
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
RNA in yeast, especially rRNA and tRNA are heavily modified to fulfill their function in protein translation. Using biosynthetic stable isotope labeled internal standards we quantified 12 modified nucleosides in tRNA from S. cerevisiae over 24 hours. We observed different quantities of modified nucleosides in dependence of the growth phase. To elucidate the underlying mechanism of the observed tRNA modification profile adaptation, it is necessary to distinguish the pre-existing tRNA pool and its modifications from newly-synthesized tRNAs. By combination of 2 differentially isotope labeled media we developed NAIL-MS, nucleic acid isotope labeling coupled mass spectrometry. During the yeast growth cycle we observe dilution of pre-existing tRNAs by newly-synthesized tRNAs by the growing number of cells. tRNA was found to be highly stable with only little degradation over the observed period. The method was further used to quantify the levels of modified nucleosides in the original and new tRNA pools. By addition of deuterium-labeled methionine, we could observe the incorporation of new methyl marks on pre-existing tRNAs. For 2′-O-methylcytidine (Cm) we observed a global increase in log phase. We identified extensive 2′-OH-cytidine methylation of the pre-existing tRNAs and the new tRNAs which masks an actual decrease of pre-existing Cm. In contrast, global 5-methylcytidine (m5C) levels decreased during growth due to a drop in m5C quantities in the original tRNA pool.
The NAIL-MS data suggests different mechanisms for tRNA modification adaptation depending on the individual modification observed. With this new tool it is possible to follow the fate of methylated RNAs during growth and potentially compare the impact of different stress conditions on the epitranscriptome.
Introduction
The central dogma of molecular biology states that DNA is the storage of the genetic code, which is transcribed into (messenger) RNA, which is translated into proteins by another (transfer) RNA. This fundamental life process is dominated by nucleic acids, which are composed of the 4 canonical nucleosides cytidine, guanosine, adenosine and thymidine in DNA and uridine in RNA, respectively. The sequence of these building blocks defines the genetic code which contains all information necessary for each organism. While DNA is the storage of the genetic code, RNAs can be found as regulators and effectors of all essential life processes. Especially tRNA (tRNA) and rRNA (rRNA) are key players in protein translation and therefore highly abundant. To fulfill their function, both tRNA and rRNA are extensively modified and across all domains of life more than 150 modified RNA nucleosides have been identified.Citation1 In recent years, the term epitranscriptomics has emerged to describe the modification level and profile of RNAs. The modification is introduced by dedicated RNA modifying enzymes which are referred to as RNA writers. Common RNA writers include methyltransferases, thiotransferases and pseudouridine-synthases and each writer has a specific substrate RNA. While modifications in tRNA and rRNA have been known for decades, modifications in mRNA have only recently been described.Citation2,3 Alongside mRNA writer enzymes, so-called RNA erasers have been identified which actively demethylate the methylated adenosine in mRNA.
Like all organisms analyzed to date, S. cerevisiae tRNAs are heavily modified as recently reviewed.Citation4 Over 25 different modified nucleosides have been identified in these tRNAs, of which 12 are mostly S-adenosyl methionine methylated derivatives of the canonical nucleosides. The quantities of tRNA modifications are known to adapt during cell growthCitation5 and during stress.Citation6 Furthermore they are essential for protein homeostasis and cell morphogenesis.Citation7 The dynamic adaptation of tRNA modifications prompts to ask the question how tRNA modification levels are changed from a mechanistic viewpoint. One possibility is an addition of modifications by tRNA writers to the already existing tRNA pool and the newly synthesized tRNA which results in increased tRNA modification levels. A decrease of tRNA modification levels might be achieved by 2 possible scenarios. The first includes the participation of tRNA erasers that actively remove existing modifications from the tRNA. While RNA erasers are known to exist for mRNAs, only recently ALKBH1 was observed in vitro to demethylate 1-methyladenosine to adenosine in certain tRNAs.Citation8 Another potential way to remove RNA modifications is degradation of the existing, modified RNA and synthesis of new, unmodified RNA. However, current methods cannot distinguish the modification profile of the existing RNA pool and the new RNA pool. Here we present a new technique, NAIL-MS (nucleic acid isotope labeling coupled mass spectrometry) which allows observation of modification dynamics in existing and newly synthesized RNAs from S. cerevisiae during growth.
Results
tRNA modification quantities fluctuate during growth
tRNA from yeast was purified from total RNA by size-exclusion chromatography (SEC) as described previouslyCitation9 and the tRNA fraction collected. shows an exemplary chromatogram of total RNA before SEC and purified tRNA after SEC. The large RNA is a mixture of rRNA and other large RNA molecules like mRNA (Fig. S1). The isolated tRNA was digested to nucleosides and subjected to liquid chromatography coupled mass spectrometry (LC-MS). An exemplary chromatogram for detection of the canonical nucleosides and 13 abundant modified nucleosides can be found in . The less common modifications like 2′-O-methyluridine (Um) or wobble-uridines like 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2U) were not included in the current study due to low detection sensitivity of uridine derivatives. Dihydrouridine was not available as a synthetic standard and could not be included in quantification analysis. Using a biosynthetic stable isotope labeled internal standard (SILIS), produced according toCitation10 and calibration with synthetic standards, we quantified the absolute level of modification per tRNA in log phase growing cells (calibration curves in Fig. S2). Fig. S3 shows a scheme how RNA modifications are quantified and which calculations and normalization steps are necessary to arrive at the number of a given modification per tRNA.
Figure 1. Workflow to determine modification levels in tRNA during yeast growth cycle. The isolated total RNA was purified by size exclusion chromatography (SEC) (*5.8S/5S rRNA peak, #unknown contamination). The tRNA fraction was then digested and the obtained nucleosides separated and quantified over LC-MS to receive the shown chromatogram on the upper right side. The average amounts of the respective modifications per molecule tRNA in log growing cells were calculated and plotted (error bars represent SD of n = 3). The quantification was repeated during growth (arrows indicate sample time points) and the quantities of the modifications Cm and m7G were plotted over time. Abbreviations: D = dihydrouridine, Ψ = pseudouridine, C = cytidine, m1A = 1-methyladenosine, U = uridine, m5C = 5-methylcytidine, m7G = 7-methylguanosine, Cm = 2′-O-methylcytidine, I = Inosine, G = guanosine, m5U = 5-methyluridine, m1G = 1-methylguanosine, Gm = 2′-O-methylguanosine, m2G = N2-methylguanosine, A = adenosine, m22G = N2,N2-dimethylguanosine, Am = 2′-O-methyladenosine.
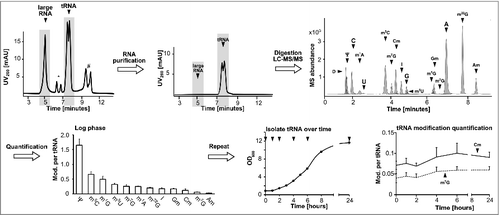
In total tRNA from log growing cells we find pseudouridine (Ψ) as the most abundant modified nucleoside, followed by the methylated nucleosides 5-methylcytidine (m5C), 1-methylguanosine (m1G) and 5-methyluridine (m5U or rT) as expected (compare Table S1 prepared fromCitation11). Here, the modification profile of 34 tRNA sequences is listed. For total tRNA the exact number of modified nucleosides depends on the ratio of the isoacceptors and isodecoders. This ratio is dynamic and used as an adaptation mechanism to overcome changes in growth conditions and external stress.Citation12,13 This adaptation and the unknown modification status of most tRNAs make it difficult to assign a quantitative number of modifications to the total tRNA pool.
During these initial experiments to quantify tRNA modifications in S. cerevisiae BY4741 we noticed fluctuations in the quantities of many modifications in dependence of the growth phase (Fig. S4). 2′-O-methylcytidine (Cm), 1-methylguanosine (m1G) and 7-methylguanosine (m7G) levels increased in log phase ( and Fig. S4). In contrast, we observed a drop in 5-methylcytidine (m5C), 5-methyluridine (m5U) and pseudouridine (Ψ) quantities in log phase. For modified nucleosides 2′-O-methylguanosine (Gm), 2′-O-methyladenosine (Am), 2-methylguanosine (m2G), 2,2-dimethylguanosine (m22G), inosine (I) and 1-methyladenosine (m1A) the levels stayed constant throughout growth (Fig. S4).
While we cannot explain this phenomenon at the moment we were curious to understand how the levels of modifications are de- or increased mechanistically. This prompts the question whether a change of modification quantity is caused by hyper- or hypomodified new tRNAs while the pre-existing tRNAs stay unaltered or by dynamic adaptation of the pre-existing tRNAs.
Developing media for nucleic acid isotope labeling
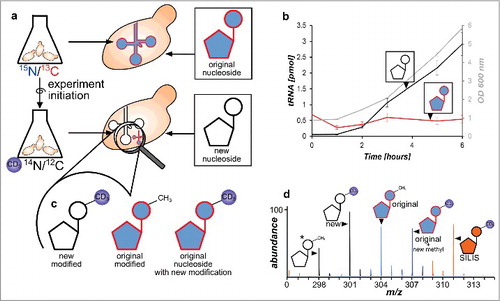
Intrigued by these results we wanted to design an isotope based assay which allows discrimination of the original tRNA pool and newly synthesized tRNAs by mass spectrometry. For this type of experiments it is necessary to have 2 growth media that result in differential labeling and therefore different masses of the nucleosides. We have tested commercial rich growth medium (Silantes, Munich, Germany) which allows efficient labeling of nucleobases with either 15Nitrogen or 13Carbon while the ribose remains unlabeled when supplemented with regular 12C-glucose (see Fig. S5). In these media it is possible to introduce additional heavy carbons in the ribose by supplementing with 13C6-glucose. Furthermore it is possible to supplement with D3-methionine which resulted in a 60–70% labeling efficiency of methylated nucleosides with D3-labeled methyl groups (see Fig. S6). The commercial medium was chosen for preparation of the stable isotope labeled internal standard (SILIS) to receive a max. labeled internal standard. The SILIS nucleosides have a +9 mass shift for uridine and cytidine, and +12 for methylated uridine and cytidine modifications (see Table S2). The purines, guanosine and adenosine, have a +10 while their methylated derivatives have a +13 mass shift (except m22G with a mass shift of +16).
For the time course experiments, the commercial media was not suitable since some modifications like 5-methylcytidine would have formed isotopomers with the same m/z as its corresponding SILIS nucleoside. Therefore, we tested the labeling efficiency in minimal medium (YNB) supplemented with isotope labeled amino acids, glucose and nucleobases. With D3-methionine, labeling of methylated nucleosides was achieved quickly (increase in deuterated over unlabeled nucleosides shown in Fig. S7). As S. cerevisiae BY4741 is a uracil auxotroph strain, we supplemented with 15N2-uracil which resulted in a complete labeling of pyrimidines and their modifications. For purine labeling we added the purine biosynthesis precursors 15N-amide labeled glutamine or 15N-aspartic acid. Although these amino acids are the nitrogen source of purines,Citation14 the incorporation efficiency was very low (Fig. S8). However, a quantitative +1 purine base label could be achieved by growth in 13C6-glucose supplemented YNB medium which results in a purine nucleoside mass increase of +6 (+1 from base and +5 from ribose, and Fig. S9) and +5 for pyrimidines (+5 from ribose). From these results a combination of 15N2-uracil with 13C6-glucose minimal medium was chosen as the first medium for our experiments. The nucleoside and nucleobase m/z values and their mass increase compared with unlabeled nucleosides for the canonicals are found in and Fig. S10.
Figure 2. Mass spectra of guanosine (G) and 7-methylguanosine (m7G) in different growth media. a unlabeled medium: [M+H]+ (G) 284, [M+H]+ (m7G) 298. b D3-methionine containing medium: [M+H]+ (G) 284, [M+H]+ (m7G) 301. c 15N2 uracil and 13C6 glucose containing medium: [M+H]+ (G) 290, [M+H]+ (m7G) 304. d 15N2 uracil, 13C6 glucose and D3 methionine: [M+H]+ (G) 290, [M+H]+ (m7G) 307. e SILIS: [M+H]+ (G) 294, [M+H]+ (m7G) 311.
![Figure 2. Mass spectra of guanosine (G) and 7-methylguanosine (m7G) in different growth media. a unlabeled medium: [M+H]+ (G) 284, [M+H]+ (m7G) 298. b D3-methionine containing medium: [M+H]+ (G) 284, [M+H]+ (m7G) 301. c 15N2 uracil and 13C6 glucose containing medium: [M+H]+ (G) 290, [M+H]+ (m7G) 304. d 15N2 uracil, 13C6 glucose and D3 methionine: [M+H]+ (G) 290, [M+H]+ (m7G) 307. e SILIS: [M+H]+ (G) 294, [M+H]+ (m7G) 311.](/cms/asset/d69d2b95-bd67-41cb-b955-e9412f6fa82b/krnb_a_1325063_f0002_oc.jpg)
Table 1. Mass transition and mass increase compared with non-labeled canonical nucleosides from “heavy” minimal medium with 15N2-uracil and 13C6 glucose label.
For the second medium we decided to use YNB minimal medium but supplemented with regular uracil (14N2-uracil) and regular 12C6-glucose which results in unlabeled canonicals (). By combination of these 2 media we can now distinguish the pre-existing nucleosides (heavy 15N/13C-label) and the newly synthesized tRNA nucleosides (unlabeled). To further allow a detailed analysis of the pre-existing tRNA modifications, we added D3-methionine to the growth medium which led to the incorporation of heavy methyl groups after medium exchange ( for newly synthesized modifications). In combination with the differential labeling of the nucleoside structure, this step allows to detect newly methylated nucleosides in the pre-existing tRNA pool by a mass increase of +3 ( and Fig. S10) compared with the pre-existing methylated nucleosides. We refer to this event as post-methylation as it happens after experiment initiation.
NAIL-MS (nucleic acid isotope labeling coupled mass spectrometry) reveals dilution of the pre-existing tRNA pool
In a next step we combined the 2 growth media in a single assay. The principle is shown in . The cells are grown overnight in 15N2-uracil/13C6-glucose supplemented YNB medium. To achieve complete labeling of all nucleosides with the heavy label the cells were centrifuged, resuspended in fresh 15N/13C YNB medium and grown overnight a second time. At this step high labeling efficiency of all nucleosides is required which was verified by high-resolution mass spectrometry (see Fig. S9 and S10). Minor signals of unlabeled nucleosides are due to impurities of the 13C6-glucose and 15N2-uracil starting material (99% ATOM 13C and 98% ATOM 15N purity) which causes <5% of unlabeled nucleosides at experiment start. For experiment initiation the cells are briefly centrifuged and suspended in D3-methionine supplemented YNB medium. Total RNA isolation and digestion to nucleosides followed by mass spectrometry revealed an increase of newly synthesized canonical nucleosides (light) from the new RNA pool during growth (Fig. S11, black line) while the original RNA was diluted (red). For modified nucleosides we performed mass spectrometry in scan mode after experiment initiation. As expected we could detect the signal of the new, D3-methylated nucleosides and the H3-methylated original nucleosides. However, we could also detect the predicted post-methylated nucleoside species with a mass increase of +3 from the original heavy isotope labeled RNA pool ( and Fig. S10). This nucleoside species is the result of additional methylation of canonicals that were not methylated in the pre-existing tRNA pool at experiment initiation. In summary, we can observe 3 different isotopomers of each methylated nucleoside () which originate from different RNA pools: 1) methylated nucleosides from the new RNA pool, 2) original nucleosides already methylated at experiment initiation and 3) original nucleosides, methylated AFTER experiment initiation.
Figure 3. Principle of the developed NAIL-MS assay and RNA species emerging over time. a Yeast is grown in heavy isotope medium which results in heavy labeled nucleosides. The experiment is initiated by medium exchange with D3-methionine labeled medium. New canonicals are unlabeled while pre-existing nucleosides remain 15N/13C labeled. b Original tRNA quantities stay constant over time (red), while new tRNAs are synthesized (black line) as cells grow (OD 600 gray line) (error bars are SD of n = 3). c Different modified nucleosides can be distinguished according to their labeling. New modified nucleosides have an unlabeled canonical building block and a D3-methyl mark, original modified nucleosides derive from labeled canonicals but carry the unlabeled methylation and original, labeled canonicals which receive a methyl mark after experiment initiation have an additional D3-methyl group. d Mass spectrum of potential isotopomers of 7-methylguanosine (m7G) in a NAIL-MS experiment: *unlabeled m7G as a reference (298), from new tRNA (301), from original tRNA with pre-existing methylation (304), from original tRNA but from post-methylation (307) and the signal of the m7G SILIS (311).
With this assay at hand we decided to grow yeast for 24 hours and observe the fate of canonical and modified nucleosides in tRNAs by sensitive triple quadrupole mass spectrometry. The mass transitions of canonical and modified nucleosides were programmed for each observed isotopomer into a multiple reaction monitoring (MRM) method (mass transitions in Table S2 and mass spectra in Fig. S9). The total RNA was isolated from 6 biologic replicates before experiment initiation (T = 0 hours), after 1, 2, 4 and 6 hours (lag phase and log phase) and 24 hours (stationary) as the last time point. Using size-exclusion chromatography we purified total tRNA and subjected all samples to the developed NAIL-MS method after digestion to nucleosides. By injection of tRNA digests we could quantify the amount of tRNA injected and separate the signal of the pre-existing and novel tRNA pool. This quantification revealed an initial degradation of the existing tRNAs in the first 60 to 120 minutes followed by constant levels thereafter (). We did not observe further tRNA degradation which is consistent with the reported long half-life of tRNA.Citation15,16 In similar pulse chase studies using tritium labeled uracil and subsequent gel electrophoretic quantification of the radioactively labeled yeast tRNA, no initial drop in tRNA levels was observed.Citation17 While this difference might be caused by technical errors during our sample collection and RNA isolation, we currently investigate the possibility of stress-induced tRNA degradation caused by the necessary pelleting of yeast cells before experiment initiation.
NAIL-MS reveals differences in the modification profile of the original and new tRNA pool
shows all signals of 7-methylguanosine (m7G) which potentially occur in a NAIL-MS experiment after medium exchange. For quantification of newly synthesized m7G, the signal for the unlabeled and D3-labeled isotopomers were summed and normalized to the amount of unlabeled tRNA injected (calculated from the signal of unlabeled adenosine). Thereby we received the number of m7G per new tRNA. Similarly we added the signal of the original m7G isotopomer and the post-methylated m7G (heavy nucleoside + heavy D3-methyl group) and normalized to the amount of pre-existing tRNA in the sample (calculated from the signal of labeled adenosine).
The plots of show the number of modifications per original tRNA or new tRNA, respectively, on the y-axis. In the unlabeled experiment () we observed growth phase dependent fluctuations for 7-methylguanosine (m7G). By NAIL-MS we identified the reason for the observed increase in a slow and steady increase of m7G in new tRNAs during log phase (). The number of m7G in the original tRNAs appears to stay constant throughout the experiment. However, a detailed analysis of the pre-existing tRNA pool reveals a constant decrease in the number of pre-existing m7G while new m7G is incorporated into the original tRNAs.
Figure 4. Levels of modifications in tRNA during a time range of 24 h discriminated by the original and newly-synthesized tRNA pool. *The quantity of modification was normalized to its respective origin (e.g., new m7G was normalized to abundance of new tRNA to receive the number of m7G per new tRNA while original m7G was normalized to original tRNA). a Level of 7-methylguanosine (m7G) in new and original tRNAs over time. The constant level of original m7G is caused by a constant increase in pre-existing m7G, while post-methylated m7G increases (box). (error bars reflect the SE of n = 6) b 2′-O-methylcytidine (Cm) increases fast in newly synthesized tRNAs, while the level of pre-existing Cm drops (box). This drop is masked by substantial post-methylation of original tRNAs. (error bars reflect the SE of n = 6) c Pseudouridine quantities are lower in newly-synthesized tRNAs while d the number of inosine per respective tRNA is constant throughout the experiment (both n = 3)
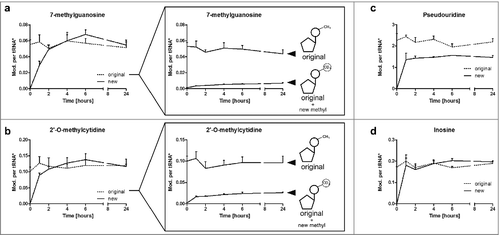
For 2′-O-methylcytidine (Cm) we observed a growth cycle dependent fluctuation of modification level ( and ). By NAIL-MS we observed an initial increase of Cm levels in pre-existing tRNAs while the new tRNA pool forms Cm rather slowly in the first 2 hours. While one explanation for the increase of Cm in the pre-existing tRNAs is degradation of those tRNAs that are not Cm modified during lag phase, the quantification of D3-labeled pre-existing cytidines revealed a substantial incorporation of new methyl marks in the pre-existing tRNAs (box of ). Curiously we observe a sudden decrease of pre-existing Cm in the original tRNA pool after entering log phase. This could be explained by several scenarios. The most likely explanation is sudden degradation of pre-existing tRNAs with Cm at a certain position (e.g. tRNAs with Cm at position 34, see Fig. S13) as these tRNAs might be no longer needed while entering log phase. In parallel, Cm might become necessary at the other positions of pre-existing tRNAs (e.g., at position 4) which leads to increased post-methylation. Another scenario for the decrease in pre-existing Cm is active demethylation of Cm by an unknown RNA eraser. Quantification of specific Cm-modified tRNAs by northern blotting might provide an answer to this open question.
The unmethylated nucleoside modifications pseudouridine and inosine are rapidly introduced into the nascent tRNA as these modifications reach their maximum abundance in the first 60 minutes ( and ). While new inosine levels in new tRNA are immediately comparable to the original tRNA inosine levels, pseudouridine levels are lower in the newly synthesized tRNAs compared with the levels in original tRNA. Furthermore, we observed a slight decrease of pseudouridine modification in the original tRNA pool after 6 hours, which might be linked to degradation of pseudouridine containing tRNAs. As both pseudouridine and inosine are non-methylated tRNA modifications, we cannot further distinguish between pre-existing modifications and those introduced after experiment initiation.
NAIL-MS results of the other methylated nucleosides are shown in Fig. S12. Like Cm, 5-methylcytidine (m5C) is found at various positions of tRNA (positions 34, 40, 48, 49) and has a pronounced drop in its abundance in the pre-existing tRNAs. This drop can be observed in both the pre-existing and post-methylated m5C molecules. The other methylated nucleosides Gm, m1A, m22G, m2G and Am are like inosine rather unresponsive with similar quantities throughout the experiment. Analysis of the incorporation speed into the nascending tRNA pool shows an almost immediate incorporation of m1G, m22G, Gm and Am while Cm, m7G, m1A, m2G, m5U and m5C are less abundant during lag phase compared with stationary and log phase.
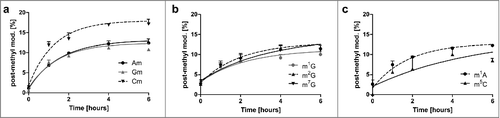
For all methylated nucleoside modifications we observed incorporation of the heavy D3-methyl mark into the pre-existing tRNAs. To compare the extent of this delayed post-methylation we formed the ratio of e.g., post-methylated Cm to all Cm in original tRNA and plotted the % of the post-methylated modifications (). After 6 hours the maximum of post-methylation is reached for all methylated nucleosides. Except Cm, all nucleosides plateau at 10% post-methylation. Cm has a 1.8-fold higher plateau of post-methylation compared with the other methylated nucleosides, indicating the necessity to quickly adapt Cm levels in tRNAs to overcome lag phase.
Figure 5. Extent of post-methylation in the original tRNA pool in %. Most modified nucleosides (Am, Gm, m1G, m2G, m22G, m1A and m5C) plateau at 10% which indicates that 10% of all modifications in the original tRNAs were made after experiment initiation (delayed methylation). Cm (graph on the left) is an exception. Here up to 18% of all Cm in original tRNA are incorporated after experiment initiation. (in all graphs the error bars reflect the SE of n = 3)
Discussion
Using stable isotope labeling mass spectrometry with a biosynthetically produced internal standard we quantified 13 modified nucleosides in total tRNA from S. cerevisiae grown in minimal medium. The detected quantities from yeast log growing cells were lower compared with the expectation from a database search (Table S1Citation11). These differences are partly explained by the low number of tRNA sequences with reported modification profile in the database (only 34 tRNA sequences available) and the general error introduced by absolute quantification approaches. In our quantification we use an “averaged” tRNA generated from the database under the assumption of equimolar contribution of each listed tRNA sequence. Therefore, absolute quantification results might slightly deviate from the true number of modification per tRNA molecule. Another explanation can be found in the dynamic nature of tRNA modifications which adapt depending on the growth phase as observed in and Fig. S4 or stress.Citation6 Especially during log phase the quantities of modified nucleosides increased for Cm, m7G and m1G, while m5C, m5U and Ψ levels decreased and Gm, Am, m2G, m22G, I and m1A stayed constant. While the biology of these changes remains unclear, we have developed a new tool to separately analyze the original and new transcripts. Using NAIL-MS (nucleic acid isotope labeling coupled mass spectrometry) we could partly identify the mechanism of these modification profile changes for methylated nucleosides ( and Fig. S12).
The mechanism, by which the modification level adapts, depends on the modified nucleoside and potentially the distribution in the RNA. For 7-methylguanosine (m7G, pos. 46) in tRNA, increased incorporation into newly synthesized tRNAs was observed. We observed a decrease in pre-existing m7G while post-methylated m7G increased, resulting in an apparent constant level of m7G in the pre-existing tRNA.
We made similar observations for 1-methylguanosine (m1G), 2,2-dimethylguanosine (m22G) and 2′O-methylguanosine (Gm) (see Fig. S12). While these methylated nucleosides showed a limited adaptation during growth, 2′O-methylcytidine (Cm, pos 34 and 4 of tRNAs) and 5-methylcytidine (m5C pos 34, 40, 48 and 49) quantities fluctuated depending on the growth phase. Cm showed an interesting modification profile over time which was partly caused by reduced modification levels in original tRNAs, high modification levels in the new tRNAs and post-methylation of the existing tRNA pool. Especially the reduced modification profile in the original tRNAs after 4 hours is intriguing (compare and Fig. S12 for m5C after 6 hours). One potential explanation is degradation of Cm and m5C modified tRNAs (e.g., tRNAGly, tRNAPhe or tRNALeu) which would result in a reduced number of these modifications in the original tRNAs. It is possible that the observed changes in the tRNA epitranscriptome reflect fluctuations in the ratio of isoaccpetors and isodecoders. Another potential mechanism that was recently described for 1-methyladenosine (m1A) in human cell culture is active removal of the methylation mark by an RNA eraser. While our current data cannot distinguish these mechanisms, it highlights m5C and Cm as potential candidates for RNA erasers. Future analysis of single modifications within their respective tRNA subtype will reveal the underlying mechanism.
We interpret methylation of original canonicals as an addition of new methyl marks into existing, intact RNA to adapt the modification profile to the physiologically needed modification status. However, it is possible that the observed signal is caused by salvage of existing heavy-labeled canonicals which are introduced into newly synthesized RNAs followed by subsequent methylation. However, if salvage was the main reason for added methylation to existing canonicals, the quantities of added methylation should be similar among the different modified nucleosides and the level would constantly increase over time as more and more RNA gets degraded and original canonicals get released. Both were not observed in our studies as the % of post-methylated nucleosides in the original tRNAs plateau after 6 hours. Although outside the scope of this manuscript, oligonucleotide mass spectrometry of e.g., a partial RNase T1 digestCitation18 of NAIL-MS samples would allow to assess the origin of these methylated nucleosides.
We used the NAIL-MS method to analyze 13 modified nucleosides, 2 of those are non-methylated tRNA modifications. For non-methylated nucleosides it is not possible to determine the quantity of added modification to the original RNA pool with the developed assay. However, we could monitor the modification extent in new and original RNAs and compare their quantities of modification.
In principle, it is possible to monitor all methylated nucleosides in original and new RNA and even observe the addition of the methyl mark to original RNA. Especially methylated uridine modifications like 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2U) or 5-methoxycarbonylmethyluridine (mcm5U) found at position 34 of tRNA would be interesting targets for such an analysis. These modifications have the strongest influence on protein translation,Citation7,19 especially during stress.Citation6,20 Due to their low abundance and low detection efficiency, large sample volumes are necessary to monitor these modifications. With NAIL-MS we now have a tool which allows a new viewpoint on the dynamics of potentially all methylated RNA nucleosides. It can be applied to heavily modified RNAs, as shown here for tRNA, but also to any other purified RNAs species like rRNA, mRNA or small non-coding RNAs.
Materials and methods
Growth medium and conditions for S. cerevisiae BY4741
S. cerevisiae BY4741 was grown in 13C labeled Yeast OD 2 (+ 10 g/L 13C6 Glucose (99% ATOM) (Euriso-top, Saclay, France)) rich medium (Silantes, Munich, Germany) for SILIS preparation or in YNB minimal medium for NAIL-MS experiments. 10x YNB (Carl Roth GmbH, Karlsruhe, Germany) was prepared according to manufacturer's manual. 1x YNB medium was supplemented with 10 g/L glucose, 0.02 g/L uracil, a mix of needed unlabeled aminoacids (final concentration: 0.02 g/L arginine, 0.02 g/L histidine, 0.06 g/L leucine, 0.03 g/L lysine, 0.05 g/L phenylalanine, 0.4 g/L serine, 0.2 g/L threonine, 0.04 g/L tryptophane, 0.03 g/L tyrosine and 0.15 g/L valine) and additional needed aminoacids used for labeling (final concentration: 0.1 g/L aspartic acid, 0.1 g/L glutamine and 0.02 g/L methionine). Depending on the desired labeling 15N2-uracil, L-aspartic-15N acid, L-glutamin-amide-15N, L-methionine-methyl-D3 or their unlabeled isotopomers were used. 15N2-uracil (98% ATOM 15N) was supplied by Euriso-top. All unlabeled aminoacids and L-aspartic-15N acid (98% ATOM 15N), L-glutamin-amide-15N (98 ATOM% 15N) and L-methionine-methyl-D3 (98% ATOM D3) were supplied by Sigma-Aldrich, Munich, Germany.
SILIS preparation
S. cerevisiae BY4741 was grown in 200 mL of 13C labeled Yeast OD 2 medium supplemented with 10 g/L 13C-glucose and 0.1 g/L D3-methionine. The cells were harvested at OD 6 by centrifugation at 3500 xg for 10 minutes. The supernatant was removed and the pellet was resuspended in 20 mL TRI reagent® (Sigma-Aldrich, Munich, Germany). For complete disruption of the cells, an amount of acid-washed beads equivalent to 200 μL (425–600 µm, Roth, Karlsruhe, Germany) were added to the suspension and vortexed for 10 minutes. RNA was isolated as specified in user manual and finally dissolved in 500 μLMilli-Q water. tRNA was isolated from total RNA and digested to nucleosides (see tRNA isolation via SEC and tRNA digestion below). The tRNA digest from 100 mL OD 6 culture was diluted in 1 mL pure water which resulted in mass spec peak areas 50–100x above the limit of quantification (LOQ) of the modified nucleosides of interest in a 1/10 dilution of the digest. As an external standard 10 mM theophylline was added to a final concentration of 1 mM in the digest solution. The resulting digest/theophylline mixture is referred to as 10x SILIS which was added to a final concentration of 1x to samples and calibration solutions.
Calibration
Synthetic modified nucleosides for preparation of calibration solutions were purchased from: Sigma-Aldrich, Munich, Germany: Nucleoside test mix: inosine (I), pseudouridine (Ψ), 5-methyluridine (m5U), 2′-O-methylcytidine (Cm), 5-methylcytidine (m5C); solid nucleosides: adenosine (A), 7-methylguanosine (m7G), 2′-O-methyladenosine (Am), 1-methyladenosine (m1A), 1-methylguanosine (m1G), 2-methylguanosine (m2G) and N2,N2-dimethylguanosin (m22G); solid nucleosides from Berry&Associates, Dexter, MI, USA: 2′-O-methyluridine (Um) and 2′-O-methylguanosine (Gm). Each nucleoside powder was weighed into a clean tube (5–10 mg per nucleoside) and dissolved in pure water to reach a final concentration of 10 mM. The nucleosides and the test mix were combined to a final concentration of 500 pmol/µL of A, 2000–500 fmol/µL of each modified nucleoside and 1x of the prepared SILIS. The calibration mix was serial diluted 1:5 and 1:10 with 1x SILIS until the lowest calibration concentration of 5 fmol/µL canonical and 5 amol/µL modified nucleoside were reached. 10 µL of each calibration solution was injected onto LC-MS/MS. After LC-MS/MS measurement the value of the integrated MS signals of the unlabeled synthetic nucleosides were set into relation to the integrated MS signals of the heavier SILIS nucleosides. The results were plotted against the nucleoside concentrations and the regression lines from the diagrams (Fig. S2) were used to calculate the respective RFN values, necessary for quantification.Citation10
NAIL-MS experiments
Five mL of 15N and 13C-Glucose labeled medium was inoculated with a single S. cerevisiae BY4741 culture from an YPD-agar plate in a 50 mL falcon tube. The cells were grown in a shaking incubator over night at 30 °C and 250 rpm. The next day 1 mL of the lysate was centrifuged, the supernatant discarded and the pellet resolved in 5 mL fresh 15N and 13C-Glucose labeld medium to achieve better labeling. After incubation over night at 30°C and 250 rpm again, the OD was determined at 600 nm using an Eppendorf Biophotometer. For experiment initiation part of the culture was centrifuged at 3000 xg for 5 minutes at room temperature. The cell pellet was resuspended in 20 mL of D3-labeled YNB medium to achieve a starting OD of 1. Using a 100 mL glass flask, the cells were incubated for 24 hours at 30 °C and 250 rpm in a shaking incubator. At set times (T = 0, 1, 2, 4, 6 and 24 hours) the OD was measured and a 2 mL sample was taken for RNA isolation. After centrifugation at 3000 xg for 3 minutes, the pellet was dissolved in 1 mL of TRI reagent®, transferred into a screw-top tube with an amount of acid-washed beads equivalent to 200 μL and vortexed for 10 minutes. Isolation of RNA was done according to user manual. The experiment was repeated with 6 biologic replicates.
tRNA isolation via SEC
Total RNA was loaded on an Agilent/HP 1100 LC system equipped with a binary pump, diode array detector set to 260 nm, autosampler, column thermostat (60 °C) and fraction collector. A size-exclusion column (Agilent Bio SEC-3, 3µm, 300Å, 7.8 × 300 mm, Agilent, Waldbronn, Germany) allowed collection of RNA fractions after isocratic elution with 100 mM ammonium acetate at pH 7. The tRNA fraction was concentrated to about 150μL in a Speed-Vac (Christ, Osterode am Harz, Germany). 5 M NH4OAc was added to a final concentration of 0.5 M and after addition of 2x Vol. ice-cold ethanol (100%) the RNA was precipitated over night at −20°C. After centrifugation at 12.000 xg for 30 minutes at 4° C the RNA pellet was subjected to an additional ethanol (80 %) wash step to verify complete removal of ammonium acetate and then resuspended in 20 μL pure water. The quality of the thus isolated tRNA was verified by analytical SEC using the same method.
tRNA digestion
tRNA was digested using 0.6 U nuclease P1 (Sigma-Aldrich) in the presence of ZnCl2, ammonium acetate pH 5.3, THU, pentostatine and BHT fromCitation21 at 50°C for one hour followed by overnight incubation at room temperature. The next day, 10 U benzonase (Sigma-Aldrich, Munich, Germany), 0.1 U phosphodiesterase I (VWR, Ismaning, Germany) and MgCl2 in a final concentration of 1 mM were added and incubated at 37°C for one hour. Then 20 U alkaline phosphatase (Sigma-Aldrich, Munich, Germany) and the proper amount to reach a final concentration of 0.1 M Tris-HCl pH 8 were added. After 2 hours at 37 °C the digestion mix was filtered through a 10 kDa MWCO filter (AcroPrep™ Advance, 350 µl, Omega™ 10K MWCO, Pall, Dreieich, Germany) at 3000 xg for 30 minutes. 18 µL of filtrate was mixed with 2 µL of the 10x SILIS and subjected to LC-MS/MS analysis.
High resolution mass spectrometry
For determination of m/z precursor and product values 5 mL cultures of each growth medium was inoculated with a colony of S. cerevisiae BY4741. Total RNA was isolated at OD 6 and digested as described for NAIL-MS experiments. The ribonucleosides were separated using a Dionex Ultimate 3000 HPLC system on an Interchim Uptisphere120–3HDO C18. Mobile phase A was 2 mM ammonium acetate and mobile phase B was 80% acetonitrile containing 2 mM ammonium acetate. Gradient elution started with 0% B and increased to 12% B after 10 minutes and to 80% after 12 minutes. After 4 minutes elution at 80% B and subsequently regeneration of starting conditions to 100% A after 5 minutes, the column was equilibrated at 100% A for 8 minutes. The flow rate was 0.2 mL/minute and the column temperature 30°C. High-resolution mass spectra of precursor and product ions were recorded by a ThermoFinnigan LTQ Orbitrap XL.
LC-MS / QQQ
Ribonucleosides were separated using a Synergy Fusion RP, 2.5 µm particle size, 100 Å pore size, 100 mm length, 2 mm inner diameter from Phenomenex (Torrance, CA), on an Agilent 1290 series HPLC system equipped with a diode array detector. Mobile phase A was 5 mM ammonium acetate adjusted to pH 5.3 with glacial acetic acid and mobile phase B was pure acetonitrile. Gradient elution started with 100% A for one minute, increase to 10% B after 10 minutes, 40% after 14 minutes and regeneration of starting conditions with 100% A for 3 additional minutes. The flow rate was 0.35 mL/minute and the column temperature 35°C. For mass spectrometric measurements an Agilent 6490 Triple Quadrupole mass spectrometer set to dynamic multiple reaction monitoring (MRM) mode was used. The MS was operated in positive ion mode with the following parameters: electro-spray ionization (ESI-MS, Agilent Jetstream), Fragmentor Voltage (set in tunefile to) 250 V, Cell Accelerator Voltage 2 V, N2-Gas temperature 150°C, N2-Gas flow 15 L/min, Nebulizer 30 psi, Sheath gas (N2) temperature 275 °C, Sheath gas flow 11 l/min, Capillary 2500 V and Nozzle Voltage 500 V. The mass transition for each modified nucleoside and its isotopomers are found in Table S2.
Data analysis
Using Agilent's Quantitative Data Analysis software, the mass signal areas of the different isotope labeled ribonucleosides were integrated. For methylated ribonucleosides the SILIS isotopmer, the 15N/13C labeled isotopomer (original), the 15N/13C/D3 labeled isotopomer (original+new methyl mark) and the D3 labeled isotopomer (new) could be identified. For the canonicals the isotopomers of the SILIS, the original and the unlabeled m/z transitions were analyzed to quantify the amount of injected tRNA. The values of the integrated signals were used for further calculations. The nucleosides were quantified by calculating the nucleoside isotope factor (NIF) and the response factors (RFN) were received by plotting the NIF over the concentration of injected synthetic standard.Citation10The signal of adenosine in each sample was divided by the response factors and the amount of injected adenosine was received. The value for adenosine was divided by the number of adenosine per tRNA molecule (see also Table S1). Thereby the injected amount of original and new tRNA molecules could be determined.
The modified nucleosides were analyzed by correcting first the original+D value under consideration of the presence of signals with the same m/z introduced by the biosynthetic SILIS (only for 5-methylcytidine and 5-methyluridine). Therefore the integral of the mass signal corresponding to original+D of an independent SILIS measurement was divided by the integrated SILIS mass signal of the same measurement. The resulting correction factor was then multiplied by the SILIS value of the actual sample and subtracted from the sample's original+D mass signal area.
Using the RFN the signal of each modified nucleoside isotopomer to SILIS ratio was converted into the amount of injected modification. The amount of new modified nucleoside was normalized by the amount of new tRNA injected and the number of the modification in new tRNA thus revealed. For original and original/post-methylated modified nucleosides the same procedure was applied but their amounts related to the amount of original tRNA injected.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental_Materials.docx
Download MS Word (2.3 MB)Acknowledgments
S.K., V.R. and M.H. thank Thomas Carell and his group for instrument time (QQQ and high-resolution mass spectrometer) and advice.
Funding
S.K. and V.R. acknowledge funding by the Fonds der chemischen Industrie. S.K acknowledges project funding by the DFG (SPP1784) and the LMU excellence program.
Related Research Data
References
- Machnicka MA, Milanowska K, Osman Oglou O, Purta E, Kurkowska M, Olchowik A, Januszewski W, Kalinowski S, Dunin-Horkawicz S, Rother KM, et al. MODOMICS: a database of RNA modification pathways–2013 update. Nucleic Acids Res 2013; 41(Database issue):D262-7; PMID:23118484; https://doi.org/10.1093/nar/gks1007
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012; 149(7):1635-46; PMID:22608085; https://doi.org/10.1016/j.cell.2012.05.003
- Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, et al. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 2014; 15(6):707-19; PMID:25456834; https://doi.org/10.1016/j.stem.2014.09.019
- Hopper AK. Transfer RNA post-transcriptional processing, turnover, and subcellular dynamics in the yeast Saccharomyces cerevisiae. Genetics 2013; 194(1):43-67; PMID:23633143; https://doi.org/10.1534/genetics.112.147470
- Sakai Y, Miyauchi K, Kimura S, Suzuki T. Biogenesis and growth phase-dependent alteration of 5-methoxycarbonylmethoxyuridine in tRNA anticodons. Nucleic Acids Res 2016; 44(2):509-23; PMID:26681692; https://doi.org/10.1093/nar/gkv1470
- Chan CT, Dyavaiah M, DeMott MS, Taghizadeh K, Dedon PC, Begley TJ. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet 2010; 6(12):e1001247; PMID:21187895; https://doi.org/10.1371/journal.pgen.1001247
- Klassen R, Ciftci A, Funk J, Bruch A, Butter F, Schaffrath R. tRNA anticodon loop modifications ensure protein homeostasis and cell morphogenesis in yeast. Nucleic Acids Res 2016; 44(22):10946-59; PMID:27496282; https://doi.org/10.1093/nar/gkw705
- Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, Ma J, Wu L. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun 2016; 7:12626; PMID:27558897; https://doi.org/10.1038/ncomms12626
- Chionh YH, Ho CH, Pruksakorn D, Ramesh Babu I, Ng CS, Hia F, McBee ME, Su D, Pang YL, Gu C, et al. A multidimensional platform for the purification of non-coding RNA species. Nucleic Acids Res 2013; 41(17):e168; PMID:23907385; https://doi.org/10.1093/nar/gkt668
- Kellner S, Ochel A, Thuring K, Spenkuch F, Neumann J, Sharma S, Entian KD, Schneider D, Helm M. Absolute and relative quantification of RNA modifications via biosynthetic isotopomers. Nucleic Acids Res 2014; 42(18):e142; PMID:25129236; https://doi.org/10.1093/nar/gku733
- Juhling F, Morl M, Hartmann RK, Sprinzl M, Stadler PF, Putz J. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res 2009; 37(Database issue):D159-62; PMID:18957446; https://doi.org/10.1093/nar/gkn772
- Pang YL, Abo R, Levine SS, Dedon PC. Diverse cell stresses induce unique patterns of tRNA up- and down-regulation: tRNA-seq for quantifying changes in tRNA copy number. Nucleic Acids Res 2014; 42(22):e170; PMID:25348403; https://doi.org/10.1093/nar/gku945
- Dong H, Nilsson L, Kurland CG. Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J Mol Biol 1996; 260(5):649-63; PMID:8709146; https://doi.org/10.1006/jmbi.1996.0428
- Michelson AM. The Chemistry of Nucleosides and Nucleotides1963. 622 p
- Nwagwu M, Nana M. Ribonucleic acid synthesis in embryonic chick muscle, rates of synthesis and half-lives of transfer and ribosomal RNA species. J Embryol Exp Morphol 1980; 56:253-67; PMID:7400745
- Karnahl U, Wasternack C. Half-life of cytoplasmic rRNA and tRNA, of plastid rRNA and of uridine nucleotides in heterotrophically and photoorganotrophically grown cells of Euglena gracilis and its apoplastic mutant W3BUL. Int J Biochem 1992; 24(3):493-7; PMID:1551462; https://doi.org/10.1016/0020-711X(92)90044-2
- Gudipati RK, Xu Z, Lebreton A, Seraphin B, Steinmetz LM, Jacquier A, Libri D. Extensive degradation of RNA precursors by the exosome in wild-type cells. Mol Cell 2012; 48(3):409-21; PMID:23000176; https://doi.org/10.1016/j.molcel.2012.08.018
- Ross R, Cao X, Yu N, Limbach PA. Sequence mapping of transfer RNA chemical modifications by liquid chromatography tandem mass spectrometry. Methods 2016; 107:73-8; PMID:27033178; https://doi.org/10.1016/j.ymeth.2016.03.016
- Nedialkova DD, Leidel SA. Optimization of Codon Translation Rates via tRNA Modifications Maintains Proteome Integrity. Cell 2015; 161(7):1606-18; PMID:26052047; https://doi.org/10.1016/j.cell.2015.05.022
- Alings F, Sarin LP, Fufezan C, Drexler HC, Leidel SA. An evolutionary approach uncovers a diverse response of tRNA 2-thiolation to elevated temperatures in yeast. RNA 2015; 21(2):202-12; PMID:25505025; https://doi.org/10.1261/rna.048199.114
- Cai WM, Chionh YH, Hia F, Gu C, Kellner S, McBee ME, Ng CS, Pang YL, Prestwich EG, Lim KS, et al. A platform for discovery and quantification of modified ribonucleosides in RNA: application to stress-induced reprogramming of tRNA modifications. Methods Enzymol 2015; 560:29-71; PMID:26253965; https://doi.org/10.1016/bs.mie.2015.03.004