
ABSTRACT
It remains a formidable challenge to characterize the diverse complexes of RNA binding proteins and their targets. While crosslink and immunoprecipitation (CLIP) methods are powerful techniques that identify RNA targets on a global scale, the resolution and consistency of these methods is a matter of debate. Here we present a comparative analysis of LIN28-pre-let-7 UV-induced crosslinking using a tandem mass spectrometry (MS/MS) and deep sequencing interrogation of in vitro crosslinked complexes. Interestingly, analyses by the two methods diverge in their identification of crosslinked nucleotide identity – whereas bioinformatics and sequencing analyses suggest guanine in mammalian cells, MS/MS identifies uridine. This work suggests the need for comprehensive analysis and validation of crosslinking methodologies.
Introduction
LIN28 is a highly conserved RNA-binding protein that was first described as the product of the heterochronic gene, LIN28, in C. elegans.Citation1-3 In mammals, the two LIN28 paralogs, LIN28A and LIN28B, play roles in a wide range of cellular processes, including stem cell self-renewal and pluripotency,Citation4-6 skeletal myogenesis,Citation7 glucose metabolism in diabetes,Citation8 and tissue repair.Citation9 These proteins are upregulated in ∼15% of human tumors and cancer cell lines, and elevated expression is associated with a poor prognosis and increased aggression in numerous malignancies, including germ cell tumors, colon cancer, and ovarian cancer.Citation10,11 Signaling components in the Wnt pathway have been shown to cooperate with LIN28 to increase the severity and invasiveness of colorectal cancer.Citation12 LIN28B also promotes metastasis of colon cancerCitation11 and tumorigenesis of the intestinal epithelium.Citation13
LIN28 exerts its profound phenotypic effects by acting as a negative regulator of let-7 miRNA biogenesis. Specifically, LIN28 binds let-7 precursors and prevents miRNA maturation, limiting cellular differentiation.Citation5,14-Citation17 High-resolution crystal structures of mouse LIN28A-let-7 complexesCitation18 and other structural and biochemical dataCitation19,20 revealed that LIN28 inhibits Dicer processing through steric hindrance and by locally unwinding the cleavage site. Furthermore, LIN28 proteins recruit the terminal uridylyltransferase, TUT4, to uridylate bound miRNAs, resulting in degradation by the Dis3l2 exonuclease.Citation20,21,22
Recent developments suggest that LIN28 also functions through let-7 independent mechanisms. Several studies using crosslinking and immunoprecipitation with deep sequencing (CLIP-seq) and photoactivatable ribonucleoside analog CLIP (PAR-CLIP) have identified thousands of potential pre-mRNA or mRNA targets of LIN28.Citation23-27 These methods rely on irradiation by UV light to generate covalent RNA-protein heteroconjugates in live cells, allowing for the isolation of RNA binding proteins (RBPs) by immunoprecipitation and subsequent high-throughput sequencing of crosslinked RNAs. CLIP was originally developed to address limitations of non-covalent RBP immunoprecipitation (RIP) methods,Citation28 such as non-specific RNA target capture, loss of lower affinity targets, and a weak signal-to-noise ratio.Citation29 Deep sequencing CLIP methods have identified global RNA targets for notable proteins such as Argonaute,Citation30 HuR,Citation31 eIF4AIII,Citation32 DDX17,Citation33 snoRNA proteins,Citation34 and splicing factors, including PTBP1 and RBFOX.Citation35 CLIP investigations of the LIN28 paralogs, combined with functional assays, have revealed that both proteins are target-specific post-transcriptional and translational regulators that alter splicing factor abundance and alternative splicing, suppress translation of secretory pathway proteins, and mildly stabilize mRNA targets.Citation23-Citation25 Several CLIP studies have seen enrichment of mRNAs containing the sequence GGAG, the signature LIN28 recognition element present in let-7 miRNAs.Citation23,24,26 Furthermore, one report identified an enrichment of pyrimidine rich binding motifs, consistent with sequences recognized by the LIN28 cold shock domain (CSD) in let-7 targets,Citation25 suggesting that LIN28 interactions with mRNA may have binding determinants that mirror those previously identified in let-7 miRNA targets.
Building on CLIP methods, crosslink induced mutation site (CIMS) analysis has emerged as a powerful bioinformatic tool for the elucidation of single nucleotide resolution crosslink interaction information derived from CLIP-seq data sets. This method aims to identify mutations, primarily deletions and substitutions,Citation36,37 that occur during reverse transcription at presumed crosslink sites within CLIP reads.Citation29,34 Despite the availability of numerous LIN28 CLIP-seq and PAR-CLIP studies, crosslink profiles from mutational analysis have been reported in only one study (see ref. Citation24), which found an increased prevalence of mutations at guanine residues that were apparently localized within a LIN28 GGAG recognition motif. To our knowledge, no other CLIP studies have been examined by CIMS, nor has the crosslinked side chain on LIN28 been concurrently elucidated.
In this work, we endeavored to characterize the products of in vitro UV-induced crosslinking within a well-characterized complex of LIN28A and a precursor let-7 fragment. Specifically, we used mass spectrometry (MS) to identify the discrete position of a crosslinked interaction on both the protein and the RNA components simultaneously. Furthermore, we performed sequencing analyses of the crosslinked RNA component to identify any crosslink-induced mutations (CIMs). Interestingly, we determined that the two methods non-redundantly determined different crosslink sites and crosslinked nucleotide identities. Overall, our work suggests that high-precision analysis methods of RNA-protein crosslinks must be cross-validated to avoid methodology-specific conclusions and gain comprehensive information concerning crosslinked interactions.
Results
Recombinant model LIN28A and pre-let-7f complexes can be crosslinked in vitro
To evaluate the in vitro protein-RNA crosslinking profile of a representative bipartite LIN28A-RNA complex, we generated complexes comprised of a previously reported, truncated mouse LIN28 construct (LIN28A-ΔΔ) and a correspondingly modified pre-element let-7f miRNA substrate, (preEM-let-7f) (, ).Citation18 LIN28A-ΔΔ consists of amino acids D33-K187 of the full-length protein and lacks the random coil N- and C- termini as well as a nine amino acid internal flexible linker between the CSD and zinc-knuckle domain (ZKD) (). PreEM-let-7f has a 5-nucleotide deletion between the AYYHY (the CSD-binding pyrimidine-rich sequence motif, where Y = C or U and H = A, C, or U),Citation18,25 and GGAG elements to accommodate the decreased space between the LIN28A binding domains (). These truncated components were previously crystallized as a complex, which reflected the interactions between the wild type full-length LIN28A and preE-let-7f, as determined by functional studies.Citation18 The binding affinity between LIN28A-ΔΔ and preEM-let-7f was comparable to the full-length LIN28A affinity for its corresponding preE-let-7f (47 – 190 nM) (), in agreement with previous data.Citation18 We also observed that purified LIN28A-ΔΔ:preEM-let-7f complexes could be crosslinked with comparable efficiency as full-length LIN28A:preE-let-7f (). This finding suggests that in vitro complexes that incorporate these truncated components are sufficient to mimic in vivo binding of the native protein with this miRNA intermediate.
Figure 1. LIN28A constructs have high affinity for and crosslink to preE-let-7f targets in vitro. (A) LIN28A constructs used in biochemistry and UV-crosslinking experiments. LIN28A-FL is full-length and LIN28A-ΔΔ is a truncated version that shortens the flexible linker between the CSD (cold-shock domain) and ZKD (zinc knuckle domain) in addition to shortening the 2 random coil termini. (B) LIN28A-FL and LIN28A-ΔΔ bind preE-let-7f and preEM-let-7f, respectively. PreEM-let-7f was shortened to accommodate the reduced interdomain linker in LIN28A-ΔΔ. The CSD and ZKD recognize AYYHY (highlighted green) and GGAG sequences (highlighted purple), respectively. (C) Gel shift binding assays with radiolabeled preEM-let-7f probe, mixed with increasing concentrations of LIN28-FL (0, 22, 44, 180, 700 nM, 2.8 μM) and LIN28A-ΔΔ (0, 24, 47, 190, 750 nM, 3 μM). (D) Corresponding SDS-PAGE gels show crosslinked complex bands following UV irradiation. (E) Sample preparation workflow for crosslinked peptide-RNA heteroconjugates for LC-MS, MS/MS and sequencing analysis.
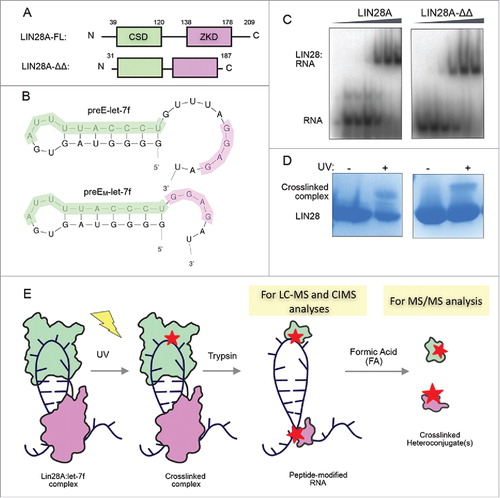
With the aim of determining crosslink sites via MS and CIMS analyses, we established a workflow to generate crosslinked heteroconjugate samples composed of the aforementioned truncated constructs. Complexes of LIN28A-ΔΔ and preEM-let-7f were exposed to 254 nm UV light, and crosslinked complexes were isolated and trypsinized to yield samples of peptide-modified RNA. The peptide-modified RNA is suitable for both liquid chromatography-mass spectrometry (LC-MS) and sequencing analyses. Subsequent RNA digestion by formic acid (FA) rendered the sample suitable for tandem mass spectrometry (MS/MS) ().
Mass spectrometry reveals a crosslinking site between the let-7 pre-element terminal loop and LIN28A cold-shock domain
To assign discrete crosslink sites in model recombinant RNA-LIN28 complexes, we used an RNA-directed enrichment approach and mass spectrometry. The full-length, unmodified preEM-let-7f RNA has an exact calculated mass of 8052.0600 Da, and crosslinked heteroconjugates were initially sought by scrutinizing species with neutral mass gains that might correspond to tryptic peptide addition. Initially, we observed a discrete species with a low resolution mass of 9268 Da, consistent with a mass gain corresponding to the predicted tryptic fragment MGFGFLSMTAR (residues 51 – 61 in full length LIN28).
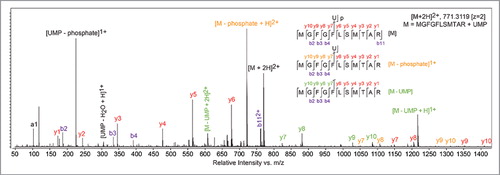
To examine this specific peptide-RNA heteroconjugate, enriched peptide-RNA samples were stringently hydrolyzed in 50% (v/v) formic acid (FA) at 80°C for 2 h. Survey spectra were searched for peptide-nucleotide heteroconjugates as either simple mass neutral conjugates or those exhibiting neutral loss of H2O. A candidate peptide-nucleotide species was observed with an accurate mass of 1540.6120 Da, which is consistent with the heteroconjugate MGFGFLSMTAR-uridine monophosphate (UMP, calculated exact mass 1540.6092 Da, 2.5 ppm mass error). The peptide sequence of the observed heteroconjugate species was confirmed by selecting the [M + 2H]2+ ion 771.31 m/z for fragmentation using collision-induced dissociation (CID, ). The fragmentation spectrum is complicated by multiple fragmentation pathways, where backbone fragment ions arising from 3 parent species exist in the tandem MS scans: the selected parent ion (, red labels), the ion resulting from loss of phosphate (, orange labels), and the ion resulting from complete loss of UMP (, green labels). These 3 species were secondarily fragmented in a manner that generates sequenceable peptide backbone y- and b-ions, giving the expected peptide sequence. Significantly, the y-ions corresponding to UMP and uridine modifications both appear for y7 – y10 but not for smaller products, indicating that the UMP is crosslinked to Phe55 in LIN28A-ΔΔ.
Figure 2. UV-crosslinking occurs between Phe55 of the LIN28A CSD and Uridine-11 of the preEM-let-7f terminal loop. Targeted tandem mass spectra identifying product ions that confirm the MGFGFLSMTAR peptide and locate the crosslinked residue as being Phe55; a uridine monophosphate was identified as the nucleotide covalently bound at Phe55.
To assign the crosslinked position within the pre-element RNA, we sought to observe crosslinked heteroconjugates composed of peptides covalently bound to larger nucleotide species: either di- or tri-nucleotides. Crosslinked heteroconjugates were generated and hydrolyzed as before, with the exception that the acid digest time was reduced (30 min) and temperature lowered (60°C) to prevent complete RNA hydrolysis. From these samples we were able to identify ions corresponding to the same peptide, MGFGFLSMTAR, neutrally conjugated to UMP, as well as to RNA fragments with the following nucleotide compositions: UU, AU, UUU, AUU and GAU (). We further confirmed the nucleotide composition and peptide sequence of these species using tandem MS (Supp. Fig. 1). Analyzing the composition of these species revealed only one consistent overlapping site in the pre-element RNA sequence: uridine-11 (). We concurrently confirmed the identity of U11 using a newly developed RNA site-specific stable isotope labeling technique.Citation38 Using pre-element RNA labeled with synthetic isotopes at either the U11 or U12 positions, we observed a mass-shifted isotope distribution exclusively for heteroconjugates arising from complexes formed from RNA labeled at U11. The identified tryptic peptide corresponds to a region within the LIN28A CSD at its binding interface with preEM-let-7f (), as observed in a high-resolution crystal structure (PDB ID: 3TS0).Citation18 Within this interface, Phe55 is oriented such that the side chain is (at closest proximity) within 3.5 Å of the uracil moiety of U11 within the pre-element terminal loop, giving a planar angle of ∼7.5 degrees between the two aromatic rings (). This contact is consistent with a strong π-π interaction between the two residues and suggests that the mass neutral crosslink identified here is physiologic.
Table 1. Masses and nucleotide composition overlap of crosslinked mono-, di- and tri-nucleotides that identify U11 as the most probable crosslinking counterpart to Phe55.
Figure 3. Crosslinking of mutant LIN28 suggests the presence of crosslink sites undetected by MS. (A)Front view surface and side view cartoon representations of the known structure of LIN28A-ΔΔ CSD (green) complexed with preEM-let-7f (gray) (PDB ID: 3TS0). Phe55, the identified crosslink site, contacts U11 of the preEM-let-7f terminal loop. The tryptic peptide MGFGFLSMTAR and Phe55 side chain are highlighted in maroon and U11 nucleotide is highlighted in red. (B) Gel shift binding assays with radiolabeled preEM-let-7f probe, mixed with increasing concentrations of LIN28A-ΔΔ mutant constructs: F55Y (0, 100, 200, 400, 800 nM, 1.6, 3.2 μM) and F55A (0, 22, 44, 180, 700 nM, 2.8 μM). (C) Corresponding SDS-PAGE gels show crosslinked complex bands following UV irradiation.
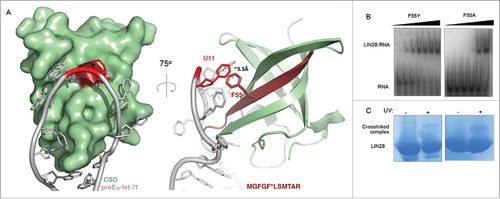
To validate the identified crosslinking site and characterize its contribution to substrate binding, we measured in vitro binding affinities of single point mutants of the determined crosslinking site, Phe55, within the LIN28A-ΔΔ construct. Using gel shift binding assays, we found that a single conservative mutation of Phe55 to tyrosine (F55Y) had a minimal effect on binding (KD: 100 – 200 nM), whereas an alanine mutation at the same position (F55A) resulted in a significant decrease in affinity (KD: ∼700 nM) (). These results demonstrate significant contribution of Phe55 to LIN28-RNA binding affinity, even though its contribution to binding specificity is small.Citation18 Despite the range of observed affinities, SDS-PAGE experiments confirmed that both mutant constructs were able to crosslink preEM-let-7f, though to varying extents (), suggesting the presence of crosslink sites undetected by MS.
CIMS analysis identifies guanine mutations in CLIP-seq data sets and within in vitro crosslinked LIN28-let-7 complexes
RNA-protein UV crosslinking causes observable mutations in CLIP sequencing reads, which are presumed to be indicative of crosslinking sites and can be mapped using CIMS analysis.Citation36,37 Thus, we generated a data processing workflow modified from previous CIMS protocols (see ref. Citation36 and Citation37) and validated our method by reanalyzing two mouse LIN28A CLIP data sets for which CIMS mutational profiles were reported (see ref. Citation24). Consistent with that work, our analysis of the monoclonal 35L33G and polyclonal antibody CLIP data sets showed that mutations arose most frequently at guanines (). Though we observed similar mutation identities and positions, our frequencies were lower, likely due to differences in filtering parameters. Nonetheless, we determined guanines make up 64% and 76% of substitution sites and 43% and 47% of deletion sites for monoclonal and polyclonal antibody data sets, respectively ().
Figure 4. CIMS analysis identifies guanines as sites of mutation. (A) Mutation frequency profiles of CLIP reads generated by CIMS analysis. Mono35L33g and Polyclonal data sets are from ref. Citation24 (M. musculus). LIN28V5 and LIN28A are data sets from ref. Citation23 (H. sapiens). WormSmall and WormBig (C. elegans) are from. ref. Citation27 Selected data sets include only CLIP experiments performed in tissue culture in the absence of photoactivatable ribonucleoside analogs (PAR-CLIP), to avoid well-known mutation bias. Percentage numbers and colors within frequency plots indicate dominant nucleotide identity and relative enrichment. (B) Mutation frequency profiles of 3 samples of preEM-let-7f: in vitro crosslinked peptide-modified preEM-let-7f (top panel), preEM-let-7f exposed to UV in the absence of LIN28 (middle panel) and untreated preEM-let-7f (bottom panel). The preEM-let-7f reference sequence is listed along the x-axis with the CSD binding motifs (AYYHY) and ZKD binding motifs (GGAG) highlighted in green and purple, respectively. The MS identified crosslink site at U11 is indicated with an asterisk (*).
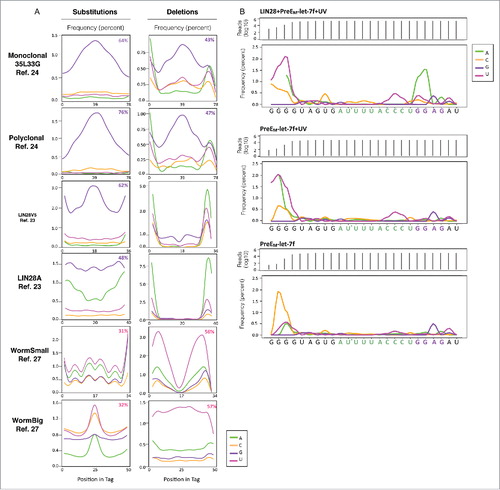
To examine the consistency of this observation across published LIN28 crosslinking studies, we applied our validated analysis to four published data sets for which crosslink-induced mutations have not yet been reported.Citation23,27 These include two data sets denoted as LIN28A and LIN28-V5 from, ref. Citation23 which identified mRNA targets of LIN28A in human H9 embryonic stem cells and HEK 293 cells, respectively, as well as two data sets from, ref. Citation27 referred to as WormSmall (CLIPseq1) and WormBig (CLIPseq2) which identified mRNA targets of LIN28 in C. elegans. Importantly, these 4 data sets are derived from CLIP-seq, rather than PAR-CLIP experiments, and therefore do not incorporate known mutation biases that occur as a result of using photoreactive nucleosides (i.e., T to C transitions in 4-thiouridine and G to A transitions in 6-thioguanosine experiments).Citation25,26
We observed variability in crosslink-induced mutation enrichment between these 4 data sets (). While the two human LIN28A CLIP data sets showed enrichments for substitutions at guanine residues, no nucleotide dominated deletion mutations. Additionally, our analysis of the C. elegans data set showed that uridine nucleotides were the most frequent points of mutation. Though the enrichment of substitutions occurring at uridines was slight, making up 31% and 32% of mutations for the two data sets, we noted a significant enrichment of uridines as the sites of deletions, which represent 56% and 57% of that type of mutation. Importantly, despite species, group and perhaps quality differences, all data sets were processed using the same analytical workflow which is likely the reason that some frequency plots appear to be irregular.
Since CIMS analysis of endogenous complexes and MS/MS of recombinant complexes point to distinct non-overlapping crosslink sites, we deep sequenced and performed CIMS analysis on recombinant complexes crosslinked in vitro. To determine relative frequencies of mutations arising from protein-RNA crosslinking, we deep sequenced and analyzed three distinct preparations of preEM-let-7f: preEM-let-7f crosslinked with recombinant LIN28A-ΔΔ, UV-exposed preEM-let-7f in the absence of protein, as well as unexposed, unmodified preEM-let-7f RNA ().
Our analysis revealed numerous points of mutation across the RNA sequence that are statistically significant (p < 0.05) (Supp. Table 1). Several sites, however, were mutated in all three RNA samples, including RNA that had no UV exposure (i.e., A22 and A24). Furthermore, a subset of sites were mutated in both the peptide-modified RNA (, top panel) and the UV-exposed RNA (, middle panel), indicating that these sites are artifacts of UV exposure and not specifically associated with protein crosslinking (i.e., C16, C17 and C18). Finally, mutation frequencies at positions G1, G2 and G3 were excluded from analytical considerations as, for reasons unclear, several orders of magnitude fewer reads could be mapped to those positions in all samples ( and Supp. Table 1). One major cluster of mutations that was unique to peptide-modified preEM-let-7f was found at positions G20 and G21 within the GGAG recognition motif. Combined mutation frequencies at positions G20 and G21 are 2.0% and 1.6%, respectively, with adenines clearly dominating the mutant nucleotide identities (, top panel). Additionally, mutations at U8 and G23 were more prevalent in the peptide-modified RNA sample compared with controls. However, their overall mutation frequencies were lower than that observed at G20 and G21 (0.53% and 0.29%, respectively).
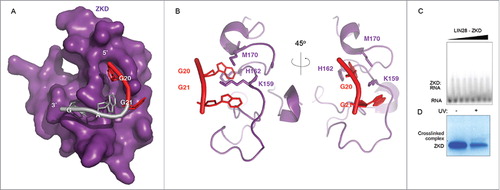
Because they fall within the well-annotated recognition motif for LIN28A, G20 and G21 participate in numerous interactions with the LIN28 ZKD (, ). However, MS analysis of crosslinked preEM-let-7f: LIN28 complexes did not detect crosslink sites within the ZKD. To determine whether crosslinking at those sites is feasible in isolation, we sought to crosslink the LIN28 ZKD in vitro, in the absence of the LIN28 CSD. To that end, a well characterized, recombinant LIN28 ZKD construct (residues 120–181) (See ref. Citation39) was overexpressed and purified from E. coli. Complexes of ZKD with a corresponding fragment of pre-let-7 RNA (sequence: UAGGAGAU) were formed in vitro and crosslinked as described previously. SDS-PAGE analysis confirmed that the ZKD alone was capable of crosslinking with a corresponding RNA fragment despite low binding affinity (KD >7.2 uM, unquantified) (, ).
Figure 5. The LIN28A ZKD can crosslink RNA in vitro. (A) Surface representation of the LIN28A ZKD in complex with the GGAG fragment of preEM-let-7f. G20 and G21, the 2 major points of mutation in the crosslinked preEM-let-7f fragment are highlighted in red. (B) Cartoon representations of the LIN28A ZKD with G20 and G21 interacting with the side chains of residues K159, H162 and M170. (C) Gel shift binding assays with the radiolabeled pre-let-7 fragment UAGGAGAU, mixed with increasing concentrations of LIN28A-ZKD (0, 56, 225, 900 nM, 1.8, 3.6, 5.4 and 7.2 μM). (D) Corresponding SDS-PAGE gel shows a crosslinked complex band following UV irradiation.
Discussion
CLIP methods have identified thousands of putative mRNA targets of LIN28, with a significant number of binding sites in both translated and untranslated regions of the transcriptome. While LIN28 unambiguously inhibits processing of the let-7 precursors it targets, reports indicate that LIN28 can both enhance and suppress translation of subsets of RNAs with numerous cellular consequences,Citation4,6,7,23,24 prompting questions about the specificity and pervasiveness of interactions, as well as concerns over reproducibility and experimental variation. The nature of LIN28 recognition of RNA likely further complicates target identification efforts as structural and biochemical studies have revealed that LIN28 recognizes let-7 precursors through a bipartite interaction mediated by two binding domains with distinct recognition characteristics.Citation18,19,40
To identify precise crosslink sites in a model recombinant LIN28A protein-RNA complex, we first used LC-MS and MS/MS to analyze covalent RNA-peptide heteroconjugates. Of the 11 ssRNA bases that mediate the LIN28A-ΔΔ:preEM-let-7f complex, our MS analysis identified only a single crosslink site. The covalent crosslink between Phe55 of the LIN28A CSD and a terminal loop uridine of preEM-let-7f, corresponds to a tight interfacial contact observable in the crystal structure of the complex.Citation18 We separately confirmed this assignment using a novel, single-nucleotide isotope labeling technique.Citation38 Aside from the single crosslink of uridine to Phe55, we were unable to identify directly any other site of modification by mass. Alternatively, our CIMS analysis of human and mouse CLIP data sets revealed an enrichment of mutations occurring at guanines, presumably indicative of crosslinking sites. Furthermore, sequencing analysis of our in vitro crosslinked peptide modified preEM-let-7 determined an enrichment of mutations at guanines (specifically at those within the GGAG motif), and in vitro crosslinking experiments confirmed that the ZKD can crosslink independently of the CSD. However, CIMS analyses did not determine a significant frequency of mutations at the uridine crosslink site detected by mass analysis.
Overall, our data indicate that the two analytical methods identify different crosslinking sites within the same UV crosslinked protein-RNA complex. Consistent with this finding, a recent report (see ref. Citation41) demonstrated that in a systematic MS/MS analysis of 124 RBPs crosslinked in vivo, MS-detectable crosslinking occurred almost exclusively at uridines. In one data set, 89% of crosslinking events were confirmed at uridines, and at least one uridine was present in crosslinked di- and tri-nucleotides in the remaining 11% of cases, although LIN28 was not included in that report. Furthermore, they noted that protein-RNA crosslinks are typically few in number (∼1–3 crosslinks per complex) and are frequently mediated by phenylalanine residues (24% of identified amino acid crosslink sites). On the other hand, the only previous report to feature CIMS profiles of LIN28 CLIP data sets (see ref. Citation24) identified primarily guanine points of mutation. Furthermore, the crosslink site mutation profile determined specifically for pre-let-7f showed mutations primarily at guanines within the GGAG motif and no mutations at the uridine site that we identified in vitro, or at any uridine within the terminal loop.
Collectively, the results of our case study of existing LIN28 binding site data and our biophysical investigation of LIN28A crosslinking to a let-7 precursor suggest that the two highest resolution crosslink site detection methods may benefit from orthogonal, complementary analyses to gain complete crosslink site identification data. Given that precise identification of protein-RNA interactions on a global scale provides an invaluable tool for mechanistic and functional studies of various cellular processes, further development of comprehensive analysis methods is needed to identify, reproducibly and with high confidence, the precise binding sites between diverse RBPs and their RNA targets. One potential opportunity might be found in the advancement of CLIP methods that examine crosslinking between individual domains of proteins and their RNA targets, such as iDo-PAR-CLIP (individual domain PAR-CLIP),Citation26 with the addition of a higher precision MS analysis step. This version of PAR-CLIP relies on a proteolytic cleavage site between two binding domains of an RBP to facilitate single domain isolation and domain specific target sequencing following UV irradiation.Citation26 Furthermore, unambiguous and comprehensive structural data generated from key techniques such as X-ray crystallography and nuclear magnetic resonance (NMR) may be used to further scrutinize or validate CLIP determined binding sites.
Material and methods
Expression and purification of LIN28A proteins
All recombinant LIN28A protein constructs were overexpressed from pET21a or pETDuet expression vectors. BL21 Rosetta cell colonies transformed with construct plasmids were used to inoculate 100ml LB starter cultures and incubated overnight (∼18–20 hrs) in a shaker incubator at 37°C. The next day, 10 ml of starter culture was used as inoculation for every 1 L of expression culture (typically 2–4 L). Expression cultures were incubated at 37°C until reaching O.D. of 0.6–0.8, at which point cultures were induced with 0.5 mM IPTG. Following induction, cultures were incubated overnight at 18°C with shaking and harvested the next day via centrifugation. Proteins were purified via Ni2+ affinity, cationic exchange and size exclusion chromatography, as described previously.Citation18
Electrophoretic mobility shift assays (EMSAs) and SDS-PAGE
For binding analyses, LIN28A was serially diluted into a low-salt binding buffer (20 mM Bis-Tris, pH 7, 100 mM NaCl, 50 μM ZnCl2, 5% glycerol and 5 mM DTT) supplemented with yeast tRNA (to a final concentration of 1 mg/ml) and the RNAse inhibitor Ribolock (Thermofisher Scientific, CAT: EO0381). Precursor let-7 RNA probes were synthesized from IDT and labeled with 32P via the T4 Polynucleotide Kinase (New England BioLabs, CT: M0201S). 32P-labeled RNA substrates (< 1 nM) were incubated with LIN28A protein dilutions of increasing concentration for ∼30 minutes at RT before samples were run on a 10% native gel. Gels were vacuum dried and pressed to radiolabel sensitive film overnight. Films were imaged the following day using scanning phosphorimager. For SDS-PAGE analyses, complexes of LIN28A proteins with pre-let-7f RNAs were exposed to UV irradiation and pre- and post-crosslinked samples were compared on SDS-PAGE gels and stained with Coomassie Blue.
UV crosslinking and hydrolysis of LIN28A and let-7 complexes
Purified complexes of LIN28A proteins and preE-let-7f targets were buffer exchanged into crosslinking buffer (20 mM Bis-Tris, pH 7, 100 mM NaCl, 50 μM ZnCl2, and 1 mM DTT), and UV crosslinking was completed by irradiating samples three times at 300 mJ/cm2 at 254 nm in a Stratalinker 1800. Crosslinked complexes were separated from non-crosslinked complexes via denaturing urea gel. Gels were stained with ethidium bromide and imaged. Crosslinked complex RNA bands (indicated by the increase in size over the free RNA bands) were excised and RNA was eluted from gel via electro-elution into a dialysis bag, 3 × 30 mins at 100V. Eluted, crosslinked complexes were concentrated and buffer exchanged into 8 M urea, 50 mM bis-Tris, pH 7. Trypsin/Lys-C (Promega, CT: V5072) was added to a final ratio of 1:25 (w/w). Digestions were incubated at 37°C for 3 hrs with shaking. Samples were diluted to lower the urea concentration to < 1 M and digestions continued overnight at 37°C.
Modified RNA was enriched from digested samples via anion exchange chromatography. Specifically, samples were loaded onto a DEAE column (GE Healthcare) with low-salt buffer (20 mM Bis-Tris, pH 6, 10% glycerol and 5 mM DTT) and eluted with a high-salt buffer fast gradient (20 mM Bis-Tris, pH 6, 10% glycerol and 5 mM DTT, 2 M NaCl). Chromatography fractions containing RNA were pooled, flash frozen, and lyophilized. Dried samples were resuspended in LC-MS grade water and quantified by their optical density at 260 nm.
LC-MS
All samples were separated on an Agilent 1200 HPLC coupled to a solvent degasser, auto sampler, diode array detector, and column oven (Agilent Technologies). Separations were performed using two solvent systems. Native and tryptic peptide-modified, full-length RNA samples were separated on a 100 mm × 1 mm i.d. Xbridge C18 column with a particle size of 3.5 μm (Waters). The solvent system was based on a previously published reverse phase ion pairing LC-MS method, and we used 200 mM HFIP with 1.25 mM trimethylamine at pH 7.0 in mobile phase A and methanol in mobile phase B.Citation42 The column was heated to 60.0°C and the flow rate was 100 μL/min. Injection volumes were 10 – 25 μL. Mobile phase B was increased from 5% to 15% from 0 – 20 minutes and then from 15% to 60% over an additional 20 minutes of run time. Absorbance was monitored at 260 nm with a reference wavelength at 380 nm and a 2 s response time. Tryptic, crosslinked peptides digested in formic acid were separated on a 150 mm × 1.0 mm i.d., micro bore rapid resolution SB-C18 column with a particle size of 3.5 um (Agilent Technologies). Mobile phase A was water with 0.1% FA and mobile phase B was acetonitrile with 0.1% FA. The column was heated to 40.0°C, and the flow rate was 150 μL/min. Injection volumes were 5 – 20 µL. Mobile phase B was increased from 5% to 45% over 0 – 20 minutes.
All samples were analyzed on an Agilent G6520A accurate-mass QTOF coupled to the LC system described above, operating in extended dynamic range mode. The system was calibrated on the same day of analysis and reference masses were continuously infused for online mass correction. Enriched RNA oligonucleotides crosslinked to tryptic peptides were separated using the HFIP/TEA solvent system and analyzed in negative ion mode from 239 – 3200 m/z with a scan rate of 1 spectrum/s using the following settings: drying gas flow, 8 L/min; drying gas temperature, 325 °C; nebulizer pressure, 30 psig; capillary voltage, 3500 V; fragmentor, 200 V; and skimmer, 65 V. Tryptic, crosslinked peptide-RNA heteroconjugates digested in formic acid were separated using the water + 0.1% FA and acetonitrile + 0.1% FA system and analyzed in positive ion mode from 104 – 3000 m/z with a scan rate of 1 spectrum/s in MS1 mode or 1.67 spectrum/s for MS acquisition and 1.2 spectrum/s for MS/MS acquisition in targeted MS/MS mode using the following settings: drying gas flow, 8 L/min: drying gas temperature, 325°C; nebulizer pressure, 35 psig; capillary voltage, 4500 V; fragmentor, 175 V; and skimmer: 65 V. In targeted MS/MS mode, ions were fragmented using collision induced dissociation with nitrogen gas, collision potentials of 10 – 30 V and an isolation width of ∼4 amu.
LC-MS data analysis and crosslink site identification
Tryptic peptides crosslinked to mono- or dinucleotides were first identified based on MS1 data using a database composed of predicted tryptic peptides with up to two missed cleavages crosslinked to all possible mono- and dinucleotides using Agilent's Find by Formula algorithm in the MassHunter software package. The analysis allowed for the loss of water so we could identify both mass neutral crosslinks and those leading to loss of water. The peptides identified in survey spectra with covalent UMP, di-, and trinucleotide modifications were then analyzed in targeted MS/MS experiments to validate the peptide sequence and characterize the species. The targeted MS/MS spectra for the heteroconjugate MGFGFLSMTAR-UMP were manually compared with theoretical product ion spectra in which the UMP was attached to different amino acids in the peptide as to determine the position of the crosslink.
Workflow for CIMS analysis
CLIP data sets for mouse (ref. Citation24) and human (ref. Citation23) LIN28 were obtained through Gene Expression Omnibus (GEO), with accessions SRR458758 (monoclonal 353L33G), SRR458760 (polyclonal), SRR531464 (LIN28A) and SRR531465 (LIN28V5). The WormBig and WormSmall data sets from ref. Citation27 were transferred via personal communication. The SRA files were downloaded and converted to FASTQ files using SRA Toolkit 2.5.2. FastQC determined the sequence quality and presence of adapters. Bowtie2 indexes were created for hg19, mm10 and ce10 for human (H. sapiens), mouse (M. musculus), and worm (C. elegans), respectively. Bowtie2 was run with the parameters -p 18 -N 1. Samtools was used to sort and convert mappings into SAM files. The SAM files were then read into R and the following filters applied: low quality reads were removed (if for any nucleotide the quality is below D then the whole read is removed) and all exact sequence duplicates were collapsed to eliminate the potential for PCR duplicates. For each mutation type (substitutions/deletions), distributions were derived separately. For substitutions, only reads with a single substitution were considered. Substitution positions were identified based on the SAM files, fields for tag MD type Z. For deletions, only reads with a single deletion were considered. The positions of deletions were identified based on CIGAR string based on the SAM files. Stringent conditions for the number of substitutions and deletions (1 per read) led to filtering out reads with adaptor sequences. Further analyses of obtained distributions were strand specific, were performed with dplyr package, and were visualized with ggplot2 package for R.
Peptide-modified preEM-let-7f sequencing library generation and analysis
Purified, lyophilized peptide-modified preEM-let-7f was resuspended in LC-MS grade water and quantified via optical density measurement at 260 nm. The QiaSeq miRNA library construction kit (Qiagen, Cat#331502) was used to generate a library from ∼100ng of peptide-modified RNA using the standard protocol, incorporating 16 cycles of PCR. For quality assurance, Tape Station High Sensitivity tape is run on the Tape Station 2200 from Agilent. Additionally, a qPCR run was completed using the Kapa Library Quantification Kit (Kapa, KK4824) with a standard 0 for normalization. Generated libraries were pooled together in equimolar proportions and were denatured per standard Illumina protocol. Sequencing was performed on an Illumina MiSeq with Nano protocol at 6 pM with 50% PhiX for a read length of PE 75 and depth of up to 1 million reads.
Sequencing analysis of peptide modified preEM-let-7f library
Reads were trimmed with cutadapt to remove the 3′ adaptor. Bowtie2 was used to create the index of the reference sequence. Then, they were aligned with bowtie2 with parameters: -D 20 -R 3 -N 0 -L 10 -i S,1,0.5 to allow short alignments. Samtools was used to sort and index the alignment file, and sambamba-depth was used to calculate the coverage along the reference sequence. Substitution ratios were calculated in R using dplyr and results were visualized with ggplot2.
Conflict of interest statement
The authors declare that there are no conflicts of interest
Abbreviated title
Comparative analysis of LIN28-RNA UV-induced crosslinks
suppl_mat_Table_Comparative_analysis_of_LIN28-RNA_binding_sites.xls
Download MS Excel (52 KB)suppl_mat_Comparative_analysis_of_LIN28-RNA_binding_sites.pptx
Download MS Power Point (535.8 KB)Acknowledgments
We would like to express our gratitude to Areum Han and Kristina Holton for preliminary analyses and troubleshooting of the bioinformatics.
Funding
This work was supported by the National Cancer Institute [R01CA163647 to P.S.]; the National Science Foundation Graduate Research Fellowship Program [DGE1144152 to E.R.]; the UNCF-Merck Graduate Research Science Initiative [E.R.]; from the Academy of Finland [A.B.]; and the Harvard Medical School Tools and Technology Committee [L.P.]. Funding for open access charge: National Institutes of Health. J.W.S. is an Investigator of the Howard Hughes Medical Institute.
Related Research Data
References
- Ambros V, Horvitz HR. Heterochronic mutants of the nematode Caenorhabditis elegans. Science. 1984;226:409–16. doi:10.1126/science.6494891.
- Ambros V. A hierarchy of regulatory genes controls a larva-to-adult developmental switch in C. elegans. Cell. 1989;57:49–57. doi:10.1016/0092-8674(89)90171-2.
- Liu ZC, Ambros V. Heterochronic genes control the stage-specific initiation and expression of the dauer larva developmental program in Caenorhabditis elegans. Genes & development. 1989;3:2039–49. doi:10.1101/gad.3.12b.2039
- Xu B, Zhang K, Huang Y. Lin28 modulates cell growth and associates with a subset of cell cycle regulator mRNAs in mouse embryonic stem cells. Rna. 2009;15:357–61. doi:10.1261/rna.1368009.
- Viswanathan SR, Daley GQ, Gregory RI. Selective blockade of microRNA processing by Lin28. Science. 2008;320:97–100. doi:10.1126/science.1154040.
- Qiu C, Ma Y, Wang J, Peng S, Huang Y. Lin28-mediated post-transcriptional regulation of Oct4 expression in human embryonic stem cells. Nucleic acids research. 2010;38:1240–8. doi:10.1093/nar/gkp1071.
- Polesskaya A, Cuvellier S, Naguibneva I, Duquet A, Moss EG, Harel-Bellan A. Lin-28 binds IGF-2 mRNA and participates in skeletal myogenesis by increasing translation efficiency. Genes & development. 2007;21:1125–38. doi:10.1101/gad.415007
- Zhu H, Shyh-Chang N, Segre AV, Shinoda G, Shah SP, Einhorn WS, Takeuchi A, Engreitz JM, Hagan JP, Kharas MG, et al. The Lin28/let-7 axis regulates glucose metabolism. Cell. 2011;147:81–94. doi:10.1016/j.cell.2011.08.033.
- Shyh-Chang N, Zhu H, Yvanka de Soysa T, Shinoda G, Seligson MT, Tsanov KM, Nguyen L, Asara JM, Cantley LC, Daley GQ. Lin28 enhances tissue repair by reprogramming cellular metabolism. Cell. 2013;155:778–92. doi:10.1016/j.cell.2013.09.059.
- Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL, Toffanin S, O'Sullivan M, Lu J, Phillips LA, Lockhart VL, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nature genetics. 2009;41:843–8. doi:10.1038/ng.392.
- King CE, Cuatrecasas M, Castells A, Sepulveda AR, Lee JS, Rustgi AK. LIN28B promotes colon cancer progression and metastasis. Cancer research. 2011;71:4260–8. doi:10.1158/0008-5472.CAN-10-4637.
- Tu HC, Schwitalla S, Qian Z, LaPier GS, Yermalovich A, Ku YC, Chen SC, Viswanathan SR, Zhu H, Nishihara R, et al. LIN28 cooperates with WNT signaling to drive invasive intestinal and colorectal adenocarcinoma in mice and humans. Genes & development. 2015;29:1074–86. doi:10.1101/gad.256693.114
- Madison BB, Liu Q, Zhong X, Hahn CM, Lin N, Emmett MJ, Stanger BZ, Lee JS, Rustgi AK. LIN28B promotes growth and tumorigenesis of the intestinal epithelium via Let-7. Genes & development. 2013;27:2233–45. doi:10.1101/gad.224659.113
- Newman MA, Hammond SM. Emerging paradigms of regulated microRNA processing. Genes & development. 2010;24:1086–92. doi:10.1101/gad.1919710
- Rybak A, Fuchs H, Smirnova L, Brandt C, Pohl EE, Nitsch R, Wulczyn FG. A feedback loop comprising lin-28 and let-7 controls pre-let-7 maturation during neural stem-cell commitment. Nature cell biology. 2008;10:987–93. doi:10.1038/ncb1759.
- Heo I, Joo C, Kim YK, Ha M, Yoon MJ, Cho J, Yeom KH, Han J, Kim VN. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell. 2009;138:696–708. doi:10.1016/j.cell.2009.08.002.
- Lehrbach NJ, Armisen J, Lightfoot HL, Murfitt KJ, Bugaut A, Balasubramanian S, Miska EA. LIN-28 and the poly(U) polymerase PUP-2 regulate let-7 microRNA processing in Caenorhabditis elegans. Nature structural & molecular biology. 2009;16:1016–20. doi:10.1038/nsmb.1675
- Nam Y, Chen C, Gregory RI, Chou JJ, Sliz P. Molecular basis for interaction of let-7 microRNAs with Lin28. Cell. 2011;147:1080–91. doi:10.1016/j.cell.2011.10.020.
- Mayr F, Schutz A, Doge N, Heinemann U. The Lin28 cold-shock domain remodels pre-let-7 microRNA. Nucleic acids research. 2012;40:7492–506. doi:10.1093/nar/gks355.
- Heo I, Joo C, Cho J, Ha M, Han J, Kim VN. Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Molecular cell. 2008;32:276–84. doi:10.1016/j.molcel.2008.09.014.
- Heo I, Ha M, Lim J, Yoon MJ, Park JE, Kwon SC, Chang H, Kim VN. Mono-uridylation of pre-microRNA as a key step in the biogenesis of group II let-7 microRNAs. Cell. 2012;151:521–32. doi:10.1016/j.cell.2012.09.022.
- Chang HM, Triboulet R, Thornton JE, Gregory RI. A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28-let-7 pathway. Nature. 2013;497:244–8. doi:10.1038/nature12119.
- Wilbert ML, Huelga SC, Kapeli K, Stark TJ, Liang TY, Chen SX, Yan BY, Nathanson JL, Hutt KR, Lovci MT, et al. LIN28 binds messenger RNAs at GGAGA motifs and regulates splicing factor abundance. Molecular cell. 2012;48:195–206. doi:10.1016/j.molcel.2012.08.004.
- Cho J, Chang H, Kwon SC, Kim B, Kim Y, Choe J, Ha M, Kim YK, Kim VN. LIN28A is a suppressor of ER-associated translation in embryonic stem cells. Cell. 2012;151:765–77. doi:10.1016/j.cell.2012.10.019.
- Hafner M, Max KE, Bandaru P, Morozov P, Gerstberger S, Brown M, Molina H, Tuschl T. Identification of mRNAs bound and regulated by human LIN28 proteins and molecular requirements for RNA recognition. Rna. 2013;19:613–26. doi:10.1261/rna.036491.112.
- Graf R, Munschauer M, Mastrobuoni G, Mayr F, Heinemann U, Kempa S, Rajewsky N, Landthaler M. Identification of LIN28B-bound mRNAs reveals features of target recognition and regulation. RNA biology. 2013;10:1146–59. doi:10.4161/rna.25194.
- Stefani G, Chen X, Zhao H, Slack FJ. A novel mechanism of LIN-28 regulation of let-7 microRNA expression revealed by in vivo HITS-CLIP in C. elegans. Rna. 2015;21:985–96
- Mili S, Steitz JA. Evidence for reassociation of RNA-binding proteins after cell lysis: implications for the interpretation of immunoprecipitation analyses. Rna. 2004;10:1692–4. doi:10.1261/rna.7151404.
- Ule J, Jensen K, Mele A, Darnell RB. CLIP: a method for identifying protein-RNA interaction sites in living cells. Methods. 2005;37:376–86. doi:10.1016/j.ymeth.2005.07.018.
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC, Munschauer M, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–41. doi:10.1016/j.cell.2010.03.009.
- Kishore S, Jaskiewicz L, Burger L, Hausser J, Khorshid M, Zavolan M. A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nature methods. 2011;8:559–64. doi:10.1038/nmeth.1608.
- Sauliere J, Murigneux V, Wang Z, Marquenet E, Barbosa I, Le Tonqueze O, Audic Y, Paillard L, Roest Crollius H, Le Hir H. CLIP-seq of eIF4AIII reveals transcriptome-wide mapping of the human exon junction complex. Nature structural & molecular biology. 2012;19:1124–31. doi:10.1038/nsmb.2420
- Moy RH, Cole BS, Yasunaga A, Gold B, Shankarling G, Varble A, Molleston JM, tenOever BR, Lynch KW, Cherry S. Stem-loop recognition by DDX17 facilitates miRNA processing and antiviral defense. Cell. 2014;158:764–77. doi:10.1016/j.cell.2014.06.023.
- Granneman S, Kudla G, Petfalski E, Tollervey D. Identification of protein binding sites on U3 snoRNA and pre-rRNA by UV cross-linking and high-throughput analysis of cDNAs. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:9613–8. doi:10.1073/pnas.0901997106.
- Li YI, Sanchez-Pulido L, Haerty W, Ponting CP. RBFOX and PTBP1 proteins regulate the alternative splicing of micro-exons in human brain transcripts. Genome research. 2015;25:1–13. doi:10.1101/gr.181990.114.
- Zhang C, Darnell RB. Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nature biotechnology. 2011;29:607–14. doi:10.1038/nbt.1873.
- Moore MJ, Zhang C, Gantman EC, Mele A, Darnell JC, Darnell RB. Mapping Argonaute and conventional RNA-binding protein interactions with RNA at single-nucleotide resolution using HITS-CLIP and CIMS analysis. Nature protocols. 2014;9:263–93. doi:10.1038/nprot.2014.012.
- Lelyveld VS, Bjorkbom A, Ransey EM, Sliz P, Szostak JW. Pinpointing RNA-Protein Cross-Links with Site-Specific Stable Isotope-Labeled Oligonucleotides. Journal of the American Chemical Society. 2015. doi:10.1021/jacs.5b10596
- Wang L, Nam Y, Lee AK, Yu C, Roth K, Chen C, Ransey EM, Sliz P. LIN28 Zinc Knuckle Domain Is Required and Sufficient to Induce let-7 Oligouridylation. Cell Reports. 2017;18:2664–75. doi.org/10.1016/j.celrep.2017.02.044.
- Piskounova E, Viswanathan SR, Janas M, LaPierre RJ, Daley GQ, Sliz P, Gregory RI. Determinants of microRNA processing inhibition by the developmentally regulated RNA-binding protein Lin28. The Journal of biological chemistry. 2008;283:21310–4. doi:10.1074/jbc.C800108200.
- Kramer K, Sachsenberg T, Beckmann BM, Qamar S, Boon KL, Hentze MW, Kohlbacher O, Urlaub H. Photo-cross-linking and high-resolution mass spectrometry for assignment of RNA-binding sites in RNA-binding proteins. Nature methods. 2014;11:1064–70. doi:10.1038/nmeth.3092.
- Apffel A, Chakel JA, Fischer S, Lichtenwalter K, Hancock WS. Analysis of Oligonucleotides by HPLC-Electrospray Ionization Mass Spectrometry. Analytical chemistry. 1997;69:1320–5. doi:10.1021/ac960916h.