
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Viroids are non-coding single-stranded circular RNA molecules that replicate autonomously in infected host plants causing mild to lethal symptoms. Their genomes contain about 250–400 nucleotides, depending on viroid species. Members of the family Pospiviroidae, like the Potato spindle tuber viroid (PSTVd), replicate via an asymmetric rolling-circle mechanism using the host DNA-dependent RNA-Polymerase II in the nucleus, while members of Avsunviroidae are replicated in a symmetric rolling-circle mechanism probably by the nuclear-encoded polymerase in chloroplasts. Viroids induce the production of viroid-specific small RNAs (vsRNA) that can direct (post-)transcriptional gene silencing against host transcripts or genomic sequences. Here, we used deep-sequencing to analyze vsRNAs from plants infected with different PSTVd variants to elucidate the PSTVd quasipecies evolved during infection. We recovered several novel as well as previously known PSTVd variants that were obviously competent in replication and identified common strand-specific mutations. The calculated mean error rate per nucleotide position was less than , quite comparable to the value of
reported for a member of Avsunviroidae. The resulting error threshold allows the synthesis of longer-than-unit-length replication intermediates as required by the asymmetric rolling-circle mechanism of members of Pospiviroidae.
| Abbreviations | ||
| NEP | = | nuclear-encoded polymerase |
| PolII | = | DNA-dependent RNA polymerase II |
| PLMVd | = | Peach latent mosaic viroid |
| PSTVd | = | Potato spindle tuber viroid |
| vsRNA | = | viroid-specific small RNA |
| C, P, TL, TR, V | = | central, pathogenicity-related, terminal left, terminal right, variable domain |
Introduction
Viroids are single-stranded circular RNA molecules that replicate autonomously in infected host plants. As non-coding RNAs, viroids do not code for any protein but possess the biologic activity and function of a minimal parasite inducing specific diseases in their hosts.Citation1-3 The severity of viroid-induced symptoms depends on the viroid variant as well as on the host and cultivar susceptibility. Members of the family Pospiviroidae, including the type species Potato spindle tuber viroid (PSTVd), are transcribed in host cell nuclei in an asymmetric rolling-circle mechanism () by the host DNA-dependent RNA polymerase II (Pol II); members of the family Avsunviroidae are replicated by a nuclear-encoded polymerase (NEP) in chloroplasts.Citation4 Viroids induce these polymerases to accept RNA instead of DNA as template with a resulting increase in error rates. Indeed, for Chrysanthemum chlorotic mottle viroid, a member of Avsunviroidae, an error rate of has been demonstrated.Citation5 Consequently, the infecting viroid molecule—the master sequence—gives rise to a sequence ensemble of master and error copies termed a ‘mutant swarm’ or quasispecies.Citation6-8
Figure 1. The asymmetric rolling-circle replication of members of the Pospiviroidae exploits the host's Pol II. The circular monomeric PSTVd genome, arbitrarily designated as (+)-strand, is transcribed into an oligomeric linear (−)-strand; the latter is template for synthesis of an oligomeric linear (+)-strand. The oligomeric (+)-strand is cleaved into monomers that are ligated to form the mature circular form. Pol II transcription introduces mutations and generates the quasispecies to explore the sequence space; the gray arrow indicate selection acting against non-viable mutations.
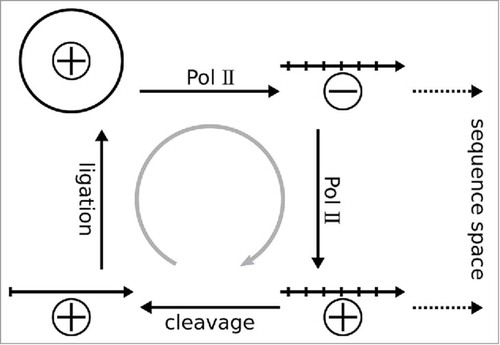
Viroids rely on both thermodynamically stable and metastable secondary structures to recruit host factors for replication, processing, ligation, and systemic trafficking.Citation9 These structures mimic host binding motifs and drive selection during replication and quasispecies formation (). Indeed, viroid infections may spontaneously, or in new hosts adaptively, generate new viable and competitive sequences.Citation10-13 Thus, viroids as minimal biologic entities with high replication and error rates provide a unique opportunity to study such quasispecies.
Viroid infections also induce the production of viroid-specific small RNAs (vsRNA) that appear to be involved in pathogenesis:Citation14,15 sequence complementarity with host transcripts results in (post-)transcriptional gene silencing and creation of a host environment favoring viroid replication. Thus far, 3 vsRNA-based pathogenic determinants have been described; the first involves a variant of Peach latent mosaic viroid (PLMVd),Citation16 the second Tomato planta macho viroid ,Citation17 and a third PSTVd.Citation18
In this context, 3 deep-sequencing small RNA data sets isolated from tomato (Solanum lycopersicum) infected with PSTVd variants are currently available. The first is from cultivar ‘Heinz 1706’ infected by PSTVd variants QFA, C3, or AS1 that induce mild, severe, and “lethal” symptoms, respectively.Citation13 The second is from cultivar ‘Rutgers’ infected by PSTVd variants M or I that induce mild and intermediate symptoms, respectively.Citation19 The third is from 4 tomato cultivars infected with PSTVd variant RG1. RG1 induces intermediate to severe symptoms in cultivars ‘Heinz 1706’ and ‘Rutgers’ but only mild symptoms in ‘Moneymaker’ and ‘UC82B’.
It is well known that the full sequence of the infecting viroid variant (or larger viruses) can be assembled from such small RNA data sets either using the viroid sequence as referenceCitation20 or even without a reference.Citation21,22 In this study, we have gone a step further; that is, we have used small RNA data to compare the mutational landscapes of different infecting viroid variants. These landscapes reflect a combination of replication efficiency, selection, and sequencing errors. We evaluated the viroids' sequence space from the deep-sequencing data following methods established for RNA virusesCitation23-27 and applied the quasispecies theoremCitation6,28 and associated concepts to viroids to estimate a lower bound for the replication fidelity of their genomes.
Results
vsRNA expression
We identified vsRNAs by mapping small RNA reads from deep-sequencing data sets Ds, Do and Dp to the respective infecting PSTVd variant with different accuracies (). In the range from 100 to 90% accuracy, only a very small number of small RNAs from mock-inoculated deep-sequenced samples mapped to the PSTVd variants (Fig. S2). Thus, we continued with vsRNAs mapped with 90% accuracy for the remaining results. That is, we tolerated up to 2 mismatches between a standard read with length < 25 nt and a viroid sequence to ensure sufficient specificity while allowing some variability.
Figure 2. Mapping of small RNA reads to PSTVd variants. (A) Frequency of reads mapping to PSTVd positions with different accuracies (see color code at top). Note the differently scaled y-axes. (B) Sequence variation (see color code at top) at 90% mapping accuracy; stacked bars represent the square root of ratio of a specific variation in relation to all variations (including the reference nucleotide) at one site. All insertions are collapsed into one count regardless of base and length. Viroid domains (see Fig. S1A) are marked close to the top. Deletions (D), insertions (I), and mutations to A, U, G, or C are marked by different colors (see legend at top).
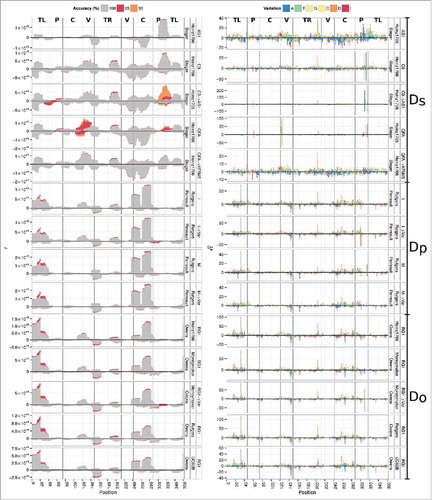
The numbers of vsRNAs from intermediate and severe PSTVd variants were similar and higher than those from mild variants (see Table S2). In data sets Dp and Do, (−)-stranded vsRNAs were less abundant than (+)-stranded vsRNAs. In Ds, vsRNA levels gradually increased from QFA to C3 to AS1. Notably, the lower pathogenicity-related domain (; see Fig. S1A) of PSTVd variants AS1 and C3 exhibited vsRNA expression peaks not seen for QFA ().
In all but one case, the numbers of mapped vsRNAs increased by about 10% (Table S2) as the mapping accuracy was decreased from 100 to 90% (). The clear exception was the vsRNA profile of variant QFA: here the additional mappings at 90% accuracy revealed sequence variations in the upper central to variable domain () and—to a minor extent—in the lower P to terminal-left domain (
). These mutations show up more clearly in a plot of differences between nucleotides in vsRNAs and the infecting PSTVd sequence (). In QFA, mutations at positions 120–125 and 303–316 are up to 10 times more abundant than the original QFA nucleotides and present in both (+)- and (−)-stranded vsRNAs. Other mutations appeared to be present only in vsRNAs derived from the respective (+)-strands; e.g., changes near position 200 visible in all panels.
With this surprising result in mind, we mapped each deep-sequencing data set to all PSTVd variants available in the subviral database.Citation29 Indeed, the reads from the QFA-infected sample were more consistent with the sequence of variant KF5M5 (AC M93685) than with QFA (see panels marked “QFA → KF5M5” in ). Results for C3 and the other variants were not as clear-cut, but this discrepancy between the infecting sequence and that of the resulting progeny assembled from the vsRNA reads in the case of variant QFA prompted us to analyze these deviations in all data sets in detail.
To elucidate the distribution of mismatched nucleotides in vsRNAs, we plotted the locations of these mismatches along the respective PSTVd sequence in and the log ratio of these mismatches to the corresponding reference nucleotide in Fig. S3. In the latter figure, the few positive values—especially in the QFA mapping—clearly show that a PSTVd variant(s) different in sequence from the originally infecting PSTVd variant was present in excess among the progeny. The same mismatch positions dominate in where a clear example is again seen at positions 61, 121, and 315 of QFA, or, less obviously, at positions 308–314 of C3.
These mutations with their relative high frequencies emerge from a background of sequence errors, visible in both polarities in all data sets independent of the infecting PSTVd variant, inoculation method, or tomato cultivars. This background makes it difficult to distinguish replication-competent variants containing complementary changes in both strands from the infecting reference sequence. To extract relevant mutations and to detect co-appearing mutations, we developed the method NETMAP.
Identification of replication-competent PSTVd variants using graph theory
NETMAP extracts viable and connected sequence positions from vsRNA data. We define mutations as viable if they are present in reads of both polarities and thus are likely to originate from replicating viroids. Mutations are connected and originate from the same genome if they are present on a single read. For further program details see Material & Methods. This network analysis revealed the presence of several potentially viable sequence variants that co-existed with the inoculated PSTVd variant.
From the reads mapping to PSTVd variant QFA in Ds, NETMAP identified 3 significantly expressed mutations. illustrates the deletion of an A in the oligo-A sequence upstream of G62; this deletion was present in about 29% of (+)-stranded reads covering this position and in about 76% of (−)-stranded reads. illustrates 2 connected mutations, an transversion and an A deletion upstream of G125; both mutations dominated the reference sequence in strands of both polarities. illustrates 5 connected mutations including 2 insertions. These mutations also dominated the reference sequence but at least part of this region (i.e., positions 303–307) was only covered by low read numbers. If all these mutations from regions
,
, and
were present in the same molecule, the inoculated PSTVd variant QFA would have evolved to variant KF5M5 (AC M93685.1).
Figure 3. Sequence networks extracted by NETMAP from reads mapping with 90% accuracy to the respective infecting PSTVd variant. Nucleotides in solid circles show sequence positions that are identical to the reference sequence; those in dashed circles show mutations. Insertions and deletions are indicated by ( ) and (
), respectively. Solid arrows connect nucleotides; dotted arrows link mutations that are located on the same read. An arrow pointing from position x to position y indicates (+)- and (−)-polarity if
and
, respectively. Read numbers (edge weights) label the arrows. Positions of mutations in the secondary structure of PSTVd are shown in Fig. S1D.
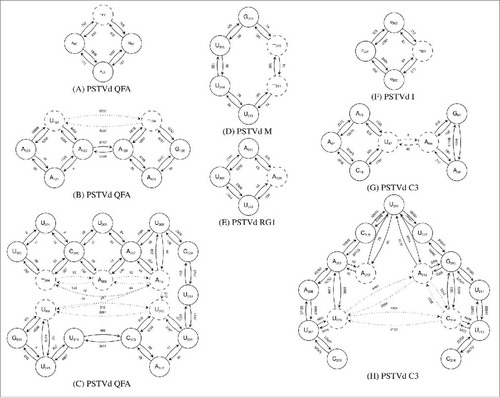
For variant C3, NETMAP identified 6 variable sites: and
in region
(), and
,
,
, and
in
(). Mutation
was only weakly supported (below 0.2% reads in both polarities) and was not connected with the neighboring mutations. The other 5 mutations were each supported by 1–15% of reads. If all 5 mutations were present in the same molecule, the sequence of the resulting progeny would be identical to that of variant AS1. Note, however, that C3 was still the dominant sequence at the time of sampling. For plants inoculated with variant AS1, NETMAP detected no sequence variations.
In Dp, network analysis revealed sequence variation in both PSTVd variants M and I. For variant M we identified 2 linked and dominant mutations (deletions of U314 and U315 in about 70% of reads; ). For variant I we identified a minor mutation (deletion of C301 in < 7% of the reads; ). Sequence comparisons revealed no known variants associated with these changes.
For Do, NETMAP detected only a single mutation (i.e., U309 → A) in about 2% of (+)- and 4% of (−)-stranded reads from ‘Moneymaker’ plants (see ). This change is present in several other PSTVd variants (e.g., KF440–2 (X58388.Equation1(1)
(1) ), X52039.1, X52040.1), but only in combination with additional mutations. No sequence variation of the infecting PSTVd variant RG1 was detected in the other tomato cultivars.
Polarity-specific mutations
Besides the potentially replication-competent mutations detected by NETMAP, several other mutations were identified in reads of only one polarity. All data sets contained a deletion at consensus position 202 in the (+)-strand with mean expression ratios (i = 204 for variants AS1, I, M, and RG1, I = 203 for C3, and i = 205 for QFA) of 0.10, 0.09, and 0.27 in Ds, Dp, and Do, respectively. This deletion could disrupt a binding motif for viroid binding protein 1 (VirP1; Fig. S1A and C), thereby resulting in loss of infectivity.Citation30,31 The Do and Dp data sets also contained a significant level of variation at position 32 in
. However, because this reflects mainly a A32 → U change at the end of reads, this variation could be a result of imperfect trimming.
We also identified a deletion at consensus position 256 that is located in the loop E motif (see Fig. S1A) in all data sets except Do. Loop E is involved, among other processes, in processing of (+) replication intermediates to circles [for review see Citationref 9] Such a deletion would destroy the tertiary structure of this loop,Citation32 which is consistent with its presence only in (+) strands. It should be noted, however, that any change not supported by the corresponding mutation in the opposite strand has a higher chance of originating from either sequencing or mapping errors.
Error rate, error copies, and error threshold
Using the ratio of sites containing a variant nucleotide to those with the reference nucleotide, we estimated the probability q for error-free reproduction of a single nucleotide in the master copy of a viroid genome during replication (see equation Equation(1)(1)
(1) ). As master copy we used the sequence of the originally infecting PSTVd variants as well as any suspected new variants that evolved during infection (see ).
Table 1. Parameters of vsRNAs mapping to their respective PSTVd genomes in both polarities.
The value of the mean error rate per nucleotide was
averaged over variants AS1, C3, QFA → KF5M5, I, M → Var, and RG1. In contrast, the calculated error rate for variant QFA was much higher, consistent with the observed accumulation of mutations and its replacement as master copy by variant KF5M5. Similarly, the calculated error rate for variant M would not allow the error-free replication of the originally infecting variant, and this sequence also tended to be overgrown by a variant.
How are these calculated error rates for viroid replication and selection affected by sequencing errors? To estimate the effect of sequencing errors, we determined the apparent error rates for 2 tomato microRNAs (see Table S3). Like viroids, microRNAs are transcribed by Pol II, but from a DNA rather than an RNA template. The calculated error rates for these microRNAs were close to and quite similar for the different data sets. Error rates for Pol II transcription of mRNAs are known to be in the range of
−
;Citation33,34 consequently, our higher-than-expected error rate for microRNA synthesis is due to sequencing error. Note, however, that this rate of sequencing error is lower than the error rate of viroid replication and selection (
) by about a factor of 10. Our data sets also contained sequences derived from several other small non-coding RNAs (e.g., 5.8S RNA, 7SL RNA, and tRNA), but their synthesis involves polymerases other than RNA Pol II. Furthermore, the number of reads recovered from such RNAs were ≥ 10-fold fewer than those derived from the 2 microRNAs (data not shown).
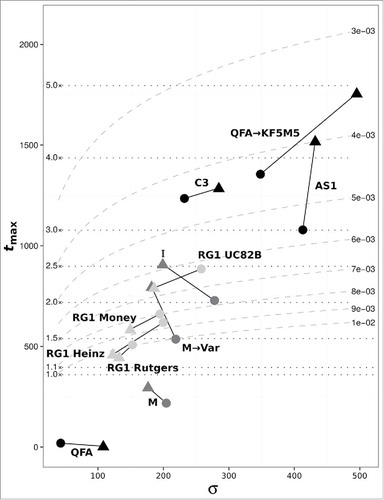
Finally, from the estimated error rates of viroid replication we calculated the probability of error-free unit-length copies .Citation6 As shown in , 20% or less of the PSTVd population per replication cycle corresponds to the original variant genome. We also calculated the error threshold based on the determined error rates to estimate the longest possible error-free viroid genome (). Error rates for the original variants M and QFA were clearly too high to support the size of their genome leading to an error catastrophe.Citation6 In contrast, the error thresholds for the evolved variants M → Var and QFA → KF5M5 were much higher, thereby allowing these sequences to co-exist with or even outcompete the respective original variants.
Figure 4. Dependence of the calculated error thresholds ( ; see equation Equation(3)
(3)
(3) ) on selective advantage ( σ ; see equation Equation(2)
(2)
(2) ) of the master copy for individual PSTVd variants based on the
values calculated from analysis of the respective vsRNA populations (see ). Solid lines connect thresholds for (+)- (circles) and (--)-strands (triangles) of the same sample. Dotted horizontal lines indicate the mean PSTVd genome length (359 nt) times the number given on the left end of lines. Dashed curved lines indicate mean error rate per nucleotide
given on the right end of lines.
Discussion
We examined 3 sets of small RNA deep-sequencing data derived from PSTVd-infected tomato plants for evidence of viroid sequence evolution/drift. As expected, we were able to reconstruct the sequences of the PSTVd variants used to initiate infection after mapping the reads with SEGEMEHL at 100% accuracy. Reducing the mapping accuracy to 90% revealed the presence of additional reads that allowed us to detect variants of the inoculated PSTVd sequences.
In the data set from plants inoculated with QFA, we detected all 8 mutations by which QFA differs from the known mild variant KF5M5. The use of cloned cDNA as inoculum and a new, viroid-free climate box to maintain the plants after inoculation (see Materials & Methods) allows us to exclude one possible explanation for such a result; i.e., contamination of the QFA inoculum by KF5M5. From the small read data alone we cannot be sure that all the mutations detected were present in the same PSTVd genome. It is highly likely, however, that at least a significant proportion of the progeny did so because only 3 of 266 unique PSTVd variants (see Fig. S4) recovered from GenBank contain all the changes between positions 302 and 312 shown in and S1D. Each of these variants (i.e., KF5M5 plus the closely related KF5M3 and KF5M4) also contain all the changes detected in the upper portions of the pathogenicity and variable domains. Thus, our analysis captured the in planta evolution of a new PSTVd quasispecies.
This uncertainty could be overcome by the analysis of quite large RNA-Seq libraries of circular PSTVd variants; to our knowledge, however, such an analysis has only been performed for PLMVd.Citation35 The uneven distribution of short reads along the PSTVd genome (“hotspots”) should not create major problems for our type of quasispecies analysis because all calculations are only based on relative count numbers (see (Equation1
(1)
(1) ) and (Equation2
(2)
(2) )).
In data sets from ‘Moneymaker’ plants inoculated with RG1 or ‘Heinz 1706’ plants inoculated with AS1, C3, or I, only minor levels of mutations were detected; that is, the inoculated variant remained the master sequence at the time of sampling. In contrast to the closing remarks in ref. Citation37 where the authors recommend the use of cloned cDNA as inoculum rather than the inherently more heterogeneous RNA transcripts, the type of inoculum used in our studies appears to have had little or no effect on sequence evolution in vivo.
PSTVd variants I and M share 97% sequence homology, and a previous comparison of cloned RT-PCR products suggested that both variants replicate stably when inoculated to tomato.Citation36 Our analysis of small RNAs extracted from an independent set of plants infected with these 2 variants indicates that this conclusion may need to be reexamined. In data set Dp a novel variant differing from PSTVd-M by a 2-nucleotide deletion in the lower P region was present at levels comparable to the inoculated variant. PSTVd-I appeared to be more stable than PSTVd-M, and the low levels (<7%) of a novel variant in the progeny could well have escaped detection by cDNA cloning.
Taken together our results fit quite well with quasispecies theory; i.e., the viroid quasispecies serves as a source of adaptation to new hosts, and changes in selective pressures are able to induce the rapid emergence of new variants.Citation11,38-43 The fitness landscape of PSTVd seems to contain a limited number of peaks,Citation12 many of which coincide with known variants. In addition to replication-competent mutants, mutations that are present in only one polarity might also be advantageous for viroid evolution. Such mutations may allow fast adaptation to new environments, as shown for example for ribozyme selection,Citation44 and may also increase the spectrum of host targets susceptible to post-transcriptional gene silencing. Thus, a PLMVd variant that contains a specific hairpin insertion sequence and induces a loss of pigmentation known as ‘peach calico’, expresses a vsRNA that targets the mRNA of chloroplastic heat-shock protein 90 leading to chloroplast malformation.Citation16 Also, PSTVd variants M and I express a scarce vsRNA from that pairs a tomato CalS11-like mRNA.Citation18 vsRNAs encoded by the mutant swarm rather than the master sequence could allow the viroid to attack additional targets, thereby creating a cellular environment more favorable for viroid replication.
The concept of an error thresholdCitation6 limits the length of a self-replicating molecule to the inverse of the error rate of its replicase. This concept also holds for viroids that utilize a host polymerase for replication. Because the host polymerase, either Pol II for members of Pospiviroidae or NEP for members of Avsunviroidae, has to act on a foreign RNA rather than its normal DNA template, it is often assumed that the error rates could well be higher than those observed with DNA templates. Indeed, the error rate of a member of Avsunviroidae has been shown to be ,Citation5 resulting in ∼1 mutation per genome replication. Considering the higher structural constraints imposed by the rod-shaped structure of members of Pospiviroidae in comparison to the branched structure of most members of Avsunviroidae, Duràn-Vila et al. have proposed that the error rate of Pospiviroidae should be 10-fold lower than that of Avsunviroidae.Citation45 Our data for PSTVd indicates, however, an upper limit of
for the error rate, a value which is quite similar to that for a member of Avsunviroidae. Additional measurements using other viroid-host combinations are needed before a firm conclusion can be drawn.
The studies described here used a collection of vsRNA samples isolated from a single host species at just one time point comparatively late in the PSTVd infection process. Despite these limitations, we have shown that vsRNA analysis can distinguish mutations that block PSTVd replication and/or movement from those that allow continued systemic infection. Future studies of carefully selected combinations of viroid variant and host plant growing under controlled conditions and repeatedly sampled throughout the course of infection should provide new insights into several aspects of viroid-host interaction.
Materials and methods
Deep-sequencing data sets of small RNAs
We analyzed 3 published sets of small RNA sequence data, which are abbreviated as Ds, Dp and Do, respectively. In the following we summarize the small RNA preparation and sequencing to point out some experimental differences and commonalities between these data sets.
DsCitation13 is from S. lycopersicum ‘Heinz 1706’ plants infected with PSTVd variants QFA (mild, GenBank AC U23059.1), C3 (severe, HE575349.1), or AS1 (“lethal,” AY518939.1). Plants containing 3 true leaves were biolistically inoculatedCitation42 with StyI-cleaved full-length cloned cDNA,Citation41 and plants were maintained in climate boxes at 28°C under a mixture of fluorescent and incandescent illumination (10,000 lx for a 16 h-day period). Samples of leaf and stem tissue were collected 30 d post infection (dpi); total RNA was prepared by phenol/chloroform extraction and ethanol precipitation, and small RNAs were enriched by denaturing gel-electrophoresis and gel-elution. Ligation of 3′ and 5′ adapters and cDNA synthesis was performed by vertis Biotechnologie (Freising, Germany); Illumina sequencing of the cDNA was done by GATC Biotech (Konstanz, Germany) on a HiSeq 2000 system.
DpCitation19 is from S. lycopersicum ‘Rutgers’ plants infected with the PSTVd variants Intermediate (I, AY937179.1) or PSTVd Dahlia (M(ild), AB623143.1), respectively. Plants having only a single true leaf were inoculated with dimeric RNA transcribed with T7 polymerase from cloned cDNA;Citation46 plants were grown in a growth chamber controlled at 25°C, with a 16-h day and high light intensity (fluorescent, 40 W × 4). Leaf samples were collected 21 dpi; total RNA was prepared by phenol/chloroform extraction and DNase I treatment; small RNAs were enriched by denaturing gel-electrophoresis and gel-elution. Ligation of adapters, cDNA synthesis and deep-sequencing was performed by Hokkaido System Science Co. (Sapporo, Japan) using a Genetic Analyzer IIx platform. Do is from 4 different tomato cultivars infected with monomeric T7 RNA transcriptsCitation47 having the sequence of the PSTVd variant RG1 (intermediate–severe, U23058.1). Cultivars ‘Heinz 1706’ and ‘Rutgers’ are PSTVd-sensitive, and the PSTVd-insensitive ‘Moneymaker’ and ‘UC82B’ are nearly asymptomatic. Plants having 3 true leaves were mechanically inoculated with T7-RNA transcripts; samples of young leaf tissue were collected 27 dpi and small RNAs were extracted using Trizol reagent (Life Technologies, Carlsbad, CA, USA) and enriched mirVana™ kit (Life Technologies). Ligation of adapters, cDNA synthesis and deep-sequencing was performed by Expression Analysis/QCitation2 Solutions (Durham, NC, USA).
For Ds, we used PrinSeqCitation48 for entropy-based removal of uninformative poly-A tails derived from the sequencing procedure. Illumina adapters were removed using TRIMMOMATIC.Citation49 We used SEGEMEHL v. 0.2.0Citation50,51 to map deep-sequenced vsRNAs to their infecting PSTVd variant reference. Parameters for use of these programs are listed in Table S1.
Sequence evolution
We followed Eigen's formalism for the evolution of sequences.Citation6 In brief, read mappings with 90% accuracy resulted in positions i deviating from the reference sequence S; consecutive insertions were handled as a single mutation at the corresponding first position. The read count with the reference nucleotide at position i is , the sum of all reads with nucleotide changes (substitutions, insertions, or deletions) at position i is denoted as
. Then the probability q for exact reproduction of a nucleotide was estimated by the mean error rate
over all sites:
(1)
(1)
Under the assumption that master and error copies exhibit the same decay rate, we estimated the average selective advantage σ of the master copy over the error copies under 90% accuracy mappingCitation52(2)
(2) and the error threshold
(3)
(3)
Sequence network mapping with NETMAP
We developed a graph-orientated method named NETMAP, a PERL script based on SAMTOOLSCitation53 and Bio::DB::Sam, to assemble the PSTVd sequence space from mapped deep-sequenced small RNA reads. A PSTVd sequence of length l is represented by vertices with
connected by weighted directed edges e. A weight w is the count of 2 adjacent vertices located in any single read; the edge direction represents (+) and (−) strandedness of reads. The graph
is constructed from short reads mapped by SEGEMEHL with 90% accuracy to a reference sequence, which is the PSTVd variant used for infection.
We applied the following filter steps to vsRNA reads to reduce noise based mainly on sequencing errors:
Two neighboring sequence positions
are only connected by an edge
Mismatching bases at the 5′ or 3′ end of a read are considered as adaptor contamination or sequencing error if no other reads embed these variations.
Only variants present in both polarities are considered competent for replication.
Two (or more) sequence variations are only considered as connected (i.e., derived from one genome) if a read includes both; that is, variations further apart than the length of a read might originate from different PSTVd genomes. If 2 variants are present on both polarities but are only connected in one polarity, we still considered them as connected.
Disclosure of potential conflicts of interest
No potential conflict of interest were disclosed.
KRNB_A_1272745_supplemental_material.pdf
Download PDF (2 MB)Funding
This work was supported by the interdisciplinary graduate school ‘Evolutionary Networks: Organisms, Reactions, Molecules’ (E-Norm), Prof. em. Dr. Dr. h. c. Detlev Riesner, and a grant from the Alexander von Humboldt Foundation to GS and JM@.
References
- Navarro B, Gisel A, Rodio M, Delgado S, Flores R, Di Serio F. Viroids: How to infect a host and cause disease without encoding proteins. Biochimie 2012; 94:1474-80; PMID:22738729; http://dx.doi.org/10.1016/j.biochi.2012.02.020
- Palukaitis P. What has been happening with viroids? Virus Genes 2014; 49:175-184; PMID:25164861; http://dx.doi.org/10.1007/s11262-014-1110-8
- Gago-Zachert S. Viroids, infectious long non-coding RNAs with autonomous replication. Virus Res 2015; 212:12-24; PMID:26319312
- Flores R, Grubb D, Elleuch A, Nohales MÁ, Delgado S, Gago S. Rolling-circle replication of viroids, viroid-like satellite RNAs and hepatitis delta virus: variations on a theme. RNA Biol 2011; 8:200-206; PMID:21358283; http://dx.doi.org/10.4161/rna.8.2.14238
- Gago S, Elena SF, Flores R, Sanjuán R. Extremely high mutation rate of a hammerhead viroid. Science 2009; 323:1308; PMID:19265013; http://dx.doi.org/10.1126/science.1169202
- Eigen M. Selforganization of matter and the evolution of biological macromolecules. Naturwissenschaften 1971; 58:465-523; PMID:4942363; http://dx.doi.org/10.1007/BF00623322
- Domingo E. Quasispecies theory in virology. J Virology 2002; 76:463-65; http://dx.doi.org/10.1128/JVI.76.1.463-465.2002
- Bull J, Meyers L, Lachmann M. Quasispecies made simple. PLoS Comput Biol 2005; 1:e61; PMID:16322763; http://dx.doi.org/10.1371/journal.pcbi.0010061
- Flores R, Serra P, Minoia S, Di Serio F, Navarro B. Viroids: from genotype to phenotype just relying on RNA sequence and structural motifs. Front Microbiol 2012; 3:217; PMID:22719735; http://dx.doi.org/10.3389/fmicb.2012.00217.
- Semancik J, Szychowski J, Rakowski A, Symons R. Isolates of citrus exocortis viroid recovered by host and tissue selection. J Gen Virol 1993; 74:2427-36; PMID:8245858; http://dx.doi.org/10.1099/0022-1317-74-11-2427
- Matoušek J, Orctová L, Ptáček J, Patzak J, Dědič P, Steger G, Riesner D. Experimental transmission of pospiviroid populations to weed species characteristic of potato and hop fields. J Virol 2007; 81:11891-99; PMID:17715233; http://dx.doi.org/10.1128/JVI.01165-07
- Elena S, Gómez G, Daròs J. Evolutionary constraints to viroid evolution. Viruses 2009; 1:241-254; PMID:21994548; http://dx.doi.org/10.3390/v1020241
- Matoušek J, Stehlík J, Procházková J, Orctová L, Wullenweber J, Füssy Z, Kováčik J, Duraisamy GS, Ziegler A, Schubert J, et al. Biological and molecular analysis of the pathogenic variant C3 of potato spindle tuber viroid (PSTVd) evolved during adaptation to chamomile (Matricaria chamomilla). Biol Chem 2012; 393:605-615; PMID:22944665; http://dx.doi.org/10.1515/hsz-2011-0286
- Papaefthimiou I, Hamilton A, Denti M, Baulcombe D, Tsagris M, Tabler M. Replicating potato spindle tuber viroid RNA is accompanied by short RNA fragments that are characteristic of post-transcriptional gene silencing. Nucleic Acids Res 2001; 29:2395-2400; PMID:11376158; http://dx.doi.org/10.1093/nar/29.11.2395
- Hammann C, Steger G. Viroid-specific small RNA in plant disease. RNA Biol 2012; 9:809-819; PMID:22617880; http://dx.doi.org/10.4161/rna.19810
- Navarro B, Gisel A, Rodio ME, Delgado S, Flores R, Di Serio F. Small RNAs containing the pathogenic determinant of a chloroplast-replicating viroid guide the degradation of a host mRNA as predicted by RNA silencing. Plant J 2012; 70:991-1003; PMID:22332758; http://dx.doi.org/10.1111/j.1365-313X.2012.04940.x
- Avina-Padilla K, Martinez de la Vega O, Rivera-Bustamante R, Martinez-Soriano J, Owens R, Hammond R, Vielle-Calzada J. In silico prediction and validation of potential gene targets for pospiviroid-derived small RNAs during tomato infection. Gene 2015; 564:197-205; PMID:25862922; http://dx.doi.org/10.1016/j.gene.2015.03.076
- Adkar-Purushothama C, Brosseau C, Giguère T, Sano T, Moffett P, Perreault J. Small RNA derived from the virulence modulating region of the Potato spindle tuber viroid silences callose synthase genes of tomato plants. Plant Cell 2015; 27:2178-94; PMID:26290537; http://dx.doi.org/10.1105/tpc.15.00523
- Adkar-Purushothama CR, Perreault JP, Sano T. Analysis of small RNA production patterns among the 2 potato spindle tuber viroid variants in tomato plants. Genom Data 2015; 25:65-66; http://dx.doi.org/10.1016/j.gdata.2015.08.008
- Li R, Gao S, Hernandez A, Wechter W, Fei Z, Ling K. Deep sequencing of small RNAs in tomato for virus and viroid identification and strain differentiation. PLoS ONE 2012; 7:e37127; PMID:22623984; http://dx.doi.org/10.1371/journal.pone.0037127
- Zhang Z, Qi S, Tang N, Zhang X, Chen S, Zhu P, Ma L, Cheng J, Xu Y, Lu M, et al. Discovery of replicating circular RNAs by RNA-seq and computational algorithms. PLoS Pathog 2014; 10:e1004553; PMID:25503469; http://dx.doi.org/10.1371/journal.ppat.1004553
- Seguin J, Rajeswaran R, Malpica-LÃ3pez N, Martin R, Kasschau K, Dolja V, Otten P, Farinelli L, Pooggin M. De novo reconstruction of consensus master genomes of plant RNA and DNA viruses from siRNAs. PLoS ONE 2014; 9:e88513; PMID:24523907; http://dx.doi.org/10.1371/journal.pone.0088513
- Tsibris AMN, Korber B, Arnaout R, Russ C, Lo CC, Leitner T, Gaschen B, Theiler J, Paredes R, Su Z, et al. Quantitative deep sequencing reveals dynamic HIV-1 escape and large population shifts during CCR5 antagonist therapy in vivo. PLoS One 2009; 4:1-12; PMID:19479085; http://dx.doi.org/10.1371/journal.pone.0005683
- Rozera G, Abbate I, Ciccozzi M, Presti AL, Bruselles A, Vlassi C, D'Offizi G, Narciso P, Giombini E, Bartolini B, et al. Ultra-deep sequencing reveals hidden HIV-1 minority lineages and shifts of viral population between the main cellular reservoirs of the infection after therapy interruption. J Med Virol 2012; 84:839-844; PMID:22996031; http://dx.doi.org/10.1002/jmv.23292
- Timm C, Akpinar F, Yin J. Quantitative characterization of defective virus emergence by deep sequencing. J Virol 2013; 88:2623-32; PMID:24352442; http://dx.doi.org/10.1128/JVI.02675-13
- Astrovskaya I, Tork B, Mangul S, Westbrooks K, Mandoiu I, Balfe P, Zelikovsky A. Inferring viral quasispecies spectra from 454 pyrosequencing reads. BMC Bioinformatics 2011; 12:S1; PMID:21989211; http://dx.doi.org/10.1186/1471-2105-12-S6-S1
- Andino R, Domingo E. Viral quasispecies. Virology 2015; 479-480:46-51; PMID:25824477; http://dx.doi.org/10.1016/j.virol.2015.03.022
- Biebricher C, Eigen M. The error threshold. Virus Res 2005; 107:117-127; PMID:15649558; http://dx.doi.org/10.1016/j.virusres.2004.11.002
- Rocheleau L, Pelchat M. The Subviral RNA Database: a toolbox for viroids, the hepatitis delta virus and satellite RNAs research. BMC Microbiol 2006; 6:24; PMID:16519798; http://dx.doi.org/10.1186/1471-2180-6-24
- Gozmanova M, Denti M, Minkov I, Tsagris M, Tabler M. Characterization of the RNA motif responsible for the specific interaction of potato spindle tuber viroid RNA (PSTVd) and the tomato protein Virp1. Nucleic Acids Res 2003; 31:5534-43; PMID:14500815; http://dx.doi.org/10.1093/nar/gkg777
- Kalantidis K, Denti M, Tzortzakaki S, Marinou E, Tabler M, Tsagris M. Virp1 is a host protein with a major role in Potato spindle tuber viroid infection in Nicotiana plants. J Virol 2007; 81:12872-80; PMID:17898061; http://dx.doi.org/10.1128/JVI.00974-07
- Zhong X, Leontis N, Qian S, Itaya A, Qi Y, Boris-Lawrie K, Ding B. Tertiary structural and functional analyses of a viroid RNA motif by isostericity matrix and mutagenesis reveal its essential role in replication. J Virol 2006; 80:8566-81; PMID:16912306; http://dx.doi.org/10.1128/JVI.00837-06
- Lynch M. Evolution of the mutation rate. Trends Genet 2010; 26:345-352; PMID:20594608; http://dx.doi.org/10.1016/j.tig.2010.05.003
- Carey L. RNA polymerase errors cause splicing defects and can be regulated by differential expression of RNA polymerase subunits. Elife 2015; 4:e09945; PMID:26652005; http://dx.doi.org/10.7554/eLife.09945
- Glouzon J, Bolduc F, Wang S, Najmanovich R, Perreault J. Deep-sequencing of the peach latent mosaic viroid reveals new aspects of population heterogeneity. PLoS ONE 2014; 9:e87297; PMID:24498066; http://dx.doi.org/10.1371/journal.pone.0087297
- Tsushima D, Tsushima T, Sano T. Molecular dissection of a dahlia isolate of potato spindle tuber viroid inciting a mild symptoms in tomato. Virus Res 2015; 214:11-18; PMID:26732488; http://dx.doi.org/10.1016/j.virusres.2015.12.018
- Podstolski W, Góra-Sochacka A, Zagórski W. Co-inoculation with 2 non-infectious cDNA copies of potato spindle tuber viroid (PSTVd) leads to the appearance of novel fully infectious variants. Acta Biochim Pol 2005; 52:87-98; PMID:15827608
- Qi Y, Ding B. Differential subnuclear localization of RNA strands of opposite polarity derived from an autonomously replicating viroid. Plant Cell 2003; 15:2566-77; PMID:14555700; http://dx.doi.org/10.1105/tpc.016576
- Góra-Sochacka A, Kierzek A, Candresse T, Zagórski W. The genetic stability of potato spindle tuber viroid (PSTVd) molecular variants. RNA 1997; 3:68-74; PMID:89904008
- Góra-Sochacka A, Candresse T, Zagórski W. Genetic variability of potato spindle tuber viroid RNA replicon. Acta Biochim Pol 2001; 48:467-476;PMID: 11732616
- Matoušek J, Orctová L, Steger G, Škopek J, Moors M, Dědič P, Riesner D. Analysis of thermal stress-mediated PSTVd variation and biolistic inoculation of progeny of viroid “thermomutants” to tomato and Brassica species. Virology 2004; 323:9-23; PMID:15165815; http://dx.doi.org/10.1016/j.virol.2004.02.010
- Matoušek J, Orctová L, Steger G, Riesner D. Biolistic inoculation of plants with viroid nucleic acids. J Virol Methods 2004; 122:153-164; PMID:15542139; http://dx.doi.org/10.1016/j.jviromet.2004.08.011
- Matoušek J, Kozlova P, Orctova L, Schmitz A, Pešina K, Bannach O, Diermann N, Steger G, Riesner D. Accumulation of viroid-specific small RNAs and increase in nucleolytic activities linked to viroid-caused pathogenesis. Biol Chem 2007; 388:1-13; PMID:17214544; http://dx.doi.org/10.1515/BC.2007.001
- Ameta S, Winz ML, Previti C, Jäschke A. Next-generation sequencing reveals how RNA catalysts evolve from random space. Nucleic Acids Res 2013; 42:1303-10; PMID:24157838; http://dx.doi.org/10.1093/nar/gkt949
- Duran-Vila N, Elena S, Daròs JA, Flores R. Structure and evolution of viroids. In: Domingo E, Parrish C, Holland J, eds., Origin and Evolution of Viruses, 43–64. London: Academic Press, 2nd edn., 2008; http://dx.doi.org/10.1016/B978-0-12-374153-0.00002-3
- Tsushima D, Adkar-Purushothama CR, Taneda A, Sano T. Changes in relative expression levels of viroid-specific small RNAs and microRNAs in tomato plants infected with severe and mild symptom-inducing isolates of Potato spindle tuber viroid. J General Plant Pathol 2015; 81:49-62; http://dx.doi.org/10.1007/s10327-014-0566-7
- Feldstein P, Hu Y, Owens R. Precisely full length, circularizable, complementary RNA: an infectious form of potato spindle tuber viroid. Proc Natl Acad Sci USA 1998; 95:6560-6565; PMID:9601006; http://dx.doi.org/10.1073/pnas.95.11.6560
- Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011; 27:863-64; PMID:21278185; http://dx.doi.org/10.1093/bioinformatics/btr026
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014; 30:2114-20; PMID:24695404; http://dx.doi.org/10.1093/bioinformatics/btu170
- Hoffmann S, Otto C, Kurtz S, Sharma CM, Khaitovich P, Vogel J, Stadler PF, Hackermüller J. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput Biol 2009; 5:e1000502; PMID:19750212; http://dx.doi.org/10.1371/journal.pcbi.1000502
- Hoffmann S, Otto C, Doose G, Tanzer A, Langenberger D, Christ S, Kunz M, Holdt L, Teupser D, Hackermüller J, et al. A multi-split mapping algorithm for circular RNA, splicing, trans-splicing, and fusion detection. Genome Biol 2014; 15:R34; PMID:24512684; http://dx.doi.org/10.1186/gb-2014-15-2-r34
- Domingo E, Biebricher C, Eigen M, Holland J, eds. Quasispecies and RNA virus evolution: principles and consequences. Austin: Landes Bioscience, 2001.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics 2009; 25:2078-79; PMID:19505943; http://dx.doi.org/10.1093/bioinformatics/btp352