
ABSTRACT
High-throughput RNA sequencing (RNA-Seq) has uncovered hundreds of small RNAs and complex modes of RNA regulation in every bacterium analyzed to date. This complexity agrees with the adaptability of most bacteria to varied environments including, in the case of pathogens, the new niches encountered in the host. Recent RNA-Seq studies have analyzed simultaneously gene expression in the intracellular pathogen Salmonella enterica and infected host cells at population and single-cell level. Distinct polarization states or interferon responses in the infected macrophage were linked to variable growth rates or activities of defined virulence regulators in intra-phagosomal bacteria. Intracellular Salmonella, however, exhibit disparate intracellular lifestyles depending the host cell, ranging from a hyper-replicative cytosolic state in epithelial cells to a non-replicative intra-phagosomal condition in varied host cell types. The basis of such diverse pathogen-host communications could be examined by RNA-Seq studies in single intracellular Salmonella cells, certainly a challenge for future investigations.
Introduction
High throughput RNA-sequencing (RNA-Seq) involves sequencing of the complete set of RNA transcripts produced by a cell in a defined moment in time.Citation1 In contrast to microarray methods, RNA-Seq technology allows definition of the transcriptome based in new generation massive sequencing methods that determine cDNA sequence. Important features of RNA-Seq include the no limitation to the detection of transcripts that correspond to an existing sequenced genome, the data accuracy, its low background noise and its relatively low cost compared with classical transcriptomic methods such as tiling microarray or Sanger sequencing of cDNA libraries.Citation1 Conceptually, RNA-Seq is performed after conversion of a population of RNA (fractionated or not for removal of highly abundant ribosomal or tRNA species) to a library of cDNA fragments with adaptors attached to one or both ends, which are subsequently sequenced from one or both ends. The reads are typically 30–400 bp, depending on the DNA-technology used.
As it has been noted for other domains of life, RNA-Seq has signified a revolution in the understanding of the transcription and regulatory modes in Prokaryotes,Citation2 including the important contribution of anti-sense RNA-mediated gene regulationCitation3 and how gene expression profiles are shaped along the infection in bacterial pathogens and their hosts.Citation4 Here, we discuss the latest advances in RNA-Seq, focusing in the analysis of the host-pathogen communications in specific cell subpopulations (infected vs. uninfected) and in single infected host cells.
RNA-Seq provides new insights into the regulatory RNA landscape
Due to the quality and type of data that RNA-Seq generates, this technology is providing a bulk of valuable data in the field of microbial pathogenesis. RNA-Seq has increased our knowledge of small non-coding RNAs (sRNA) produced by the pathogen in laboratory media and infection conditions. One of the most extensively-analyzed bacterial pathogens in this regard is Salmonella enterica, a facultative intracellular pathogen that invades all known host cell types. S. enterica causes a variety of diseases in animals and humans, ranging from self-limited gastroenteritis to systemic infection involving penetration of the intestinal epithelium and colonization of deeper tissues.Citation5,6 This pathogen is considered a model in the microbial pathogenesis field given the availability of powerful animal models such as those of typhoid fever or gastroenteritis in mice and the diversity of in vitro infection models based on distinct host cell types. Using in vitro infection models, investigators have deciphered at the cellular level sophisticated mechanisms of invasion, phagosome remodeling and autophagy evasion.Citation7 S. enterica is also easily manipulable at the genetic level, which favors the rapid generation of derivate mutants to test in virulence assays.
In a recent study, Hinton and colleagues used RNA-Seq to determine the transcriptome of S. enterica serovar Typhimurium (S. Typhimurium) in 22 different infection-relevant growth conditions, many of them involving specific stresses encountered by the pathogen in the host.Citation8 Some of these conditions included acidic pH, high osmolarity, low oxygen, limited availability of essential cations as magnesium and iron, and exposure to bile salts and compounds that impose oxidative and nitrosative stresses. This work, performed entirely with bacteria grown in laboratory media, made also use of a differential approach for RNA sequencing (dRNA-seq), designed for mapping of 5′ ends in primary transcripts.Citation9 dRNA-seq exploits terminal 5′ exonuclease to digest those RNAs lacking the 5′-triphosphate present in primary transcripts. The compendium of data obtained by Hinton and colleagues allowed to identify experimentally at the level of single nucleotide resolution the transcriptional start sites (TSS) for most genes, detecting expression in 86% of the genes previously annotated in the S. Typhimurium genome. The study also provided relevant information supporting transcriptional signatures linked to the distinct pathogenicity islands. This RNA-Seq analysis uncovered a total of 280 regulatory RNA encompassing sRNA located in intergenic locations (168 of these 280), in 5′ or 3′ ends (69 cases), antisense RNAs (41 cases) and intragenic RNA (2 cases).Citation8 The authors were able to classify known sRNA attending to their responsiveness in defined growth conditions as well as assign a precise level of response in the infection-relevant conditions for a few novel sRNA species.Citation8 Some of the changes in sRNA expression were validated by alternative methods such as Northern blotting. Despite the value of these data, we should be cautious when interpreting these changes in sRNA expression since they involve average data obtained at the population level. Future investigations should address whether variability in sRNA expression exists at the single-cell level among individuals of the bacterial community subjected to analysis.
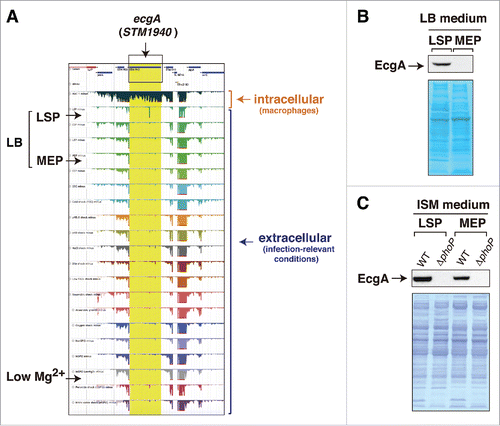
Subsequent studies by the same group focused on the isolation of RNA from intracellular S. Typhimurium following infection of macrophages.Citation10 RNA-seq performed on this material identified up to 31 genes that are strongly expressed by the pathogen inside the macrophage and not by extracellular bacteria. A total of 3,583 TSS were unequivocally identified in the study. Interestingly, this RNA-seq approach showed that 88% of the 280 sRNA previously identified in infection-relevant microbiological mediaCitation8 are expressed by S. Typhimurium inside the macrophage.Citation10 Thirty-four of these 280 sRNA were upregulated and 119 downregulated by intracellular bacteria. Some of these upregulated sRNA includes the Hfq-associated sRNA STnc440, STnc470 and STnc3750, which had not been previously characterized. Two aspects of this study that merit consideration for future studies involve the infection conditions and the comparison of RNA-seq data obtained in intracellular bacteriaCitation10 regarding the previous 22-infection relevant conditions.Citation8 Macrophage infection conditions included a multiplicity of infection (MOI) of 100:1 and an incubation period of 30 min. At a first glance, these conditions seem to exceed what it is needed to obtain relatively few bacteria per macrophage at early infection times. Due to their high phagocytic activity, it is probable that such high MOI could result in over-infection of the macrophages with undesirable cytotoxic effects. For comparison, recent RNA-seq single-cell studies involving S. Typhimurium and macrophages used instead a MOI of 1:1, precisely to ensure that macrophages were generally infected with only one bacterium.Citation11 Regarding gene expression profiles, the available data point to a necessity for prioritizing the analysis of RNA isolated from intracellular bacteria instead of focusing in extracellular conditions that probably mimic only partially the environment encountered by the pathogen inside the host cell. A remarkable example in S. Typhimurium is that involving the STM1940 (SL1344_1873) gene, renamed by us as ecgA, which encodes a peptidoglycan hydrolase.Citation12 The RNA-seq data obtained in S. Typhimurium using infection-relevant conditionsCitation8 and in intracellular bacteriaCitation10 indicate that ecgA is a gene specifically up-regulated inside macrophages, with expression levels barely detected in any of the extracellular conditions tested (). At the protein level, EcgA is however detected not only in intracellular bacteria but also in extracellular bacteria grown in LB medium at stationary phase (), ISM medium () and PCN medium at pH 5.8,Citation12 this latter is a medium used by Hinton and colleagues in the RNA-seq study covering infection-relevant conditions.Citation8 Moreover, production of EcgA protein by S. Typhimurium was also shown to respond to low magnesium concentrations,Citation12 a regulation that remained “invisible” at the RNA-seq data level ().Citation8 Although we cannot a priori discard differences in the exact growth conditions used by different laboratories, these comparisons alert us about the requirement of performing, when possible, parallel studies at the protein level when interpreting RNA-Seq data. In the specific case of EcgA, it is tempting to test whether post-transcriptional regulation (perhaps involving some sRNA?) could favor production of the protein in the acidic pH or low-magnesium conditions recreated in laboratory media, even when a low number of ecgA transcript molecules is produced.
Figure 1. Disparity between RNA-Seq data and those obtained at the protein level for the ecgA (STM1940) gene of S. Typhimurium. (A) expression profiles obtained by RNA-Seq for the genomic region in which ecgA is located. These profiles were obtained from intracellular bacteria following infection of macrophages (see Srikumar et al.)Citation10 and from bacteria grown in media mimicking different infection-relevant conditions (see Kröger et al.)Citation8 The image was mounted from the publicly available S. Typhimurium gene expression compendium (http://bioinf.gen.tcd.ie/cgi-bin/salcom.pl?db = salcom_mac_HL); (B) EcgA protein levels detected in bacteria grown in LB medium up to late stationary phase (LSP) or medium exponential phase (MEP); (C) EcgA levels in bacteria grown in ISM minimal medium to LSP and MEP. Protein extracts were prepared from wild-type (WT) and a ΔphoP mutant. Note the absence of reads in the RNA expression profile for ecgA (STM1940) in the LB medium-LSP and in low Mg2+ medium, conditions in which the EcgA protein is produced (see also Rico-Pérez et al.).12 In concordance with transcriptomic data obtained from intracellular S. Typhimurium isolated from fibroblastsCitation31 and the RNA-Seq data obtained in bacteria infecting macrophages,Citation10 EcgA protein levels increase notoriously in intracellular bacteria by a regulation mediated by the PhoP-PhoQ two-component system (see Rico-Pérez et al.)Citation12.
Another aspect for discussion are the different states that S. Typhimurium can reach inside macrophages ranging from effective proliferation to persistence as viable non-replicating bacteria.Citation13,14 Such heterogeneity was not represented in the RNA-seq data obtained from a mixed population of intracellular bacteria. This aspect has been successfully addressed in a recent single-cell study by Vogel and colleagues (see below).Citation15
Pathogen and host variability during infection
The last decade has accumulated increasing evidence for disparate heterogeneous behaviors in bacterial pathogens upon host encounter.Citation16 In addition to the intrinsic anatomic and physiologic complexity of host tissues and organs, distinct studies have shown varied type of lesions that progress to either high pathogen densities or resolution of the infection foci.Citation16 In some cases, these phenomena are observed simultaneously in the same tissue, as for S. Typhimurium following infection of mice.Citation14 In this infection model, Bumann and colleagues observed subsets of slow-growing pathogen cell coexisting with fast-growing subsets that ensured progression of the infection.Citation14 Phenotypic variation in the pathogen and host responses is also well documented in infections by Mycobacterium tuberculosis, which associate to distinct types of granulomas and other anatomic lesions associated to varied populations of immune cells.Citation17
Histological studies have therefore proved that heterogeneous responses are prominent at the tissue level in defined type of infections. In the case of S. Typhimurium, microscopy analyses have certified that such heterogeneity also translates at the cellular level. As the infection progresses in epithelial cells, subpopulations of cytosolic and intra-vacuolar intracellular bacteria evolve having distinct expression profiles for virulence determinants encoded by Salmonella-pathogenicity islands 1 and 2 (SPI1 and SPI2).Citation18 These two intracellular lifestyles have been reported both in cultured epithelial cells and in vivo in enterocytes of infected mice.Citation18,19 Remarkably, in some instances both lifestyles are observed in the same infected cell.Citation18 The exact signals that drive S. Typhimurium to colonize the cytosol of the infected epithelial or to remain enclosed in a phagosomal compartment, remain unknown. Factors that may potentially predispose to a precise intracellular lifestyle in the pathogen include the phase of cell cycle in the host cell at the time it is invaded and/or an intrinsic variability in gene expression in the invading bacterium. These are challenging ideas for which robust technologies such as RNA-Seq may certainly be of value.
RNA-seq applied to both the pathogen and the host
The infection process normally involves a continuous interchange of signals between the invading pathogen and the infected cell. This communication has been approached by the so called “dual” RNA-Seq technology.Citation20 In a recent study, Vogel and colleagues afforded this goal by using GFP-expressing S. Typhimurium to infect HeLa epithelial cells.Citation21 Infected cells were sorted at different post-inoculation times (2, 4, 8, 16 and 24 h) and RNA-Seq was applied to get simultaneous information of transcripts from the infected eukaryotic cells and intracellular bacteria in a population of infected cells previously sorted by fluorescence-activated cell sorting (FACS). RNA used in the RNA-Seq step was previously depleted of both bacterial and eukaryotic ribosomal rRNA. The study uncovered the function of a novel sRNA, STnc440 (renamed as PinT), in controlling expression of virulence determinants related to the pathogenicity islands SPI1 and SPI2. The authors proposed PinT as a timer sRNA with capacity to fine-tune the activity of these two pathogenicity islands, including some of their cognate translocated effectors (SopE/SopE2, both SPI1 effectors). Assays with mutants either lacking or overexpressing PinT served as proof of principle for variation in the expression profile of their targets (validated up to the protein level) with consequences to the infected epithelial cell. These effects included changes in the activation status of the JAK-STAT signaling pathway and the relative expression levels of numerous long noncoding RNAs.Citation21 This study is a breakthrough in the field although it did not address the intrinsic heterogeneity existing in subpopulations of cytosolic and intra-phagosomal S. Typhimurium that coexist in epithelial cells.Citation22 Different genetic programs may operate in each subpopulation since cytosolic bacteria express SPI1 and flagellar genes at high levels whereas intra-phagosomal bacteria up-regulate SPI2 genes.Citation18 A recent study has also provided evidence of genes used by S. Typhimurium to specifically modulate the growth rate in the cytosol.Citation23 Based on these observations, and the dual RNA-Seq data obtained by Vogel and colleagues,Citation21 it would be of much interest to determine whether the two subpopulations (cytosolic and intra-phagosomal) of intracellular S. Typhimurium express in a selective manner specific regulatory sRNA such as PinT. Appropriate reporters fused to the pinT promoter could provide valuable insights by direct examination at the microscope. The same approach could be in principle applicable to any other gene(s) of interest to be monitored in cytosolic and intra-phagosomal intracellular bacteria.
The advent of single-cell analyses and RNA-Seq technologies: A great combination
The studies discussed above, involving RNA-Seq in macrophages and epithelial cells infected with S. Typhimurium,Citation10,21 manifest the difficulty of analyzing transcriptome differences in subpopulations of intracellular bacteria that display distinct phenotypes. This is especially relevant concerning variable growth rates in the pathogen and adaptation to lifestyles in niches differing in their subcellular location. On top of that, one should also consider the variability in the population of the infected cells exposed to the pathogen.
In an elegant study using single-cell RNA-seq, Shalek et al. demonstrated that seemingly identical mammalian cells can respond differently to the same stimulus.Citation24 These authors sequenced single-cell RNA-Seq libraries from over 1,700 primary dendritic cells exposed to several experimental conditions, involving different stimuli and incubation times. Important messages of the study are that a few precocious cells react at early times to the stimulus; that variations exist in the fraction of cells expressing a defined mRNA and its relative levels; that expression profiles vary overtime; and, that stimulation of bystander cells occurs essentially by paracrine effects.Citation24
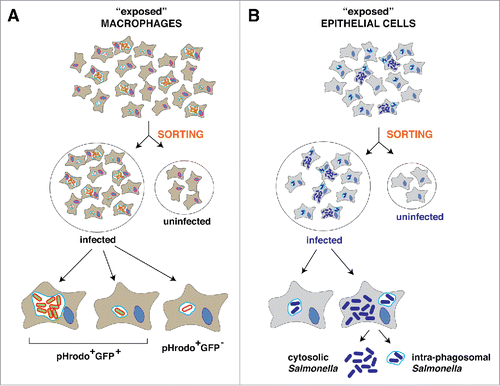
Following a similar experimental set-up, Hung and colleagues addressed this cellular variability in the context of infection by exposing macrophage cultures to S. Typhimurium.Citation11 These authors used GFP-expressing S. Typhimurium pre-incubated with pHrodo, a dye that binds to the cell wall and fluoresces red only when external pH becomes acidic. Thus, extracellular bacteria were green whereas intracellular bacteria should appear with both colors (green, red) if remain viable and enclosed in acidic compartments. Those intracellular bacteria killed by the phagocytic activity of the macrophage should be only red. Control experiments demonstrated the validity of this experimental set-up to sort different macrophage subpopulations, which included: exposed to the pathogen but uninfected (green-red-); infected and harboring live bacteria (red+, green+); and, infected and harboring dead bacteria (red+, green-) (). A fourth population of naïve non-exposed macrophages was also included in the analyses. The authors performed several types of RNA-seq: i) on individual single cells, ii) on sorted cell populations; and, iii) on bulk RNA-Seq libraries from entire exposed populations.Citation11 Principal component analysis (PCA) of several gene clusters in the distinct macrophage subpopulations did not infer differences between the two infected populations, which were merged in further analyses.
Figure 2. Experimental conditions in which dual RNA-Seq has been applied to analyze the communication of S. Typhimurium with eukaryotic cells. (A) studies in macrophages. The scheme highlights the experimental set-up used by Hung and colleagues based on the use of pH-rodo to detect dead and live intracellular bacteria and distinct cell sorting procedures (see text).Citation11 See text for other studies in macrophages as those of Hinton and colleagues,Citation10 based on dual-RNA-Seq in unsorted cultures exposed to the pathogen, and Vogel and colleagues,Citation15 which applied RNA-Seq of eukaryotic poly-A+ transcripts in single cells from uninfected and infected populations. (B) studies in epithelial cells. In this case, Vogel and colleagues applied dual RNA-Seq to sorted populations of infected and uninfected HeLa epithelial cells exposed to the pathogen.Citation21 Gene expression profiles have not yet been yet defined for cytosolic and intra-phagosomal bacterial populations colonizing epithelial cells.
Remarkably, data obtained from the 96 single cells analyzed by RNA-Seq revealed that uninfected macrophages showed stimulation of some cluster genes, probably due to exposure to pathogen components recognized by TLR4. Another cluster gene, named as cluster III and encompassing genes related to the type I IFN response, was however modulated at a higher extent by the presence of intracellular bacteria. This cluster III changed significantly in expression in the infected macrophage subpopulation. Three additional observations of the study have major implications. First, type I IFN genes showed a bimodal distribution in the infected cell population, with some infected cells showing higher expression levels than others, also infected.Citation11 Second, this bimodal expression pattern was not observed in macrophages exposed to inert bead particles containing conjugated LPS, which resulted in high expression of cluster III genes in all the exposed cells. These differences probably accounted for a subpopulation of infected macrophages that harbored bacteria actively attenuating the type I IFN defense response. Similar observations have recently been found in S. Typhimurium-infected fibroblasts, in which infected and uninfected cell subpopulations were sorted and examined separately for analysis of the NF-κB defense response.Citation25 Third, and most importantly, the authors were able to correlate the bimodal induction of type I INF response genes in infected macrophages with differences in the activity of PhoP-PhoQ regulatory system in intracellular bacteria. This latter finding was possible by FACS sorting of the infected macrophages populations using a fluorescent reporter to an IFN-stimulated response element (ISRE). RNA-seq of single cells in the ISRE+ and ISRE- infected populations revealed higher expression of PhoP-PhoQ-dependent genes in the ISRE+ (positive) cells. This correlation between induction of type IFN response and intracellular bacteria expressing the PhoP-PhoQ regulatory system was further validated using specific null and constitutive bacterial mutants (ΔphoP, phoPc), and beads in which LPS having (or not) modifications promoted by the PhoP-PhoQ system was conjugated. Note that this correlation between type I IFN response and a specific physiologic state of the intracellular bacteria was discovered due to dual RNA-Seq technology in which RNA from both the macrophage and the pathogen were simultaneously sequenced.
A minor aspect of this work not fully resolved is whether the coating of invading bacteria with pHrodo had consequences in the behavior of intracellular bacteria. Control experiments involving infections with coated and non-coated bacteria should resolve this issue. With the data that have been collected, authors could also exploit in future studies specific markers for the distinct macrophage subpopulations analyzed, avoiding in this manner the usage of pHrodo-coated bacteria.
In another recent and elegant study, Vogel and colleagues addressed whether a distinct growth rate of intracellular S. Typhimurium associates to a particular physiologic state in the infected macrophage.Citation15 These authors sorted infected from exposed uninfected macrophages based on fluorescence derived from bacteria expressing constitutively red mCherry fluorescent protein. These infected macrophages were further differentiated for distinct bacterial loads based on a conditionally-expressed green GFP fluorescent protein under an arabinose-dependent promoter, an inducer that is removed at the onset of the infection. Importantly, the authors noted a gradation of distinct bacterial loads which led them to focus on extremes for the RNA-Seq, i.e. with either very low or very high bacterial loads.Citation15 Using this experimental set-up, they performed single RNA-Seq in 64 cells, including 4 groups of 16 cells each representing the four cell populations: i) naïve macrophages (non-exposed to bacteria); ii) exposed but not infected (bystanders); iii) infected with low bacterial load; and, iv) infected with high bacterial load. The RNA-Seq libraries involved poly(A) capture, implicating the exclusive analysis of host transcripts. This approach proved to be fundamental to unravel a distinct polarization state of the macrophage depending the bacterial load. Those macrophages heavily infected displayed an anti-inflammatory, M2-state while those bearing low bacterial numbers exhibited an expression profile consistent with a pro-inflammatory M1 polarization state. The study invoked to postulate specific mechanisms of resistance in those non-growing bacteria that will be directed to evade host defense responses active in the intracellular environment. Future studies, in which single-cell dual RNA-Seq is applied to comparable experimental set-up, could provide new valuable insights into this phenomenon. As Vogel and colleagues discuss in their work, it will also of major interest to decipher whether changes in the macrophage polarization status are cause or consequence of the diverse growth rates exhibited by intracellular S. Typhimurium. It will be also important to define in a temporal basis if there are transitions in the polarization states as the infection progresses, i.e., as bacterial loads increase or decrease over time.
Concluding remarks and future perspectives
The studies of Hang and colleaguesCitation11 and Vogel and colleaguesCitation15 have certainly paved the way to a new era of analysis of the host-pathogen interaction and point to a tremendous variability in responses that take place over time. These variable responses might result from multiple encounters occurring in a host cell primed (or not) to confront a bacterium that intrudes the niche following a precise infection program (e.g., prone to integrate signals activating PhoP-PhoQ or SPI2). The available data support the idea of many possible encounters with large differences in the outcome of such interactions.Citation16 Such scenario is sustained, at least for S. Typhimurium, by in vivo data showing distinct populations of infected cells with variable potentials to combat the infection.Citation14,26
Many other technical questions remain open regarding our current capabilities to obtain RNA-Seq data in complex systems. For example, following infection of macrophages by S. Typhimurium, infected cells normally harbor distinct loads of viable bacteria, an aspect recently addressed by Vogel and colleagues.Citation15 Other macrophages however kill effectively the intracellular bacteria and these cells could be erroneously added to the subpopulation of infected macrophages when they do not harbor any more viable bacteria. Gene expression dynamics in both the macrophage and the pathogen unraveled by RNA-Seq are major goals for future investigations. Interestingly, the study of Hung and colleaguesCitation11 showed that there was no correlation in the interferon response of infected macrophages bearing high and low bacterial loads. This result favors the idea of a requirement for a PhoP-PhoQ “ON” status in the invading pathogen to trigger a defined response in the macrophage rather than reaching a certain load of intracellular bacteria.
Future single RNA-Seq-based investigations should also consider more homogeneous infection models in which undesirable effects for RNA purifications, such as that introduced by infected apoptotic host cells or infected cells harboring dead bacteria, do not occur. These variables are of special concern in macrophages and epithelial cells, in which phenomena of pyroptosis or cell death linked to bacterial infection or overgrowth, have been reported. Unlike these infection models, fibroblasts infected with S. Typhimuirum show a long-term stability in culture without denoting any signs of cytotoxicity. Part of this stability has been recently shown to reside in the unique capacity of fibroblasts to selectively digest by a specialized autophagy process only part of the population of intracellular S. Typhimurium.Citation27 At late infection times (> 16 hpi), only persistent intracellular bacteria are observed in otherwise healthy infected cells, which seems an ideal homogeneous scenario for applying single-cell dual RNA-Seq and uncovering features related to persistent infections.
An additional challenge imposed by the virulence traits of S. Typhimurium is the analysis of gene expression profiles in cytosolic and intra-phagosomal bacteria that, in some instances, co-exist in the same epithelial cell. Cytosolic bacteria are accessible to antibodies when infected cells are permeabilised with mild detergents such as digitonin. So, it may be worth to optimize isolation conditions to purify separately RNA from cytosolic and intra-phagosomal intracellular bacteria to get insights into the transcriptomes associated to these disparate lifestyles. The fact that cytosolic and intra-phagosomal bacteria express differently genes of pathogenicity islands SPI1 and SPI222 or that bacteria use defined genes to proliferate in the cytosol,Citation23 account for physiologic differences that RNA-Seq could potentially unravel in more detail. Single-bacterium RNA-Seq has been recently performed in the cyanobacterium Synechocystis sp.,Citation28 showing changes in individual cells in response to nitrogen starvation. Importantly, this study demonstrated a high coverage of all genes of the isolate used (ca. 85% at early times post-starvation) and an apparent high heterogeneity in the response between individual cells when comparing genes located in mobile elements versus core genes.Citation28
Finally, although the study of Hung and colleagues listed relative expression of all S. Typhimurium coding genes in bacteria located within ISRE+ and ISRE− macrophage subpopulations, there was no report on expression of regulatory sRNA. This information was however reported by Vogel and colleagues in their dual RNA-Seq analysis using epithelial cells.Citation21 Optimization of these technologies, directed to the simultaneous profiling of host and bacterial transcripts from minute amounts of material from single cells, will be of outstanding value in future studies. The study in Synechocystic cells allow us to be optimistic for the success of future single-cell RNA-Seq applied to the host-pathogen communication.Citation29 Protocols addressing these key aspects in the context of infection and exploiting novel RNA-Seq methodology have been recently published.Citation30 These approaches highlight as a major limitation the low abundance of bacterial mRNA with respect to total RNA of infected cells. High sequencing depth and dedicated algorithms for data analysis may overcome these limitations and allow to obtain a full transcriptomic landscape in individual intracellular bacteria.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
Work in our laboratories is supported by grants BIO2014–55238-R (to M.G.P.) and BIO2013-46281-P (to F.G.-dP.) from the Spanish Ministry of Economy and Competitiveness.
References
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 2009; 10:57-63; PMID:19015660; http://dx.doi.org/10.1038/nrg2484
- Sorek R, Cossart P. Prokaryotic transcriptomics: a new view on regulation, physiology and pathogenicity. Nat Rev Genet 2010; 11:9-16; PMID:19935729; http://dx.doi.org/10.1038/nrg2695
- Sesto N, Wurtzel O, Archambaud C, Sorek R, Cossart P. The excludon: a new concept in bacterial antisense RNA-mediated gene regulation. Nat Rev Microbiol 2013; 11:75-82; PMID:23268228; http://dx.doi.org/10.1038/nrmicro2934
- Caldelari I, Chao Y, Romby P, Vogel J. RNA-mediated regulation in pathogenic bacteria. Cold Spring Harb Perspect Med 2013; 3:a010298; PMID:24003243; http://dx.doi.org/10.1101/cshperspect.a010298
- Rivera-Chavez F, Baumler AJ. The pyromaniac inside you: Salmonella metabolism in the host gut. Annu Rev Microbiol 2015; 69:31-48; PMID:26002180; http://dx.doi.org/10.1146/annurev-micro-091014-104108
- Haraga A, Ohlson MB, Miller SI. Salmonellae interplay with host cells. Nat Rev Microbiol 2008; 6:53-66; PMID:18026123; http://dx.doi.org/10.1038/nrmicro1788
- LaRock DL, Chaudhary A, Miller SI. Salmonellae interactions with host processes. Nat Rev Microbiol 2015; 13:191-205; PMID:25749450; http://dx.doi.org/10.1038/nrmicro3420
- Kroger C, Colgan A, Srikumar S, Handler K, Sivasankaran SK, Hammarlof DL, Canals R, Grissom JE, Conway T, Hokamp K, et al. An infection-relevant transcriptomic compendium for Salmonella enterica serovar Typhimurium. Cell Host Microbe 2013; 14:683-95; PMID:24331466; http://dx.doi.org/10.1016/j.chom.2013.11.010
- Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, Chabas S, Reiche K, Hackermüller J, Reinhardt R, et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 2010; 464:250-5; PMID:20164839; http://dx.doi.org/10.1038/nature08756
- Srikumar S, Kroger C, Hebrard M, Colgan A, Owen SV, Sivasankaran SK, Cameron AD, Hokamp K, Hinton JC. RNA-seq brings new insights to the intra-macrophage transcriptome of Salmonella Typhimurium. PLoS Pathog 2015; 11:e1005262; PMID:26561851; http://dx.doi.org/10.1371/journal.ppat.1005262
- Avraham R, Haseley N, Brown D, Penaranda C, Jijon HB, Trombetta JJ, Satija R, Shalek AK, Xavier RJ, Regev A, et al. Pathogen cell-to-cell variability drives heterogeneity in host immune responses. Cell 2015; 162:1309-21; PMID:26343579; http://dx.doi.org/10.1016/j.cell.2015.08.027
- Rico-Pérez G, Pezza A, Pucciarelli MG, de Pedro MA, Soncini FC, García-del Portillo F. A novel peptidoglycan D,L-endopeptidase induced by Salmonella inside eukaryotic cells contributes to virulence. Mol Microbiol 2016; 99:546-56; PMID:26462856; http://dx.doi.org/10.1111/mmi.13248
- Helaine S, Holden DW. Heterogeneity of intracellular replication of bacterial pathogens. Curr Opin Microbiol 2013; 16:184-91; PMID:23485258; http://dx.doi.org/10.1016/j.mib.2012.12.004
- Claudi B, Sprote P, Chirkova A, Personnic N, Zankl J, Schurmann N, Schmidt A, Bumann D. Phenotypic variation of Salmonella in host tissues delays eradication by antimicrobial chemotherapy. Cell 2014; 158:722-33; PMID:25126781; http://dx.doi.org/10.1016/j.cell.2014.06.045
- Saliba AE, Li L, Westermann AJ, Appenzeller S, Stapels DA, Schulte LN, Helaine S, Vogel J. Single-cell RNA-seq ties macrophage polarization to growth rate of intracellular Salmonella. Nat Microbiol 2016; 2:16206; PMID:27841856; http://dx.doi.org/10.1038/nmicrobiol.2016.206
- Bumann D. Heterogeneous host-pathogen encounters: act locally, think globally. Cell Host Microbe 2015; 17:13-9; PMID:25590757; http://dx.doi.org/10.1016/j.chom.2014.12.006
- Mattila JT, Ojo OO, Kepka-Lenhart D, Marino S, Kim JH, Eum SY, Via LE, Barry CE 3rd, Klein E, Kirschner DE, et al. Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. J Immunol 2013; 191:773-84; PMID:23749634; http://dx.doi.org/10.4049/jimmunol.1300113
- Knodler LA, Vallance BA, Celli J, Winfree S, Hansen B, Montero M, Steele-Mortimer O. Dissemination of invasive Salmonella via bacterial-induced extrusion of mucosal epithelia. Proc Natl Acad Sci U S A 2010; 107:17733-8; PMID:20876119; http://dx.doi.org/10.1073/pnas.1006098107
- Sellin ME, Muller AA, Felmy B, Dolowschiak T, Diard M, Tardivel A, Maslowski KM, Hardt WD. Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host Microbe 2014; 16:237-48; PMID:25121751; http://dx.doi.org/10.1016/j.chom.2014.07.001
- Westermann AJ, Gorski SA, Vogel J. Dual RNA-seq of pathogen and host. Nat Rev Microbiol 2012; 10:618-30; PMID:22890146; http://dx.doi.org/10.1038/nrmicro2852
- Westermann AJ, Forstner KU, Amman F, Barquist L, Chao Y, Schulte LN, Müller L, Reinhardt R, Stadler PF, Vogel J. Dual RNA-seq unveils noncoding RNA functions in host-pathogen interactions. Nature 2016; 529:496-501; PMID:26789254; http://dx.doi.org/10.1038/nature16547
- Knodler LA. Salmonella enterica: living a double life in epithelial cells. Curr Opin Microbiol 2015; 23:23-31; PMID:25461569; http://dx.doi.org/10.1016/j.mib.2014.10.010
- Wrande M, Andrews-Polymenis H, Twedt DJ, Steele-Mortimer O, Porwollik S, McClelland M, Knodler LA. Genetic determinants of Salmonella enterica serovar Typhimurium proliferation in the cytosol of epithelial cells. Infect Immun 2016; 84:3517-26; PMID:27698022; http://dx.doi.org/10.1128/IAI.00734-16
- Shalek AK, Satija R, Shuga J, Trombetta JJ, Gennert D, Lu D, Chen P, Gertner RS, Gaublomme JT, Yosef N, et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 2014; 510:363-9; PMID:24919153; http://dx.doi.org/10.1038/nature13437
- Ramos-Marquès E, Zambrano S, Tierrez A, Bianchi ME, Agresti A, García-Del Portillo F. Single-cell analyses reveal an attenuated NF-kappaB response in the Salmonella-infected fibroblast. Virulence 2016:1-22; PMID:27575017; http://dx.doi.org/10.1080/21505594.2016.1229727
- Burton NA, Schurmann N, Casse O, Steeb AK, Claudi B, Zankl J, Schmidt A, Bumann D. Disparate impact of oxidative host defenses determines the fate of Salmonella during systemic infection in mice. Cell Host Microbe 2014; 15:72-83; PMID:24439899; http://dx.doi.org/10.1016/j.chom.2013.12.006
- López-Montero N, Ramos-Marquès E, Risco C, García-Del Portillo F. Intracellular Salmonella induces aggrephagy of host endomembranes in persistent infections. Autophagy 2016; 12:1886-901; PMID:27485662; http://dx.doi.org/10.1080/15548627.2016.1208888.
- Wang J, Chen L, Chen Z, Zhang W. RNA-seq based transcriptomic analysis of single bacterial cells. Integr Biol (Camb) 2015; 7:1466-76; PMID:26331465; http://dx.doi.org/10.1039/c5ib00191a
- Avraham R, Hung DT. A perspective on single cell behavior during infection. Gut Microbes 2016; 7:518-25; PMID:27658058; http://dx.doi.org/10.1080/19490976.2016.1239001
- Avraham R, Haseley N, Fan A, Bloom-Ackermann Z, Livny J, Hung DT. A highly multiplexed and sensitive RNA-seq protocol for simultaneous analysis of host and pathogen transcriptomes. Nat Protoc 2016; 11:1477-91; PMID:27442864; http://dx.doi.org/10.1038/nprot.2016.090
- Nuñez-Hernández C, Tierrez A, Ortega AD, Pucciarelli MG, Godoy M, Eisman B, Casadesús J, García-del Portillo F. Genome expression analysis of nonproliferating intracellular Salmonella enterica serovar Typhimurium unravels an acid pH-dependent PhoP-PhoQ response essential for dormancy. Infect Immun 2013; 81:154-65; PMID:23090959; http://dx.doi.org/10.1128/IAI.01080-12