain mutant sRNA was markedly increased, and removed mutant Md from further analysis. As shown in and S5A, although S-20 inhibited bacterial growth, these inhibitory effects were partially ameliorated in all the mutants except Md, in which the OD600 was 0.7-0.9 after incubation for 12 h in the presence of IPTG. The stronger amelioration roughly mapped to 2 ribonucleotide regions in the S-20 sequence: (i) ribonucleotide positions 4G, 5G, and 6A (mutants Ma, Mab, and Mg), and (ii) ribonucleotide positions 9U, 10A, 11A, and 12G (mutant Mf). Based on the results for the first 3 mutants, we concluded that the guanines (Gs) at nucleotide positions 4 and 5 in the random nucleotide region of S-20 were at least partially responsible for the growth inhibition of E. coli. These results also show that osmC is not responsible for this growth inhibition, and suggest that S-20 targets multiple mRNAs. The gel shift analysis of the mutant S-20 Ma abolished its binding activity to trpB, but not to osmC. Moreover, based on the results for the second region mutant Mf, which was designed not to hybridize with trpB mRNA and was expressed at the same level as the original S-20, the partial amelioration of E. coli growth inhibition by the Mf mutant suggests that trpB is one of the candidate proteins responsible for E. coli growth. This observation is also supported by the gel shift analysis of mutant S-20 Mf (). Mutations in the middle region of S-20 (mutant Mb) or in the 3′ half of the sequence (mutant Me) still inhibited E. coli growth. According to the analysis of all these mutants, it is possible that the interrupted translation of either glyQ mRNA or ygiM mRNA may also be responsible for this bacterial growth inhibition.
To summarize the results of this study, a possible explanation of the S-20-mediated inhibition of E. coli growth is described. S-20 may target several mRNAs, encoding proteins such as trpB and GlyRS and inhibiting their translation. Therefore, the expression of these proteins is reduced. Because trpB and trpA are important enzymes, supplying the amino acid tryptophan to the cell, we speculate that the depletion of this important enzyme may be the main reason for the inhibition of E. coli growth. Indeed, it has been reported that trp mutant E. coli strains (trpB8 or trpA2) do not grow in minimal medium or under tryptophan-limited conditions. Citation33 Furthermore, E. coli growth was previously inhibited by the expression of RNA mini-helices directed against GlyRS.Citation34 Because we observed a reduction in GlyRS protein in both a proteome analysis and an in vitro translation analysis, we also infer that the depletion of GlyRS is involved in the inhibition of E. coli growth, although in this case, we did not reconstitute the direct RNA-RNA interaction in vitro. Simultaneously, S-20 may also indirectly target other enzymes involved in nucleic acid metabolism, such as RNase II and exodeoxyribonuclease 7 (Fig. S3). Although we have no exact data to explain how these enzymes are downregulated by S-20, the depletion of these enzymes may also inhibit E. coli growth. Pertinently, it has been reported that RNase II is associated with the known degradation component of the RNA degradosome in E. coli.Citation35 Therefore, the depletion of RNase II may increase the levels of other sets of mRNAs, and the expression of the corresponding proteins. Alternatively, it is also true that we only focused on the mRNA candidates that were selected by the bioinformatics analysis (predicted by TargetRNA2), although we identified more than 30 other candidate targets of S-20 (Table S2). Therefore, to determine the exact mRNA set that is responsible for E. coli growth inhibition, another set of experiments may be required, including a series of mutational analyses of endogenous candidate target mRNAs.
sRNAs are usually only conserved within closely related species of prokaryotes.Citation8,15,36,37 For example, most sRNAs identified in E. coli are restricted to 2 genera, Escherichia and Shigella.Citation8 In other words, the genes for sRNAs may evolve more rapidly than protein-coding genes.Citation15 In this study, we identified an artificial sRNA, S-20, that directly affects the metabolic systems of E. coli, just like endogenous sRNAs, based on the following points: (i) S-20 may target several mRNAs by its partial hybridization with each target; and (ii) the downregulation of each target protein occurs at the translation level. Therefore, it is conceivable that this artificial sRNA mimics the regulatory mechanisms of endogenous sRNAs. Our results suggest that although the artificial sRNA was not originally associated with any biologic regulatory system in E. coli, it could be used as a functional biomolecule once selected in the cell. Escherichia coli-genome-derived sequences may be better than random sequences for constructing an artificial sRNA library with which to efficiently isolate artificial antisense RNAs. However, our data suggest that at least the S-20 sRNA sequence was selected to hybridize with a group of target mRNAs from the partially randomized sRNA library. We believe that this is an important feature of sRNA selection (evolution). Furthermore, although there is no S-20-like sequence in the E. coli genome, some endogenous sRNAs are reportedly involved in bacterial cell growth under certain stress or nutritional conditions. Citation38,39 Our finding implies that E. coli has the capacity to use artificial sRNAs for its urgent regulation, and we speculate that this capacity may be associated with the presumed rapid evolution of bacterial sRNAs. However, we could not isolate any sRNA clones that conferred extra abilities on E. coli, such as heat resistance, UV resistance, or drug resistance, despite several rounds of rigorous screening. Appropriate screening and selection systems will be required to resolve this problem. It is also important to use a non-leaky inducible promoter for the next step. The establishment of these systems should open new opportunities for synthetic RNA biology.
Materials and methods
Construction of an expression plasmid library of artificial sRNAs
To prepare the expression plasmid (pASRII) library of artificial sRNAs, the plasmid pET-28DEL () was initially constructed with the following 2 steps. (i) To remove an 88-bp sequence between the NotI restriction site and the T7 terminator in the pET-28b expression vector (Novagen, #69865-3), PCR was used to amplify the external region of the 88-bp sequence using 5′-phosphorylated primers, DEL-S0 and DEL-A0. After the self-ligation of the amplified fragment, the plasmid (designated pET-28DEL0) was generated with a conventional cloning method. (ii) To remove a further nucleotide sequence between the T7 promoter and the NotI restriction site, and to insert an XbaI restriction site immediately downstream of the T7 promoter sequence in pET-28DEL0, PCR was performed again using 5′-phosphorylated primers, DEL-S and DEL-A, and pET-28DEL was generated in a similar way. To construct the expression plasmid pASRII (), the inserted DNAs were first amplified with PCR from a synthetic 111-bp DNA template containing 30 bp of random nucleotide sequence (purchased from Hokkaido System Science, Japan) using primers 30S and 30A. The resulting DNA fragments were sub-cloned into the XbaI-NotI sites in the pET-28DEL vector. Within the insert, a sequence of approximately 30 bp, corresponding to the Hfq-RNA-chaperone-binding region (Hfq-BR: the sequence was originally derived from DsrA sRNACitation40,41), was placed next to the random sequence.
Construction of pASRII-S-20 mutant plasmids and luciferase reporter plasmids
To construct the pASRII-S-20 mutant plasmids, each annealed oligonucleotide containing a mutation (see Table S1 and ) was sub-cloned into the XbaI-NotI sites in the pET-28DEL vector. To construct the luciferase reporter plasmids, the synthetic Renilla luciferase gene (hRluc) was first amplified from the psiCHECK-2 plasmid (Promega, #C8021) with tagged PCR primers, and sub-cloned into the HindIII-XhoI sites in the pET-23b vector (Novagen, #69746-3). Each annealed oligonucleotide containing possible S-20 target regions (see Table S1, and ) was then sub-cloned into the XbaI-HindIII sites in each resulting plasmid.
Expression and screening of artificial sRNAs that inhibit E. coli growth
E. coli HMS174(DE3) (Novagen, #69453-3) was transformed with the pASRII library. Immediately before the transformants were spread on 90 mm Luria-Bertani (LB) agar plates containing 30 μg/ml kanamycin, 100 μl of IPTG (40 μM) was applied to each plate. The transformants were grown at 37°C for 16 h. To isolate the plasmid clones containing sRNAs that inhibited E. coli growth, the smaller single colonies were selected. The plasmids were isolated and the same E. coli strain was transformed with each of the plasmids, and grown at 37°C for 16 h on plates containing 30 μg/ml kanamycin. Single colonies picked from the plates were used to inoculate 1 ml samples of LB medium containing 30 μg/ml kanamycin. After incubation at 37°C for 7 h, the cultures were diluted (1:44) with fresh LB medium containing 30 μg/ml kanamycin, and 200 μl aliquots were placed in a 96-well plate (TPP, Switzerland). The growth of each E. coli culture at 37°C with slight agitation, with or without 40 μM IPTG, was monitored directly at OD600 on a spectrophotometer (SpectraMax Plus 384; Molecular Devices, Inc., USA). The OD600 values were used to construct the growth curves (Figs 2A, S2, and S5A). Clones showing highly inhibitory effects on the growth of E. coli were selected, and the nucleotide sequence of each DNA insert was determined.
E. coli culture conditions
Escherichia coli was cultured with the following 2 protocols. (i) To monitor E. coli growth (Figs 2A, S2, and S5A), a single E. coli colony carrying pASRII or the control vector (pET-28DEL) was used to inoculate 1 ml of LB medium containing 30 μg/ml kanamycin. After incubation at 37°C for 7 h, the culture was diluted (1:44) with fresh LB medium containing both 30 μg/ml kanamycin and 40 μM (or the indicated concentration of) IPTG, and then cultured in 200 μl aliquots in a 96-well plate at 37°C with slight agitation for 16 h to monitor E. coli growth. (ii) To prepare the total RNA for RNA gel blot hybridization (Figs 2B and S5B) and for the qRT-PCR analysis (Fig. S4), or to express the proteins used in the proteomic analysis (Table S2 and Figs 3B and S3), a single E. coli colony carrying pASRII or the control vector (pET-28DEL) was used to inoculate 2 ml of LB medium containing 30 μg/ml kanamycin. After incubation overnight at 37°C, the culture was diluted to OD600 = 0.3 with fresh LB medium containing 30 μg/ml kanamycin and then cultured at 37°C for 1 h. The expression of the artificial sRNAs was induced with 40 μM IPTG at 37°C. These cultured cells were harvested after induction for 1 h for RNA gel blot hybridization or at each indicated time point for qRT-PCR analysis and proteomic analysis. E. coli was also cultured at 37°C without IPTG as the control. The samples were immediately stored at −80°C for later analysis.
Northern blot hybridization and qRT-PCR analysis
Total RNA was extracted with the RNeasy Midi Kit (Qiagen, #75144), with slight modification. Instead of using an RNeasy Midi column, we used phenol-chloroform extraction to efficiently collect the complete set of RNAs, including the low-molecular-weight RNAs.
To analyze the sRNAs (20-500 nt) with RNA gel blot hybridization, the total RNA (0.8 μg per lane) was separated on a denaturing 6% polyacrylamide gel containing 8 M urea and then transferred onto Hybond-N+ membrane (GE Healthcare, USA) with electroblotting. After UV cross-linking, the membrane was pre-hybridized manually in hybridization buffer containing 1× Denhardt's solution (0.2 μg/ml Ficoll 400, 0.2 μg/ml polyvinylpyrrolidone, and 0.2 μg/ml bovine serum albumin), 4× saline sodium citrate (SSC) diluted from UltraPure 20× SSC (Invitrogen, #15557-036), 0.1 mg/ml UltraPure Salmon Sperm DNA Solution (Invitrogen, #15632-011), and 0.5% sodium dodecyl sulfate (SDS). The biotin-labeled antisense oligodeoxynucleotide was prepared with the Biotin 3′ End DNA Labeling Kit (Pierce Biotechnology, #89818), and hybridization was performed overnight at 37°C in the same buffer as the labeled antisense oligodeoxynucleotide. The membrane was then washed with buffer containing 4× SSC and 0.5% SDS at 37°C. The non-isotopic blots were visualized with ECF Substrate (GE Healthcare, #RPN5785). In the qRT-PCR analysis, reverse transcription was performed with the TaKaRa PrimeScriptRT-PCR Kit (Takara Bio Inc., #RR014A), according to the manufacturer's protocol. The qRT-PCR analysis was conducted using the StepOnePlus Real-Time PCR System (Applied Biosystems, USA) with SYBR Premix Ex Taq II, Tli RNaseH Plus (Takara Bio Inc., #RR820A), according to the manufacturer's protocol. The relative amounts of mRNAs were normalized to the gyrB transcript in each sample. P values were calculated with a t test (n = 3).
Proteomic analysis
The protein mixture was extracted from pelleted E. coli with 100 mM triethylammonium bicarbonate (TEAB; pH 8.5) containing 12 mM sodium deoxycholate, 12 mM sodium N-dodecanoyl sarcosinate, and 1% protease inhibitor cocktail (Sigma-Aldrich Co., #P8340). The resulting protein mixture was reduced with 10 mM dithiothreitol at room temperature for 30 min, and alkylated with 47 mM iodoacetamide at room temperature for 30 min in the dark. The samples were diluted 5-fold with 100 mM TEAB (pH 8.5) and digested overnight with sequencing-grade lysyl endopeptidase (Lys-C) (Wako, #129-02541) and trypsin (Promega, #V5113) at room temperature. An LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific, USA) equipped with a hand-made spray needle column (100 µm i.d., 5 µm tip i.d., 130 mm length) packed with Reprosil-Pur C18 material (3 µm, 120 Å; Dr. Maish, Germany), a Dionex UltiMate 3000 pump with an FLM-3000 Flow Manager (Dionex Softron GmbH, Germany), and an HTC-PAL Autosampler (CTC Analytics, Switzerland) was used for the nanoLC-MS/MS measurements. The mobile phase, composed of (A) acetic acid:water (0.5:100, v/v) and (B) acetic acid:water:acetonitrile (0.5:20:80, v/v/v), was added at a flow rate of 500 nl/min. The digested material was dissolved in acetic acid:water:acetonitrile (0.5:95:5, v/v/v) and injected into the LC/MS system. The composition of the mobile phase was (B) 5-10% (0-5 min), 10-40% (5-65 min), 40-100% (65-70 min), 100% (70-80 min), and 5% (80-110 min). The eluted peptide fragments were ionized with electrospray ionization in positive ion mode, and the strongest 10 peaks derived from multiple charged peptide ions were subjected to MS/MS with collision-induced dissociation. The spray needle voltage was 2400 V, the capillary voltage 35 V, and the tube lens voltage 100 V. The capillary temperature was 200°C. The MS scan was performed in a scan range of m/z 300-1500 at a resolution of 60,000. The MS/MS scan was performed under the following conditions: isolation width, 2; normalized collision energy, 35 V; activation Q, 0.25; activation time, 30 s.
The mass spectrum data generated with nanoLC/ESI-MS/MS were screened against UniProtKB/Swiss-Prot entries (http://www.uniprot.org/) with the Mascot program (Matrix Science, UK), under the following conditions: taxonomy, E. coli K-12; peptide ion tolerance, 3 ppm; product ion tolerance, 0.8 Da; enzyme, trypsin; missed coverage, 2; fixed modification, carbamidomethylation at Cys; variable modification, oxidation at Met. The peptides identified with a MASCOT score of >95% reliability were considered in this study. The expression levels of the proteins were calculated as follows. The maximal signal intensities of all the peptide ions identified on the mass spectra were analyzed with Mass Navigator ver. 1.2 (Mitsui Knowledge Industry, Japan), and then each intensity value was adjusted for the total ion intensity of the mass spectra at m/z 300-1500 and a retention time of 30-80 min, during which most peptides were detected, to normalize the intensity values between the different LC-MS runs. The relative expression levels of the peptides were then calculated as the intensity of each LC-MS run divided by the mean intensity of the peptide with the same sequence on all LC-MS runs. The expression level of each protein was calculated as the mean of the relative expression levels of those peptides without outlier values, determined with Thompson's outlier test (P < 0.1).
Prediction of S-20 target mRNAs
The web server TargetRNA232 was used to predict the mRNAs targeted by S-20, with the following parameters: replicon, E. coli str. K-12 sub-strain MG1655; P value threshold, 1.0; nucleotides in the interaction region, 20 or 30 nt. The target sites located within the mRNAs were selected based on information from the BioCyc Database Collection (http://biocyc.org/).
In vitro transcription/translation and Renilla luciferase reporter assay
Two kits were used for the in vitro transcription reactions, depending on the size of the RNA synthesized: the MEGAshortscript T7 Transcription Kit (Ambion, #AM1354) for sRNAs (approximately 100 bases; ) and the MEGAscript T7 Transcription Kit (Ambion, #AM1334) for longer RNAs (approximately 1,000 bases; ). Recombinant RNase Inhibitor (Takara Bio Inc., #2313A) was added to each reaction mixture, which was then incubated at 37°C for 3 h. The resulting products were treated with RNase-free DNase I (Takara Bio Inc., #2270A), and then extracted with phenol-chloroform and purified with CHROMA SPIN-30 Columns (BD Biosciences Clontech, #636087). The S30 T7 High-Yield Protein Expression System, which is an E. coli-lysate-based translation system made by Promega (Promega, #L1115) was used for the in vitro translation reaction, according to the manufacturer's protocol. The Renilla Luciferase Assay System (Promega, #E2810) was used to estimate the amount of Renilla luciferase translated. Luciferase activity was measured with a luminometer (Lumat LB9507; Berthold, Germany).
RNA gel shift analysis
5′-Fluorescein isothiocyanate (FITC)-labeled synthetic oligoribonucleotides (purchased from Hokkaido System Science, Japan) were used as the artificial small RNAs. For the target mRNAs, in vitro-transcribed 111-nt oligoribonucleotides containing each S-20-binding site (60 nt) fused to part of the hRluc mRNA (as shown in ) were used. Binding reactions containing the 5′-FITC-labeled oligoribonucleotide (6 pmol) and target mRNA oligoribonucleotide (0-20 pmol) in 20 μl of RNA binding buffer (10 mM Tris-HCl [pH 7.2], 100 mM NaCl, 0.5 mM EDTA and 2.5 mM MgCl2) were incubated at 70°C for 7 min, and then at room temperature for 1 h. The RNA-RNA complexes were promptly analyzed with electrophoresis on a 4% (w/v) non-denaturing polyacrylamide gel at room temperature.Citation42 The RNA-RNA complexes were quantitated by scanning the fluorescent image with the Molecular Imager FX Pro (Bio-Rad Laboratories, USA).
Disclosure of potential conflicts of interest
The authors declare that they have no potential conflicts of interest regarding this study.
Supplemental_Data.zip
Download Zip (5.6 MB)Acknowledgments
We thank Ms. Yukari Sato (Keio University, Japan) for technical assistance with the qRT-PCR analysis. We also thank all the members of the RNA Group at the Institute for Advanced Biosciences, Keio University, Japan, for their insightful discussions.
Funding
This work was supported, in part, by research funds from the Yamagata Prefectural Government and Tsuruoka City, Japan.
Related Research Data
References
- Wagner EG, Romby P. Small RNAs in bacteria and archaea: who they are, what they do, and how they do it. Adv Genet 2015; 90:133-208; PMID:26296935; http://dx.doi.org/10.1016/bs.adgen.2015.05.001
- Desvignes T, Batzel P, Berezikov E, Eilbeck K, Eppig JT, McAndrews MS, Singer A, Postlethwait JH. miRNA Nomenclature: A view incorporating genetic origins, biosynthetic pathways, and sequence variants. Trends Genet 2015; 31:613-26; PMID:26453491; http://dx.doi.org/10.1016/j.tig.2015.09.002
- Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet 2016; 17:47-62; PMID:26666209; http://dx.doi.org/10.1038/nrg.2015.10
- Storz G, Vogel J, Wassarman KM. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell 2011; 43:880-91; PMID:21925377; http://dx.doi.org/10.1016/j.molcel.2011.08.022
- Durand S, Tomasini A, Braun F, Condon C, Romby P. sRNA and mRNA turnover in Gram-positive bacteria. FEMS Microbiol Rev 2015; 39:316-30; PMID:25934118; http://dx.doi.org/10.1093/femsre/fuv007
- Raghavan R, Groisman EA, Ochman H. Genome-wide detection of novel regulatory RNAs in E. coli. Genome Res 2011; 21:1487-97; PMID:21665928; http://dx.doi.org/10.1101/gr.119370.110
- Babski J, Maier LK, Heyer R, Jaschinski K, Prasse D, Jager D, Randau L, Schmitz RA, Marchfelder A, Soppa J. Small regulatory RNAs in Archaea. RNA Biol 2014; 11:484-93; PMID:24755959; http://dx.doi.org/10.4161/rna.28452
- Shinhara A, Matsui M, Hiraoka K, Nomura W, Hirano R, Nakahigashi K, Tomita M, Mori H, Kanai A. Deep sequencing reveals as-yet-undiscovered small RNAs in Escherichia coli. BMC Genomics 2011; 12:428; PMID:21864382; http://dx.doi.org/10.1186/1471-2164-12-428
- Hoe CH, Raabe CA, Rozhdestvensky TS, Tang TH. Bacterial sRNAs: regulation in stress. Int J Med Microbiol 2013; 303:217-29; PMID:23660175; http://dx.doi.org/10.1016/j.ijmm.2013.04.002
- Durand S, Braun F, Lioliou E, Romilly C, Helfer AC, Kuhn L, Quittot N, Nicolas P, Romby P, Condon C. A nitric oxide regulated small RNA controls expression of genes involved in redox homeostasis in Bacillus subtilis. PLoS Genet 2015; 11:e1004957; PMID:25643072; http://dx.doi.org/10.1371/journal.pgen.1004957
- Chao Y, Vogel J. A 3′ UTR-derived small RNA provides the regulatory noncoding arm of the inner membrane stress response. Mol Cell 2016; 61:352-63; PMID:26805574; http://dx.doi.org/10.1016/j.molcel.2015.12.023
- Bobrovskyy M, Vanderpool CK. Diverse mechanisms of post-transcriptional repression by the small RNA regulator of glucose-phosphate stress. Mol Microbiol 2016; 99:254-73; PMID:26411266; http://dx.doi.org/10.1111/mmi.13230
- Sabharwal D, Song T, Papenfort K, Wai SN. The VrrA sRNA controls a stationary phase survival factor Vrp of Vibrio cholerae. RNA Biol 2015; 12:186-96; PMID:25826569; http://dx.doi.org/10.1080/15476286.2015.1017211
- Jaschinski K, Babski J, Lehr M, Burmester A, Benz J, Heyer R, Dörr M, Marchfelder A, Soppa J. Generation and phenotyping of a collection of sRNA gene deletion mutants of the haloarchaeon Haloferax volcanii. PLoS One 2014; 9:e90763; PMID:24637842; http://dx.doi.org/10.1371/journal.pone.0090763
- Peer A, Margalit H. Evolutionary patterns of Escherichia coli small RNAs and their regulatory interactions. RNA 2014; 20:994-1003; PMID:24865611; http://dx.doi.org/10.1261/rna.043133.113
- Skippington E, Ragan MA. Evolutionary dynamics of small RNAs in 27 Escherichia coli and Shigella genomes. Genome Biol Evol 2012; 4:330-45; PMID:22223756; http://dx.doi.org/10.1093/gbe/evs001
- Mellin JR, Cossart P. Unexpected versatility in bacterial riboswitches. Trends Genet 2015; 31:150-6; PMID:25708284; http://dx.doi.org/10.1016/j.tig.2015.01.005
- Peselis A, Gao A, Serganov A. Cooperativity, allostery and synergism in ligand binding to riboswitches. Biochimie 2015; 117:100-9; PMID:26143008; http://dx.doi.org/10.1016/j.biochi.2015.06.028
- Apostolidi M, Saad NY, Drainas D, Pournaras S, Becker HD, Stathopoulos C. A glyS T-box riboswitch with species-specific structural features responding to both proteinogenic and nonproteinogenic tRNAGly isoacceptors. RNA 2015; 21:1790-806; PMID:26276802; http://dx.doi.org/10.1261/rna.052712.115
- Pedrolli D, Langer S, Hobl B, Schwarz J, Hashimoto M, Mack M. The ribB FMN riboswitch from Escherichia coli operates at the transcriptional and translational level and regulates riboflavin biosynthesis. FEBS J 2015; 282:3230-42; PMID:25661987; http://dx.doi.org/10.1111/febs.13226
- Purnick PE, Weiss R. The second wave of synthetic biology: from modules to systems. Nat Rev Mol Cell Biol 2009; 10:410-22; PMID:19461664; http://dx.doi.org/10.1038/nrm2698
- Sharma V, Yamamura A, Yokobayashi Y. Engineering artificial small RNAs for conditional gene silencing in Escherichia coli. ACS Synth Biol 2012; 1:6-13; PMID:23651005; http://dx.doi.org/10.1021/sb200001q
- Liang JC, Bloom RJ, Smolke CD. Engineering biological systems with synthetic RNA molecules. Mol Cell 2011; 43:915-26; PMID:21925380; http://dx.doi.org/10.1016/j.molcel.2011.08.023
- Sindhu A, Arora P, Chaudhury A. Illuminating the gateway of gene silencing: perspective of RNA interference technology in clinical therapeutics. Mol Biotechnol 2012; 51:289-302; PMID:21947958; http://dx.doi.org/10.1007/s12033-011-9456-9
- Pestka S, Daugherty BL, Jung V, Hotta K, Pestka RK. Anti-mRNA: specific inhibition of translation of single mRNA molecules. Proc Natl Acad Sci U S A 1984; 81:7525-8; PMID:6438637; http://dx.doi.org/10.1073/pnas.81.23.7525
- Coleman J, Green PJ, Inouye M. The use of RNAs complementary to specific mRNAs to regulate the expression of individual bacterial genes. Cell 1984; 37:429-36; PMID:6202422; http://dx.doi.org/10.1016/0092-8674(84)90373-8
- Man S, Cheng R, Miao C, Gong Q, Gu Y, Lu X, Han F, Yu W. Artificial trans-encoded small non-coding RNAs specifically silence the selected gene expression in bacteria. Nucleic Acids Res 2011; 39:e50; PMID:21296758; http://dx.doi.org/10.1093/nar/gkr034
- Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014; 346:1258096; PMID:25430774; http://dx.doi.org/10.1126/science.1258096
- Barrangou R, Marraffini LA. CRISPR-Cas systems: Prokaryotes upgrade to adaptive immunity. Mol Cell 2014; 54:234-44; PMID:24766887; http://dx.doi.org/10.1016/j.molcel.2014.03.011
- Komasa M, Fujishima K, Hiraoka K, Shinhara A, Lee BS, Tomita M, Kanai A. A screening system for artificial small RNAs that inhibit the growth of Escherichia coli. J Biochem 2011; 150:289-94; PMID:21546360; http://dx.doi.org/10.1093/jb/mvr055
- O'Donnell SM, Janssen GR. The initiation codon affects ribosome binding and translational efficiency in Escherichia coli of cI mRNA with or without the 5′ untranslated leader. J Bacteriol 2001; 183:1277-83; PMID:11157940; http://dx.doi.org/10.1128/JB.183.4.1277-1283.2001
- Kery MB, Feldman M, Livny J, Tjaden B. TargetRNA2: identifying targets of small regulatory RNAs in bacteria. Nucleic Acids Res 2014; 42:W124-9; PMID:24753424; http://dx.doi.org/10.1093/nar/gku317
- Zhao GP, Somerville RL. Genetic and biochemical characterization of the trpB8 mutation of Escherichia coli tryptophan synthase. An amino acid switch at the sharp turn of the trypsin-sensitive “hinge” region diminishes substrate binding and alters solubility. J Biol Chem 1992; 267:526-41; PMID:1309752
- Hipps D, Schimmel P. Cell growth inhibition by sequence-specific RNA minihelices. EMBO J 1995; 14:4050-5; PMID:7664744
- Lu F, Taghbalout A. The Escherichia coli major exoribonuclease RNase II is a component of the RNA degradosome. Biosci Rep 2014; 34:e00166; PMID:25299745; http://dx.doi.org/10.1042/BSR20140113
- Hershberg R, Altuvia S, Margalit H. A survey of small RNA-encoding genes in Escherichia coli. Nucleic Acids Res 2003; 31:1813-20; PMID:12654996; http://dx.doi.org/10.1093/nar/gkg297
- Irnov I, Sharma CM, Vogel J, Winkler WC. Identification of regulatory RNAs in Bacillus subtilis. Nucleic Acids Res 2010; 38:6637-51; PMID:20525796; http://dx.doi.org/10.1093/nar/gkq454
- Antal M, Bordeau V, Douchin V, Felden B. A small bacterial RNA regulates a putative ABC transporter. J Biol Chem 2005; 280:7901-8; PMID:15618228; http://dx.doi.org/10.1074/jbc.M413071200
- Sun Y, Vanderpool CK. Physiological consequences of multiple-target regulation by the small RNA SgrS in Escherichia coli. J Bacteriol 2013; 195:4804-15; PMID:23873911; http://dx.doi.org/10.1128/JB.00722-13
- Lease RA, Belfort M. A trans-acting RNA as a control switch in Escherichia coli: DsrA modulates function by forming alternative structures. Proc Natl Acad Sci U S A 2000; 97:9919-24; PMID:10954740; http://dx.doi.org/10.1073/pnas.170281497
- Sledjeski DD, Whitman C, Zhang A. Hfq is necessary for regulation by the untranslated RNA DsrA. J Bacteriol 2001; 183:1997-2005; PMID:11222598; http://dx.doi.org/10.1128/JB.183.6.1997-2005.2001
- Morita T, Maki K, Aiba H. Detection of sRNA-mRNA interactions by electrophoretic mobility shift assay. Methods Mol Biol 2012; 905:235-44; PMID:22736008; http://dx.doi.org/10.1007/978-1-61779-949-5_15