
ABSTRACT
RNA decay and RNA maturation are important steps in the regulation of bacterial gene expression. RNase J, which is present in about half of bacterial species, has been shown to possess both endo- and 5′ to 3′ exo-ribonuclease activities. The exonucleolytic activity is clearly involved in the degradation of mRNA and in the maturation of at least the 5′ end of 16S rRNA in the 2 Firmicutes Staphylococcus aureus and Bacillus subtilis. The endoribonuclease activity of RNase J from several species has been shown to be weak in vitro and 3-D structural data of different RNase J orthologs have not provided a clear explanation for the molecular basis of this activity. Here, we show that S. aureus RNase J1 is a manganese dependent homodimeric enzyme with strong 5′ to 3′ exo-ribonuclease as well as endo-ribonuclease activity. In addition, we demonstrated that SauJ1 can efficiently degrade 5′ triphosphorylated RNA. Our results highlight RNase J1 as an important player in RNA turnover in S. aureus.
Introduction
Antiobiotic resistance is a major health concern and intensive search for new drugs must be a priority, in particular against bacterial pathogens such as Staphylococcus aureus. Giving the importance of RNA decay in gene expression in bacteria, it has been proposed that RNases could be targets for antiobiotic development.Citation1 RNA half-lives varies between minutes to hours, although most mRNA have half-lives that are in the minute range.Citation2 The signals which determine the stability of a specific RNA are not well understood, but are probably multifactorial such as RNA structure, RNA protein binding, efficiency of ribosome binding sequence, accessibility to RNase and RNase specificity.Citation3 Nonetheless, mRNA degradation is in general thought to be regulated at the initiation step, which typically involves an RNA endonucleolytic cleavage.Citation3-5 Although RNA decay is universal in bacteria, there is a surprising diversity in the genetic organization of the bacterial RNases.Citation4,Citation6,Citation7 In Escherichia coli, the initiating endonucleolytic cleavage is achieved by the essential RNase E, whereas RNA decay was proposed to be initiated by the endonuclease RNase Y in the gram-positive model organism Bacillus subtilis, which lacks a homolog of RNase E.Citation8 The phenotype of RNase Y deletion in B. subtilis is quite severe and pleiotropic.Citation9 More significantly, several studies show that RNA abundance is strongly affected in a strain that has been depleted for RNase Y.Citation10-12 However, phenotypes of RNase Y deletion vary in different organisms containing an ortholog of RNase Y. Importantly, RNase Y (encoded by the cvfa/rny gene) can be deleted in both S. aureus and Streptococcus pyogenes with only modest to no increase in doubling time.Citation13-15 Altogether, these results suggest that the importance of RNase Y for growth and RNA turnover is not conserved in all species containing an ortholog of RNase Y. Indeed, our laboratory showed that deleting RNase Y significantly increased the stability (more than 2-fold) of only about 6% of the S. aureus RNA in cells grown at 37 °C.Citation14 This result raises the question of which RNase initiates RNA decay of most of S. aureus RNAs.
In addition to RNase Y, Firmicutes such as B. subtilis, S. aureus and S. pyogenes also possess 2 additional RNases, which are absent in E. coli, the paralogs RNase J1 and RNase J2. These 2 proteins form heterotetramers in vitroCitation16 and this complex has both an endo- and a 5′ to 3′ exo-ribonuclease activities.Citation16,Citation17 The 5′ to 3′ exoribonuclease activity originally thought to be absent in bacteria was first established in B. subtilis and was shown to reside in the RNase J1 protein (the B. subtilis protein will henceforth be referred to as BsuJ1).Citation17 RNase J2 by itself had a very weak endo- and exo-ribonucleolytic activity and its function in vivo remains obscure.Citation4,Citation16,Citation18 Note that the in vitro activity of RNase J1 does not require the presence of RNase J2 in the complex and many bacteria do not even contain a RNase J2 ortholog in their genome.Citation4,Citation5
RNase J1 (encoded by the rnjA gene), first reported as an essential gene in B. subtilis,Citation19 was then demonstrated to be non-essential emphasizing the importance of the strategy used to knock out a gene for identifying essential genes in bacteria.Citation9 Although non-essential, this gene has been reported to be important for cell growth, cell morphology and sporulation.Citation9 More importantly, it has been reported that the abundance of about 20% mRNA is increased at least 2 folds in a B. subtilis strain expressing about 30-fold less BsuJ1,Citation12 a phenotype very similar to the one observed with the depletion of RNase Y12. One of the reasons that RNase J1 was not proposed to be the major endonuclease in B. subtilis was based mainly on the observation that a depletion of RNase Y has a stronger effect on bulk mRNA stability than a RNase J1 depletion.Citation10,Citation20 Moreover, structural data of RNase J1 do not allow a straightforward explanation for a strong endonuclease activity (see below and in ref. Citation4 and Citation21).
To gain further insight into the molecular basis of the dual activities of RNase J, several laboratories crystalized RNase J from different bacteria. Crystal structures of RNase J from Streptomyces coelicolor, B. subtilis, Thermus thermophilus and Deinococcus radiodurans reveal that RNase J is a homotetrameric or a homodimeric enzyme in the crystal, and adopts a 3-domain architecture with a β-lactamase domain flanked by a β-CASP and a C-terminal domainCitation22-26 The catalytic site is buried into a deep inter-domain cleft between the β-lactamase and β-CASP domains and contain 2 zinc ions, which are coordinated to His and Asp residues present in conserved signature motif I to IV, Motif A, and Motif C, respectively.Citation25,Citation26 The well-conserved Motif II (HxHxDH: in which x represents any amino acid) coordinates both zinc ions and is well conserved in the metallo β-lactamase nuclease family.Citation27 These 2 metal ions have been proposed to activate a water molecule to perform a nucleophilic attack on the 5′ terminal phosphodiester bond to initiate RNA hydrolysis.Citation27 Although RNase J was proposed to be zinc-dependent based on structural data, it clearly needs other metal ions, in most cases magnesium, to be active.Citation20,Citation22,Citation26
Structural studies of D. radiodurans RNase J demonstrated that manganese was important to promote homodimerization.Citation25 However, no divalent metal ions were found at the dimer interface of S. coelicolor RNase J, indicating that the role of the manganese in promoting homodimerization of RNase J is not conserved across bacterial species.Citation26 Interestingly it has been recently proposed that magnesium (or manganese) associates transiently with the catalytic site and/or the RNA substrate to optimize the proximity and orientation of the activated water molecule with the scissile phosphate.Citation22,Citation26 Nevertheless, the exact role in catalysis of this extra metal remains obscure.
Mutagenesis showed clearly that RNase J contains a single catalytic site for both endo- and exonucleolytic activities (reviewed in.Citation4,Citation21). The tertiary and quaternary structures of different RNase J orthologues were compatible with the 5′ to 3′ exoribonuclease activities. However, the endonuclease activity needs to invoke a partial (or a complete) dissociation of the RNase J homotetramer to monomer and a major structural rearrangement to load the RNA substrate into the catalytic site (Citation4,Citation21). Alternatively, it has been proposed that the RNA can pass though the catalytic site without being cleaved (sliding endonuclease mode).Citation4,Citation28
In vitro studies on BsuJ1 revealed that the 5′ to 3′ exoribonuclease of RNase J is more active on a 5′ monophosphate RNA compared with a 5′ triphosphorylated RNA.Citation17,Citation22 The greater preference for a 5′ monophosphate (or 5′ hydroxyl) over triphosphate ends has been explained by difficulties of modeling a triphosphorylated RNA in the catalytic site of RNase J Citation22,26. Binding of the gamma phosphate in the phosphate-binding site would be incompatible with a phosphodiester bond cleavage.Citation22,Citation26 However, the endonuclease activity, the first activity described for RNase J1, is capable of cleaving a triphosphorylated RNA.Citation16,Citation20,Citation22,Citation28 Biochemical studies showed that the preferential degradation of a 5′ monophosphate compared with 5′ triphosphate RNA substrates by RNase J varies from 3-fold to more than 10-fold depending on RNase J ortholog, the RNA substrate, and the experimental conditions. Citation17,Citation22,Citation26,Citation28-32 Thus, different orthologs of RNase J seem to have different inherent 5′ end specificities.
In S. aureus, the RNase J1 gene rnjA was shown to be non-essentialCitation18 but its deletion prevented growth outside a narrow range of temperature and media.Citation18 By using the EMOTE (the exact mapping of transcriptome ends) assay we demonstrated that RNase J1 activity is important for both normal degradation of bulk RNA and RNA maturation. In fact, 5′ end maturation of both ribosomal 16S and the RNase P ribozyme are dependent on the activity of RNase J1.Citation18 Our recent unpublished RNA-seq data demonstrated that most RNA fragments (more than 70%) are more abundant in a RNase J1 deletion strain (manuscript in preparation). This is in sharp contrast with the lack of phenoptypes observed for a RNase Y deletion.Citation14 Therefore, our in vivo data are consistent with a major role of RNase J1 in RNA turnover in S. aureus. Remarkably, Redko et al., (Citation2016) recently suggested that RNase J has a crucial role in RNA degradation in Helicobacter pylori. In fact, the abundance of 80% of mRNAs is increased 2-fold in a RNase J depleted H. pylori strain.Citation33
Here we conducted a biochemical characterization of the RNase J1 ortholog from S. aureus (SauJ1). The rnjA gene encodes a predicted 565-amino acid polypeptide related to the BsuJ1; amino acid sequence alignment of SauJ1 to BsuJ1 sequences revealed high sequence conservation (66% of amino acid identities). As in B. subtilis, SauJ1 shows in vivo interactions with RNA helicase CshA with the component of a putative degradosome.Citation34 To better understand the enzymology of RNase J1 in S. aureus and its role in RNA turnover, we produced and purified SauJ1. We found that SauJ1 is a homodimer in solution and is a manganese dependent enzyme at the concentration used in our enzymatic assay. As expected, SauJ1 possesses a robust 5′ to 3′ exoribonuclease activity on a short single strand RNA. However, in sharp contrast to other studies, we report that SauJ1 is able to efficiently degrade exo- and endonucleotically a triphosphophorylated RNA.
Materials and methods
S. aureus strains, growth conditions, and electron microscopy
S. aureus strains, growth medium, and spotting dilutions to determine growth defect are described in ref. Citation18. The primers used for vector construction (J1 deletion and B. subtilis J1 plasmids) are shown in Table S1. Plasmids used in this study are listed in Table S2. For electron microscopy, wild-type and ΔRNase J1 strains were grown on Mueller-Hinton broth (MH) (Becton-Dickinson) with uracil (20 mg/l) at 37°C under shaking conditions until OD600 = 0.4. The fixation was performed by adding 5% (v/v) glutaraldehyde to 900 µl of exponential phase bacteria for 30 min. Cells were pelleted by low speed centrifugation and the fixed cells were further treated as described in ref. Citation35
Bacterial expression vectors for Staphylococcus aureus ribonuclease SauJ1
The open reading frame encoding S. aureus J1 (SauJ1) was amplified from Staphylococcus aureus DNA (strain SA564) by PCR with primers designed to introduce a BamHI site at the start codon and an NotI site 3′ of Step-Flag. The primary structure of the 565-aa polypeptide encoded by the resulting cDNA was identical to that of SauJ1 deposited in NCBI database under accession WP_00811375.1. The double alanine mutation (H74A-H76A; referred as 2XAla in this manuscript) was introduced into the SauJ1 gene fragment by PCR amplification with mutagenic primers. The PCR products were digested with BamHI and NotI and then ligated into BamHI/NotI-cut plasmid pET28-His10Smt3-SF to fuse the SauJ1 proteins in-frame with an amino-terminal His10Smt3 tagCitation36 and a C-terminal Strep-Flag. The plasmid inserts were fully sequenced to exclude the acquisition of mutations during amplification and cloning.
Recombinant S. aureus J1
The pET28-His10Smt3-J1-SF plasmids were transformed into E. coli BL21(DE3). Cultures (500 ml) derived from single transformants were grown at 37°C in LB medium containing 50 µg/ml kanamycin until the OD600 reached 0.6. The cultures were adjusted to 0.2 mM IPTG and 2% (v/v) ethanol and incubation was continued for 20 h at 17°C. Cells were harvested by centrifugation and stored at −80°C. All subsequent procedures were performed at 4°C. Thawed bacteria were resuspended in 25 ml of buffer A (50 mM Tris-HCl, pH 8.0, 200 mM NaCl, 10% glycerol) and supplemented with one tablet of protease inhibitor cocktail (Roche). The suspension was adjusted to 0.1 mg/ml lysozyme and incubated on ice for 30 min. Imidazole was added to a final concentration of 10 mM and the lysate was sonicated to reduce viscosity. Insoluble material was removed by centrifugation. The soluble extracts were mixed for 30 min with 3 ml of Ni2+-NTA-agarose (Qiagen) that had been equilibrated with buffer A containing 10 mM imidazole. The resins were recovered by centrifugation, resuspended in buffer A with 10 mM imidazole, and poured into columns. The column was washed with 15 ml aliquots of 20 mM imidazole in buffer A and then eluted step-wise with 5 ml aliquots of buffer A containing 50, 100, 250, and 500 mM imidazole, respectively. The elution profiles were monitored by SDS-PAGE (not shown). The recombinant protein His10Smt3-SauJ1-SF was recovered predominantly in the 250 mM imidazole fraction. 1 mg of His10Smt3-J1-SF polypeptides from the 250 mM imidazole were cleaved with 25 µg of His6ULP1 (prepared as described inCitation36) for 60 min at 4 °C and applied to 1-ml Strep-Tactin column (IBA), that had been equilibrated with buffer A. The column was washed with 10 ml of buffer A and then eluted step-wise with 1 ml buffer B containing (100 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 10% glycerol, 2 mM D-biotin). The polypeptide compositions of the column fractions were monitored by SDS-PAGE. The recombinant SauJ1-SF was recovered predominantly in the fraction 2, 3, 4 and 5. These fractions were pooled, concentrated with a centrifugal filter (Amicon Ultra 4; 10′000 MWCO) and then stored at −80°C. The protein concentration was determined using the Bio-Rad dye reagent with BSA as the standard. SauJ1-SF concentration was calculated by interpolation to the BSA standard curve. H74A-H76A was produced and purified as described above for the wild-type SauJ1-SF.
Recombinant FlagStrep-Smt3-SauJ1 used the glycerol gradient sedimentation were produced into E. coli BL21(DE3) and purified from soluble bacterial lysates by Strep-tactin chromatography as described above for SauJ1-SF.
RNA substrates
RNA oligonucleotides are purchased from MicroSynth (PAGE purified quality). Synthetic RNA oligonucleotides were 5′ 32P labeled with T4 polynucleotide kinase phosphatase minus (NEB) and [γ-32P]ATP (32P-labeled nucleotides used to label RNA are all from Hartmann Analytic GmbH). Concentration of ATP is adapted to obtain at least 95% phosphorylation at the 5′ ends by PNK phosphatase minus. Alternatively RNA oligonucleotides were 3′ [5′ 32P] pCp labeled by reaction with T4 RNA ligase (NEB) following the manufacturer's protocol. Unlabeled 135-mer RNA substrate, which is the positive control of the MEGAshortscript kit (Ambion), were synthesized according to the manufacturer's protocol. For uniformly labeling this 135-mer RNA, the T7 polymerase reaction of the MEGAshortscript kit reaction was modified by including 40 µCi of [α-32P]UTP to the 20 µl reaction mixtures. For gamma 5′ end labeling, the manufacturer's protocol was modified by reducing GTP concentration from 7.5 mM to 1.5 mM GTP and including 60 µCi [γ-32P]GTP in the 20 µl reaction mixture. Radioactive reaction mixtures were passed though a NucAway spin columns (Ambion) and purified by using the miRNeasy kit from Qiagen according the manufacturer's protocol. 5′ end monophosphate labeling was performed as follows: Aliquot (50 µl) containing 20–40 µg of 5′ triphosphorylated unlabeled 135-mer RNA was dephosphorylated with 50 U of alkaline phosphatase (Roche). To check that 100% of the RNA are dephosphorylated by alkaline phosphatase, we included in a parallel reaction a mixture of cold triphosphorylated RNA and gamma-labeled RNA and we followed the release of the gamma phosphate on PEI-cellulose TLC plates. Alkaline phosphatase reaction mixtures were purified with miRNaesy kit and labeled with PNK phosphatase minus and [γ-32P]ATP. Radioactive reaction mixtures were passed though a NucAway spin columns (Ambion) and purified by using the miRNeasy kit from Qiagen according the manufacturer's protocol.
Ribonuclease Assay
Reaction mixtures (15 µl) containing 50 mM Tris-HCl, pH 8.0, 1 mM MnCl2, RNA substrate as specified, and enzyme as specified were incubated for 15 min at 37°C. Reactions were quenched with an equal volume of 95% formamide, 20 mM EDTA, 0.05% bromophenol blue. The reaction products were then analyzed by electrophoresis through a 20% (or 12.5%) polyacrylamide gel (PAGE) containing 8 M urea and the products were visualized by autoradiography. Radioactive product formation was quantified by scanning the urea-PAGE with laser Scanner Typhoon FLA 7000 (General Electric). Alternatively, reactions were quenched with 3.8 µl formic acid. Aliquots (2µl) were spotted on PEI-cellulose TLC plates (Merck KgaA), which were developed with 1 N formic acid and 0.5 M lithium chloride. The radiolabeled material was visualized by autoradiography, and 32P-UMP formation was quantified by scanning the TLC plate with laser Scanner Typhoon FLA 7000 (General Electric).
Non-denaturing gel
Reaction mixtures (10 µl) containing 50 mM Tris-HCl pH8, 25 mM NaCl, 10% glycerol and either no metal or 1 mM metal as indicated were incubated at room temperature for 20 min. A control with 1 mM EDTA was also included. The mixtures were adjusted to 20% glycerol and then analyzed by native PAGE 4–16% Bis-Tris protein gel (Novex, life technology). This gel was run with 1X Native PAGE cathode buffer additive according the protocol's manufacturer (Novex life technologies).
Glycerol gradient sedimentation
An aliquot (40 µg) of the nickel-agarose preparation of Flag-Step-Smt3-SauJ1 was mixed with catalase (45 µg), bovine serum albumin (45 µg), and cytochrome c (45 µg). The mixture was applied to a 4.8-ml 15–30% glycerol gradient containing 50 mM Tris-HCl (pH 8.0), 0.25 M NaCl, 1 mM EDTA, and 2 mM DTT. The gradient was centrifuged for 20 h at 4 °C in a Beckman SW50 rotor at 48,000 rpm. Fractions (0.17 ml) were collected from the bottom of the tube. Aliquots (15 µl) of odd-numbered gradient fractions were analyzed by SDS-PAGE.
Results
In vivo activity of SauJ1
We showed that a deletion of S. aureus RNase J1 was viable at 37°C on Mueller-Hinton (MH) plate although its growth was slowed, and almost no growth was observed at 25°C.Citation18 Moreover, this strain did not grow on a defined RPMI medium. We equally showed that the wild-type rnjA gene on a low copy number plasmid under the control of its own promoter is fully capable of complementing growth of an S. aureus ΔJ1 strain at 25, 30, 37, and 42 °C ( and Citation18). Here, we found that neither a C-terminal domain deletion (a C-terminal 68 amino acids deletion; SauJ1Δ498–565) or a short N-terminal deletion (the first 30 amino acids; SauJ1Δ2–30) complemented growth of ΔJ1 cells when expressed from a plasmid (). It is noteworthy that the C-terminal domain of B. subtilis of RNase J1 (450–555) has also been shown essential for the in vitro activity.Citation22 It has been postulated that this domain is essential for homodimerization and that the quaternary structure is required for ribonuclease activity.Citation22 Curiously, we observed that the expression of an N-terminally truncated version of SauJ1 from a plasmid, inhibits growth of the ΔJ1 strain compared with a strain with an empty plasmid at all tested conditions (, compare ΔJ1 + pEB01 and ΔJ1 + pSauJ1Δ2–30). However, it is unclear why removing the N-terminal 30 amino acids should render the protein toxic. We also found that B. subtilis RNase J1 is capable of fully complementing an S. aureus ΔJ1 strain (), which was not surprising given the fact RNase J1 from S. aureus and B. subtilis have 83% amino acid identities/similarities.
Figure 1. Complementation and electronic microscopy. (A) Transformants of wild-type and ΔRNase J1 strains with the indicated plasmids were grown overnight, diluted to 10−5 and 10−6 respectively, spotted on MH, RH, and LB agar-plates at the indicated temperatures as described Citation18 pEB01 is the empty plasmid. (B) Cultures of wild-type and ΔRNase J1 strains were harvested at an OD600 of 0.4 and subjected to transmission electron microscopy as described in Materials and Methods. The two upper panels show wild-type cells, and the two lower panels show ΔJ1 cells, with white arrows indicating non-canonical divisions.
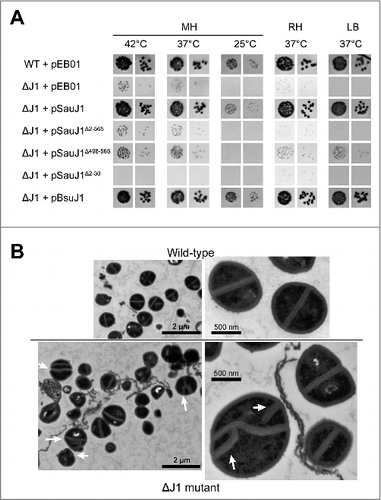
The reason for why RNase J1 mutants grow at a very restrictive temperature range and on a narrow range of medium plate is unknown, and is likely a combination of mis-regulating several genes. However, electron microscopic analysis of negatively stained S. aureus ΔJ1 cells clearly indicate significant division defects, although not every single cell appears to exhibit these ().
Ribonuclease activity of SauJ1
The open reading frame encoding SauJ1 was cloned into a bacterial expression vector to fuse the SauJ1 protein to an N-terminal His10-Smt3 domain and a C-terminal Strep-Flag (-SF), respectively (see Materials and Methods). The His10-Smt3- SauJ1-SF protein was purified from a soluble bacterial extract by adsorption to nickel-agarose and subsequent elution with imidazole. SDS-PAGE analysis of the 250 mM imidazole eluates revealed one major species corresponding to His10-Smt3-SauJ1-SF (, lane 1). The His10-Smt3 domain was removed by treatment with purified His6-tagged Ulp1, a Smt3-specific protease that hydrolyzes the polypeptide chain at the junction between His10-Smt3 and the downstream protein.Citation36 The recombinant SauJ1-SF was separated from the His10-Smt3 tag by adsorption to Strep-tactin, and subsequent elution with buffer containing 2 mM D-biotin. SDS-PAGE analysis of the peak eluate fraction showed that the original His10- Smt3-SauJ1-SF (calculated to be a 82-kDa polypeptide) was converted to a ∼65 kDa species (calculated to be a 67 kDal polypeptide) corresponding to SauJ1-SF (; lane 3). Further characterization of recombinant SauJ1-SF was performed with this fraction and will henceforth be referred to as SauJ1. In parallel we produced and purified a mutant version of SauJ1 in which His 74 and His 76 within the active site were replaced by alanines (referred thereafter as 2XAla; see , lane 2, and lane 4). In B. subtlis, His74 and His76 are part of motif II of the β-lactamase domain and binds one of the metal inside the catalytic site.Citation23
Figure 2. Ribonuclease activity of recombinant SauJ1. (A) Protein preparations. Aliquots (2.5 µg) of either the nickel-agarose preparations of His10Smt3-SauJ1-SF (lane 1), and His10Smt3-SauJ1 (2XAla)-SF (lane 2) or the Step-Tactin preparations of SauJ1-SF (lane 3) and SauJ1 (2XAla)-SF (lane 4) were analyzed by SDS-PAGE. The polypeptides were visualized by staining with Coomassie Blue dye. The positions and sizes (kDa) of marker polypeptides are indicated on the left. The polypeptide composition of the elution fraction of the tagged-SauJ1 is shown on the right. (B) Divalent cation requirement. Reaction mixtures (15 µl) containing 50 mM Tris-HCl (pH 8.0), 8 nM 5′ end 32P labeled 26-mer RNA (*pRCitation26nt), 100 ng of wild-type SauJ1 and either 10 mM EDTA, no added divalent cation (-) or divalent cation (chloride salt) as specified were incubated for 15 min at 37 °C. The reaction products were analyzed on a 20% urea PAGE and the products were visualized by autoradiography. (C) Determination of the predominant SauJ1 products. Reaction mixtures (15 µl) containing 50 mM Tris-HCl (pH 8.0), 1.3 U RNasin, 8 nM 26-mer RNA (*pRCitation26nt), 10 mM MgCl2, and 1 Unit of XRN1 (New England Biolabs) were incubated for 15 min at 37 °C, and analyzed on a 20% urea PAGE in parallel with the reaction products of SauJ1 obtained as described in panel B. D. Analysis of the SauJ1 products by PEI-cellulose TLC. Reaction mixtures (15 µl) containing 50 mM Tris-HCl (pH 8.0), 8 nM 26-mer RNA (*pRCitation26nt), 1 mM MnCl2, and 100 ng of SauJ1, 100 ng of 2XAla or 1 U of XRN1 were incubated for 15 min at 37 °C. Reactions were quenched by adding formic acid and aliquots (2µl) of the mixtures were applied to a PEI-cellulose TLC plate, which was developed with 1 M formic acid, 0.5 M LiCl. 32P-labeled material was visualized by autoradiography. (E) Protein titration. Reaction mixtures (15 µl) containing 50 mM Tris-HCl, 1 mM MnCl2, 1.3 U RNasin, 25 nM 26-mer RNA (*pRCitation26nt) and either SauJ1 or 2xAla proteins as indicated were incubated for 15 min at 37 °C. The reaction products were analyzed on a 20% urea PAGE and the products were visualized by autoradiography. Minor degradation products are indicated by dashed arrows.
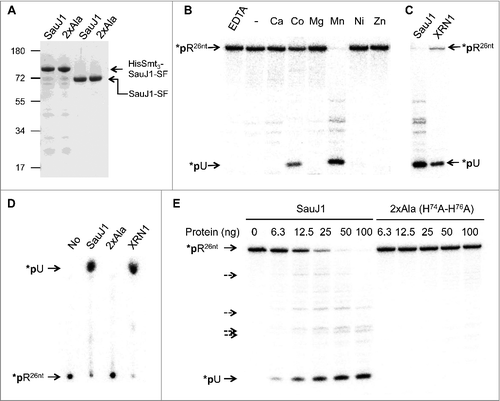
Although RNase J1 contains a β-lactamase domain, known to be a zinc binding domain,Citation27 the activity of B. subtilis RNase J1 (BsuJ1) has been shown to be magnesium dependent.Citation16,Citation17 However, we were unable to observe nuclease activity of SauJ1 in the presence magnesium, although a variety of buffers were tested. In an attempt to detect activity, we incubated 100 ng of SauJ1 with 25 nM 5′ 32P-labeled 26-mer RNA (*pRCitation26nt) and a range of different cation cofactors at 37 °C for 15 minutes. The reactions were quenched with EDTA-formamide and the products were analyzed by urea-PAGE. We found that recombinant SauJ1 was able to degrade, in a quantitative manner, the 5′ 32P-labeled RNA substrate in the presence of 1 mM manganese (). Whereas cobalt could replace manganese as the requisite metal (about 30% activity), calcium, magnesium, nickel or zinc could not (). No RNase activity was detected when either metal was omitted or 10 mM EDTA was added indicating that the activity was strictly metal dependent. Yeast XRN1, a highly processive 5′ to 3′ exoribonuclease purified from E. coli, was used to confirm that the product released by SauJ1 in presence of 1 mM manganese was an uridine 5′ monophosphate (*pU) as expected for a 5′ to 3′ exoribonuclease (26-mer RNA starts with an uridine; Table 1), and could show that the products generated by the XRN1 and SauJ1 co-migrated on both a 20% denaturing PAGE () and on a PEI-cellulose TLC ().
The major product detected was always *pU, when a range of SauJ1 concentrations were tested (), so the main SauJ1 activity was indeed 5′ to 3′ exoribonuclease activity. However, it is noteworthy that several minor products (less than 5% of the total products) were detected with sizes between the full-length 26-mer 5′ labeled RNA and the *pU product. Since the RNA is labeled at the 5′ end, these minor products could not arise from a 5′ to 3′ RNA degradation but are compatible with a endonuclease activity of SauJ1 (see below), and some of these endo-nucleolytic products even appeared to be protected from the 5′ to 3′ exo-nucleolytic activity of SauJ1, even at high concentrations (, lanes 50 ng and 100 ng). Importantly, the RNase activity observed is inherent to the recombinant protein SauJ1 at the concentrations used in this RNase assay, since the generation of both the *pU exo-nucleolytic product and the endo-nucleolytic products were completely absent with the 2xAla mutants enzyme ( and ).
Quaternary structure of SauJ1
The manganese-dependent dimerization described for D. radiodurans RNase J Citation25, prompted us to gauge whether the quaternary structure of SauJ1 could be influenced by the presence of different metal cofactors in the reaction mixtures. We observed that SauJ1 would run as a mix of homo-dimer (∼70%) and homo-tetramer (∼30%) on a 4–16% gradient non-denaturing PAGE (lane (-) in Fig. S1). Adding 10 mM EDTA, or either 1 mM calcium, cobalt, magnesium, manganese, nickel, or zinc didn't alter the percentage of SauJ1 homo-dimer and homo-tetramer, suggesting that the quaternary structure of SauJ1 is not influenced by metals (Fig. S1). We furthermore analyzed the native size of recombinant FlagStrep-Smt3-J1 by sedimentation of the protein through a 15–30% glycerol gradient. Marker proteins were included as internal standards in the same gradient. After centrifugation, the gradient fractions were analyzed by SDS-PAGE (data not shown). FlagStrep-Smt3-J1 (calculated to be a 79 kDa) sedimented as a discrete peak 2 fractions heavier than BSA (66 kDa). A plot of the S values of the 3 standards versus fraction number yielded a straight line and allowed us to calculate a S value of 6.1 for the tagged J1 protein (data not shown). This value is very similar to the theoretical S-value of 6.19 found for the BsuJ123, and is compatible with the protein being a homodimer in solution.
Optimal enzymatic conditions
Optimal reaction conditions were delineated via systematic variations of the concentrations of divalent cation cofactors, salt, and pH. The optimal concentration of manganese, determined at non-saturating level of SauJ1, was between 63 µM and 1 mM; note that even a manganese concentration as low as 8 µM only reduced the nuclease activity about 3-fold (). The presence of 30, 60, and 125 mM NaCl reduced the *pU formation from a 26-mer RNA substrate to 20%, 16% and 5%, respectively, compared with the absence of added NaCl (data not shown), showing that increased ionic strength exerted a strong inhibitory effect on SauJ1 activity. Finally, SauJ1 RNase activity displayed a bell-shaped pH profile with an optimum at pH 7.0–8.0 (). The BsuJ1 structure contained 2 zinc ions in the active site, although both endo- and 5′ to 3′ exo-ribonuclease activities have been demonstrated in the presence of 8–10 mM magnesium chloride.Citation16,Citation17 Surprisingly, magnesium did not support activity of SauJ1 between 0.08 and 10 mM magnesium (), and zinc and calcium were completely ineffective as RNase cofactors from 250 µM up to 10 mM (data not shown).
Figure 3. Characterization of SauJ1 RNase activity. (A) Manganese titration. Reaction mixtures (15 µl) containing 50 mM Tris-HCl (pH 8.0), 100 nM 26-mer RNA (*pRCitation26nt), 12.5 ng of SauJ1, and MnCl2 as specified were incubated for 15 min at 37 °C. The reaction products were analyzed on PEI-cellulose TLC plate as shown in panel 1B of . The percent of uridine 5′ phosphate (5′pU) release is plotted as a function of manganese concentration. (B) pH dependence. Reaction mixtures containing either 50 mM Tris acetate (pH 5.0 to 7.0), or Tris-HCl (pH 7.5 to 9.5), 1 mM MnCl2, 8 nM 26-mer RNA (*pRCitation26nt) and 25 ng of SauJ1 were incubated for 15 min at 37 °C. The percent of uridine 5′ phosphate (5′pU) release is plotted as a function of pH. (C) Metal titration at saturating level of SauJ1. Reaction mixtures (15 µl) containing 50 mM Tris-HCl (pH 8.0), 8 nM 26-mer RNA (*pRCitation26nt), 100 ng of SauJ1, and MgCl2 or MnCl2 as specified were incubated for 15 min at 37 °C. The percent of uridine 5′ phosphate (5′pU) release is plotted as a function of divalent cation concentration. (D) Mixing of divalent cations. Reaction mixtures (15µl) 50 mM Tris-HCl (pH 8.0), 100 nM 26-mer RNA (*pRCitation26nt), 2 mM MnCl2, 50 ng of SauJ1, and either 1 mM EDTA, or 1mM of the indicated divalent cations were incubated for 15 min at 37 °C. The percent of uridine 5′ phosphate (5′pU) release is shown. A control reaction lacking divalent cation is indicated in the first lane. The data are the averages of 3 experiments plus standard errors of the mean (SEM).
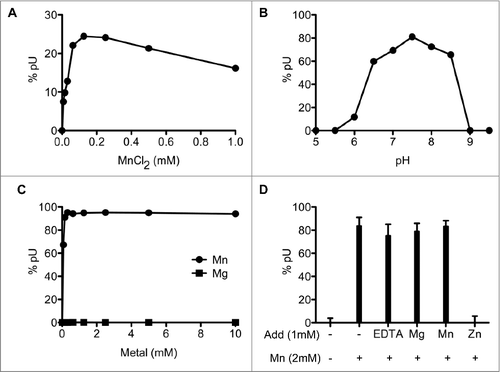
All published crystal structures of RNase J proteins contain zinc-ions, and it is therefore surprising that zinc ions do not permit activity of SauJ1. To verify this results in the most rigorous manner, a mixing experiment was performed with saturating level of SauJ1, where the reaction contained 2 mM manganese and was mixed with 1 mM of either EDTA, magnesium, manganese, or zinc (). RNase activity was unaffected when we added 1 mM EDTA, magnesium or manganese, however 1 mM zinc caused complete inhibition of activity (). This RNase inhibition was equally observed when we used a mixture of 1 mM manganese and either 5 µM or 1 mM zinc at non-saturating level of SauJ1 (data not shown). This result suggests that zinc can bind avidly either SauJ1 or the RNA substrate and inhibits the role of the manganese. From this experiment it is obvious that zinc cannot substitute for the activation of SauJ1 by manganese, however, it remains to be determined how manganese acts to activate SauJ1.
5′ end specificity of SauJ1
To examine the 5′ end specificity of SauJ1, we labeled, either a 5′ hydroxyl or a 5′ phosphate 26-mer RNA at the 3′ end with 32pCp using T4 RNA ligase to generate a 27-mer RNA with 3′ phosphate ends. The kinetics of RNase activity of the 5′ hydroxyl substrate under conditions of a 2-fold excess substrate over enzyme are shown in . Approximately 30% of the input substrate was degraded after 2 min; no intermediates were detected at this time-point, implying that the enzyme is highly processive. Product release increased with time and reached a plateau at 15 min, at which time about 95% of the input substrate had been digested. The time course of the 27-mer RNA mono-phosphorylated at its 5′ end gave an almost identical profile. By plotting the extent of the main products released at 2 and 5 min respectively for both RNA substrates, we estimated that 5′ hydroxyl 27-mer RNA was degraded ∼1.4-fold faster than its 5′ monophosphate counterpart, suggesting that SauJ1 had no significant preferences for a 5′ hydroxyl or a 5′ monophosphate end.
Figure 4. SauJ1 degrades efficiently a 27-mer RNA with either a 5′ monophosphate or a 5′ hydroxyl ends. Reaction mixtures (60 µl) containing 50 mM Tris-HCl, 1 mM MnCl2, 1.3 U RNasin, 20 nM of SauJ1 and 40 nM of either 5′ phopshate 26-mer RNA or 5′ hydroxyl 26-mer RNA (which had been labeled with radioactive 32pCp as described in material and methods; depicted at the bottom of ) were incubated at 37 °C for the indicated time. The reaction products were analyzed on a denaturing PAGE and the products were visualized by autoradiography. Radioactive *pCp is included as a size marker. (B) Characterization of SauJ1 products. A reaction mixture containing 50 mM Tris-HCl (pH8), 40 nM 5′ hydroxyl 26-mer RNA labeled with *pCp (sequences are shown at the bottom with the RNase T1 cleavage site) and 5000 U of RNase T1 were incubated for 60 min at 37 °C and supplemented for 30 min with either T4 PNK (phosphatase minus; T4PNK-), T4 PNK or nothing (lane T1) as described in Material and Methods. The reaction products were analyzed on a 20% urea-PAGE and the products were visualized by autoradiography. *pCp, and SauJ1 products obtained as described in panel A at 30 min, are also shown.
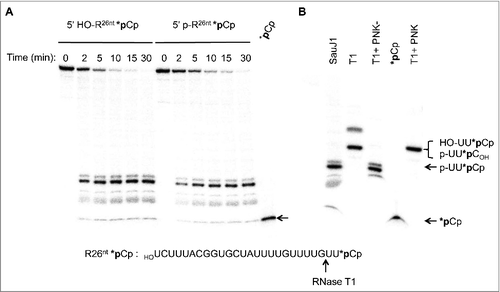
Unexpectedly, we found that the major product generated by SauJ1 with both substrates migrated slower than pure 32pCp (; *pCp). To accurately identify the product generated by SauJ1, we analyzed this reaction product by 20% urea-PAGE in parallel with a digest by RNase T1 of the 3′-end-labeled 5′ hydroxyl 27-mer RNA. RNase T1 cleaves 3′ of guanosines to yield a 32P trinucleotide (5′ HOUU32pCp; B). The product generated by RNase T1 clearly migrated slower than the product generated by SauJ. Since the migration of a 3-mer oligoribonucleotide on a 20% urea-PAGE is greatly influenced by the net charge of the 3-mer (for an example seeCitation37), we phosphorylated the 5′ hydroxyl ends of the RNase T1 products using T4 polynucleotide kinase (3′ phosphatase minus mutant enzyme) to obtain a 32P-labeled trinucleotide (5′ pUU32pCp) (Lane T1+PNK-). This product co-migrated with the major SauJ1 product. The mutant T4 polynucleotide kinase was needed, since the wild-type enzyme has a strong 3′ phosphatase activity and therefore removed 3′ phosphate from the trinucleotide. The RNase T1 product treated with wild-type T4 polynucleotide kinase thus gives a 32P-labeled trinucleotide pUU32pC (with no 3′ phosphate) that co-migrated with the products of RNase T1 (5′ HOUU32pCp; lanes T1 and T1+PNK). All together, these experiments suggest that degradation of a single-strand RNA substrate by SauJ1 is strictly metal dependent and occurs with a 5′ to 3′ polarity to end with a 5′ phosphate trinucleotide product. Moreover, the nature of the 5′ ends (5′ phosphate vs. 5′ hydroxyl ends) does not influence the efficiency of SauJ1 5′ to 3′ exoribonuclease activity or the length of the final product.
Effect of triphosphorylated substrates
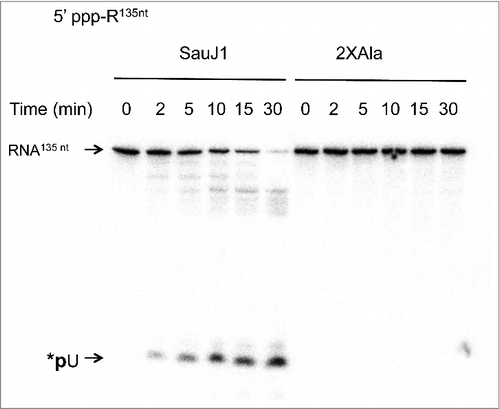
Because RNase J from various organisms was strongly inhibited by 5′ triphosphate ends Citation4, we tested the capacity of SauJ1 to degrade a uniformly labeled 135-mer triphosphorylated RNA (see Fig. S3 for the sequence and the putative structure of this RNA). The time course of SauJ1 with the 5′ triphosphate internally labeled RNA with an approximately 8-fold excess substrate over enzyme showed that SauJ1 degraded more than 90% of the substrate bearing a triphosphate ends in a 30-min reaction, and generated a major product that co-migrated with *pU (; left panel). The active site mutation (2XAla) abolished the RNase activity (; right panel) confirming that the RNase activity observed was inherent to the recombinant protein SauJ1 at the concentration used in the assay.
Figure 5. SauJ1 digests a 5′ triphosphorylated RNA. Reaction mixtures (60 µl) containing 50 mM Tris-HCl, 1 mM MnCl2, 1.3 U RNasin, 250 nM of 135-mer internally labeled with α-32P-UTP and 30 nM SauJ1 or 2xAla mutant were incubated at 37 °C for the indicated time. The reaction products were analyzed on a 12.5% urea PAGE and the products were visualized by autoradiography.
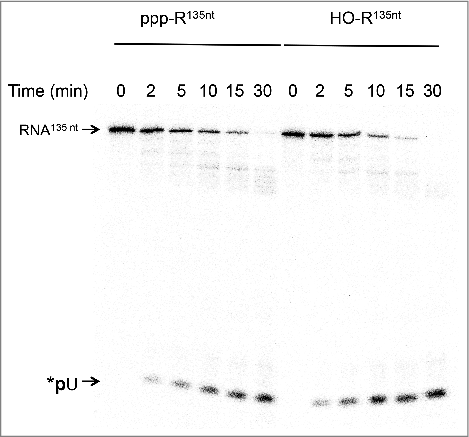
We next compared the efficiency of SauJ1 to degrade a 5′ triphosphorylated RNA vs. a 5′ hydroxyl RNA. The uniformly labeled 135-mer triphosphorylated RNA was first treated by alkaline phosphatase as described in material and methods. We found that the pattern of RNA degradation of a 5′ hydroxyl RNA was very similar to the pattern of digestion of the 5′ triphosphate RNA (). The percentage of uridine monophosphate release from the 5′ hydroxyl substrate at 2, 5, and 10 min was 1.7, 1.7 and 1.3 times higher than with the 5′ triphosphate RNA counterpart, respectively. This result implies only a modest inhibition by the 5′ triphosphate end. We also noticed that several minor products (around 60–90 nucleotides as judged with a labeled DNA marker; not shown) were also detected from 10 min to 30 min, irrespective of whether the substrate had 5′ triphosphate or 5′ hydroxyl ends ( and ; see below for an explanation).
Figure 6. SauJ1 degrades equally well a RNA bearing a 5′ triphosphate or a 5′ hydroxyl ends. Reaction mixtures (20 µl) containing 50 mM Tris-HCl, 1 mM MnCl2, 1.3 U RNasin, 38 nM SauJ1 and 250 nM of a 135-mer internally labeled RNA (with α-32P-UTP) bearing a either a 5′ triphosphate or a 5′ monophosphate ends were incubated at 37 °C for the indicated time. The reaction products were analyzed on a 12.5% urea PAGE and the products were visualized by autoradiography.
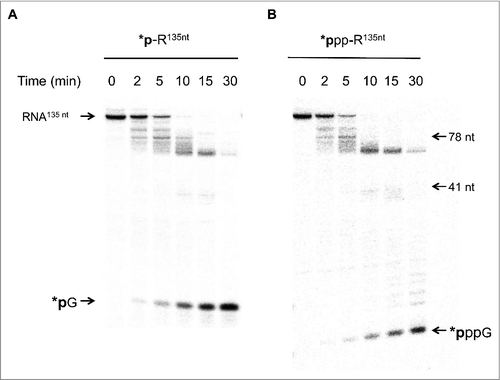
The evolution of the product distribution during the 30-min reaction on either a 5′ phosphate or a 5′ triphosphate end uniformly labeled RNA implies that either: (i) SauJ1 degrades both substrates in a pure 5′ to 3′ exoribonucleolytic mode of action but can be blocked by a RNA structure; or (ii) SauJ1 possesses in addition to the 5′ to 3′ exonucleolytic activity an endonuclease activity which could be modulated by the nature of the 5′ ends. To discriminate between these possibilities, we prepared a gamma-labeled triphosphate and a 5′ α-labeled monophosphate RNA substrate. As the input 135-mer 5′ labeled RNA substrates were degraded to end products of lower mass, the results allowed us to gauge the distribution of products and to estimate the 5′ to 3′ exoribonuclease vs. endoribonuclease activities. The kinetic profile of the SauJ1 reaction with 5′ gamma-labeled triphosphate RNA under conditions of ∼2.5-fold substrate excess revealed that at early time points (2 and 5 min) before all input had been degraded, and before the appearance of GTP (or GMP), several 32P labeled products could be observed (estimated to be between 60 and 134 nucleotides). These products must be generated by an initial endoribonucleolytic activity since 5′ exonucleolytic activity should exclusively generate GMP or GTP. Note that after 30-min reaction, there was no residual triphosphorylated RNA substrate and that 90% of the product corresponded to GTP (less than 10% corresponded to products about 60 to 70 nucleotides; ; right panel). Digestion of a 5′ monophosphate labeled 135-mer RNA by SauJ1 gave a similar product distribution (; left panel). Quantification of the kinetic profile of both RNA substrates is plotted in Fig. S2. We observed that release of GMP at 5 and 10 min is 6 and 2-fold faster than the release of GTP, respectively, implying that SauJ1 was not very sensitive to the phosphorylation state of the 5′ ends. It is important to note that this RNA has a putative structure at the 5′ end, making it partially resistant to a 5′ to 3′ exoribonuclease XRN1 (data not shown). These results implied that in vitro endoribonuclease activity can be strong in presence of 1 mM manganese if the RNA is longer than 26-mer. Note also that weak endonuclease activity was always detected, even with the 26-mer RNA substrate ( and ).
Figure 7. SauJ1 has both an endonucleotic and a 5′ to 3′ exonucleotic activities. Reaction mixtures (60 µl) containing 50 mM Tris-HCl, 1 mM MnCl2, 1.3 U RNasin, and 75 nM of WT SauJ1, and 300 nM 135-mer 5′ labeled monophosphate RNA (panel A) or 200 nM 135-mer gamma-labeled triphosphorylated RNA (panel B) or were incubated at 37 °C for the indicated time. The reaction products were analyzed on a 12.5% urea PAGE and the products were visualized by autoradiography. The indicated size markers are: radioactive guanosine monophosphate (*pG was produced by reacting XRN1 with 135-mer 5′ labeled monophosphate RNA), gamma-labeled guanosine triphosphate (*pppG), 5′-end labeled 41-mer RNA and 5′ ends labeled 78-mer DNA were included as size markers. Panels A and B are from the same gel, but panel B has been exposed much longer, since gamma-labeled RNA had a low specific activity.
Discussion
The finding that SauJ1 activity was strictly dependent on manganese distinguishes SauJ1 from all known RNase J orthologs characterized previously. It is noteworthy that RNase J shares structural and mechanistic similarities with the zinc metallo-hydrolases of the β-lactamase superfamily that are found in all three domains of life.Citation27,Citation38 As members of this superfamily are known to use zinc ions in their catalytic site (to activate the nucleophile) it has been assumed that zinc is the metal necessary for catalysis. Indeed, zinc ions have been found in every structure solved to date, and our current understanding of the reaction mechanism of RNase J relied on 2 zinc ions in the active site that are crucial to activate a water molecule. However, with the exception of 2 Thermococcal enzymes,Citation30 RNase J orthologs need an extra metal ion, either magnesium or manganese, to be fully active. A role for the additional divalent metals has been recently proposed for D. radiodurans RNase J. Manganese ion was shown crucial for homodimerization of D. radiodurans RNase J25. An alanine mutation at the putative metal binding site at the dimer interface resulted in monomeric enzyme. Importantly, size-exclusion chromatography of wild-type RNase J in the presence of EDTA revealed that RNase J is a mixture of monomer and homodimer that is converted entirely to homodimer in the presence of manganese. However, the role of the manganese in homodimerization seems not to be universal since Pei et al. (2015)Citation26 were unable to detect manganese ions at the dimer interface of S. coelicolor RNase J even when the co-crystals of RNase J/11-mer RNA were soaked in a manganese buffer.
We surmised via velocity sedimentation and non-denaturing gel that SauJ1 has a homodimeric native structure (Fig. S1). This is consistent with the findings of Newman et al., (2011) for the BsuJ123, which they judged via gel filtration to be an homodimer in solution. However, the quaternary structure of BsuJ1 has also been reported to be a homotetramer in vitro by gel filtration.Citation16 The reasons for these conflicting results are still unclear.
RNase activity of BsuJ1, the closest ortholog of SauJ1, was shown to be strictly dependent on 5–10 mM magnesium although high enzyme concentrations and a large excess of enzyme over substrate was needed to detect this activity (reviewed inCitation39). Our results confirm that adding an extra metal ion is crucial for RNase catalysis in vitro, although surprisingly we were not able to substitute manganese by magnesium. Many enzymes involved in DNA and RNA metabolism are magnesium dependent (reviewed inCitation40). These enzymes generally use 2 magnesium ions to coordinate the substrate and the nucleophile (a water molecule or 2′ hydroxyl) to catalyze the phosphodiester bond cleavage. There are many examples that manganese can substitute for the magnesium in the nuclease reaction buffer, and even in some cases it may improve enzyme catalysis.Citation40,Citation41 The reason for this improvement may reside in the fact that manganese function better to reduce the activation barrier.Citation41 Therefore, a trivial explanation is that SauJ1 would be active with magnesium if we were to use higher enzyme concentrations. In attempting to test this hypothesis, we noticed that 250 nM of 2XAla mutant (H74A-H76A) had non-specific activity. This prevented us from testing the metal dependence of the wild-type enzyme. It is clear the magnesium is much more abundant than manganese in the cellular environment, however, even a 100 fold excess of magnesium compared with the optimal manganese concentration did not allow any detectable activity in our assay (), suggesting that manganese is indeed the preferred natural ligand.
It is important to note that only very few enzymes are strictly manganese dependent (reviewed inCitation42). We can also speculate that the level of cellular manganese could regulate the virulence of S. aureus in the case that manganese is not the main physiologic ion. In fact, manganese transport has been shown to regulate the bacterial virulence of several bacteria.Citation42 Alternatively, different enzymes of the β-CASP family of β-metallo-lactamases in bacteria may vary in their responsiveness to metal in vitro and/or in vivo. Importantly the in vivo physiologic ion of RNase J remains unknown.
We found that 5 µM zinc by itself is unable to support catalysis, and even inhibits SauJ1 activity in the presence of 1 mM manganese. It is possible that zinc indeed binds avidly the RNase J catalytic site (as published in several structural studies), but this ion is not able to correctly orient the molecule of water to attack the scissile phosphate. We suggest that manganese can compete for zinc in the catalytic site and once one (or 2) manganese ions are bound, then cleavage of the phosphodiester bond can proceed. However, our observations cannot rule out that manganese (or magnesium) might interact transiently or weakly with the RNA substrate and/or the enzyme to facilitate the alignment of the activated water with the scissile phosphate as proposed by Pei et al., (2015).Citation26 However, this hypothesis is difficult to reconcile with our mixing experiment, where zinc inhibited enzymatic activity, even in the presence of excess manganese ().
The results presented here show that SauJ1 has strong 5′ to 3′ exonucleolytic activity on a short 26-mer single strand RNA substrate, and rapidly removes the first nucleotide. Unexpectedly, the enzyme was unable to degrade this oligonucleotide all the way to the 3′-end but stops when a tri-nucleotide remains. This is in contrast to studies of RNase J's from other organisms, where the final nucleotide is released.Citation17,Citation29,Citation30 The reason for this difference is unclear but could be due to the identity of the divalent metal ion used in the in vitro assay (manganese vs. magnesium). Nevertheless, we should emphasize that failure to complete digestion should not be problematic for the cell, since S. aureus encodes an ortholog of the B. subtilis nano RNase A (YtqI gene), which has been shown to be a functional analog of the E. coli oligoribonuclease (orn gene).Citation43
Early work indicated that the exoribonuclease activity of B. subtilis, Mycobacterium. smegmatis, T. thermophilus, and S. coelicolor RNase J was inhibited by a triphosphorylated RNA.Citation17,Citation22,Citation26,Citation28,Citation29,Citation31,Citation44 However, in those studies endoribonuclease activity were detected with a triphosphorylated RNA. It has been proposed that the relative endo- vs. exo-ribonuclease activities is determined by the 5′ phosphorylation state.Citation22,Citation26,Citation28 Endoribonuclease activity is favored when the RNA has a 5′ triphosphate whereas exoribonuclease activity prevails with a 5′ monophosphate ends.Citation22,Citation26,Citation28 We should emphasize that the observation of endonuclease activity is complicated by the fact that both endo- and exo-ribonuclease activities share the same catalytic site and an endoribonucleotic cleavage generates a very potent substrate for the 5′ to 3′ exoribonuclease activities. Unexpectedly we found that SauJ1 has a strong dual activity on a 135-mer RNA independently of the 5′ phosphorylation status ().
It is noteworthy that biochemical data showed the 5′ to 3′ exoribonuclease activity of RNase J on a gamma labeled triphosphorylated RNA has been demonstrated until now to be either weakCitation26,Citation31 or absent.Citation22,Citation29 Those results were rationalized by modeling a triphosphorylated RNA into the catalytic site and observing that the scissile phosphate is not correctly oriented to be attacked by a water molecule.Citation22,Citation26 However, our results established that SauJ1 is able to orient a 5′ triphosphorylated RNA in the catalytic site, insofar as we observed an efficient release of gamma GTP at early time points ( and Fig. S2). The difference in the specificity of BsuJ1 and SauJ1 could be due to either the metal difference preferences or an inherent property of SauJ1. Nevertheless, it should be noted that with the exception of D. radiodurans RNase J, no biochemical studies examined the RNase J activity/specificity in the presence of manganese.
We found that SauJ1 endonuclease activity is not very sensitive to the 5′ phosphorylation state of the RNA ( and ) as demonstrated with B. subtilis RNase J1 with thr leader RNA transcript.Citation20 By using 3 different uniformly labeled RNA, we found that 5′ phosphorylation state do not significantly influence SauJ1 activity. The structural data of B. subtilis RNase J1 indicates that an endoribonuclease mode will either require that the homodimer dissociates and reassembles internally on the RNA or that RNA passes through the catalytic site and is then cleaved endonucleotically at a downstream site (sliding endonuclease model).Citation26 It has been shown that M. smegmatis RNase J can endonucleotically cleave an RNA substrate very close to the 5′ end.Citation28 However it should be noted that these authors also detected trace of GTP during the time course suggesting that this nucleotide was produced by a second endonucleolytic cleavage of the short 5′ end the short 5′ fragment formed.Citation28 Our results favor the hypothesis that GTP release arises from the exoribonuclease activity of SauJ1 since the release of GTP appears mostly at early times (detectable after 5-min reaction, see and Fig. S2). Therefore, ours results are more compatible with RNase J dissociating and re-associating internally on the RNA since the detected endonuclease cuts are found far from the 5′ end.
It is possible that the length of RNA is important to trigger endoribonuclease activity since very weak activity is detected with a short oligoribonucleotide (26-mer) whereas stronger endoribonuclease activity is observed with 135-mer RNA (compare and ). However, several other factors might influence which of the 2 activities acts on a given RNA, and further studies are needed to examine whether for example the 5′ end structure and/or RNA sequences can influence the strength of the endo-vs. exoribonuclease activities.
We would emphasize here that orthologous RNase J from different species cannot be presumed to have the same inherent biochemical properties. It is therefore possible that in S. aureus RNase J1 is an important enzyme to initiate RNA degradation. Our in vitro data for SauJ1 presented here and our unpublished RNaseq data on an S. aureus strain deleted for RNase J1 suggests a model where both RNase J1 and RNase Y are responsible for initiating RNA decay in this organism. We have previously shown that a tagged version of RNase J1 would pull down CshA and ribosomal proteins, and we equally established that a tagged version of CshA interacted with both RNase Y and RNase J1.Citation34 This is intriguing, as RNase Y has been shown to be anchored to the membrane whereas RNase J1 is not, and we therefore infer that 2 different RNA degradation initiation machineries co-exist in S. aureus, one with CshA and RNase Y as central factors, and one with CshA and RNase J1. Due to the endo-nucleolytic activity of RNase J1 shown here, this hypothesis is consistent with an endonucleotic cut as an initiating event for RNA degradation in S. aureus, similar to the role of RNase E in E.coli.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
SH PR and PL developed the project. SH performed biochemical analysis of SauJ1. VG performed the complementation analysis and electron microscopy. SH PR DG and PL analyzed the data. SH PR and PL wrote the paper.
Supplemental_Tables_and_figures.docx
Download MS Word (1.6 MB)Acknowledgments
We are grateful to Joshua Armitano and Vanessa Khemici for continuous discussion.
Funding
This work was supported by grants from the Swiss National Science Foundation (PL), the Boninchi Foundation (PL) and by the Canton of Geneva.
Related Research Data
References
- Eidem TM, Roux CM, Dunman PM. RNA decay: a novel therapeutic target in bacteria. Wiley Interdiscip Rev RNA 2012; 3:443-54; PMID:22374855; https://doi.org/10.1002/wrna.1110
- Anderson KL, Dunman PM. Messenger RNA Turnover Processes in Escherichia coli, Bacillus subtilis, and Emerging Studies in Staphylococcus aureus. Int J Microbiol 2009; 2009:525491; PMID:19936110; https://doi.org/10.1155/2009/525491
- Mohanty BK, Kushner SR. Regulation of mRNA Decay in Bacteria. Annu Rev Microbiol 2016; 70:25-44; PMID:27297126; https://doi.org/10.1146/annurev-micro-091014-104515
- Laalami S, Zig L, Putzer H. Initiation of mRNA decay in bacteria. Cell Mol Life Sci 2014; 71:1799-828; PMID:24064983; https://doi.org/10.1007/s00018-013-1472-4
- Hui MP, Foley PL, Belasco JG. Messenger RNA degradation in bacterial cells. Annu Rev Genet 2014; 48:537-559; PMID:25292357; https://doi.org/10.1146/annurev-genet-120213-092340
- Durand S, Tomasini A, Braun F, Condon C, Romby P. sRNA and mRNA turnover in Gram-positive bacteria. FEMS Microbiol Rev 2015; 39:316-330; PMID:25934118; https://doi.org/10.1093/femsre/fuv007
- Condon C, Putzer H. The phylogenetic distribution of bacterial ribonucleases. Nucleic Acids Res 2002; 30:5339-46; PMID:12490701; https://doi.org/10.1093/nar/gkf691
- Lehnik-Habrink M, Lewis R.J, Mader U, Stulke J. RNA degradation in Bacillus subtilis: an interplay of essential endo- and exoribonucleases. Mol Microbiol 2012; 84:1005-17; PMID:22568516; https://doi.org/10.1111/j.1365-2958.2012.08072.x
- Figaro S, Durand S, Gilet L, Cayet N, Sachse M, Condon C. Bacillus subtilis mutants with knockouts of the genes encoding ribonucleases RNase Y and RNase J1 are viable, with major defects in cell morphology, sporulation, and competence. J Bacteriol 2013; 195:2340-8; PMID:23504012; https://doi.org/10.1128/JB.00164-13
- Shahbabian K, Jamalli A, Zig L, Putzer H. RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J 2009; 28:3523-33; PMID:19779461; https://doi.org/10.1038/emboj.2009.283
- Laalami S, Bessieres P, Rocca A, Zig L, Nicolas P, Putzer H. Bacillus subtilis RNase Y activity in vivo analysed by tiling microarrays. PLoS One 2013; 8:e54062; PMID:23326572; https://doi.org/10.1371/journal.pone.0054062
- Durand S, Gilet L, Bessieres P, Nicolas P, Condon C. Three essential ribonucleases-RNase Y, J1, and III-control the abundance of a majority of Bacillus subtilis mRNAs. PLoS Genet 2012; 8:e1002520; PMID:22412379; https://doi.org/10.1371/journal.pgen.1002520
- Kang SO, Caparon MG, Cho KH. Virulence gene regulation by CvfA, a putative RNase: the CvfA-enolase complex in Streptococcus pyogenes links nutritional stress, growth-phase control, and virulence gene expression. Infect Immun 2010; 78:2754-67; PMID:20385762; https://doi.org/10.1128/IAI.01370-09
- Khemici V, Prados J, Linder P, Redder P. Decay-Initiating Endoribonucleolytic Cleavage by RNase Y Is Kept under Tight Control via Sequence Preference and Sub-cellular Localisation. PLoS Genet 2015; 11:e1005577; PMID:26473962; https://doi.org/10.1371/journal.pgen.1005577
- Marincola G, Schafer T, Behler J, Bernhardt J, Ohlsen K, Goerke C, Wolz C. RNase Y of Staphylococcus aureus and its role in the activation of virulence genes. Mol Microbiol 2012; 85:817-32; PMID:22780584; https://doi.org/10.1111/j.1365-2958.2012.08144.x
- Mathy N, Hebert A, Mervelet P, Benard L, Dorleans A, Li de la Sierra-Gallay I, Noirot P, Putzer H, Condon C. Bacillus subtilis ribonucleases J1 and J2 form a complex with altered enzyme behaviour. Mol Microbiol 2010; 75:489-98; PMID:20025672; https://doi.org/10.1111/j.1365-2958.2009.07004.x
- Mathy N, Benard L, Pellegrini O, Daou R, Wen T, Condon C. 5′-to-3′ exoribonuclease activity in bacteria: role of RNase J1 in rRNA maturation and 5′ stability of mRNA. Cell 2007; 129:681-92; PMID:17512403; https://doi.org/10.1016/j.cell.2007.02.051
- Linder P, Lemeille S, Redder P. Transcriptome-wide analyses of 5′-ends in RNase J mutants of a gram-positive pathogen reveal a role in RNA maturation, regulation and degradation. PLoS Genet 2014; 10:e1004207; PMID:24586213; https://doi.org/10.1371/journal.pgen.1004207
- Kobayashi K, Ehrlich SD, Albertini A, Amati G, Andersen KK, Arnaud M, Asai K, Ashikaga S, Aymerich S, Bessieres P, et al. Essential Bacillus subtilis genes. Proc Natl Acad Sci U S A 2003; 100:4678-83; PMID:12682299; https://doi.org/10.1073/pnas.0730515100
- Even S, Pellegrini O, Zig L, Labas V, Vinh J, Brechemmier-Baey D, Putzer H. Ribonucleases J1 and J2: two novel endoribonucleases in B.subtilis with functional homology to E.coli RNase E. Nucleic Acids Res 2005; 33:2141-52; PMID:15831787; https://doi.org/10.1093/nar/gki505
- Richards J, Belasco JG. Ribonuclease J: how to lead a double life. Structure 2011; 19:1201-3; PMID:21893280; https://doi.org/10.1016/j.str.2011.08.004
- Li de la Sierra-Gallay I, Zig L, Jamalli A, Putzer H. Structural insights into the dual activity of RNase J. Nat Struct Mol Biol 2008; 15:206-12; PMID:18204464; https://doi.org/10.1038/nsmb.1376
- Newman JA, Hewitt L, Rodrigues C, Solovyova A, Harwood CR, Lewis RJ. Unusual, dual endo- and exonuclease activity in the degradosome explained by crystal structure analysis of RNase J1. Structure 2011; 19:1241-51; PMID:21893285; https://doi.org/10.1016/j.str.2011.06.017
- Dorleans A, Li de la Sierra-Gallay I, Piton J, Zig L, Gilet L, Putzer H, Condon C. Molecular basis for the recognition and cleavage of RNA by the bifunctional 5′-3′ exo/endoribonuclease RNase J. Structure 2011; 19:1252-61; PMID:21893286; https://doi.org/10.1016/j.str.2011.06.018
- Zhao Y, Lu M, Zhang H, Hu J, Zhou C, Xu Q, Ul Hussain Shah AM, Xu H, Wang L, Hua Y. Structural insights into catalysis and dimerization enhanced exonuclease activity of RNase J. Nucleic Acids Res 2015; 43:5550-9; PMID:25940620; https://doi.org/10.1093/nar/gkv444
- Pei XY, Bralley P, Jones GH, Luisi BF. Linkage of catalysis and 5′ end recognition in ribonuclease RNase J. Nucleic Acids Res 2015; 43:8066-76; PMID:26253740; https://doi.org/10.1093/nar/gkv732
- Dominski Z, Carpousis AJ, Clouet-d'Orval B. Emergence of the beta-CASP ribonucleases: highly conserved and ubiquitous metallo-enzymes involved in messenger RNA maturation and degradation. Biochim Biophys Acta 2013; 1829:532-51; PMID:23403287; https://doi.org/10.1016/j.bbagrm.2013.01.010
- Taverniti V, Forti F, Ghisotti D, Putzer H. Mycobacterium smegmatis RNase J is a 5′-3′ exo-/endoribonuclease and both RNase J and RNase E are involved in ribosomal RNA maturation. Mol Microbiol 2011; 82:1260-76; PMID:22014150; https://doi.org/10.1111/j.1365-2958.2011.07888.x
- Bralley P, Aseem M, Jones GH. SCO5745, a bifunctional RNase J ortholog, affects antibiotic production in Streptomyces coelicolor. J Bacteriol 2014; 196:1197-205; PMID:24415725; https://doi.org/10.1128/JB.01422-13
- Clouet-d'Orval B, Rinaldi D, Quentin Y, Carpousis AJ. Euryarchaeal beta-CASP proteins with homology to bacterial RNase J Have 5′- to 3′-exoribonuclease activity. J Biol Chem 2010; 285:17574-83; PMID:20375016; https://doi.org/10.1074/jbc.M109.095117
- Richards J, Liu Q, Pellegrini O, Celesnik H, Yao S, Bechhofer DH, Condon C, Belasco JG. An RNA pyrophosphohydrolase triggers 5′-exonucleolytic degradation of mRNA in Bacillus subtilis. Mol Cell 2011; 43:940-9; PMID:21925382; https://doi.org/10.1016/j.molcel.2011.07.023
- Levy S, Portnoy V, Admon J, Schuster G. Distinct activities of several RNase J proteins in methanogenic archaea. RNA Biol 2011; 8:1073-83; PMID:21955587; https://doi.org/10.4161/rna.8.6.16604
- Redko Y, Galtier E, Arnion H, Darfeuille F, Sismeiro O, Coppee JY, Medigue C, Weiman M, Cruveiller S, De Reuse H. RNase J depletion leads to massive changes in mRNA abundance in Helicobacter pylori. RNA Biol 2016; 13:243-53; PMID:26726773; https://doi.org/10.1080/15476286.2015.1132141
- Giraud C, Hausmann S, Lemeille S, Prados J, Redder P, Linder P. The C-terminal region of the RNA helicase CshA is required for the interaction with the degradosome and turnover of bulk RNA in the opportunistic pathogen Staphylococcus aureus. RNA Biol 2015; 12:658-74; PMID:25997461; https://doi.org/10.1080/15476286.2015.1035505
- Renzoni A, Andrey DO, Jousselin A, Barras C, Monod A, Vaudaux P, Lew D, Kelley WL. Whole genome sequencing and complete genetic analysis reveals novel pathways to glycopeptide resistance in Staphylococcus aureus. PLoS One 2011; 6:e21577; PMID:21738716; https://doi.org/10.1371/journal.pone.0021577
- Mossessova E, Lima CD. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol Cell 2000; 5:865-76; PMID:10882122; https://doi.org/10.1016/S1097-2765(00)80326-3
- Das U, Shuman S. Mechanism of RNA 2′,3′-cyclic phosphate end healing by T4 polynucleotide kinase-phosphatase. Nucleic Acids Res 2013; 41:355-65; PMID:23118482; https://doi.org/10.1093/nar/gks977
- Redko Y, Li de la Sierra-Gallay I, Condon C. When all's zed and done: the structure and function of RNase Z in prokaryotes. Nat Rev Microbiol 2007; 5:278-86; PMID:17363966; https://doi.org/10.1038/nrmicro1622
- Condon C. What is the role of RNase J in mRNA turnover? RNA Biol 2010; 7:316-21; PMID:20458164; https://doi.org/10.4161/rna.7.3.11913
- Yang W, Lee JY, Nowotny M. Making and breaking nucleic acids: two-Mg2+-ion catalysis and substrate specificity. Mol Cell 2006; 22:5-13; PMID:16600865; https://doi.org/10.1016/j.molcel.2006.03.013
- Kehres DG, Maguire ME. Emerging themes in manganese transport, biochemistry and pathogenesis in bacteria. FEMS Microbiol Rev 2003; 27:263-90; PMID:12829271; https://doi.org/10.1016/S0168-6445(03)00052-4
- Papp-Wallace KM, Maguire ME. Manganese transport and the role of manganese in virulence. Annu Rev Microbiol 2006; 60:187-209; PMID:16704341; https://doi.org/10.1146/annurev.micro.60.080805.142149
- Mechold U, Fang G, Ngo S, Ogryzko V, Danchin A. YtqI from Bacillus subtilis has both oligoribonuclease and pAp-phosphatase activity. Nucleic Acids Res 2007; 35:4552-61; PMID:17586819; https://doi.org/10.1093/nar/gkm462
- Daou-Chabo R, Condon C. RNase J1 endonuclease activity as a probe of RNA secondary structure. RNA 2009; 15:1417-25; PMID:19458035; https://doi.org/10.1261/rna.1574309
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 2003; 31:3406-15; PMID:12824337; https://doi.org/10.1093/nar/gkg595
- Oun S, Redder P, Didier JP, Francois P, Corvaglia AR, Buttazzoni E, Giraud C, Girard M, Schrenzel J, Linder P. The CshA DEAD-box RNA helicase is important for quorum sensing control in Staphylococcus aureus. RNA Biol 2013; 10:157-65; PMID:23229022; https://doi.org/10.4161/rna.22899