
ABSTRACT
Trans-encoded sRNA154 is exclusively expressed under nitrogen (N)-deficiency in Methanosarcina mazei strain Gö1. The sRNA154 deletion strain showed a significant decrease in growth under N-limitation, pointing toward a regulatory role of sRNA154 in N-metabolism. Aiming to elucidate its regulatory function we characterized sRNA154 by means of biochemical and genetic approaches. 24 homologs of sRNA154 were identified in recently reported draft genomes of Methanosarcina strains, demonstrating high conservation in sequence and predicted secondary structure with two highly conserved single stranded loops. Transcriptome studies of sRNA154 deletion mutants by an RNA-seq approach uncovered nifH- and nrpA-mRNA, encoding the α–subunit of nitrogenase and the transcriptional activator of the nitrogen fixation (nif)-operon, as potential targets besides other components of the N-metabolism. Furthermore, results obtained from stability, complementation and western blot analysis, as well as in silico target predictions combined with electrophoretic mobility shift-assays, argue for a stabilizing effect of sRNA154 on the polycistronic nif-mRNA and nrpA-mRNA by binding with both loops. Further identified N-related targets were studied, which demonstrates that translation initiation of glnA2-mRNA, encoding glutamine synthetase2, appears to be affected by sRNA154 masking the ribosome binding site, whereas glnA1-mRNA appears to be stabilized by sRNA154. Overall, we propose that sRNA154 has a crucial regulatory role in N-metabolism in M. mazei by stabilizing the polycistronic mRNA encoding nitrogenase and glnA1-mRNA, as well as allowing a feed forward regulation of nif-gene expression by stabilizing nrpA-mRNA. Consequently, sRNA154 represents the first archaeal sRNA, for which a positive posttranscriptional regulation is demonstrated as well as inhibition of translation initiation.
Introduction
Microorganisms achieve survival under periods of nutrient starvation or stress, due to drastic environmental changes, by regulating the uptake and assimilation of different N-sources.
Particularly, regulation of N2-fixation in bacterial diazotrophes, in response to environmental fluctuations is tightly controlled on both, the transcriptional and post-translational level (reviewed inCitation1-4). In contrast to bacteria, little is known on the regulation of N-metabolism and N2-fixation in archaea. Moreover, as the archaeal transcription and translation machineries have more similar features to their eukaryotic than their bacterial counterparts, novel non-bacterial like regulatory mechanism appear to be likely.Citation4-7
Methanosarcina mazei strain Gö1, a representative mesophilic, methylotrophic methanogenic archaeon of the order Methanosarcinales, is able to fix and use molecular nitrogen (N2) as sole N-source.Citation8 In M. mazei, as well as other methanogenic archaea, regulation of N-metabolism, including N2-fixation, has been shown to be governed by the global nitrogen regulator NrpR, representing a heterooligomeric transcriptional repressor.Citation9-12 NrpR binding to its corresponding operator under N-sufficiency inhibits RNA polymerase recruitment to the promoter, causing transcriptional repression of the respective NrpR-regulated promoters. Under N-limitation however, operator-binding of NrpR is antagonized by increasing 2-oxoglutarate levels, providing the intracellular signal for N-limitation, resulting in RNA polymerase recruitment to the promoter and transcription initiation.Citation4,11 Recently we demonstrated that in M. mazei an additional newly identified regulatory protein, NrpA - which itself is under direct NrpR control - represents and acts as nif-specific transcriptional activator required for maximal nif-induction under N-limiting conditions.Citation13
The last decades revealed a plethora of small RNAs (sRNAs) with major roles in cellular environments in all domains of life. Whereas eukaryotic miRNAs and siRNAs were predominantly involved in post-transcriptional regulation of gene expression by targeting the 3′ end of a cognate mRNAs,Citation14 the regulatory repertoire of bacterial sRNAs is diverse. In bacteria, translational repression of target mRNAs is achieved by often short and imperfect base-pairing within the 5′ UTR rather than targeting the mRNA's 3′end, ultimately resulting in ribosome binding site (RBS) sequestration by the sRNA and consequently translational repression. On the other hand, translational activation, modulation of protein activity, or RNA mimicry have also been described (for review seeCitation15-17). The vast majority of bacterial sRNAs demonstrated to be transcribed and highly regulated in response to various external stimuli and certain growth and / or stress conditions,Citation18-22 e.g. modulation of porin composition upon envelope stress (MicA and RybB).Citation23-26 Surprisingly, sRNAs directly participating in response to environmental N-fluctuations or particularly in regulation to N2-fixation have not been reported until very recently, though the expression of nif-genes and proteins is tightly controlled on the transcriptional and (post-) translational level (for review seeCitation3,4). However, several indirect involvements of sRNAs in N-metabolism have been previously observed. For instance, the heterocyst-specific sRNA NsiR1 in cyanobacteria,Citation27 CyaR of E. coli, which inhibits translation of nadE, an ammonium-dependent NAD synthethase,Citation28 or ArrF of Azotobacter vinelandii, which is involved in FeSII regulation, a protein which protects the key enzyme of N2-fixation (nitrogenase) under oxidative conditions.Citation29 In addition, Mitschke et al. performed RNA-seq analysis of the cyanobacterium Anabena sp. PCC7120 in response to N-availability identifying ∼600 transcriptional start sites potentially corresponding to cis- or trans-encoded sRNAs (including a homolog of the aforementioned NsiR1), strongly suggesting a prominent role of sRNAs in regulating nitrogen assimilation.Citation30 The first sRNA confirmed to be directly involved in N-regulation has been recently reported by Klähn et al.,Citation31 demonstrating that sRNA NsiR4, which is under the direct control of the global N-transcriptional regulator NtcA, plays a crucial role in regulating glutamine synthetase (GS) activity in cyanobacteria. By targeting the 5′UTR of the mRNA encoding the GS inactivating factor 7 (IF7), NsiR4 is affecting IF7 expression and consequently GS activity.
RNA-seq analysis of M. mazei grown under different N-availability, identified 18 differentially expressed small RNAs, further corroborated by northern blot analysis, implying a potential involvement in N-stress response.Citation32 One of those, sRNA154, has been demonstrated to be highly upregulated (approx. 20 fold) when cells were grown with N2 as sole N-source.Citation32 Examining the promoter revealed the presence of the NrpR operator,Citation11,12,32 demonstrating that sRNA154 is under direct control of the global N-repressor NrpR. A potential regulatory function in the global N-regulatory network has been obtained by characterizing the respective deletion mutant (ΔsRNA154), which displayed significantly reduced growth under N-limitation compared to the wild type (wt), while under N-sufficiency no obvious phenotype was detectable.Citation33 Aiming to unravel its potential regulatory function in the N metabolism, we here present functional analysis of sRNA154 and provide insight into potential targets using genetic, biochemical approaches and bioinformatics target predictions. Overall, our results demonstrate direct and indirect involvements of sRNA154 in the N-regulatory network of M. mazei under N-limitation.
Results
sRNA154 characterization
The sRNA154 gene is localized in the intergenic region (IGR) between MM3337 and MM3338, both encoding hypothetical proteins. The transcriptional start site (TSS) and termination site (TT) were determined by 5′-RACE and 3′-RACE analysis, revealing the native transcript length of 134 nucleotides (nt) and identifying the binding sites for the general transcription factors - the BRE and TATA box – as well as the operator of the global nitrogen repressor NrpR close to the TSS (). Newly performed RNA-seq approaches using the Illumina technique verified our previous finding,Citation32 a significant upregulation of sRNA154 under N-limitation (). Direct binding of NrpR to the sRNA154 promotor was confirmed by in vitro electrophoretic mobility shift assays, incubating radioactively 5′end labeled PCR-products of the respective promotor with various amounts of purified NrpR (Fig. S1), strongly indicating a direct regulation of sRNA154 expression by the global N-repressor.
Figure 1. Characterization of sRNA154 (A) Genomic context of sRNA154, promotor and terminator region of sRNA154. Potential TATA- and BRE box, the transcriptional start site (TSS) (+1), as well as the termination site (TT) are indicated. The 5´and 3´end of sRNA154 was determined by RACE analysis (Ambion, Thermo Scientific, Darmstadt, Germany). (B) RNA-seq analysis of total RNA of M. mazei wt - grown under nitrogen sufficient (+NH4+) and fixing (N2) conditions - using the Illumina technique revealed an absence of sRNA154 transcript under +NH4+ and high induction under N2-conditions. (C) Conservation of promotor regions of sRNA154 homologues from those various Methanosarcina isolates described in . The regions upstream of the TSS were aligned using the ClustalW multiple alignment tool.Citation71
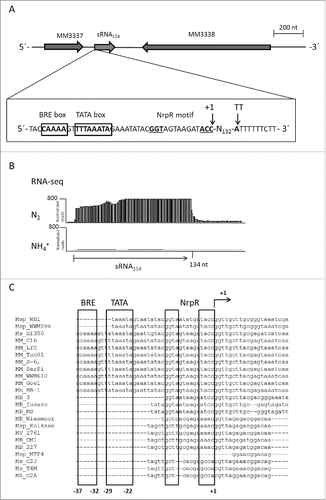
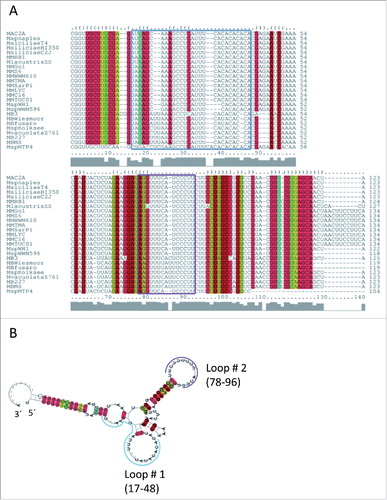
BLAST analysis identified 24 homologs of sRNA154 in genomes of Methanosarcina strains newly isolated by Metcalf and colleagues,Citation34 which showed high sequence conservation (). In order to assess structure conservation, we generated a multiple sequence alignment of all homologs using LocARNA.Citation35 Then, we applied RNAlifold,Citation35 which predicted a very stable consensus structure showing high structure conservation (), pointing toward an important function of sRNA154 in Methanosarcina strains. Besides, a variable 3′ end of 10 nucleotides only present in several homologs, two highly conserved single stranded RNA regions – loop 1 (nt 17 – 48, blue box) and loop 2 (nt 78 – 96, purple box) were identified, which occasionally showed deletions of one or two nucleotides in the different homologs (). Notably, loop 1 contained an unusual (CA)5 stretch at the 3′end, whereas loop 2 represents an C/U rich stretch potentially containing anti-ribosome binding sites (). Interestingly, despite the sRNA sequences also the promoter regions of the different homologs show high conservation including the TATA and BRE-box as well as the NrpR operator ().
Figure 2. Conservation of sRNA154 in Methanosarcina species Multiple secondary structure alignment of sRNA154 homologues in related Methanosarcina species performed with LocARNA,Citation72 MM, Methanosarcina mazei strains S-6, Go1, WWM610, TMA, SarPi, LYC, C16, Tuc01; MMHB, Methanosarcina horonobensis HB1; MA, Methanosarcina acetivorans strain C2A; Msp, Methanosarcina sp strains Naples 100, WWM596, WH1, MTP4, Kolksee; Msiciliae, Methanosarcina siciliae strains T4M, HI350, C2J; Mvacuolata, Methanosarcina vacuolata Z761; MB, Methanosarcina barkeri strains fusaro, Wiesmoor, MS, 227, 3; Mlacustris, Methanosarcina lacustris strains ZS, Z7289.Citation34 (B) Consensus secondary structure prediction by RNAlifold.Citation73 Conserved single stranded loop RNA regions are indicated in blue (loop 1) and purple (loop 2).
Identification of potential sRNA154 targets by genetic approaches
To elucidate the potential regulatory function of sRNA154 in M. mazei a strain ectopically overexpressing sRNA154 under its native promoter was constructed, leading to approximately 40-fold higher transcript levels under N-limitation in comparison to the wild type (wt) as demonstrated by RNA gel blot analysis (Fig. S2A). Whereas slower growth has been observed for the sRNA154 deletion mutant (ΔsRNA154) using a markerless exchange strategy,Citation33 overexpression of sRNA154 did not affect growth under N-limitation. However, in the course of continuous culturing of this deletion mutant under strict N-limitation, the growth phenotype was lost over time. Taking the polyploidy of M. mazei into account this is most likely due to reversion of the deletion by homologous recombination with a remaining not detected wt chromosome (first indication see Fig. S2A), strongly pointing to a crucial role of sRNA154 under N-limitation. Thus, a new deletion mutant was generated simultaneously inserting the puromycin resistance marker gene (ΔsRNA154::pac) (see Fig. S2B). This strain showed a stable growth phenotype under N-limitation, when cultured in the presence of puromycin (Fig. S2C), while no phenotype under N-sufficiency was observed.
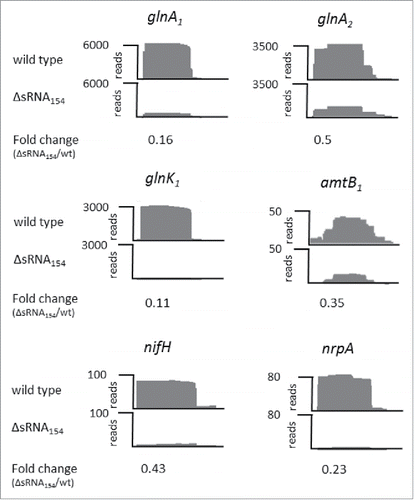
For target identification by evaluating the transcript patterns in the sRNA154 deletion mutant, an RNA-seq approach was performed analyzing RNA of two independently constructed mutant clones (ΔsRNA154::pac) and two wt clones grown under N-limitation with methanol as sole energy and carbon source (see Materials and Methods). The results demonstrated that approximately 62 of all genes showed higher or lower transcript levels (fold change ≥ 2, see Table S1). Those included seven genes of which the products are crucial components of the N-metabolism under N-limitation: nifH encoding α–subunit of nitrogenase, two genes encoding a glutamine synthetase (glnA136 and glnA2), a gene encoding a PII-like protein (glnK1),Citation36 two genes encoding ammonium transporter (amtB1 and amtB2) and most interestingly, the gene encoding the nif-specific transcriptional activator NrpA.Citation13 All of those N-related genes showed lower transcript levels in the absence of sRNA154, (summarized in , see also ), which was confirmed by quantitative (q)RT-PCR except for glnA2, the transcript of which appears not to be reduced when evaluated with qRT-PCR. Overall, the finding of reduced transcript levels of those N-related genes in the absence of sRNA154 is in agreement with the obtained growth phenotype under N-limiting conditions.
Table 1. Differential gene expression analysis. Selected genes involved in N-metabolism analyzed by differential RNA sequencing (DEseq2) in M. mazei ΔsRNA154 (ΔsRNA154::pac) in comparison to the wt, both grown exponentially under N limitation, for each strain two biological replicates were analyzed and further verified by qRT PCR (as described in Materials and Methods). The calculation is based on three independent biological replicates.
Figure 3. Transcript patterns of a sRNA154 chromosomal deletion mutant using an RNA-seq approach RNA sequence analysis (using the Illumina technique) was performed using RNA isolated from M. mazei wt and sRNA154 chromosomal deletion mutants (ΔsRNA154:: pac) growing under N-fixing conditions. For each strain two biological replicates were analyzed, representing two independent wt clones and two independent generated mutant clones (ΔsRNA154:: pac). Visualization of the distribution of cDNA reads of selected genes involved in the N-metabolism (glnA1, glnA2, glnK1, amtB1, nifH and nrpA) are exemplarily shown for one biological replicate. The fold change (ΔsRNA154:: pac vs. wt) indicated below represent the average change of both independent biological replicates.
Stabilization of target mRNA by sRNA154
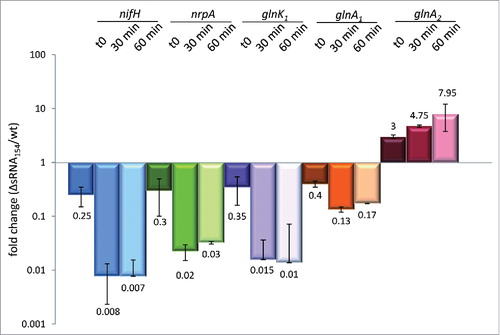
To elucidate whether sRNA154 directly affects the stability of the potential N-related target mRNAs, identified by the RNA-seq approach, we evaluated the respective transcript stability in vivo. Actinomycin D was added to exponentially growing cultures of M. mazei wt and ΔsRNA154::pac deletion strain (final conc. 100 µg/ml) to inhibit transcription (see Materials and Methods). Total RNA was isolated at time point t0, 30 and 60 min after adding actinomycin D. Relative transcript levels of predicted sRNA154 targets, nifH, glnA1, glnK1, glnA2 and nrpA, were determined in the chromosomal sRNA154 deletion mutant by qRT-PCR in comparison to the wt. Transcript stability of nifH, nrpA and glnK1 is highly affected in the absence of sRNA154 (see ), thus sRNA154 appears to stabilize the transcripts most likely by protecting them from degradation by a yet unknown RNase. sRNA154 also appears to be important for the stabilization of glnA1.The glnA2 transcript is the only RNA that appears slightly more stable in the absence of sRNA154 (see ).
Figure 4. mRNA stability assay comparing M. mazei Δ sRNA154 with the parental strain. To validate the stabilizing effects of sRNA154 on its target mRNAs we performed an mRNA half-life assay, using 100 µg/ml actinomycin D to inhibit transcription. Cells were harvested before (at time point zero) and after 30 and 60 min supplementing actinomycin D, followed by RNA isolation and qRT-PCR analysis to verify mRNA decay in the chromosomal deletion strain compared to the wt (for primer sets see Table S3). Fold changes in the sRNA154 deletion mutant vs. wt are given by mean values of two biologically independent experiments.
Verification of transcript stabilization due to sRNA154 by a complementation approach
In order to validate the observed stabilizing effects of sRNA154 on its targets and distinguish between the two potential interacting ssRNA loops, we constructed three mutant derivatives of sRNA154, resulting in the (partial) deletion of either loop 1 (mut1) or loop 2 (mut2 and 3) (see Fig. S3A). The respective mutant derivatives as well as the wt of sRNA154 encoded on a plasmid under the control of the native promoter were transformed into the deletion strain (ΔsRNA154::pac) to evaluate their ability for functional complementation. Northern analysis verified the overproduction of the sRNA154 derivatives and wt under N-limiting conditions as shown in Fig. S3 B. Analyzing the complementary effects on the transcript level of the identified target genes by qRT-PCR indicated that in case of nrpA and glnA1 both loops of sRNA154 are required for transcript stabilization. In contrast for nifH mRNA mainly loop 2 appears to increase stability, and loop1 appears to have larger impact on stabilization of glnK1 mRNA than loop 2 (see ). In contrast, glnA2 transcript level is slightly more stable in the absence of sRNA154 and particularly in the absence of loop 2 (see and ). Overall, the results of the complementation assays are in agreement and confirm the stability data observed ( and ).
Table 2. sRNA154 effects on target stability. Complementation assays of ΔsRNA154 mutant strain (ΔsRNA154::pac) complemented with various plasmid based derivatives of sRNA154. qRT PCR analysis was performed to identify the transcript levels of target genes. The fold change (FC) of ΔsRNA154 complemented with sRNA154 wt or derivatives (see Fig. S3A) versus wt were determined, data and respective standard deviations are representatives of at least three independent biological replicates.
In vivo target validation on the protein level
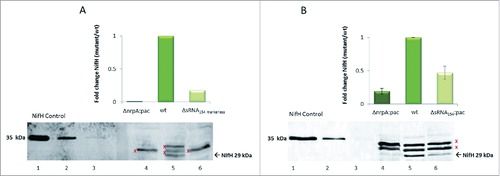
Quantitative western blot analysis was performed comparing protein patterns of identified targets in the sRNA154 mutant and the wt background during growth under N-limitation. Only a small positive effect of sRNA154 presence was evident in case of GlnA1 (Fig. S4A), whereas in case of GlnA2, a clear increase of the protein was detectable in the absence of sRNA154. Concordantly, the amounts of GlnA2 decreased in the presence of higher amounts of sRNA154 when overexpressed from a plasmid (see Fig. S4B), strongly indicating that the interaction of loop 2 of sRNA154 possibly targeting the ribosome binding site of glnA2 mRNA (see , glnA2) leads to significant lower translation initiation. Unfortunately, the cellular amount of the nif-specific transcriptional activator NrpA under N-limitation does not appear to be sufficient to allow detection by western blot analysis. However, in the absence of sRNA154 a negative effect on expression of the down-stream target gene of NrpA, the nifH gene, was detected. The relative amounts of NifH (α subunit of nitrogenase, 29 kDa) are significantly reduced in the absence of sRNA154 in the two independent deletion mutant strains (ΔsRNA154::markerless and ΔsRNA154::pac) resulting in a similar low amount of NifH as seen in a nrpA deletion background (ΔnrpA::pac) (see ). Taking the reduced transcript levels of nifH as well as nrpA in the absence of sRNA154 into account ( and and ), these findings strongly indicate that on the one hand, higher amounts of NifH and thus nitrogenase are expressed due to direct stabilizing the nifH transcript by sRNA154. Moreover, sRNA154 stabilizing the nrpA-mRNA - resulting in higher amounts of the transcriptional activator subsequently leading to higher nifH transcript levels – also results in additional expression of NifH.
Figure 5. NifH protein expression patterns in the absence of sRNA154 under N- limitation Cell extracts were prepared from exponentially growing cultures of M. mazei wt, M. mazei sRNA154::pac-mutant, M. mazei nrpA::pac mutant and M. mazei sRNA154::markerless mutant strains under N-limitation. Defined amounts of cell extracts were separated by SDS PAGE followed by western blot analysis using polyclonal antibodies generated against NifH. Relative amounts of NifH in the M. mazei sRNA154 deletion and nrpA deletion-mutant strain compared to M. mazei wt strain were calculated using the Aida image analyzer for three independent biological replicates. The average fold-expression changes are depicted, the lower panel represents one exemplarily chosen original western blot. A): lane 1–3, His-NifH standards (20 ng, 10 ng, 5 ng); lane 4, M. mazei ΔnrpA::pac-mutant (100 µg); lane 5, M. mazei wt cell extract (100 µg); lane 6, M. mazei ΔsRNA154::markerless-mutant strain (100 µg); B lane 1–3, His-NifH standards (20 ng, 10 ng, 5 ng); lane 4, M. mazei ΔnrpA::pac-mutant strain (50 µg); lane 5, M. mazei wt cell extract (50 µg); lane 6, M. mazei ΔsRNA154::pac-mutant (50 µg). X, protein bands which are also present under NH4+ sufficient growth conditions under which NifH protein is not translated.
Computational target predictions of sRNA154
In a parallel approach to the identification of potential targets by a genetic approach (RNA-seq), in silico target predictions were performed using the IntaRNA toolCitation37 which predicts putative interactions between sRNAs and the respective transcripts of each gene. This tool, originally designed for bacterial sRNAs, has been successfully used in the past to identify the target of the first identified archaeal sRNA.Citation38 First, we applied IntaRNA to predict interactions between sRNA154 and all identified 3,287 genes of M. mazei (data not shown). Most of the mRNAs encoding components of the N-metabolism are below the 50 with highest probability. In silico predicted interaction sites within the sRNA154 were mainly located in the highly conserved single stranded loops 1 and 2 (see Fig. S5 showing the 25 predicted most probable targets), which is in agreement with fact that interaction sites tend to be accessible.Citation39 In the genetic approach down regulation of mRNAs encoding components of the N-metabolism crucial under nitrogen limitation were observed in the absence of sRNA154 (glnA1, nifH, glnK1, amtB1, amtB2, and nrpA, see ). Thus, additional individual predictions for several of those genes using the IntaRNA tool were performed. Strikingly, these individual predictions revealed with significant probability that the majority of those N-metabolism related target mRNAs are predicted to interact several times with the two conserved single stranded loop regions of the sRNA (three or four predicted interaction sites). Details of the predicted interactions including the binding energies and pairing are shown in and summarized in . Based on the predicted interactions, the targets can be classified into two separate classes. class I targets are predicted to interact with both loops of sRNA154. The respective interaction sites are located in the 5′UTR as well as within the coding sequence, and frequently the RBS it is predicted to be targeted by loop 2 (see ). For mRNA-nrpA, loop 1 is predicted to interact in the coding region, whereas loop 2 is predicted to interact once within the first 8 nt of the 5′UTR and twice within the coding region (see ). For nifH-mRNA, four sRNA154 interaction sites are predicted with the first one located within the 5′ UTR, where loop 2 covers the RBS and the start codon of nifH. Furthermore, three interactions with loop 1 are predicted within the coding sequence of nifH (). Most interestingly, in addition to the predicted interactions within the nifH part, several additional interactions were predicted in the downstream part of the polycistronic mRNA of the 7 kbp nif-operon encoding the three structural subunits of nitrogenase, NifHDK as well as accessory proteins NifI1I2EN (see Fig. S6). This finding strongly suggests a stabilization of the complete 7 kbp polycistronic mRNA by sRNA154. class II targets are predicted to interact several times with only one of the loops, for glnK1 loop 1 is predicted to exclusively interact within the coding region (see ).
Figure 6. Target predictions for sRNA154 (IntaRNA) In silico prediction of potential interactions between sRNA154 and mRNA targets performed with IntaRNA.Citation37,67 Selected predicted interactions of genes involved in N-metabolism are classified into two classes. class I: mRNA targets interacting with both loops of sRNA154, targeting several sites of the mRNA including the ribosome binding site (RBS). class II: mRNA targets interacting with one loop of sRNA154 at several positions of the target mRNA. Loop1 of sRNA154 = indicated in turquoise, Loop2 of sRNA154 = indicated in purple.
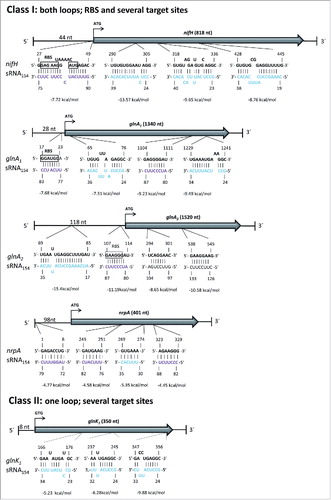
Table 3. Target predictions for sRNA154 using the IntaRNA tool. Selected predicted interactions of genes involved in N-metabolism. Following of the genome wide prediction by IntaRNA, selected N-regulated genes were analyzed again using IntaRNA. Additional interactions with sRNA154 are listed in Table S1. UTR = untranslated region; RBS = ribosome binding site; cds = coding sequence.
Overall, the global initial prediction resulted in a high number of pot. targets (∼3200), however most of the mRNAs, encoding components of the N-metabolism are below the 50 “best” targets (highest probability). The respective individual target predictions for those mRNAs showed multiple target sites with sRNA154. This finding is in agreement with the RNA-seq results observed for the sRNA154 mutant in comparison to the wt (, ) and supports the proposed stabilization effect of sRNA154 on the target mRNAs nif, nrpA and glnA1.
In vitro verification of direct sRNA154/ mRNA target interactions
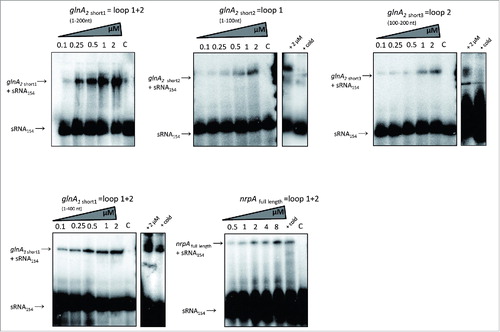
Electrophoretic mobility shift assays were performed to verify the predicted interaction sites between sRNA154 and the identified target mRNAs. Using various in vitro synthesized fragments of the identified mRNA targets and in vitro synthesized 5′, labeled full length sRNA154 demonstrated that transcripts of glnA1, glnA2 and nrpA bind to sRNA154 (). In case of glnA2, from the three different fragments used, the 200 nt fragment including both predicted interaction sites with loop 1 and 2 (glnA2 short1, see also ) showed highest binding capability to sRNA154 and a significant shift detected at concentrations ≥ 0.25 µM, whereas shorter fragments including only one interaction site (glnA2 short 2 ( = loop 1)) and short3 ( = loop 2)) showed significantly lower binding capability. The glnA1 fragment (1-400 nt) including the two first predicted interaction sites with loop 1 and 2 (see ) showed nearly comparable binding capability to sRNA154 as the glnA2 short1 fragment. Significant binding was also detected for the full length nrpA mRNA target, which showed lower binding capability compared to the glnA mRNA fragments. Overall, low binding capabilities were obtained in vitro which might indicate the requirement of a M. mazei scaffold protein(s) for maximal binding. However, the obtained interactions were significantly diminished when equal amounts of non-labeled sRNA154 were used as competitor RNA, confirming specificity of the binding (, compare lanes with highest amount of target and ‘+ cold’). In contrast, glnK1 mRNA (full length 360 nt) and nifH mRNA (1-100 nt), did not significantly affect the mobility of sRNA154 (see Fig. S7). The nifH fragment used in electrophoretic mobility shift assays contains only the first predicted interaction sites covering the RBS and the start codon of nifH (). This finding of no direct interaction between the 5′ UTR of nifH and sRNA154 most likely excludes that the translation of nifH is directly affected by sRNA154 and thus supports the proposed stabilization of the complete polycistronic mRNA of the nif-operon by sRNA154.
Figure 7. Electrophoretic mobility shift assays Electrophoretic mobility shift assays (EMSAs) were performed using approximately 5 nM of radioactively 5′end labeled sRNA154 or additionally added 2 µM cold sRNA154 (retardation experiment). The assays were performed with increasing concentrations of unlabeled target mRNAs. After 15 min incubation, samples were run on a native 6% PAA gel. The respective autoradiographs of the gels are shown for: glnA1 short fragment of the first 400 nt; for glnA2 short fragments from 1–100 nt, 100–200 nt and from 1–200 nt; nrpA full length transcript. The respective retardation of sRNA154 is indicated on the left site of the corresponding EMSAs.
The independent binding assay using a pulldown approach with biotinylated sRNA154 bound to streptavidin coated magnetic beads confirmed a significant binding of glnA2-mRNA to sRNA154 which is strong enough to pull glnA2 mRNA out of total RNA isolated from cells grown under N-limitation. However, the amount of nrpA-, nifH- and glnA1-mRNA in total RNA was apparently too low for detection by this pull down approach (see Fig. S8).
Discussion
sRNA154 is directly involved in regulation of N2-fixation
A recent study reported the first sRNA directly involved in regulation of N-metabolism in bacteria, sRNA NsiR4 of Synechocystis sp PCC6803, which inhibits translation of GS inactivating factor 7 under conditions of N-limitation and thus induces GS activity.Citation31 Besides, in bacteria several sRNAs reported or predicted to be involved in N-regulation act on mRNA targets, which are mostly indirectly linked to N-metabolism or N2-fixation e.g.Citation27-29,40 Very recently the sRNA NfiS was reported to be involved in regulation of nitrogen fixation in Pseudomonas stutzeri A1501. It was shown to enhance translation efficiency and the half-life of nifK transcript by binding to a single site of nifK mRNA potentially unfolding an inhibitory structure just after the nifK start codon.Citation41 Nevertheless, the archaeal sRNA154 presented in this study, is to our knowledge the first sRNA directly affecting regulation of N2-fixation on two levels as well as several other components of the central N-metabolism in M. mazei.
While investigating the potential regulatory function and molecular mechanisms of sRNA154 in N-metabolism by means of biochemical and genetic approaches, we obtained strong evidence that sRNA154 directly increases the stability of the mRNA encoding nitrogenase, the key enzyme of nitrogen fixation (see and ). Based on the fact that in contrast to P. stutzeriCitation41 multiple interaction sites within the coding sequence of the polycistronic 7 kbp mRNA are predicted, particularly with loop 2 (Fig. S6), we propose that sRNA154 most likely stabilizes the polycistronic mRNA by masking several endonucleolytic cleavage sites of RNases by loop 2. This agrees with the complementation assay which clearly demonstrated that predominantly loop 2 increases stability of nifH (). The mechanism of targeting multiple sites located in one mRNA has been previously described by Sharma et al. 2011 for the small RNA GcvB in Salmonella enterica serovar Typhimurium,Citation42 which shows a similar mechanism of interaction with its targets as predicted for sRNA154 and nif-mRNA.
Furthermore, there is strong evidence indicating that in addition to the post transcriptional regulation of the nif-operon expression by sRNA154 targeting and stabilizing the polycistronic nif-transcript directly there is as well post-transcriptional regulation by sRNA154 of the nif-specific transcriptional activator NrpA.Citation13 Quantification of nrpA-transcript levels in independently generated sRNA154 deletion mutants showed significant reduction of nrpA transcripts (, ). In accord, stability and complementation assays clearly demonstrated that in the absence of sRNA154 nrpA transcripts were significantly faster degraded than in the wt (see and ). In silico target predictions identified four potential interactions with loop 1 and 2 of sRNA154 which target the 5′ UTR and the coding region of nrpA-mRNA (). Based on those findings we hypothesize that binding of both loops of several sRNA154 molecules stabilizes nrpA-mRNA most likely inhibiting endonucleolytic cleavage by masking specific recognition sites for RNases. Considering the low binding capability determined in EMSAs, an additional small scaffold protein might be required to effectively stabilize those interactions in vivo.
mRNA stabilization by sRNAs in archaea
mRNA stabilization and degradation are important regulatory features to quickly respond to changes in the cellular environment. From bacteria it is known that generation of dsRNA due to sRNA-mRNA interaction can mediate RNA cleavage by the double-strand specific RNase III or prevent cleavage by RNase E (recently reviewed in.Citation17,43 A well-studied example in bacteria is the rpoS mRNA encoding the stationary sigma factor σS in E. coli. Three independent small RNAs are able to target specific sites of the 5′ UTR and affect RNase E cleavage of the rpoS mRNA.Citation44,45 There are numerous other bacterial examples for sRNA mediated stabilization of target mRNAs (e.g.Citation44,46) but significantly less is known on RNA stabilization and degradation in archaea. In Sulfolobus solfataricus RNA degradation preferentially occurs at 5′monophosphates of processed RNAs mediated by the 5′-3´ exonuclease RNase J, although the dephosphorylation mechanism at the 5′ triphosphate end is still unknown. S. solfataricus also contains the archaeal exosome which cleaves specifically ssRNA at the 3´end when modified by polyA tails.Citation47,48 In contrast, in halophilic archaea, which do not contain the archaeal exosome, transcripts often contain long 3´UTRs,Citation49 which are proposed to have a regulatory function. For several methanogenic archaea the presence of RNase J homologs has been reported, which have shown to contain endonucleolytic as well as 5′- 3´exonucleolytic cleavage activity.Citation50 The respective homologous proteins of M. mazei strain Gö1 recently identified and crystalized by Mir-Montazeri et al. 201151 might be responsible for the proposed enhanced target degradation in the absence of sRNA154.
Further targets of sRNA154 in the central N-metabolism of M. mazei
Besides nifH- and nrpA-mRNA, RNA-seq analysis, stabilization and complementation assays identified several other potential targets representing components of the N-metabolism, e.g., glnK1 as well as glnA1 and glnA2 (and ). Further evidence suggests that glnA1-mRNA is directly stabilized by sRNA154, including both loops, whereas glnA2 mRNA appears slightly more stable in the absence of sRNA154 particularly in the absence of loop 2 (, ). Moreover, significantly increased protein levels of GlnA2 have been detected in the absence of sRNA154 in accord with reduced protein levels in the presence of additional sRNA154 (see ). Since loop 2 is predicted to interact with the RBS of glnA2 mRNA and significant binding of sRNA154 to glnA2 transcripts was detected with two independent approaches ( and Fig. S8), we propose that the RBS of glnA2-mRNA is masked by loop 2 of sRNA154 resulting in inhibition of translation initiation.
Molecular mechanisms of post-transcriptional regulation by sRNAs in archaea
Until today, only a few regulatory mechanisms of identified archaeal sRNAs of have been explained at a molecular level. The first reported mechanism was for an archaeal sRNA, namely sRNA162 of M. mazei, which targets the 5′UTR of its trans-target (MM_2441) and consequently masks the RBS as well as the translational start site leading to inhibition of translation initiationCitation38 arguing that this particular archaeal sRNA acts similar as its bacterial counterparts. Moreover, it was shown that the same sRNA targets a second cis-encoded mRNA target challenging the paradigm of a strict border between cis- and trans- encoded sRNAs.Citation38 Beside 5′ UTR targeting by sRNA162, sRNA257 from S. solfataricus has been shown to act in trans on the 3´UTR of the ORF_1183 mRNA (encoding a putative phosphate transporter) inducing degradation under phosphate-rich growth conditions.Citation52 Further, first indications were obtained that sRNAs of M. mazei might also target 3′UTRs of transcripts, which have been shown to be unexpectedly long, often in the range of 88 +/− 42 nt.Citation53
Consequently, sRNA154 not only represents the first archaeal sRNA for which a positive posttranscriptional regulation is demonstrated by enhancing the stability of its mRNA targets, e.g., nifH, nrpA and glnA1, but it also downregulates translation initiation of glnA2 by interacting with the 5′UTR and masking its RBS.
Conclusion and proposed working model for sRNA154 as regulator in N2-fixation
On the basis of our findings we propose the following working model for the physiological role of sRNA154 in regulating N2-fixation: Under N-limitation, transcription of the nif-operon, glnA1, glnK1amtB1 operon, nrpA and sRNA154 is induced caused by NrpR dissociation from the respective operators. The nifHI1I2DKEN and nrpA transcripts are further stabilized by sRNA154 leading to enhanced nitrogenase amounts due to the direct stabilization of the nif-transcript and due to higher nif-transcript level based on higher NrpA protein level. Thus, we propose that beside a direct stabilization of the nif-transcript, sRNA154 facilitates a feed forward regulation of nif-gene expression by stabilizing the nrpA-transcript. In addition, sRNA154 only transcribed under N-limitation has a pleitropic effect on several components of the N-metabolism in M. mazei, which agrees with the phenotype under N-limitation in the absence of sRNA154. Overall, this tight network of nitrogenase expression regulation on the transcriptional and post-transcriptional level, involving two transcriptional regulators (NrpR and NrpA) and a central N-regulated sRNA (sRNA154) facilitates a fast response under changing N-availabilities, and allows fine tuning on almost every level of gene expression.
Considering the high sequence and structural conservation of sRNA154, including also the transcriptional control by NrpR (see and ), direct effects on N2-fixation is most likely also true for various other nitrogen fixing Methanosarcina strains encoding sRNA154. This is supported by the presence of sRNA154 correlating with the simultaneous presence of the gene encoding the nif-specific transcriptional regulator NrpA, showing high sequence conservation including the 5′UTR and the predicted interaction sites with loop 1 and 2 of sRNA154 (see nrpA alignment Fig. S9).
Material and methods
Strains and plasmids
Strains and plasmids, which were used in this study, are listed in Table S2. Plasmid DNA was transformed into M. mazei as described by.Citation54
Growth
Cells of M. mazei wild type and mutant strains were grown under nitrogen limitation as described in Weidenbach et al. 2014Citation13 and harvested at mid-exponential phase (OD600 = 0.15 – 0.2) at 4 °C.
Construction of M. mazei mutants
All primers used in this study are listed in Table S3. sRNA154 overproduction strain was constructed by PCR amplification of sRNA154 including its native promoter from genomic M. mazei DNA using primers s154-XhoI-for and s154-KpnI-rev. The 338 bp PCR-fragment was inserted into the multiple cloning site of pWM321.Citation55 The resulting plasmid pR723 was transformed into a M. mazei strain with higher plating efficiency, M. mazei* by liposome-mediated transformation as previously described.Citation54,55 Puromycin-resistant transformants were selected as colonies that grew on minimal medium plates with trimethylamine as the carbon and energy source plus 5 µg puromycin/ml during incubation. sRNA154 chromosomal deletion strain was constructed as follows: the pac-cassette was cut out of the plasmid pRS207 and digested with mung bean nuclease. The plasmid pRS631,Citation33 containing the flanking regions of the small RNA154, was linearized with XhoI and also digested with mung bean nuclease. Next, the blunt ended pac cassette was ligated into linearized pRS631, resulting in plasmid pRS927. pRS927 was transformed into M. mazei* by liposome-mediated transformation and inserted into the chromosome by homologous recombination. All constructs were verified by sequence analysis. Southern blot analyses of genomic DNA from puromycin-resistant transformants was used to verify pac insertion as described by Ehlers et al.Citation54 Complementation assays were performed using plasmids containing sRNA154 derivatives under its native promoter, which were transformed in M. mazei ΔsRNA154. Plasmids were constructed as follows: sRNA154 was amplified with gene specific primers (see Table S3) from genomic M. mazei wt DNA and the resulting PCR product was TOPO-TA cloned into the vector pCRII (Invitrogen, Thermo Fisher Scientific, Darmstadt, Germany). Deletion versions of sRNA154 (schematically shown in Fig. S3) were constructed by site directed mutagenesis. The respective constructs were cut out of the pCRII derivative using SacI and KpnI and ligated into linearized shuttle-vector pWM-neoCitation56 resulting in plasmids pRS953, pRS954, pRS974 and pRS1194. These plasmids were transformed into M. mazei by liposome-mediated transformation as described by Ehlers et al. 2005.Citation54
RNA isolation
RNA isolation was performed using TRI reagent (5 PRIME, Hilden, Germany) following the manufacturer´s protocol followed by DNaseI treatment and phenol-chloroform precipitation as described in.Citation57
Northern blot analysis
Northern Blot analysis was performed using the recently described protocol.Citation32 RNAs were detected with 5′-32P labeled ssDNA oligo probes (see Table S3).
RACE (rapid amplification of cDNA ends) analysis
RACE analysis was performed to determine the transcriptional start site (TSS) as well as the transcript termination site of sRNA154 as recently described in Prasse et al.,Citation58 using 5′-RLM-sRNA154in and 5′-RLM-sRNA154out (see Table S3).
A hammerhead ribozyme transcriptional fusion with sRNA154
As described in Jäger et al. 201238 we constructed and synthesized a DNA template of sRNA154 for T7 polymerase, where the promotor was fused to a hammerhead ribozyme. The fusion guarantees transcription at the native +1 site of sRNA154, and can be engineered for any template choice, thereby providing an efficient method for the preparation of native RNA transcripts. Detailed description of the method is published in Jäger et al. 2012.Citation38
In vitro T7 transcription, purification and 5′end labeling of RNA
T7 templates were amplified with gene specific primers from genomic M. mazei wild type DNA (see Table S2 and 3). The forward gene specific primers contain an artificial T7 promotor and the revers gene specific primers contain a SmaI restriction site. The obtained PCR fragments were ligated in the pCRII vector using the TOPO-TA cloning kit (Invitrogen, Darmstadt, Germany). Both, the resulting vectors linearized by SmaI digestion resulting in run-off plasmids, as well as PCR fragments, amplified from the plasmids, were used as templates, depending on transcription efficiency. In vitro transcription was performed using the TranscriptAid high yield kit (Thermo Scientific, Thermo Fisher Scientific, Darmstadt, Germany) according to the manufacturer's instructions followed by DNase I treatment and RNA extraction using the RNA clean-up and concentrator-25 kit (Zymo Research, Freiburg, Germany). RNA quality and integrity were checked on a 1% agarose gel. The in vitro transcribed RNA was dephosphorylated for 10 min with FastAP (Thermosensitive Alkaline Phosphatase, Thermo Scientific, Darmstadt, Germany) and radioactively labeled at the 5′ end as described in Jäger et al. 2012,Citation38 2µl of RNase Inhibitor was additionally added to the labeling reaction (Thermo Scientific, Darmstadt, Germany).
Construction of cDNA libraries for Illumina sequencing and differential gene expression analysis
M. mazei wild type and sRNA154 deletion mutant (Δ154::pac) were grown under N-fixing conditions with 80% N2 and 20% CO2 in the gas phase in 50 ml in closed tubes as described.Citation59,60 Cells were harvested at a turbidity of 0.15-0.2 at 600 nm followed by RNA isolation as described above. To construct RNA-seq libraries two biological replicates of isolated total RNA were used (described above). cDNA library preparation were described previously.Citation61 For Illumina sequencing of cDNA molecules, the libraries were constructed by vertis Biotechnology AG (Freising, Germany) as described previously for eukaryotic microRNA librariesCitation62 but without a RNA size-fractionation step before the cDNA synthesis. The cDNA libraries were sequenced using a HiSeq 2500 machine (Illumina) in single-read mode. The Illumina reads in FASTQ format were trimmed based on a cut-off phred score of 20 by the program fastq_quality_trimmer from FASTX toolkit version 0.0.13 (http://hannonlab.cshl.edu/fastx_toolkit/). The following steps were performed using the subcommand “create,” “align” and “coverage” of the tool READemptionCitation63 version 0.3.0. The poly(A)-tail sequences introduced in the library preparation were removed and a size filtering step was applied in which sequences shorter than 12 nt were eliminated. The collections of remaining reads were mapped to the reference genome sequences (NC_003901.1 - downloaded from the NCBI ftp server) using segemehl version 0.1.7.Citation64 Coverage plots in wiggle format representing the number of aligned reads per nucleotide were generated based on the aligned reads and visualized in the Integrated Genome BrowserCitation65 or the gene expression quantification annotation files in GFF3 format were retrieved from the NCBI ftp server and extended by manually curated sRNA and UTR entries. Reads per genes were quantified with READemption's subcommand “gene_quanti” and pairwise expression comparison based on these gene quantifications was performed with the subcommand “deseq” which applied DESeq2 version 1.4.5. Gene with a fold-change of equal or higher than 2.0 and an adjusted p-value below 0.1 were considered as differentially expressed. The RNA-seq data discussed in this publication have been deposited in NCBI's Gene Expression OmnibusCitation66 and are accessible through GEO Series accession number GSE85456 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE85456). A shell script that can be used to reproduce the RNA-seq analysis can be retrieved from Zenodo at https://zenodo.org/record/59989 (DOI: 10.5281/zenodo.59989).
Computational target predictions
In silico predictions were performed using IntaRNACitation37,67 as recently described by Jäger et al. 2012.Citation38 For genome-wide target predictions the IntaRNA web-server 1.2.5 (wrapper 1.0.7.1) was used,Citation68 using the given default settings (hybridization temperature: 37.0 °C; window size: 150 nt; base pair distance: 100 nt) but extending the prediction area around the translational start site of the mRNAs from 75 nt to 100 nt. For the individual target predictions settings of the IntaRNA webserver were varied as follows: for interactions between sRNA154 and nifH-mRNA, glnA1-mRNA, glnA2-mRNA, nrpA-mRNA and glnK1-mRNA: number of suboptimal interactions: 3; Minimal number of basepairs in seed: 6; Maximal number of mismatches in seed: 3; for interactions with the nifHDEKN operon the given default settings, as described above, were used.
Quantitative reverse transcriptase (RT)-PCR analysis
Quantitative reverse transcriptase PCR analysis was performed as described in,Citation69,70 but using 200 ng of total M. mazei RNA per reaction and the ViiA 7 Real-Time PCR System from Applied Biosystems (Thermo Fisher Scientific, Darmstadt, Germany). Ct values were normalized in respect to the corresponding Ct values obtained from the same RNA for three genes (MM 1621, MM 2181, MM 1215; see Table S3), which were shown to be transcribed to the same amount irrespective of the nitrogen or carbon availability in microarray experiments (K. Veit and R. A. Schmitz, unpublished data).Citation69,70 Primer sets used including the control genes are listed in the Table S3.
Electrophoretic mobility shift assays
Electrophoretic mobility shift assays (EMSAs) were conducted in a total volume of 10 µl in the presence of 1X structure buffer (Ambion, Thermo Scientific, Darmstadt, Germany) and 1 mg yeast RNA (Ambion, Thermo Scientific, Darmstadt, Germany). 20 pmol of in vitro transcribed RNA were dephosphorylated and radioactively 5′ labeled as described earlier. 5nM of the labeled RNA were incubated in presence with increasing amounts of the target RNA for 15 min at 37 °C and subsequently separated on native 6–8% poly acrylamide gel in a 0.5 × Tris–borate buffer system (0.45 M, pH 8.0). Gels were analyzed using a phosphoimager (FLA-5000 Series, Fuji). Protein-DNA EMSAs were performed as described in Weidenbach et al. 2014,Citation13 using 4ng PCR product of sRNA154 gene and purified MBP-tagged NrpR protein.
RNA-RNA pulldown
For in vivo detection of interactions between sRNA154 and target mRNAs, we used the Pierce magnetic RNA-protein pulldown Kit (Thermo Scientific, Darmstadt, Germany). First, the in vitro synthesized sRNA154 was biotinylated at the 3´end using the Pierce RNA 3′ End Desthiobiotinylation Kit (Thermo Scientific, Darmstadt, Germany) according to the manufacturer's instructions. Next, the biotinylated sRNA154 was bound to streptavidin magnetic beads, subsequently the respective protocol of the Pierce magnetic RNA-protein pulldown Kit was followed with the modification that purified total RNA from M. mazei wt cells, grown under N-limiting conditions was used instead of protein extract. To determine the potential sRNA - mRNA interactions northern analysis (dot blots) was performed as follows: 25 µl of the pulldown eluate and 10µg of total RNA as well as 5 pmol of the respective in vitro transcribed RNA as internal positive controls were applied on a nitrocellulose membrane. After UV cross-linking, the membrane was hybridized with10 pmol [−32P]-ATP end-labeled oligodeoxynucleotides for 2 h (Table S3). After washing 3 times for 15 min in 5 x, 1 x and 0.5 × SSC–0.1% SDS solutions (42 °C), signals were visualized using a phosphorimager (FLA-5000 Series, Fuji) and quantified with AIDA software (Raytest).
Transcription inhibition by actinomycin D
Actinomycin D was dissolved in DMSO (100%) and added to exponentially growing M. mazei cultures with a final concentration of 100 μg/ ml to the samples. Cultures (15 ml) were grown in closed serum bottles as described above. At time point zero 5 ml were harvested as reference and actinomycin D was added to the remaining culture (10 ml). After 30 and 60 min of incubation 5 ml cultures were harvested and centrifuged for 30 min at 4,000 rpm and 4°C, followed by RNA isolation as described.
Western blot analysis
Polyclonal rabbit antiserum directed against his-tagged M. mazei proteins were generated as described in Weidenbach et al. 2014.Citation13 For Western blot analysis crude extracts of wt and sRNA154 deletion mutant (Δ154::pac) were prepared from cells growing exponentially under N-limitation. Further procedures were performed as described in detail inCitation11,13 using purified his-tagged proteins as controls and for quantification.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary_Material.docx
Download MS Word (2.8 MB)Acknowledgments
We thank the members of our laboratory for useful discussions on the manuscript, as well as Claudia Kießling and Cornelia Goldberg for technical assistance. We particularly thank Prof. Dr. William Metcalf (University of Illinois) for sequence information of sRNA154 homologs before publishing the Methanosarcina genomes and Dr. Claudia Ehlers for performing the NrpR-sRNA154 EMSA.
Funding
This work was supported by the German Research Council (DFG) priority program (SPP) 1258 ‘Sensory and Regulatory sRNAs in Prokaryotes’ [Schm1052/9-1, Schm1052/9-2)].
Related Research Data
References
- Huergo LF, Pedrosa FO, Muller-Santos M, Chubatsu LS, Monteiro RA, Merrick M, Souza EM. PII signal transduction proteins: pivotal players in post-translational control of nitrogenase activity. Microbiology 2012; 158:176-90; PMID:22210804; https://doi.org/10.1099/mic.0.049783-0
- Masepohl B, Hallenbeck PC. Nitrogen and molybdenum control of nitrogen fixation in the phototrophic bacterium Rhodobacter capsulatus. Adv Exp Med Biol 2010; 675:49-70; PMID:20532735; https://doi.org/10.1007/978-1-4419-1528-3_4
- Dixon R, Kahn D. Genetic regulation of biological nitrogen fixation. Nat Rev Microbiol 2004; 2:621-31; PMID:15263897; https://doi.org/10.1038/nrmicro954
- Dodsworth JA, Leigh JA. Regulation of nitrogenase by 2-oxoglutarate-reversible, direct binding of a PII-like nitrogen sensor protein to dinitrogenase. Proc Natl Acad Sci U S A 2006; 103:9779-84; PMID:16777963; https://doi.org/10.1073/pnas.0602278103
- Cohen-Kupiec R, Blank C, Leigh JA. Transcriptional regulation in Archaea: in vivo demonstration of a repressor binding site in a methanogen. Proc Natl Acad Sci U S A 1997; 94:1316-20; PMID:9037050
- Kessler PS, Blank C, Leigh JA. The nif gene operon of the methanogenic archaeon Methanococcus maripaludis. J Bacteriol 1998; 180:1504-11; PMID:9515920
- Leigh JA. Transcriptional regulation in Archaea. Curr Opin Microbiol 1999; 2:131-4; PMID:10322164; http://doi.org/10.1016/S1369-5274(99)80023-X
- Ehlers C, Veit K, Gottschalk G, Schmitz RA. Functional organization of a single nif cluster in the mesophilic archaeon Methanosarcina mazei strain Gö1. Archaea 2002; 1:143-50; PMID:15803652
- Lie TJ, Leigh JA. A novel repressor of nif and glnA expression in the methanogenic archaeon Methanococcus maripaludis. Mol Microbiol 2003; 47:235-46; PMID:12492867
- Veit K, Ehlers C, Ehrenreich A, Salmon K, Hovey R, Gunsalus RP, Deppenmeier U, Schmitz RA. Global transcriptional analysis of Methanosarcina mazei strain Gö1 under different nitrogen availabilities. Mol Genet Genomics 2006; 276:41-55; PMID:16625354; https://doi.org/10.1007/s00438-006-0117-9
- Weidenbach K, Ehlers C, Kock J, Schmitz RA. NrpRII mediates contacts between NrpRI and general transcription factors in the archaeon Methanosarcina mazei Gö1. FEBS J 2010; 277:4398-411; PMID:20875081; https://doi.org/10.1111/j.1742-4658.2010.07821.x
- Weidenbach K, Ehlers C, Kock J, Ehrenreich A, Schmitz RA. Insights into the NrpR regulon in Methanosarcina mazei Go1. Arch Microbiol 2008; 190:319-32; PMID:18415079; https://doi.org/10.1007/s00203-008-0369-3
- Weidenbach K, Ehlers C, Schmitz RA. The transcriptional activator NrpA is crucial for inducing nitrogen fixation in Methanosarcina mazei Go1 under nitrogen-limited conditions. FEBS J 2014; 281:3507-22; PMID:24930989; https://doi.org/10.1111/febs.12876
- Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009; 136:642-55; PMID:19239886; https://doi.org/10.1016/j.cell.2009.01.035
- Beisel CL, Storz G. Base pairing small RNAs and their roles in global regulatory networks. FEMS Microbiol Rev 2010; 34:866-82; PMID:20662934; https://doi.org/10.1111/j.1574-6976.2010.00241.x
- Brantl S, Jahn N. sRNAs in bacterial type I and type III toxin-antitoxin systems. FEMS Microbiol Rev 2015; 39:413-27; PMID:25808661; https://doi.org/10.1093/femsre/fuv003
- Wagner EG, Romby P. Small RNAs in bacteria and archaea: who they are, what they do, and how they do it? Adv Genet 2015; 90:133-208; PMID:26296935; https://doi.org/10.1016/bs.adgen.2015.05.001
- Balbontin R, Villagra N, Pardos de la Gandara M, Mora G, Figueroa-Bossi N, Bossi L. Expression of IroN, the salmochelin siderophore receptor, requires mRNA activation by RyhB small RNA homologues. Mol Microbiol 2016 Apr; 100(1):139-55; PMID:26710935; http://doi.org/10.1111/mmi.13307
- Adnan F, Weber L, Klug G. The sRNA SorY confers resistance during photooxidative stress by affecting a metabolite transporter in Rhodobacter sphaeroides. RNA Biol 2015; 12:569-77; PMID:25833751; https://doi.org/10.1080/15476286.2015.1031948
- Billenkamp F, Peng T, Berghoff BA, Klug G. A cluster of four homologous small RNAs modulates C1 metabolism and the pyruvate dehydrogenase complex in Rhodobacter sphaeroides under various stress conditions. J Bacteriol 2015; 197:1839-52; PMID:25777678; http://doi.org/10.1128/JB.02475-14
- Hoe C-H, Raabe CA, Rozhdestvensky TS, Tang T-H. Bacterial sRNAs: regulation in stress. Int J Med Microbiol 2013; 303:217-29; PMID:23660175; https://doi.org/10.1016/j.ijmm.2013.04.002
- Storz G, Vogel J, Wassarman Karen M. Regulation by small RNAs in bacteria: Expanding frontiers. Mol Cell 2011; 43:880-91; PMID:21925377; https://doi.org/10.1016/j.molcel.2011.08.022
- Bouffartigues E, Moscoso JA, Duchesne R, Rosay T, Fito-Boncompte L, Gicquel G, Maillot O, Bénard M, Bazire A, Brenner-Weiss G, et al. The absence of the Pseudomonas aeruginosa OprF protein leads to increased biofilm formation through variation in c-di-GMP level. Front Microbiol 2015; 6:630; PMID:26157434; https://doi.org/10.3389/fmicb.2015.00630
- Guo MS, Updegrove TB, Gogol EB, Shabalina SA, Gross CA, Storz G. MicL, a new sigmaE-dependent sRNA, combats envelope stress by repressing synthesis of Lpp, the major outer membrane lipoprotein. Genes Dev 2014; 28:1620-34; PMID:25030700; https://doi.org/10.1101/gad.243485.114
- Fröhlich KS, Papenfort K, Berger AA, Vogel J. A conserved RpoS-dependent small RNA controls the synthesis of major porin OmpD. Nucleic Acids Res 2012; 40:3623-40; PMID:22180532; http://doi.org/10.1093/nar/gkr1156
- Papenfort K, Bouvier M, Mika F, Sharma CM, Vogel J. Evidence for an autonomous 5 target recognition domain in an Hfq-associated small RNA. Proc Natl Acad Sci USA 2010; 107:20435; PMID:21059903; https://doi.org/10.1073/pnas.1009784107
- Ionescu D, Voss B, Oren A, Hess WR, Muro-Pastor AM. Heterocyst-specific transcription of NsiR1, a non-coding RNA encoded in a tandem array of direct repeats in cyanobacteria. J Mol Biol 2010; 398:177-88; PMID:20227418; http://doi.org/10.1016/j.jmb.2010.03.010
- De Lay N, Gottesman S. The Crp-activated small noncoding regulatory RNA CyaR (RyeE) links nutritional status to group behavior. J Bacteriol 2009; 191:461-76; PMID:18978044; https://doi.org/10.1128/JB.01157-08
- Jung YS, Kwon YM. Small RNA ArrF regulates the expression of sodB and feSII genes in Azotobacter vinelandii. Curr Microbiol 2008; 57:593-7; PMID:18830664; https://doi.org/10.1007/s00284-008-9248-z
- Mitschke J, Vioque A, Haas F, Hess WR, Muro-Pastor AM. Dynamics of transcriptional start site selection during nitrogen stress-induced cell differentiation in Anabaena sp. PCC7120. Proc Natl Acad Sci U S A 2011; 108:20130-5; PMID:22135468; https://doi.org/10.1073/pnas.1112724108
- Klähn S, Schaal C, Georg J, Baumgartner D, Knippen G, Hagemann M, Muro-Pastor AM, Hess WR. The sRNA NsiR4 is involved in nitrogen assimilation control in cyanobacteria by targeting glutamine synthetase inactivating factor IF7. Proc Natl Acad Sci U S A 2015; 112:E6243-52; PMID:26494284; https://doi.org/10.1073/pnas.1508412112
- Jäger D, Sharma CM, Thomsen J, Ehlers C, Vogel J, Schmitz RA. Deep sequencing analysis of the Methanosarcina mazei Gö1 transcriptome in response to nitrogen availability. Proc Natl Acad Sci U S A 2009; 106:21878-82; PMID:19996181; https://doi.org/10.1073/pnas.0909051106
- Ehlers C, Jäger D, Schmitz RA. Establishing a markerless genetic exchange system for Methanosarcina mazei strain Gö1 for constructing chromosomal mutants of small RNA genes. Archaea 2011; 2011:439608; PMID:21941461; https://doi.org/10.1155/2011/439608
- Youngblut ND, Wirth JS, Henriksen JR, Smith M, Simon H, Metcalf WW, Whitaker RJ. Genomic and phenotypic differentiation among Methanosarcina mazei populations from Columbia River sediment. ISME J 2015; 9:2191-205; PMID:25756680; https://doi.org/10.1038/ismej.2015.31
- Smith C, Heyne S, Richter AS, Will S, Backofen R. Freiburg RNA Tools: a web server integrating INTARNA, EXPARNA and LOCARNA. Nucleic Acids Res 2010; 38:W373-7; PMID:20444875; https://doi.org/10.1093/nar/gkq316
- Ehlers C, Weidenbach K, Veit K, Forchhammer K, Schmitz RA. Unique mechanistic features of post-translational regulation of glutamine synthetase activity in Methanosarcina mazei strain Go1 in response to nitrogen availability. Mol Microbiol 2005; 55:1841-54; PMID:15752204; https://doi.org/10.1111/j.1365-2958.2005.04511.x
- Busch A, Richter AS, Backofen R. IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics 2008; 24:2849-56; PMID:18940824; https://doi.org/10.1093/bioinformatics/btn544
- Jäger D, Pernitzsch SR, Richter AS, Backofen R, Sharma CM, Schmitz RA. An archaeal sRNA targeting cis- and trans-encoded mRNAs via two distinct domains. Nucleic Acids Res 2012 Nov; 40(21):10964-79; PMID: 22965121; http://doi.org/10.1093/nar/gks847
- Richter AS, Backofen R. Accessibility and conservation: General features of bacterial small RNA-mRNA interactions? RNA Biol 2012 9:954-65; PMID:22767260; https://doi.org/10.4161/rna.20294
- Hao Y, Updegrove TB, Livingston NN, Storz G. Protection against deleterious nitrogen compounds: role of sigmaS-dependent small RNAs encoded adjacent to sdiA. Nucleic Acids Res 2016; 44:6935-48; PMID:27166377; https://doi.org/10.1093/nar/gkw404
- Zhan Y, Yan Y, Deng Z, Chen M, Lu W, Lu C, Shang L, Yang Z, Zhang W, Wang W, et al. The novel regulatory ncRNA, NfiS, optimizes nitrogen fixation via base pairing with the nitrogenase gene nifK mRNA in Pseudomonas stutzeri A1501. Proc Natl Acad Sci U S A 2016; 113:E4348-56; PMID:27407147; https://doi.org/10.1073/pnas.1604514113
- Sharma CM, Papenfort K, Pernitzsch SR, Mollenkopf HJ, Hinton JC, Vogel J. Pervasive post-transcriptional control of genes involved in amino acid metabolism by the Hfq-dependent GcvB small RNA. Mol Microbiol 2011; 81:1144-65; PMID:21696468; https://doi.org/10.1111/j.1365-2958.2011.07751.x
- Belasco JG. All things must pass: contrasts and commonalities in eukaryotic and bacterial mRNA decay. Nat Rev Mol Cell Biol 2010; 11:467-78; PMID:20520623; https://doi.org/10.1038/nrm2917
- Papenfort K, Vanderpool CK. Target activation by regulatory RNAs in bacteria. FEMS Microbiol Rev 2015; 39:362-78; PMID:25934124; https://doi.org/10.1093/femsre/fuv016
- McCullen CA, Benhammou JN, Majdalani N, Gottesman S. Mechanism of positive regulation by DsrA and RprA small noncoding RNAs: pairing increases translation and protects rpoS mRNA from degradation. J Bacteriol 2010; 192:5559-71; PMID:20802038; https://doi.org/10.1128/JB.00464-10
- Ramirez-Pena E, Trevino J, Liu Z, Perez N, Sumby P. The group A Streptococcus small regulatory RNA FasX enhances streptokinase activity by increasing the stability of the ska mRNA transcript. Mol Microbiol 2010; 78:1332-47; PMID:21143309; http://doi.org/10.1111/j.1365-2958.2010.07427.x
- Hou L, Klug G, Evguenieva-Hackenberg E. The archaeal DnaG protein needs Csl4 for binding to the exosome and enhances its interaction with adenine-rich RNAs. RNA Biol 2013; 10:415-24; PMID:23324612; https://doi.org/10.4161/rna.23450
- Evguenieva-Hackenberg E, Blasi U. Attack from both ends: mRNA degradation in the crenarchaeon Sulfolobus solfataricus. Biochem Soc Trans 2013; 41:379-83; PMID:23356315; https://doi.org/10.1042/BST20120282
- Babski J, Maier L-K, Heyer R, Jaschinski K, Prasse D, Jäger D, Randau L, Schmitz RA, Marchfelder A, Soppa J. Small regulatory RNAs in Archaea. RNA Biol 2014; 11:484-93; PMID:24755959; https://doi.org/10.4161/rna.28452
- Levy S, Portnoy V, Admon J, Schuster G. Distinct activities of several RNase J proteins in methanogenic archaea. RNA Biol 2011; 8:1073-83; PMID:21955587; https://doi.org/10.4161/rna.8.6.16604
- Mir-Montazeri B, Ammelburg M, Forouzan D, Lupas AN, Hartmann MD. Crystal structure of a dimeric archaeal cleavage and polyadenylation specificity factor. J Struct Biol 2011; 173:191-5; PMID:20851187; https://doi.org/10.1016/j.jsb.2010.09.013
- Märtens B, Manoharadas S, Hasenöhrl D, Manica A, Bläsi U. Antisense regulation by transposon-derived RNAs in the hyperthermophilic archaeon Sulfolobus solfataricus. EMBO Rep 2013; 14:527-33; PMID:23579342; https://doi.org/10.1038/embor.2013.47
- Dar D, Prasse D, Schmitz RA, Sorek R. Widespread formation of alternative 3′ UTR isoforms via transcription termination in archaea. Nat Microbiol 2016; 1:16143; PMID:27670118; https://doi.org/10.1038/nmicrobiol.2016.143
- Ehlers C, Weidenbach K, Veit K, Deppenmeier U, Metcalf WW, Schmitz RA. Development of genetic methods and construction of a chromosomal glnK(1) mutant in Methanosarcina mazei strain Gö1. Mol Genet Genomics 2005; 273:290-8; PMID:15824904; http://doi.org/10.1007/s00438-005-1128-7
- Metcalf WW, Zhang JK, Apolinario E, Sowers KR, Wolfe RS. A genetic system for Archaea of the genus Methanosarcina: liposome-mediated transformation and construction of shuttle vectors. Proc Natl Acad Sci U S A 1997; 94:2626-31; PMID:9122246
- Mondorf S, Deppenmeier U, Welte C. A novel inducible protein production system and neomycin resistance as selection marker for Methanosarcina mazei. Archaea 2012; 2012:973743; PMID:22851906; https://doi.org/10.1155/2012/973743
- Nickel L, Weidenbach K, Jager D, Backofen R, Lange SJ, Heidrich N, Schmitz RA. Two CRISPR-Cas systems in Methanosarcina mazei strain Go1 display common processing features despite belonging to different types I and III. RNA Biol 2013; 10:779-91; PMID:23619576; http://doi.org/10.4161/rna.23928
- Prasse D, Thomsen J, De Santis R, Muntel J, Becher D, Schmitz RA. First description of small proteins encoded by spRNAs in Methanosarcina mazei strain Go1. Biochimie 2015; 117:138-48; PMID:25890157; https://doi.org/10.1016/j.biochi.2015.04.007
- Deppenmeier U, Blaut M, Mahlmann A, Gottschalk G. Reduced coenzyme F420: heterodisulfide oxidoreductase, a proton- translocating redox system in methanogenic bacteria. Proc Natl Acad Sci U S A 1990; 87:9449-53; PMID:11607121
- Ehlers C, Grabbe R, Veit K, Schmitz RA. Characterization of GlnK1 from Methanosarcina mazei strain Go1: complementation of an Escherichia coli glnK mutant strain by GlnK1. J Bacteriol 2002; 184:1028-40; PMID:11807063
- Dugar G, Herbig A, Forstner KU, Heidrich N, Reinhardt R, Nieselt K, Sharma CM. High-resolution transcriptome maps reveal strain-specific regulatory features of multiple Campylobacter jejuni isolates. PLoS Genet 2013; 9:e1003495; PMID:23696746; https://doi.org/10.1371/journal.pgen.1003495
- Berezikov E, Thuemmler F, van Laake LW, Kondova I, Bontrop R, Cuppen E, Plasterk RH. Diversity of microRNAs in human and chimpanzee brain. Nat Genet 2006; 38:1375-7; PMID:17072315; https://doi.org/10.1038/ng1914
- Forstner KU, Vogel J, Sharma CM. READemption-a tool for the computational analysis of deep-sequencing-based transcriptome data. Bioinformatics 2014; 30:3421-3; PMID:25123900; https://doi.org/10.1093/bioinformatics/btu533
- Hoffmann S, Otto C, Kurtz S, Sharma CM, Khaitovich P, Vogel J, Stadler PF, Hackermüller J. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput Biol 2009; 5:e1000502; PMID:19750212; https://doi.org/10.1371/journal.pcbi.1000502
- Freese NH, Norris DC, Loraine AE. Integrated genome browser: visual analytics platform for genomics. Bioinformatics 2016; 32:2089-95; PMID:27153568; https://doi.org/10.1093/bioinformatics/btw069
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002; 30:207-10; PMID:11752295
- Richter AS, Schleberger C, Backofen R, Steglich C. Seed-based INTARNA prediction combined with GFP-reporter system identifies mRNA targets of the small RNA Yfr1. Bioinformatics 2010; 26:1-5; PMID:19850757; https://doi.org/10.1093/bioinformatics/btp609
- Wright PR, Georg J, Mann M, Sorescu DA, Richter AS, Lott S, Kleinkauf R, Hess WR, Backofen R. CopraRNA and IntaRNA: predicting small RNA targets, networks and interaction domains. Nucleic Acids Res 2014; 42:W119-23; PMID:24838564; https://doi.org/10.1093/nar/gku359
- Veit K, Ehlers C, Schmitz RA. Effects of nitrogen and carbon sources on transcription of soluble methyltransferases in Methanosarcina mazei strain Gö1. J Bacteriol 2005; 187:6147-54; PMID:16109956; https://doi.org/10.1128/JB.187.17.6147-6154.2005
- Weidenbach K, Gloer J, Ehlers C, Sandman K, Reeve JN, Schmitz RA. Deletion of the archaeal histone in Methanosarcina mazei Go1 results in reduced growth and genomic transcription. Mol Microbiol 2008; 67:662-71; PMID:18086209; https://doi.org/10.1111/j.1365-2958.2007.06076.x
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, McWilliam H, Valentin F, Wallace IM, Wilm A, et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007; 23:2947-8; PMID:17846036; https://doi.org/10.1093/bioinformatics/btm404
- Heyne S, Will S, Beckstette M, Backofen R. Lightweight comparison of RNAs based on exact sequence-structure matches. Bioinformatics 2009; 25:2095-102; PMID:19189979; https://doi.org/10.1093/bioinformatics/btp065
- Bernhart SH, Hofacker IL, Will S, Gruber AR, Stadler PF. RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinformatics 2008; 9:474; PMID:19014431; https://doi.org/10.1186/1471-2105-9-474