
ABSTRACT
When given an option to choose among a set of alternatives and only one selection is right, one might stop and reflect over which one is best. However, the ribosome has no time to stop and make such reflections, proteins need to be produced and very fast. Eukaryotic translation initiation is an example of such a conundrum. Here, scanning for the correct codon match must be fast, efficient and accurate. We highlight our recent computational findings, which show how the initiation machinery manages to recognize one specific codon among many possible challengers, by fine-tuning the energetic landscape of base-pairing with the aid of the initiation factors eIF1 and eIF1A. Using a recent 3-dimensional structure of the eukaryotic initiation complex we have performed simulations of codon recognition in atomic detail. These calculations provide an in-depth energetic and structural view of how discrimination against near-cognate codons is achieved by the initiation complex.
Introduction
To start the synthesis of proteins on the ribosome, the small ribosomal subunit first has to be prepared and loaded with mRNA so that it can later couple uniquely with a correct (cognate) anticodon present in an initiator tRNA. The way in which bacteria and eukaryotes carry out this codon-anticodon recognition process is quite different. In bacteria, 3 initiation factors (IF1, IF2 and IF3) are required, as is the positioning of the mRNA onto the small subunit by pairing the Shine-Dalgarno sequenceCitation1 with the complementary sequence of the 16S rRNA. Between this sequence and the start codon there are usually 8 base pairs, which properly positions the start codon at the ribosomal P-site so that initiator tRNA in complex with IF2 can bind.
In eukaryotes, on the other hand, the initiation process is considerably more complex. The initiation factors eIF1, eIF1A and eIF3 bind to the small 40S ribosomal subunit followed by initiator tRNA, eIF2•GTP and eIF5, creating the 43S pre-initiation complex (PIC). The PIC is then loaded near the 5’-cap of an activated complex formed between mRNA and different proteins (eIF4A, eIF4B, eIF4E, eIF4G and poly(A)-binding protein).Citation2 In eukaryotes, in contrast to bacteria where the start codon is already positioned in the P-site, the initiator tRNA is the molecule already positioned at the P-site but the start codon is not. Correct positioning of the start codon instead occurs via a scanning mechanism where the mRNA is threaded base by base through the PIC. During scanning the initiation complex will scan lots of near cognate start codons with the initiator tRNA switching between two conformations, a closed Pin state and an open Pout state.Citation3 In the Pin conformation one mRNA triplet at a time is either accepted or rejected as a start codon by the initiator tRNA. If rejected, the initiator tRNA goes to the Pout state and allows a new triplet to be set up in the P-site. This scanning process continues until the correct AUG start codon is found (for a structural overview, see ).
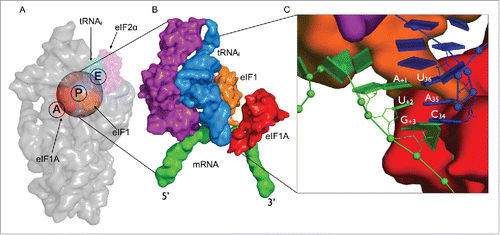
Figure 1. Structural overview of the eukaryotic initiation complex (A) Small subunit of the eukaryotic ribosome (gray) showing the initiation factors eIF1 (red), eIF1A (orange) and eIF2-α (purple) in complex with initiator tRNA (blue) bound to the AUG start codon (green). The transparent sphere on top of the P-site is the volume where the simulations are focused. (B) A close-up view onto the mRNA-tRNAi complex in surface representation illustrating the proximity of the initiation factors to the codon-anticodon minihelix. (C) A zoomed in view of the codon-anticodon triplet is represented as blocks using the standard reference frame for nucleic acids. eIF1 can be seen to bind closer to the first codon position toward the E-site while eIF1A is located in the A-site closer to the third codon position. The rRNA and proteins are omitted for clarity.
To show how the initiator tRNA distinguishes one nucleotide from another, which only differ by a few atoms, we have done detailed molecular dynamics (MD) computer simulations of the yeast PIC.Citation4 To explore how the initial codon selection occurs we used free energy perturbation (FEP) calculationsCitation5,6 where the relative free energy of binding can be computed between two different states. In practice, one base is then changed into another during the MD simulations and free energy differences can be calculated. With this method one can carefully explore the preference of the initiator tRNA for binding the cognate start codon in comparison to near-cognate codons, which as we show are energetically penalized. We calculated the binding free energies relative to AUG for the near-cognate codons GUG, CUG, ACG, AAG, AUA and AUC in three different cases. That is, when both eIF1 and eIF1A are present, when only eIF1 is bound and the case with neither of the two initiation factors bound. The results show that the presence of both eIF1 and eIF1A is fundamental for distinguishing correct from incorrect codons, but that the 2 factors have different roles.Citation7
Discrimination of near-cognate codons
We tried to examine to what extent a highly regulated process such as eukaryotic translation initiation, which differs significantly from its bacterial counterpart, can go wrong in reading the specific start codon. Not surprisingly, calculations on the initiation complex, including initiator tRNA and eukaryotic initiation factors 1 and 1A, show that the AUG start codon is strongly preferred over all other tested codons (we tested 6 out of 9 possible single base changes from the AUG codon). The striking result is that the discrimination strength by which initiator tRNA manages to distinguish right from wrong dramatically decreased once the initiation factors were left out of the calculations. This effect was larger for near-cognate mismatches in the flanking mRNA nucleotides at the 1st and 3rd codon positions than for the middle codon nucleotide, presumably since the latter is more constrained at the center of the codon-anticodon mini-helix. That is, from a structural viewpoint, binding of eIF1 and eIF1A appears to remedy the intrinsically higher conformational freedom of the two flanking base pair positions of the codon-anticodon complex when near-cognate mismatches are sensed. Moreover, in presence of the two initiation factors, a near-cognate 1st position G-U mismatch and C-A mismatches in the 2nd and 3rd position show a uniform strength of discrimination. The presence of eIF1 was found to mainly affect the 1st position fidelity, while eIF1A mainly acts on the 3rd position. The three near-cognate start codons GUG, ACG and AUA also show an equal efficiency of translation initiation in vivo.Citation8 The calculations further predict that a 2nd position A-A mismatch will have the highest energetic penalty of the examined codons (8.4 kcal/mol), in agreement with experiments that clearly show AAG to be non-functional as initiation codon.Citation8,9 With these computational results we can get a precise view of how the codon-anticodon binding energetics are fine-tuned due to the presence of the initiation factors eIF1 and eIF1A in the close vicinity of the codon recognition site. The resulting energetics show the crucial importance of eIF1 and eIF1A for correct start codon selection.
The basic principle of boosting the discriminatory power of codon-anticodon matching by binding of additional elements resembles that used in codon reading during the regular peptide elongation cycle, although the mechanisms are different. In that case, computer simulations showed that the conformational change of the monitoring bases (nucleotides A1492 and A1493 - E. coli numbering) on the small ribosomal subunit increase codon reading fidelity, primarily by excluding solvent from the codon-anticodon minihelix.Citation10 Likewise, the ms2i6A tRNAPhe modification at position 37 was shown to exclusively increase the penalty against a 1st position codon mismatch, essentially by lying on top of the mismatched base pair and preventing its relaxation to a more favorable conformation.Citation10 In the context of codon reading and the interplay between proteins and nucleic acids, computer simulations have also turned out to be very useful for determining the precise energetics, fidelity and structural mechanisms of stop codon reading, both by bacterial and mitochondrial release factors.Citation11,12
For the present review, we also decided to test the intrinsic discrimination strength that a bare tRNA would bring to start codon recognition, that is, for a complex of just mRNA and initiator tRNA in water. We thus performed free energy perturbation calculations, analogous to the ones described in Citationref. 7 to examine single purine to purine and pyrimidine to pyrimidine base changes. In this case the simulations were done in a smaller water sphere of radius 20 Å merely containing the tRNA, mRNA and counter ions. We can see from that the intrinsic penalty against near-cognate mismatches in the first two positions is similar with a value of about 4 kcal/mol, while that of the last codon position is smaller. Interestingly, the ribosomal P-site itself (without eIF1 and eIF1A) does not change this energetic picture much. The results are also very similar to those obtained for codon reading in the A-site when the monitoring bases (A1492 and A1493) do not interact with the codon-anticodon minihelix.Citation10 The calculated intrinsic penalty against the first position G-U mismatch is surprisingly high, but this may be due to the fact that the tRNA-mRNA conformation from the ribosome complex was used, which imposes structural constraints on the codon-anticodon pair that would not be found in a regular RNA duplex structure.
Table 1. Binding free energy penalties (kcal/mol) for mutating the canonical AUG start codon to near-cognate codons at the three codon positions.Footnotea
Overall, this underscores the fact that although there is an intrinsic and well-known discriminatory power against incorrect codon triplets, where that of the 3rd position is weaker, other factors are of major importance for improving fidelity on the ribosome. Such factors involve both tRNA modifications,Citation13,14 binding of proteins such as eIF1 and eIF1A, as well as conformational changes of the monitoring rRNA bases.
Codon selection in translation is not entirely universal
Even though the genetic code is often seen as universal, it has been known for a long timeCitation15 that there are numerous deviations from it throughout known life forms.Citation16,17 As far as initiation of translation in the eukaryotic cell is concerned, many reports indicate that non-AUG codons are and can be used as alternative initiation codons.Citation18 It might be the case that Met-tRNAi is not the only initiator tRNA. For example, Starck et al. showed that Leu-tRNA can be the initiator at CUG start codonsCitation19 but possibly not as efficiently as AUG.Citation20,21 Moreover, there are various reports as to which alternative start codon would be most preferred. Kolitz et al. rank CUG as the best option based on their experiments using a luciferase reporter assay.Citation8 Peabody et al. obtain ACG as their best candidate based on their study of translation of dihydrofolate reductase in mammalian cellsCitation9 and Donahue and Cigan find that AUA is preferred in S. cerevisiae with a HIS4-lacZ fusion construct.Citation22 There can be various reasons for why different alternative initiation codons are reported and perhaps there is no preferred alternative start codon at all. Here, the details of how the experiments are set up may be an explanation for the possible discrepancies. For instance, the lengths of the 5’UTR's can vary, or there might be deviations in the sequences making the Kozak context suboptimal for the tested start codon in the different studies.Citation23 Specifically, both CUG and ACG have been reported to be used in initiation with up to 15% of the efficiency of AUG.Citation8,9 Our calculations, however, suggest that with the standard eukaryotic initiation complex with eIF1 and eIF1A present, the selection for AUG is extremely precise. If these initiation factors are not present it is possible to point out a few codons that could probably be read by Met-tRNAi. Hence, the reading of GUG, CUG and AUA is predicted to be considerably easier in the absence of the initiation factors. Looking into other translational systems, that do not have the same level of regulation as the eukaryotic cytosolic initiation process, we can see that a wide range of alternative start codons are being used. In yeast mitochondria, the AUA codon is reassigned from the standard isoleucine to methionine and it is decoded by Met-tRNA, thus making it a suitable alternative start codon. In fact, in some vertebrate species AUA and GUG are used as an alternative initiation codon in mitochondrial translation.Citation17,24
The symbiosis of biochemistry, structural biology and computer simulations
A little over a hundred years ago, the first X-ray diffraction patterns produced by crystals enabled solution of their 3-dimensional structures.Citation25 Development in the field of structural biology is ongoing at an even faster and wider reaching pace than before.Citation26 Today structures can be solved relatively fast and, in favorable cases, true atomic detail can be attained for very complex biologic structures and assemblies. However, the 3D structures essentially only provide static pictures of continuously dynamic biologic machines. The methods developed in the computational field are among the most suitable tools for analyzing both conformational transitions in large systems and for analysis of the detailed chemistry and energetics of reactions and intermolecular interactions.
The main purpose of the ribosome is to take in mRNA and translate its message to produce functional proteins. However, from the beginning to the end of translation, a whole plethora of different events take place. The 3D structures determined for ribosomes and their complexes can only give snapshots of the overall process and they often lack true atomic detail which only emerges at around 2 Å resolution. Hence, the direct connection to the biochemistry and kinetics of translation is often far from obvious, in the sense that detailed chemical mechanisms are not immediately apparent from the structures. This is the case for specific events such as translation initiation using different start codonsCitation8,9,22 or addition of amino acids in elongation by different tRNAs.Citation27-31 Computational chemistry techniques definitely have a role to play here, by allowing us to piece together the structural and biochemical interpretations of what is going on. However, in order for computations to be reliable they dependent strongly on the availability of sufficiently accurate 3D structures. Due to the relatively modest resolution of many of these structures, the univocal positioning of water molecules, counter ions and even key functional protein or RNA groups may be impaired. This situation often requires rather tedious initial computer simulations to verify that structural and energetic results really make sense. However, MD simulations certainly also have the potential to make relevant predictions ahead of experiments. Here, one can mention prediction of the hydrogen bonding network in the peptidyl transferase center that appears to explain the unusually favorable activation entropy for the peptidyl transfer reaction.Citation32 Another case in point is the prediction that the conformational change of the monitoring bases is associated with GTPase activation in the initial selection of elongator tRNAs,Citation10 a conclusion also reached in recent experimental work.Citation33
Hence it is clear that computer simulations have become very useful for addressing some of the key problems of the ribosomal translational machinery, ranging from large movements involving the full ribosomeCitation34 to the mechanisms of tRNA selection,Citation10, 35-37 peptide elongation,Citation27,32,38-40 translocationCitation41,42 and mechanisms of translation termination.Citation11,12,43-45 The whole toolbox of computational chemistry is powerful and includes quantum mechanical calculations, molecular dynamics free energy simulations, coarse-grained methods and elastic network models, as well as molecular dockingCitation44 and homology modeling.Citation12 The future deluge of structural information from cryoEM is further likely to increase the demand for ribosome computer simulation efforts to piece together the information from such experiments as well as traditional X-ray crystallography.
As far as translation initiation in eukaryotes is concerned, it can be further explored with the help of computer simulations. For example, does the initial codon recognition just involve the codon-anticodon base pairs, or is there any influence from the other leaves of the tRNA clover?Citation46 Another interesting issue that could be addressed is the influence of Kozak's optimum sequence context,Citation23 which could be computationally analyzed by mutating upstream nucleotides and examining the effects on start codon recognition. Furthermore, scanning must be done one base at a time so that reading of each possible codon is not missed. Here, one could use MD simulations to explore the ATP-driven mechanism of mRNA threading, and how this couples with the conformational changes between the Pin and Pout states.Citation47
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Shine J, Dalgarno L. The 3'-terminal sequence of Escherichia coli 16S ribosomal RNA: Complementarity to nonsense triplets and ribosome binding sites. Proc Natl Acad Sci U S A 1974; 71:1342-6; PMID:4598299; https://doi.org/10.1073/pnas.71.4.1342
- Hinnebusch AG. The scanning mechanism of eukaryotic translation initiation. Annu Rev Biochem 2014; 83:779-812; PMID:24499181; https://doi.org/10.1146/annurev-biochem-060713-035802
- Llácer JL, Hussain T, Marler L, Aitken CE, Thakur A, Lorsch JR, Hinnebusch AG, Ramakrishnan V. Conformational differences between open and closed states of the eukaryotic translation initiation complex. Mol Cell 2015; 59:399-412; PMID:26212456; https://doi.org/10.1016/j.molcel.2015.06.033
- Hussain T, Llácer JL, Fernández IS, Munoz A, Martin-Marcos P, Savva CG, Lorsch JR, Hinnebusch AG, Ramakrishnan V. Structural changes enable start codon recognition by the eukaryotic translation initiation complex. Cell 2014; 159:597-607; PMID:25417110; https://doi.org/10.1016/j.cell.2014.10.001
- Brandsdal BO, Osterberg F, Almlöf M, Feierberg I, Luzhkov VB, Åqvist J. Free energy calculations and ligand binding. Adv Protein Chem 2003; 66:123-58; PMID:14631818; https://doi.org/10.1016/S0065-3233(03)66004-3
- Zwanzig RW. High-temperature equation of state by a perturbation method. I. Nonpolar gases. J Chem Phys 1954; 22:2099; https://doi.org/10.1063/1.1740409
- Lind C, Åqvist J. Principles of start codon recognition in eukaryotic translation initiation. Nucleic Acids Res 2016; 44:8425-32; PMID:27280974; https://doi.org/10.1093/nar/gkw534
- Kolitz SE, Takacs JE, Lorsch JR. Kinetic and thermodynamic analysis of the role of start codon/anticodon base pairing during eukaryotic translation initiation. RNA 2009; 15:138-52; PMID:19029312; https://doi.org/10.1261/rna.1318509
- Peabody DS. Translation initiation at non-AUG triplets in mammalian cells. J Biol Chem 1989; 264:5031-5; PMID:2538469.
- Satpati P, Sund J, Åqvist J. Structure-based energetics of mRNA decoding on the ribosome. Biochemistry 2014; 53:1714-22; PMID:24564511; https://doi.org/10.1021/bi5000355
- Sund J, Andér M, Åqvist J. Principles of stop-codon reading on the ribosome. Nature 2010; 465:947-50; PMID:20512119; https://doi.org/10.1038/nature09082
- Lind C, Sund J, Åqvist J. Codon-reading specificities of mitochondrial release factors and translation termination at non-standard stop codons. Nat Commun 2013; 4:2940; PMID:24352605; https://doi.org/10.1038/ncomms3940
- Björk GR, Ericson JU, Gustafsson CE, Hagervall TG, Jönsson YH, Wikström PM. Transfer RNA modification. Annu Rev Biochem 1987; 56:263-87; PMID:3304135; https://doi.org/10.1146/annurev.bi.56.070187.001403
- Grosjean H, de Crécy-Lagard V, Marck C. Deciphering synonymous codons in the three domains of life: Co-evolution with specific tRNA modification enzymes. FEBS Lett 2010; 584:252-64; PMID:19931533; https://doi.org/10.1016/j.febslet.2009.11.052
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, et al. Sequence and organization of the human mitochondrial genome. Nature 1981; 290:457-65; PMID:7219534; https://doi.org/10.1038/290457a0
- Osawa S, Jukes TH, Watanabe K, Muto A. Recent evidence for evolution of the genetic code. Microbiol Rev 1992; 56:229-64; PMID:1579111.
- Jukes TH, Osawa S. Evolutionary changes in the genetic code. Comp Biochem Physiol, B 1993; 106:489-94; PMID:8281749; https://doi.org/10.1016/0300-9629(93)90243-W
- Elzanowski AA, Ostell J. The genetic codes. National Center for Biotechnology Information; 2016 Nov 18 [accessed 2017 Jan 30]. https://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi.
- Starck SR, Jiang V, Pavon-Eternod M, Prasad S, McCarthy B, Pan T, Shastri N. Leucine-tRNA initiates at CUG start codons for protein synthesis and presentation by MHC class I. Science 2012; 336:1719-23; PMID:22745432; https://doi.org/10.1126/science.1220270
- Starck SR, Ow Y, Jiang V, Tokuyama M, Rivera M, Qi X, Roberts RW, Shastri N. A distinct translation initiation mechanism generates cryptic peptides for immune surveillance. PLoS One 2008; 3:e3460; PMID:18941630; https://doi.org/10.1371/journal.pone.0003460
- Starck SR, Shastri N. Nowhere to hide: Unconventional translation yields cryptic peptides for immune surveillance. Immunological Rev 2016; 272:8-16; PMID:27319338; https://doi.org/10.1111/imr.12434
- Donahue TF, Cigan AM. Genetic selection for mutations that reduce or abolish ribosomal recognition of the HIS4 translational initiator region. Mol Cell Biol 1988; 8:2955-63; PMID:3043200; https://doi.org/10.1128/MCB.8.7.2955
- Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell 1986; 44:283-92; PMID:3943125; https://doi.org/10.1016/0092-8674(86)90762-2
- Desjardins P, Morais R. Nucleotide sequence and evolution of coding and noncoding regions of a quail mitochondrial genome. J Mol Evol 1991; 32:153-61; PMID:1706782; https://doi.org/10.1007/BF02515387
- Bragg WL. The structure of some crystals as indicated by their diffraction of X-rays. Proc R Soc A 1913; 89:248-77; https://doi.org/10.1098/rspa.1913.0083
- Berman HM, Coimbatore Narayanan B, Di Costanzo L, Dutta S, Ghosh S, Hudson BP, Lawson CL, Peisach E, Prlić A, Rose PW, et al. Trendspotting in the protein data bank. FEBS Lett 2013; 587:1036-45; PMID:23337870; https://doi.org/10.1016/j.febslet.2012.12.029
- Johansson M, Ieong K-W, Trobro S, Strazewski P, Åqvist J, Pavlov MY, Ehrenberg M. pH-sensitivity of the ribosomal peptidyl transfer reaction dependent on the identity of the A-site aminoacyl-tRNA. Proc Natl Acad Sci U S A 2011; 108:79-84; PMID:21169502; https://doi.org/10.1073/pnas.1012612107
- Zhang J, Ieong K-W, Johansson M, Ehrenberg M. Accuracy of initial codon selection by aminoacyl-tRNAs on the mRNA-programmed bacterial ribosome. Proc Natl Acad Sci U S A 2015; 112:9602-7; PMID:26195797; https://doi.org/10.1073/pnas.1506823112
- Gromadski KB, Rodnina MV. Kinetic determinants of high-fidelity tRNA discrimination on the ribosome. Mol Cell 2004; 13:191-200; PMID:14759365; https://doi.org/10.1016/S1097-2765(04)00005-X
- Gromadski KB, Daviter T, Rodnina MV. A uniform response to mismatches in codon-anticodon complexes ensures ribosomal fidelity. Mol Cell 2006; 21:369-77; PMID:16455492; https://doi.org/10.1016/j.molcel.2005.12.018
- Kuhlenkoetter S, Wintermeyer W, Rodnina MV. Different substrate-dependent transition states in the active site of the ribosome. Nature 2011; 476:351-4; PMID:21804565; https://doi.org/10.1038/nature10247
- Trobro S, Åqvist J. Mechanism of peptide bond synthesis on the ribosome. Proc Natl Acad Sci U S A 2005; 102:12395-400; PMID:16116099; https://doi.org/10.1073/pnas.0504043102
- Fischer N, Neumann P, Bock LV, Maracci C, Wang Z, Paleskava A, Konevega AL, Schröder GF, Grubmüller H, Ficner R, et al. The pathway to GTPase activation of elongation factor SelB on the ribosome. Nature 2016; 540:80-5; PMID:27842381; https://doi.org/10.1038/nature20560
- Sanbonmatsu KY, Joseph S, Tung C-S. Simulating movement of tRNA into the ribosome during decoding. Proc Natl Acad Sci U S A 2005; 102:15854-9; PMID:16249344; https://doi.org/10.1073/pnas.0503456102
- Allner O, Nilsson L. Nucleotide modifications and tRNA anticodon-mRNA codon interactions on the ribosome. RNA 2011; 17:2177-88; PMID:22028366; https://doi.org/10.1261/rna.029231.111
- Satpati P, Bauer P, Åqvist J. Energetic tuning by tRNA modifications ensures correct decoding of isoleucine and methionine on the ribosome. Chem Eur J 2014; 20:10271-5; PMID:25043149; https://doi.org/10.1002/chem.201404016
- Zeng X, Chugh J, Casiano-Negroni A, Al-Hashimi HM, Brooks CL. Flipping of the ribosomal A-site adenines provides a basis for tRNA selection. J Mol Biol 2014; 426:3201-13; PMID:24813122; https://doi.org/10.1016/j.jmb.2014.04.029
- Wallin G, Åqvist J. The transition state for peptide bond formation reveals the ribosome as a water trap. Proc Natl Acad Sci U S A 2010; 107:1888-93; PMID:20080677; https://doi.org/10.1073/pnas.0914192107
- Sharma PK, Xiang Y, Kato M, Warshel A. What are the roles of substrate-assisted catalysis and proximity effects in peptide bond formation by the ribosome? Biochemistry 2005; 44:11307-14; PMID:16114867; https://doi.org/10.1021/bi0509806
- Świderek K, Martí S, Tuñón I, Moliner V, Bertrán J. Peptide bond formation mechanism catalyzed by ribosome. J Am Chem Soc 2015; 137:12024-34; PMID:26325003; https://doi.org/10.1021/jacs.5b05916
- Bock LV, Blau C, Schröder GF, Davydov II, Fischer N, Stark H, Rodnina MV, Vaiana AC, Grubmüller H. Energy barriers and driving forces in tRNA translocation through the ribosome. Nat Struct Mol Biol 2013; 20:1390-6; PMID:24186064; https://doi.org/10.1038/nsmb.2690
- Nguyen K, Whitford PC. Steric interactions lead to collective tilting motion in the ribosome during mRNA–tRNA translocation. Nat Commun 2016; 7:10586; PMID:26838673; https://doi.org/10.1038/ncomms10586
- Kazemi M, Himo F, Åqvist J. Peptide release on the ribosome involves substrate-assisted base catalysis. ACS Catalysis 2016; 6:8432-8439; https://doi.org/10.1021/acscatal.6b02842
- Trobro S, Åqvist J. A model for how ribosomal release factors induce peptidyl-tRNA cleavage in termination of protein synthesis. Mol Cell 2007; 27:758-66; PMID:17803940; https://doi.org/10.1016/j.molcel.2007.06.032
- Acosta-Silva C, Bertran J, Branchadell V, Oliva A. Quantum mechanical study on the mechanism of peptide release in the ribosome. J Phys Chem B 2013; 117:3503-15; PMID:23442058; https://doi.org/10.1021/jp3110248
- Kapp LD, Kolitz SE, Lorsch JR. Yeast initiator tRNA identity elements cooperate to influence multiple steps of translation initiation. RNA 2006; 12:751-64; PMID:16565414; https://doi.org/10.1261/rna.2263906
- Kozak M. Role of ATP in binding and migration of 40S ribosomal subunits. Cell 1980; 22:459-67; PMID:7448869; https://doi.org/10.1016/0092-8674(80)90356-6