
ABSTRACT
Post-testicular sperm maturation and storage within the epididymis is a key determinant of gamete quality and fertilization competence. Here we demonstrate that mouse spermatozoa possess a complex small non-protein-coding RNA (sRNA) profile, the composition of which is markedly influenced by their epididymal transit. Thus, although microRNAs (miRNAs) are highly represented in the spermatozoa of the proximal epididymis, this sRNA class is largely diminished in mature spermatozoa of the distal epididymis. Coincident with this, a substantial enrichment in Piwi-interacting RNA (piRNA) abundance in cauda spermatozoa was detected. Further, features of cauda piRNAs, including; predominantly 29–31 nts in length; preference for uracil at their 5' terminus; no adenine enrichment at piRNA nt 10, and; predominantly mapping to intergenic regions of the mouse genome, indicate that these piRNAs are generated by the PIWIL1-directed primary piRNA production pathway. Accordingly, PIWIL1 was detected via immunoblotting and mass spectrometry in epididymal spermatozoa. These data provide insight into the complexity and dynamic nature of the sRNA profile of spermatozoa and raise the intriguing prospect that piRNAs are generated in situ in maturing spermatozoa. Such information is of particular interest in view of the potential role for paternal sRNAs in influencing conception, embryo development and intergenerational inheritance.
Introduction
The production of fertilization competent mammalian spermatozoa represents the culmination of an extraordinary sequence of cytological differentiation and functional maturation events. These processes are initiated within the testes, whereby spermatogonial stem cells undergo sequential phases of mitosis, meiosis and spermiogenesis.Citation1 During the latter of these phases, the developing germ cells experience a striking morphological transformation in which they expel the majority of their cytoplasm and concurrently remodel their nuclear content in such a way that the cells are rendered both transcriptionally and translationally quiescent.Citation2 Despite the accompanying loss of a substantial portion of stored RNA cargo, the highly specialized spermatozoa that leave the testis do however retain a complex RNA payload, including both protein-coding messenger RNAs (mRNA) and numerous classes of non-protein-coding RNAs, including the small RNA (sRNA) species; microRNAs (miRNAs), endogenous small-interfering RNA (endo-siRNAs), interference RNAs (iRNAs), transfer RNA-derived sRNAs (tsRNA), antisense RNAs (asRNAs), and the piwi-interacting RNAs (piRNAs).Citation3-8
A precise biological function remains to be assigned for a majority of the thousands of RNA transcripts carried by sperm.Citation5 Nevertheless, the presence of such a diverse RNA profile takes on added significance in view of their potential to be delivered to the oocyte, along with the paternal genome, whereupon they may contribute to fertilization and embryo development and fitness.Citation3,Citation9-11 This implies that, rather than simply being a legacy of the spermatogenic process, these transcripts may actually have fitness consequences and potentially adaptive value.Citation12 As an extension of this hypothesis, it has recently been proposed that sperm RNA could encode information about the paternal environment, with males having the capacity to modify the RNA content of their sperm under different conditions to prime the embryo to develop appropriately within the prevailing environment.Citation13 Although unequivocal evidence of whether such effects are adaptive is presently lacking,Citation14 the premise that environmental experience can influence the RNA profile of spermatozoa is gaining growing acceptance.Citation15,Citation16 Indeed, compelling evidence now exists that exposure of the paternal linage to a wide range of environmental stressors, including: dietary perturbations,Citation17-19 corticosterone administration,Citation20 psychological stress,Citation21-23 cigarette smoke,Citation24 and environmental pollutants such as sodium fluoride,Citation25 can alter the RNA profile that the exposed spermatozoa carry, with significant implications for post-fertilization functions, including the transmission of transgenerational inheritance phenotypes to the offspring.
Despite emerging and repeated demonstration that the sperm RNA profile displays considerable plasticity,Citation5,Citation26-28 our current understanding of the timing and mechanistic basis responsible for such alterations remains poor. The possibility that these changes are imposed on the developing germ cells within the testis is seemingly incompatible with the strict regulation imposed by sRNAs on germ cell differentiation,Citation29-31 and the lack of overt morphological defects in the spermatozoa of males exposed to environmental stresses.Citation32 Rather, there is mounting consensus that such changes may be communicated to spermatozoa after they have left the protective environment of the testes during their post-testicular maturation en route through the several meters of male reproductive tract (epididymis).Citation17,Citation28,Citation33 Indeed, the combined phases of sperm transit and storage within the epididymal tubule, provides a protracted developmental window of several weeksCitation34,Citation35 to effect changes in the sperm epigenome.Citation32,Citation36 This agrees with our own evidence that the sperm miRNA profile is dynamically modified as the cells migrate through the epididymis,Citation28 and that epididymosomes (small membrane bound vesicles secreted by the epididymal soma) can serve as a vehicle to mediate soma-spermatozoa intercellular communication.Citation17,Citation37 However, the magnitude of such changes, and whether they extend beyond the widely studied miRNA class of small regulatory RNA, to include other forms of developmentally important sRNA species has yet to be explored under normal physiological conditions.
Here, we interrogate our existing deep sequencing datasets to systematically survey the complete sRNA profile of mouse spermatozoa at the different stages of their post-testicular epididymal maturation. Further, we compared the sperm profiles to those of the epididymal epithelium and the epididymosomes they secrete in order to determine whether this form of soma-spermatozoa intercellular communication could account for the dynamic changes in the sperm-borne sRNA profile. Such an approach revealed that sperm acquire an abundance of piRNAs as these cells transit the epididymis. Interestingly, elevated piRNA abundance was determined to be specific to the sperm, and further, immunolocalization demonstrated that sperm harbor key machinery proteins required for piRNA production. Taken together, our data raises the intriguing possibility that maturing sperm are functionally competent in the production of this developmentally important class of small regulatory RNA.
Results
The composition of the small RNA profile of spermatozoa, epididymosomes and epithelial cells is both complex and dynamic
To assess the spatio-temporal distribution of the major species of sRNA across the mouse epididymis, and maturing spermatozoa during epididymal transit, the abundance of each sRNA species was assessed in our existing datasets [described in[Citation28,Citation37-39]] sampled from the soma, epididymosomes and spermatozoa derived from the caput, corpus and cauda epididymal segments. illustrates the dynamic nature of the spatio-temporal accumulation of each of the major sRNA species between spermatozoa, epididymosomes, and epididymal epithelial cells, and between each segment of the epididymis. For example, miRNAs are enriched in the epithelial cells, epididymosomes, and spermatozoa isolated from the caput segment relative to their abundance in the more distal segments of the epididymis (). More specifically, miRNAs accounted for 39, 61 and 50% of the global sRNA population of caput soma, epididymosomes and sperm, respectively. The dynamic reduction in miRNA accumulation across the epididymis regions is not a surprise as it is well documented that sperm become transcriptionally and translationally inert as they mature (migrate). Therefore, the requirement for miRNAs to regulate target gene expression is removed as the sperm progress to maturity. A number of other trends were also readily evident in the sRNA profiles between assessed datasets. Notably, the tRNA component of the spermatozoa sRNA landscape increased dramatically during post-testicular maturation, with tRNA abundance increasing from 24% in caput spermatozoa to 65% in cauda spermatozoa (). Given that tRNA-derived fragments (tsRNAs) are associated with epigenetic regulation and intergenerational inheritance in mice,Citation17 this finding is of interest for future study.
Figure 1. The dynamic nature of the sRNA landscape of the epididymal caput, corpus and cauda segments. Following filtering and normalization, sRNA reads were mapped to known sRNA populations retrieved from the mouse Ensembl non-coding RNA database to determine the contribution of each of the major sRNA species to the epigenome of mouse spermatozoa, epididymosomes, and epithelial cells. (A) Proportion of sRNAs belonging to each of the six major sRNA subclasses: piRNAs, miRNAs, tsRNAs, rRNAs, snoRNAs and snRNAs. (B) piRNA abundance and size distribution in epididymal spermatozoa, epididymosomes, and epithelial cells. Notably, the majority of piRNAs within cauda spermatozoa are between 29 and 31nt in size, suggesting generation via the PIWIL1-directed primary production pathway.
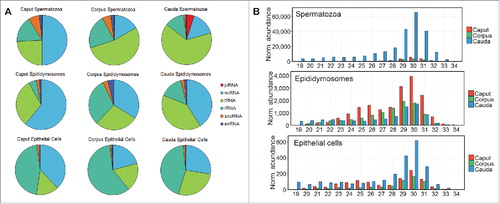
Profiling also demonstrated that the sRNA composition of epididymosomes is surprisingly complex given their relatively simple structure (). Multiple studies have demonstrated that the sRNA payload of epididymosomes, expelled from the epididymal epithelium, is delivered to maturing spermatozoa during epididymal transit.Citation17,Citation37 So far, epididymosomes have been associated with transfer of miRNAsCitation37 and tsRNAs.Citation17 However, the presence of considerable amounts of rRNA fragments, snoRNA, and snRNA in epididymosomes, in addition to the miRNA and tsRNA component, indicates that these epididymis-specific exosomes play a more central role in facilitating transfer of multiple sRNA species to maturing spermatozoa ().
Cauda spermatozoa are remarkably enriched in piRNA sequences
One of the most striking findings of our dataset profiling was the observed enrichment of piRNAs within cauda spermatozoa relative to the sperm fraction isolated from the more proximal (caput and corpus) regions of the epididymal tract (). More specifically, piRNAs were determined to comprise equal to or less than 0.05% of the sRNA pool in epididymal epithelial cells and epididymosomes, irrespective of which epididymal segment from which they were sampled (). In contrast, piRNAs are comparatively enriched in caput (0.22%) and corpus (0.25%) spermatozoa, with the greatest enrichment evident in cauda spermatozoa (4.87%; ). The substantial enrichment of piRNAs in cauda spermatozoa in comparison to other spermatozoa datasets was surprising, particularly as epididymosomes and epithelial cells failed to return a comparable accumulation trend (). As epididymosomes, proposed transporters of sRNA (and other RNA and protein cargo) produced in epididymal soma to maturing spermatozoa, did not return a comparable piRNA abundance, this result provided an early indication that the production of piRNAs in caudal spermatozoa may be autonomous in nature and thereby independent of an epididymosome-mediated transfer pathway.
Having identified that cauda spermatozoa harbor an elevated abundance of piRNAs relative to other cell types and regions, we focused on uncovering the distribution and features of the piRNA population within each dataset (). The length and abundance of piRNAs was assessed for each of the nine datasets to gain additional insight into the epididymal sperm piRNA landscape (). From this analysis, it is apparent that piRNAs of all size classes were enriched in cauda spermatozoa compared to spermatozoa isolated from the caput or corpus segments (). Further, this analysis again confirmed that piRNAs are substantially more abundant in spermatozoa than in either epididymosomes or the epididymal soma ().
Previous studies have highlighted that piRNA length is an indicator of the mechanism by which they are produced, that is; generation by the primary or secondary (“ping-pong”) piRNA production pathway.Citation40 PIWIL1 preferentially associates with piRNAs 29–31 nucleotides (nt) in length, whereas PIWIL2-associated piRNAs are typically slightly shorter in length ranging from 26 to 28 nt.Citation41,Citation42 Therefore, documenting the length of piRNAs that accumulate in each dataset provided a preliminary indication of the pathway responsible for piRNA production. Across the nine datasets assessed, most piRNAs were 29–31 nt in length, and with approximately 60% of cauda spermatozoa piRNAs falling within this size range (). Based on this analysis, we suggest that the majority of piRNAs detected in cauda sperm appear derived from the primary PIWIL1-directed production pathway, rather than the secondary pathway directed by PIWIL2/PIWIL4 ().
In addition to an overall enrichment in the abundance of piRNA in caudal spermatozoa, another interesting finding was that the largest number of unique piRNA sequences, compared to the caput and corpus datasets, was profiled in caudal spermatozoa. More specifically, a total of 316,867 unique piRNA sequences, identified via proTRAC analysis, were detected in cauda spermatozoa. In comparison, the caput and corpus spermatozoa returned piRNA pools at least 10-fold less heterogeneous in composition, consisting of 30,454 and 18,295 unique piRNA sequences, respectively. Further, the number of unique piRNA sequences identified in caput spermatozoa was substantially higher than the total number of piRNA sequences identified in our epithelial cell or epididymosome datasets. This finding clearly indicates that piRNA enrichment in caudal spermatozoa does not simply reflect enhanced production of a subset of piRNAs, but rather, that the primary piRNA production pathway is stimulated in sperm as they enter the distal segment of the tract. The apparent activation of the primary piRNA pathway in caudal sperm was highly curious, and raised intriguing questions on how such a diverse and distinct population of piRNAs is produced at this late stage of sperm maturation.
Characteristics of piRNA clusters active in mouse cauda spermatozoa
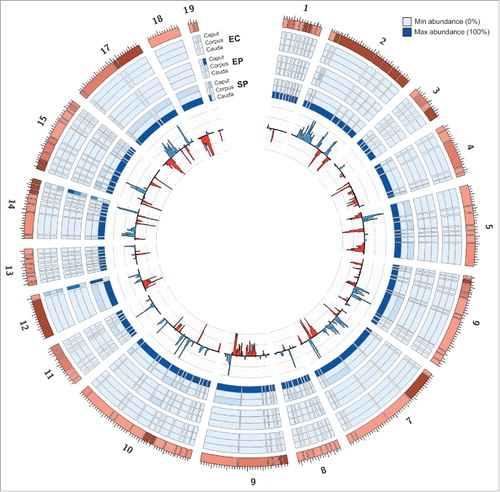
To investigate the potential role of the piRNA population in cauda sperm, the identity of the piRNA clusters present in epididymal spermatozoa, epithelial cells, and epididymosomes was assessed. A total of 116 piRNA clusters were identified across all datasets (Supplementary Table S1). The length of piRNA clusters ranged from 5,000 bp to 76,860 bp, with a mean cluster size of 16,948 bp ± 1731 bp (Supplementary Table S1). As a number of piRNAs clusters have previously been described and annotated during spermatogenesis, we first set out to determine whether the piRNA clusters identified here represented known or novel clusters. A list of known piRNA cluster coordinates was retrieved from an online piRNA cluster database which aggregates piRNA clusters from multiple tissues and developmental stages.Citation43 The piRNA cluster database employed in this study describes over two thousand mouse piRNA clusters, the majority of which were found to arise during spermatogenesis in the testis.Citation43 Of the 116 clusters identified in this study, 114 clusters overlapped “known” clusters from which piRNAs have been demonstrated to accumulate from during pachytene and/or pre-pachytene spermatogenesis (Supplementary Table S1). Notably, the piRNA clusters identified in this study (n = 116) are significantly less numerous than previously reported for spermatogenesis, with some studies indicating the presence of several hundred piRNA clusters in a single tissue.Citation43,Citation44 Taken together, piRNA cluster analysis showed that the overwhelming majority of piRNAs produced in mouse sperm during epididymal transit arise from piRNA clusters also active during the initial stages of spermatogenesis in the mouse testis.
Next, the abundance of piRNAs arising from each of the 116 identified piRNA clusters was assessed across the nine datasets to document piRNA origin across the mouse epididymis. Average abundance was calculated between biological replicates to determine the relative abundance of piRNAs originating from each cluster. illustrates the proportion of piRNAs arising from each cluster across the nine assessed datasets with fold change calculated relative to the dataset harboring the highest abundance. This analysis clearly demonstrates that piRNA abundance is almost uniformly enhanced across all piRNA clusters (enhanced in 113/116 clusters) in caudal spermatozoa. In comparison, the average piRNA abundance in spermatozoa harvested from caput and corpus regions is consistently less than 10% of their levels in cauda sperm (). This finding further confirmed that piRNA production is globally upregulated in caudal spermatozoa, and that the proximal-distal trend of enhanced piRNA production during sperm maturation is distinct to epithelial cells and epididymosomes. Therefore, unlike the miRNAs that have been previously demonstrated to be specifically trafficked from the epididymal soma to maturing sperm via epididymosomes, the data presented here suggests that piRNAs are produced within the caudal spermatozoa ().
Figure 2. piRNAs are globally enriched in the cauda spermatozoa dataset. Outer Rings: The outer rings display a heat-map, indicating the relative enrichment of piRNAs from each cluster between datasets (EC, epithelial cell; ES, epididymosome; SP, spermatozoa). Each blue tile in the outer rings represents one of the 116 piRNA clusters identified in this study. The width of each tile represents the relative size of the piRNA cluster. The abundance of piRNAs in each dataset was calculated relative to the dataset which possessed the maximal abundance for each cluster. The shade of the tile reflects the abundance of piRNAs in the dataset. Inner Ring: The inner ring displays the normalized abundance of piRNAs arising from each cluster in the cauda spermatozoa dataset. Blue bars indicate piRNAs produced from the sense strand, while red bars indicate piRNAs produced from the antisense strand.
The hypothesis that caudal spermatozoa piRNA production is independent of epididymosomes raises a number of questions regarding the underlying biogenesis reactions that must occur within the maturing sperm during epididymal transit. Currently it is understood that primary piRNA production initiates via synthesis of a long non-protein-coding RNA substrate from each piRNA cluster, and that this piRNA precursor transcript is subsequently processed via PIWIL1 into piRNA sRNAs in either a mono- or bi-directional manner.Citation45,Citation46 As epididymal spermatozoa are believed to be transcriptionally quiescent, piRNA production poses a perplexing question regarding the origin of the abundant piRNA sRNA detected in cauda sperm. Therefore, in order to provide further insight into how piRNAs are produced in caudal sperm, the genomic origin and sequence-based features of the cauda sperm piRNA population was subsequently assessed.
piRNA production in caudal spermatozoa
Two principal pathways for the production of piRNA sRNAs have been described previously; (1) the primary piRNA production pathway catalyzed by PIWIL1, and; (2) the secondary, or ping-pong, production pathway catalyzed by PIWIL2/PIWIL4. Distinction between the two documented piRNA production pathways is achieved via analysis of piRNA sequence features, namely; piRNAs generated in the primary pathway display an enriched proportion of sRNAs with a uracil (U) residue at their 5' terminus (sRNA nt position 1). In contrast, piRNAs produced in the secondary pathway are additionally enriched for an adenine (A) residue at position 10 of the piRNA sRNA. Therefore, in order to provide further insight into the molecular mechanism of piRNA production in cauda spermatozoa, the nucleotide composition was assessed across the length of each detected piRNA sRNA. This analysis, , clearly shows that the majority (> 80%) of piRNAs detected in cauda sperm harbored a U residue at their 5' terminus and no significant enrichment for an A residue at piRNA sRNA nt position 10. This finding, taken together with the piRNA size length analysis (), provides further evidence that cauda sperm piRNAs are predominantly produced via the primary production pathway.
Figure 3. Features of the cauda spermatozoa piRNA population indicate generation via the PIWIL1-directed primary production pathway. The characteristics of the pool of piRNAs present in cauda spermatozoa (n = 316,867) was assessed to provide insight into the mechanism through which this piRNA population was generated during sperm transit of the epididymis. (A) Nucleotide frequency at each position for all detected cauda sperm piRNA sRNAs. Notably, >80% of piRNA sRNAs were determined to possess a U residue at their 5' terminus, and no enrichment for an A residue at piRNA position 10. (B) Proportion of piRNAs which map uniquely to the mouse genome compared to piRNAs which map to multiple genomic locations. (C) Genomic origin of the piRNA population was determined via BEDtools. piRNAs were determined to belong to genic or TE regions if they intersected with previously described features. The remaining pool of piRNAs were classed as intergenic. (D) Of the subset of piRNAs which aligned to transposons and other repeat-associated sequences, the superfamily from which the cauda sperm piRNA originated was also assessed. (E) Of the subset of piRNAs found to arise from specific sequence based features of a gene body, we next determined whether the piRNA mapped to the 5' UTR, CDS, or 3' UTR of the gene body.
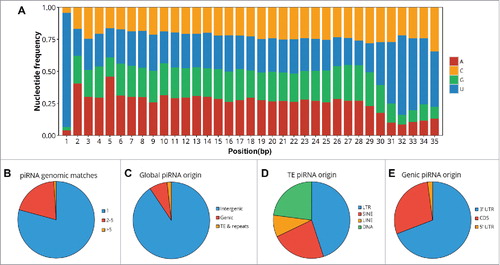
The origin of each piRNA sequence was next assessed to provide further understanding of both their mechanism of production and likely in vivo function. PIWIL2/PIWIL4-generated piRNAs commonly map to transposons and other repeat associated sequences and are involved in repressing the activity of such sequences via either transcriptional or post-transcriptional silencing mechanisms. As piRNA clusters are typically long sequences, usually greater than five kilobases (kb) in length, these precursors tend to span multiple genomic features. Therefore, rather than characterizing piRNA origin on a per-cluster basis, the origin of each unique piRNA sequence was assessed via identifying overlapping genomic features. This was achieved via intersection of piRNA genomic coordinates with those of known genes, transposons, and other repeat-associated elements across the mouse genome. The overwhelming majority, 81%, of cauda spermatozoa piRNAs were found to map to, and therefore assumed to have originated from, a single genomic position (). Further, approximately 18% of piRNAs mapped to two to five genomic positions, while only 1% of analyzed piRNAs mapped to more than five genomic locations. Interestingly, more than 90% of the caudal sperm piRNAs were determined to have originated from an intergenic region. This result indicates that these piRNAs did not originate from a region of the mouse genome previously annotated as a gene or other genic-based feature (). Only 7.6% of caudal sperm piRNAs appeared derived from transposons to provide further evidence that piRNAs in cauda sperm are generated via the PIWIL1-directed primary pathway, rather than the PIWIL2/PIWIL4-directed secondary pathway. The piRNAs determined to have originated from repetitive regions of the mouse genome were subsequently mapped to multiple transposable element families, including the DNA, LINE, SINE, and LTR transposon families. It is important to note that no enrichment to a specific class of repetitive element was observed for this subpopulation of caudal sperm piRNA (). Considerably less cauda sperm piRNAs of unique sequence mapped to gene rich regions of the mouse genome (). Previous research has demonstrated that piRNAs produced during spermatogenesis in mice disproportionally accumulate to the 3' untranslated region (UTR) of protein-coding genes.Citation47,Citation48 To investigate whether our data aligned with that previously reported, piRNAs mapping to genic features were further sub-categorized on the basis of their location within each specific feature of a gene body, including the 5′ UTR, coding sequence (CDS) and 3′ UTR. Approximately 69% of genic piRNAs were determined derived from 3′ UTRs, compared to 29% aligning to the CDS region of protein coding genes and 2% to the 5' UTR (). The 3′ UTR bias of gene-specific piRNAs in mouse cauda sperm was confirmed via a Mann-Whitney U test which revealed that, on a per-gene basis, piRNAs were enriched to this genic feature compared to either the 5' UTR or CDS of protein coding genes (p < 0.05).
Experimental validation of piRNA enrichment in cauda spermatozoa
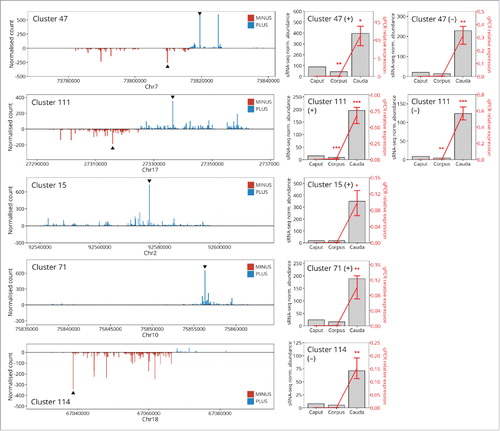
Bioinformatic assessment across our datasets repeatedly indicated that piRNA accumulation is enhanced in sperm sampled from the distal cauda segment of the epididymis. This enrichment in piRNA abundance is specific to the sperm as we failed to observe a similar trend in the sRNA profiles generated for the epithelial cells and epididymosomes sourced from the same region of the epididymis. This specific enrichment to piRNA abundance in cauda sperm has not been described previously, and raises a number of interesting questions regarding the molecular origin of this sRNA species. To experimentally validate the bioinformatic data presented here, piRNA abundance was assessed via a stem-loop RT-qPCR (SL-qPCR)-based approach. The most highly abundant piRNA sequences from selected loci (both bidirectional and unidirectional piRNA generating loci) were included in this analysis. Bidirectional clusters formed the minority of identified piRNA clusters, with only 15% of clusters displaying bidirectional formation. Stem-loop qPCR assays were performed on RNA isolated from epithelial cells, epididymosomes, and spermatozoa harvested from the caput, corpus, and cauda segments of the mouse epididymis. displays the distribution of piRNA sRNAs from both bidirectional and unidirectional piRNA clusters. This SL-qPCR approach repeatedly demonstrated that piRNA sRNA abundance is highly elevated in cauda spermatozoa, compared to spermatozoa harvested from either the caput or corpus segment of the epididymis (p < 0.05, ). This experimental validation of our bioinformatic data further indicates that a previously uncharacterized piRNA production pathway becomes highly activated in spermatozoa during their epididymal transit. In contrast, we confirmed minimal accumulation of piRNA in either epididymal epithelial cells or epididymosomes (Supplementary Figure S1).
Figure 4. Experimental validation of sRNA-seq data confirms that piRNA accumulation is significantly more abundant in the cauda than the caput or corpus spermatozoa. To experimentally confirm the observation that cauda spermatozoa are enriched in piRNAs, SL-qPCR was performed on highly abundant piRNA sequences from two bidirectional (Cluster 47 and Cluster 111) and three monodirectional (Cluster 15, Cluster 71, and Cluster 114) piRNA clusters. The distribution of piRNAs arising from each clusters is presented on the left hand side of each panel. Blue bars indicate piRNAs corresponding to the sense (“plus”) strand, while red bars indicate piRNAs arising from the antisense (“minus”) strand. Black triangles indicate highly abundant piRNAs validated by SL-qPCR. The right side of each panel presents the results of the validation experiments; cauda spermatozoa are significantly enriched for each assessed piRNA relative to caput or corpus sperm. Error bars represent SEM of three biological replicates. Asterisks indicate significant difference (p < 0.05 = *, p < 0.01 = **, p < 0.001 = ***).
Localization of piRNA sRNA machinery protein in epithelial cells and spermatozoa of the mouse epididymis
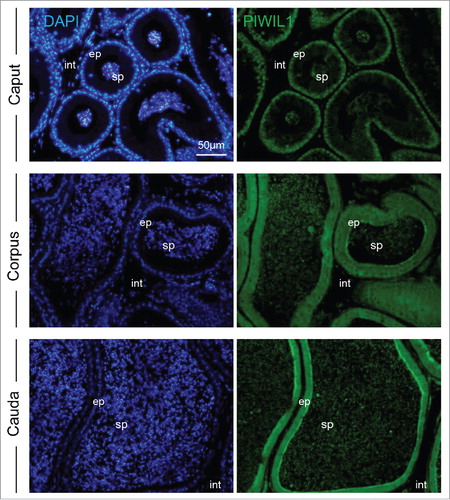
Considering the substantial abundance of piRNA that specifically accumulate in cauda epididymal spermatozoa, we examined the localization of the PIWIL1 protein implicated in catalysis of the primary piRNA production pathway, within the epithelial cells and spermatozoa of the mouse epididymis. Immunolocalization of PIWIL1 within the proximal (caput) segment of the epididymis revealed that this protein is predominantly localized to the perinuclear domain within the majority of epithelial cells and on the spermatozoa within the lumen of the duct (). In the downstream segments of the epididymis, the corpus and cauda, PIWIL1 displayed a more uniform labeling of the entire cytosolic region of the epithelial cells and was again strongly detected in the luminal spermatozoa. The specificity of these immunolocalization patterns was confirmed through the absence of staining in epididymal sections labeled with secondary antibodies alone (data not shown). Similarly, the interstitial tissue surrounding the epididymal duct consistently failed to label with the anti-PIWIL1 antibodies used in this study.
Figure 5. Immunolocalization of the PIWIL1 protein required for piRNA production in mouse epididymal tissue. Mouse epididymal sections were labeled with anti-PIWIL1 antibodies followed by an appropriate AlexaFluor-488 conjugated secondary antibodies. All tissue sections were counterstained with 4',6-diamidino-2-phenylindole (DAPI) and viewed using confocal microscopy. ep = epididymal epithelium, sp = spermatozoa residing in the epididymal lumen, int = interstitial tissue. Scale bar = 50 μm. These experiments were replicated three times using independent samples from three mice and representative images are shown.
Considering the detection of immunostaining on epididymal spermatozoa, these cells were isolated from the caput, corpus and cauda epididymal segments and subjected to immunocytochemistry to assess the precise domains to which PIWIL1 localizes. As shown in , PIWIL1 proteins was clearly detected in isolated sperm cells but appeared to undergo characteristic changes in localization during sperm transport through the epididymal tubule. Thus, PIWIL1 displayed strong labeling of the anterior region of the sperm head in most epididymal regions that was accompanied by variable staining of either the mid and/or principal piece of the flagellum ().
Figure 6. Immunolocalization of the PIWIL1 protein in mouse spermatozoa. (A) Mouse spermatozoa recovered from the caput, corpus, and cauda epididymis were labeled with anti-PIWIL1 antibodies followed by appropriate AlexaFluor-488 conjugated secondary antibodies. The cells were then viewed using confocal microscopy. Scale bar = 10 μm. (B) Antibody specificity was confirmed through immunoblotting of whole mouse testis lysate with anti-PIWIL1 antibodies followed by appropriate HRP-conjugated secondary antibodies. All experiments were replicated three times using independent samples and representative images are shown. (C) Additional supporting evidence for the presence of PIWIL1 was sought through the use of mass spectrometry. This analysis returned a high number of peptides that uniquely mapped to the PIWIL1 protein (underlined, red font).
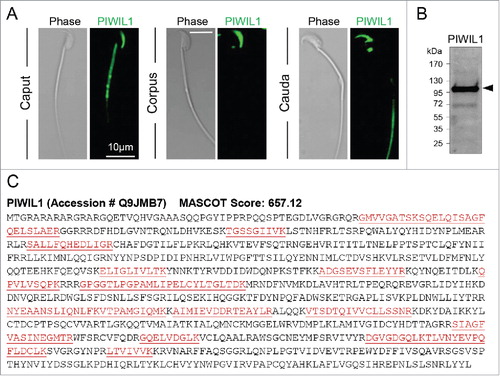
Immunoblotting of whole testis lysates confirmed the specificity of the anti-PIWIL1 antibodies for detection of the cognate mouse protein (, arrowheads). Specifically, a predominant band of the appropriate molecular weight (∼95 kDa) was detected. Importantly, no such labeling was observed for the secondary antibody only controls (data not shown). Similarly, pre-absorption of the anti-PIWIL1 antibodies with excess immunizing peptide also effectively eliminated all immunolabeling of this protein in sperm (Supplementary Figure S2A). Further, mass spectrometry analysis of spermatozoa returned a high confidence identification of the PIWIL1 protein (). Together, these data provide additional evidence supporting the presence and putative activity of the PIWIL1-mediated primary piRNA production pathway in mouse spermatozoa.
Discussion
Here we sought to construct comprehensive profiles of the global sRNA population for epithelial cells, epididymosomes and spermatozoa sampled from the mouse epididymis. Further, we aimed to characterize the extent to which the sperm sRNA profiles altered as the sperm gain functional maturity during their transit of the epididymis. Interrogation of next generation sequencing datasets revealed several novel insights into the complexity of the sRNA pool carried by maturing spermatozoa under normal physiological conditions. Firstly, we uncovered a remarkable level of plasticity in the sperm sRNAome, confirming that the sRNA population harbored by mature spermatozoa is unlikely to simply result from the legacy of the spermatogenic cycle. Rather, we detected profound changes both in terms of the relative abundance and overall proportion of the different species of sRNA that collectively contribute to the global sRNA population of mature sperm. As an important caveat to this finding, we acknowledge that at least a portion of the sRNA changes we document may be attributed to the loss of the cytoplasmic droplet, an entity that originates during the morphological differentiation of spermatids before being progressively shed during epididymal transit. Secondly, we were able to again demonstrate the potential role played by epididymosomes in mediating intercellular trafficking of many of the sRNA species that accumulate in the tract between the epididymal soma and luminal spermatozoa. However, we also identified a substantial subset of sRNA whose production is highly unlikely to be attributed to the surrounding epididymal soma. Chief among these was the piRNA, as this class of sRNA was essentially absent in immature caput spermatozoa, before representing ∼5% of the total sRNA population of mature cauda spermatozoa. This result was not expected, with the weight of research indicating that nuclear gene expression is completely repressed in the latter phases of germ cell development within the testes. Given the novelty of such findings, characterization of the sperm-borne piRNAs became the focus of this study.
Analysis of the sperm piRNA population revealed a suite of evidence to suggest that maturing cauda sperm generate piRNAs via the PIWIL1-directed primary production pathway. Firstly, piRNAs isolated from epididymal spermatozoa are predominantly 29–31 nt in length (), which is a hallmark of PIWIL1-mediated piRNA production.Citation49 In addition, piRNAs identified in cauda spermatozoa did not display the characteristic enrichment of an A residue at sRNA nt position 10 to suggest that the detected piRNAs are not generated in the secondary production pathway via a ping-pong mode of action. Further evidence to support that the cauda sperm piRNA detected here predominantly result from the primary piRNA production pathway was provided by the mapping of the piRNA sRNAs to their genomic origins (). This analysis clearly showed that >80% of piRNA sRNA mapped to a single genomic location, and were primarily derived from intergenic regions (). Additionally, piRNAs arising from within genes showed a strong bias for piRNAs mapping to the 3′ UTR of the gene body, as previously reported.Citation50 This is in marked contrast to piRNAs arising from the secondary piRNA pathway, in which piRNAs are typically produced from transposons or other repeat-associated regions of the genome and subsequently function in maintenance of genomic integrity via repression of repetitive DNA activity. Together, these multiple pieces of evidence indicate strongly that the piRNA population identified in spermatozoa are products of a PIWIL1-directed production pathway, a finding that aligns closely with those previously described for piRNA production during spermatogenesis.Citation50 In this context, previous studies have established that mouse piRNAs are not overtly enriched for transposon-encoding sequences with only 17% reportedly mapping to repetitive elements in the mouse genome.Citation51,Citation52 These data support the notion piRNAs may hold central roles in regulation of male germ cell development that extend beyond the repression of retrotransposon elements to include functions such as piRNA-directed repression of protein coding gene expression.Citation53,Citation54
Notably, this study also indicates that piRNA production in cauda spermatozoa may be autonomous, and thereby independent of epididymosome-mediated sRNA trafficking. Epididymosomes have recently been shown to supplement the sRNA population of maturing spermatozoa during transit through the epididymal lumen; epididymosomes released by the epididymal epithelial cells are enriched in miRNAs and tsRNAs, which are delivered to spermatozoa and thereby regulate the epigenome of the immature sperm.Citation17,Citation37 Therefore, we initially postulated that similarly, epididymosomes may serve as a cargo vessel in the delivery of piRNAs to spermatozoa during their protracted storage in the cauda region of the epididymis. However, the abundance of piRNAs in the epididymosome sRNA pool is dwarfed by the size of the cauda sperm piRNA population, indicating that epididymosomes are unlikely to be involved, either directly or indirectly, with the dramatic enhancement of piRNA abundance observed in cauda spermatozoa.
These data accord with the detection, in mouse spermatozoa, of PIWIL1, a member of the murine PIWI subfamily of Argonaute nucleases that is required for primary piRNA production.Citation45 Prior research has established that PIWI proteins are predominantly, but not exclusively, restricted to the germline, where they function across virtually all phases of the germline cycle. Indeed, PIWI proteins have been implicated in crucial roles associated with the earliest stages of germline fate specification, maintenance/renewal of the germline stem cell population, and regulation of the later stages of gametogenesis encompassing spermiogenesis, oocyte activation, and fertilization.Citation55 While such functions appear highly conserved across metazoan species, our findings do contrast those of early reports suggesting that neither PIWI proteins, nor piRNAs, are detectable in the cauda epididymis of mice.Citation55,Citation56 The reason for such a dichotomy is not immediately apparent, yet our detection of PIWIL1 in mature caudal spermatozoa is supported by several lines of evidence including immunocytochemical labeling and high confidence mass spectrometric identification.
The activation of a putative PIWIL1-mediated primary piRNA production pathway in cauda spermatozoa raises a number of questions that challenge the existing paradigm regarding the transcriptional activity of post-testicular spermatozoa.Citation57 Indeed, the prevailing paradigm indicates that, following spermatogenesis, spermatozoa are rendered both transcriptionally and translationally quiescent.Citation1,Citation2 As such, the bulk of the processes that control the functional maturation of these cells are considered to be driven by extrinsic factors present in the epididymal luminal milieu, rather than acting at the transcriptional level.Citation58 However, the observation that the primary piRNA production pathway may be active in cauda spermatozoa clearly challenges this view. The current understanding of the primary piRNA production pathway in mice suggests that piRNA clusters are transcribed into long non-protein-coding RNAs, which are subsequently processed into 29–31 nt piRNAs via PIWIL1 activity.Citation46 At present, it remains to be determined whether precursor piRNA transcripts are present, but remain latent, in spermatozoa throughout epididymal transit or whether piRNA loci are actively transcribed and processed in cauda spermatozoa. Based on current understanding of sperm chromatin packaging, and a lack of experimental evidence that these cells possess the transcriptional machinery necessary for piRNA production, we favour the processing of piRNA precursors in cauda spermatozoa as a more plausible explanation. If this mechanism does prove correct then it poses the intriguing question of how the primary piRNA processing pathway is selectively stimulated within spermatozoa stored in the cauda epididymis. Since the PIWIL1 protein was detected in populations of caput, corpus and cauda epididymal sperm it is unlikely that the catalyst for such activation resides with the uptake of this protein from the epididymal fluids. Rather, in keeping with well-established models of epididymal maturation,Citation59 it is conceivable that the activity of PIWIL1 is dynamically regulated via post-translational modification of either the protein itself or one of its many interacting regulatory partners.
Irrespective of their mode of production, the extreme diversity and abundance of piRNAs within cauda spermatozoa, relative to other datasets analyzed, raises an array of further questions regarding their function in vivo. Over the last decade, piRNAs have been ascribed diverse roles that largely center on the repression of transposable elements to prevent their mobilization and thus maintain the integrity of the germline.Citation60 The active involvement of piRNAs in the protection of the paternal genome during spermatogenesis is highlighted by the dysregulation of sperm production and resulting sterility that is precipitated by loss-of-function mutation in key PIWI-associated proteins.Citation55 However, the mechanistic role of piRNAs in the reservoir of spermatozoa held within the cauda epididymis remains less certain. One intriguing possibility is that they serve as an additional tier of innate defense to protect the mature sperm population against the horizontal transfer of retrotransposon sequences via exosomes. In this regard, it has recently been postulated that blood-borne exosomes could facilitate the transmission of retrotransposon sequences from either somatic cells or parasitic vectors (such as ticks) to the germline.Citation61-63 Although the validity of such a model awaits further investigation, the elaboration of piRNAs in mature spermatozoa could conceivably mitigate the risk of such transfer leading to permanent changes in the paternal genome and/or their transfer to offspring. The impetus to investigate this possibility is given credence by the fact that piRNAs also feature prominently among the multiple classes of sRNA identified in ejaculated human spermatozoa. Indeed, the interrogation of next-generation sequencing data has revealed that piRNAs may constitute as much as 17% of the complex pool of sRNA harbored by the spermatozoa of fertile normozoospermic individuals.Citation5
Conclusions
In summary, our study has provided important new insight into the complexity and dynamic nature of the global sRNA profile of maturing spermatozoa following their production within the testes. These data have revealed a dramatic increase in piRNA abundance in spermatozoa in the distal cauda segment of the epididymis that occurs independently of commensurate elevation in the abundance of this sRNA species in either the surrounding epididymal soma or epididymosomes sampled from the same epididymal segment. Further, we report the detection of the PIWIL1 protein implicated in primary piRNA production in cauda spermatozoa. Together, the data presented here raises the intriguing possibility that spermatozoa are functionally competent in the production of this developmentally important class of regulatory sRNA. While we remain uncertain what role the detected piRNA play in mature spermatozoa, our data take on added clinical significance with the recognition that abundant populations of piRNA are harbored in the mature spermatozoa of our own species. Further investigation of both piRNA production, and function, are thus likely to afford significant advances in our understanding of the mechanisms that underpin the functional maturation of spermatozoa and preserve the integrity of the paternal genome that they carry.
Materials and methods
Ethics statement
All experimental procedures were carried out with the approval of the University of Newcastle's Animal Care and Ethics Committee (approval number A-2013–322), in accordance with relevant national and international guidelines. Inbred Swiss mice were housed under a controlled lighting regime (16L: 8D) at 21–22°C and supplied with food and water ad libitum. Prior to dissection, animals were euthanized via CO2 inhalation.
Reagents
All reagents used were of research grade, and unless specified, were obtained from Sigma Aldrich or Thermo Fisher Scientific. Rabbit polyclonal anti-PIWIL1 (AB12337) was purchased from Abcam. Alexa Fluor 488-conjugated goat anti-rabbit (A-11008) antibodies were purchased from Thermo Fisher Scientific.
Epididymal sperm isolation
Immediately after 8 week old adult male mice were euthanized, their vasculature was perfused with pre-warmed PBS to reduce the possibility of blood contamination. The epididymides were then removed, separated from fat and overlying connective tissue, and carefully dissected into three anatomical segments corresponding to the caput, corpus, and cauda. Caudal spermatozoa were collected from the lumen via retrograde perfusion with water-saturated paraffin oil as previously described.Citation28 In contrast, caput and corpus spermatozoa were recovered by placing the tissue in a 500 μL droplet of modified Biggers, Whitten, and Whittingham media (BWW) composed of 91.5 mM NaCl, 4.6 mM KCl, 1.7 mM CaCl2·2H2O, 1.2 mM KH2PO4, 1.2 mM MgSO4·7H2O, 25 mM NaHCO3, 5.6 mM D-glucose, 0.27 mM sodium pyruvate, 44 mM sodium lactate, 5 U/mL penicillin, 5 μg/mL streptomycin and 20 mM Hepes buffer (pH 7.4; osmolarity 300 mOsm/kg).Citation64 After multiple incisions were made with a razor blade, the spermatozoa were gently washed into the medium with mild agitation. All sperm preparations were then subjected to centrifugation (400 × g for 15 minutes (min)) on a 27% Percoll density gradient. The pellet, consisting of an enriched population of > 95% spermatozoa was resuspended in BWW and again centrifuged at 400 × g for 2 min to remove any excess Percoll.Citation28 A subset of the sperm population was set aside for labelling with SYTOX Green vitality stain, as previously described, to ensure that membrane integrity was not compromised during sperm isolation. Sperm populations with >85% vitality were then washed with sterile PBS and stored at -80°C for subsequent RNA isolation. Notably, the isolation protocols employed herein generated sperm populations in which approximately 30 – 40% of the cells had shed their cytoplasmic droplet.
Epididymal epithelial cell isolation
Following recovery of spermatozoa, epididymal tissue was utilized for epithelial cell isolation using methodology originally described by Zuo et al.Citation65 In brief, the tissue was washed by subjecting it to agitation, prior to being minced with forceps and washed a further three times in sterile PBS. The washed tissue was then digested in 100 μg/mL trypsin (Promega) at 37°C for 30 min with vigorous shaking. Epididymal tissue was pelleted by centrifugation at 800 × g for 5 minutes and then digested in 1 mg/mL collagenase for 30 min with shaking at 37°C. The epithelial cells were subjected to a further centrifugation at 800 × g for 5 minutes and the supernatant discarded. The cell pellet was resuspended in Dulbecco's Modified Eagle Medium (DMEM) culture medium containing sodium pyruvate (1 mM), 10% FBS (v/v), penicillin (100 IU/mL), and streptomycin (100 μg/mL), before being filtered through a 70 μm cell strainer and incubated in 6-well plates at 32°C. After 4 h of incubation, all non-epithelial cells (fibroblasts and muscle cells) were found attached to the base of the plate while the epithelial cells remained in suspension.Citation39 These populations of isolated epithelial cells were washed in sterile PBS and frozen at -80°C prior to RNA isolation.
Epididymosome isolation
Epididymides were dissected from adult mice as described above and after separation into caput, corpus and caudal segments, the tissue was placed in 500 µL of BWW and subjected to multiple incisions with a razor blade while being gently agitated. The resultant epididymal fluid suspensions were subsequently filtered through 70 μm membranes and subjected to sequential centrifugation with increasing velocity (500 × g, 2,000 × g, 4,000 × g, 8,000 × g, 17,000 × g) to eliminate all cellular debris. The supernatant was then layered onto a discontinuous OptiPrep density gradient (40%, 20%, 10%, and 5%), created by diluting 60% OptiPrep with a solution comprising 0.25 M sucrose and 10 mM Tris. The gradient was ultracentrifuged at 100,000 × g for 18 hours (h) at 4°C after which twelve equivalent fractions were collected. Fractions 9 and 10 were pooled and diluted in PBS prior to a final ultracentrifugation step at 100,000 × g for 3 h at 4°C to pellet purified epididymosomes.Citation37
RNA extraction and next-generation sequencing of the small RNA fraction
Total RNA was extracted from purified spermatozoa, epididymal epithelial cells, and epididymosomes using a Direct-zol RNA MiniPrep Kit (Zymo Research Corporation) according to manufacturer's instructions. Briefly, each sample was lysed by homogenization in Tri-Reagent for 1 min on ice prior to the removal of cellular debris by centrifugation at 12,000 × g for 1 min. Samples were then diluted in absolute ethanol and bound to a Zymo-Spin IIC column. RNA was further purified via a number of stringent washes prior to being eluted from the column into DNase/RNase-free water. Genomic DNA contamination was eliminated via incubation in 1% DNase (Promega). The purity of all samples was then assessed via determination of their A280/260 ratio and only those samples with values >1.8 were used for sequencing analysis.
Total RNA from each epididymal segment (caput, corpus, cauda) was pooled from a minimum of nine animals to generate a single biological replicate. Three such biological replicates of epididymosomes, and two biological replicates of epithelial cells and spermatozoa were subjected to the Illumina TruSeq small RNA sample preparation protocol as per the manufacturers’ instructions (Illumina Inc.) at the Australian Genome Research Facility (AGRF).Citation28,Citation37-39 The sRNA libraries generated from these samples were sequenced using an Illumina Hiseq-2000 RNA-seq platform as 50 bp single end chemistry at AGRF as previously described.Citation28,Citation37-39 Briefly, the sequence reads from all samples were analyzed for quality control on an Agilent 2100 Bioanalyser (Agilent Technologies) and screened for the presence of contaminants by matching against the contaminant database (containing PhiX, ChrM, rDNA and Illumina small RNA adaptor sequences) using cutadaptCitation66 and Bowtie aligner. Following adapter removal and quality validation, the cleaned sequence reads were subsequently normalized to allow comparisons between each condition employed in the study. Size factors for normalization were estimated via DESeq2,Citation67 and were calculated using the raw count of all sRNA sequences present in ≥ 10 reads in each dataset.
Once the normalized count data for each sequence in the dataset was generated, the identity of each sRNA sequence was assessed by aligning sequences to sRNA references databases with Bowtie 2. Sequences were classified as rRNA, tRNA, snoRNA or snRNA by alignment to the Ensembl mouse non-coding RNA database, while known miRNAs were identified via standard alignment to miRBase (version 21). Piwi-interacting RNAs were detected using proTRAC.Citation68 In brief, known sRNAs were filtered from the datasets before alignment of the remaining sequences to the mm10 genome using sRNAmapper.Citation68 The resulting map files were input into proTRAC, which yielded a list of putative piRNA clusters and piRNA sequences for each dataset. To ensure that all piRNAs were successfully identified in each dataset, the complement of piRNAs identified via proTRAC was used as a database for alignment via Bowtie 2. The genomic origin of each piRNA was determined based on their location within the mm10 genome. The coordinates of each unique piRNA sRNA sequence were compared with known genome features, including pseudogenes, coding genes, non-protein-coding genes, transposons, and repeat-associated elements; coordinates of genome features were retrieved from the mm10 genome annotation file (gff), and were intersected with piRNA coordinates using BEDtools.Citation69
The datasets analyzed here have previously been deposited in the Gene Expression Omnibus at NCBI and are publically accessible through GEO Series accession numbers GSE70197 (epididymal epithelial cells), GSE70198 (epididymal spermatozoa), GSE79500 (epididymosomes) (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE70197; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE70198; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE79500).
Quantitative real time PCR validation of piRNA read data
Validation of the sequencing data used to generate the piRNA profiles was conducted using quantitative real time PCR (RT-qPCR) with (non-locked nucleic acid modified) custom TaqMan sRNA assay reagents according to the manufacturer's instructions (Thermo Fisher Scientific). This approach only detects and amplifies the mature forms of each piRNA under assessment. The piRNAs selected for RT-qPCR quantification were;
piRNA_ch17_272(+) (TAGTAGTCTGAAATGCAAGTGCTTCGGCCCT), piRNA_ch17_272(-) (TAAAAGAGAATAACATGAACCCCTGAACAGA), piRNA_ch7_737(+) (TATGTGAATTTCAACTGTTGGCTCCACAGG), piRNA_ch7_737(-) (TAAATGGACTTTGGCCCCAGGTCAATTCTGGCC), piRNA_ch9_676(+) (TTGATGGTAATTGGAAGAGCTTCTGGGTGT), piRNA_ch18_670 (TAACATTAGGCACGTAGAGTCACAAATCGA), piRNA_ch15_746 (TAGGACCTGAGAACTTAACCTTGTTATGGGG), piRNA_ch10_758 (TGAAGTGAACCTGAGCAATTCCATGTGGAAC), piRNA_ch2_925 (TAGTAGAATCAAACAGCAAGAGAACCTTAGT), piRNA_ch11_103 (TACGAGATCGTCCCTTAAAGCTCAAAGGAC).
RT-qPCR was performed using a Light Cycler 96 SW 1.1 (Roche, Castle Hill, Australia). All piRNA RT-qPCR quantification data was normalized against the U6 snRNA (snRNA; assay ID 001973), as this endogenous snRNA was identified as evenly abundant across each epididymis segment, and relative abundance was calculated using the 2−ΔCt method.Citation70 All sRNA RT-qPCR analyses were performed in triplicate using pooled biological samples (6–9 mice/sample). However, due to limitations in generating the volume of epididymosome material required for total RNA extraction for sRNA sequencing, the cDNA used for these RT-qPCR analyses was synthesised from a separate pool of animals to that used for sequencing.
Immunofluorescent localization of PIWIL1 protein in spermatozoa
Purified epididymal spermatozoa were assessed for the presence of PIWIL1 protein, a key element of the cellular machinery required for the primary piRNA synthesis pathway. In brief, spermatozoa were settled onto poly-L-lysine-coated coverslips via overnight incubation at 4°C. All subsequent incubations were performed in a humidified chamber, and all antibody dilutions and washes were conducted in PBS containing 0.1% Tween-20 (PBST). Fixed cells were permeabilized in 0.2% Triton X-100/PBS for 10 min and blocked in 3% (w/v) BSA in PBST for 1 h. Cells were then labeled with anti-PIWIL1 antibodies (diluted 1:50) overnight at 4°C followed by three washes and subsequent incubation in goat anti-rabbit Alexa Fluor 488 (diluted 1:400) secondary antibodies for 1 h at room temperature. Cells were then washed and mounted in antifade reagent (Mowiol 4–88). Labeled cells were viewed on a Zeiss LSM510 laser scanning confocal microscope (Carl Zeiss MicroImaging, Inc.). In addition to a secondary antibody only control, spermatozoa were also labeled with antibodies that had previously been pre-absorbed with excess immunizing peptide (PIWIL1 peptide, cat# AB13827) to ensure specificity of labeling.
Immunofluorescent localization of PIWIL1 in epididymal tissue
Whole mouse epididymal tissue was fixed in Bouins solution, embedded in paraffin and sectioned. Embedded tissue was dewaxed, rehydrated and subjected to antigen retrieval by incubating in a water bath at 95°C in TE buffer (10 mM Tris, 1 mM EDTA, pH 9.0) containing 0.1% Tween-20 for 40 min. Following antigen retrieval, tissue sections from three individual mice were labeled with anti-PIWIL1 antibodies (diluted 1:50 in 1% BSA/PBS) overnight at 4°C. Post incubation, slides were washed three times and then incubated in goat anti-rabbit Alexa Fluor 488 secondary antibodies (diluted 1:400 in 1% BSA/PBS) for 1 h at room temperature per secondary antibody. Sections were then washed and counterstained in DAPI before mounting in antifade reagent. Labeled epididymal sections were viewed on an Axio Imager A1 microscope (Carl Zeiss MicroImaging) equipped with epifluorescence optics and images captured with an Olympus DP70 microscope camera (Olympus).
SDS-PAGE, immunoblotting and protein identification
Proteins were extracted from mouse testes in a modified SDS-PAGE sample buffer (2% w/v SDS, 10% w/v sucrose in 0.1875 M Tris, pH 6.8, supplemented with a protease inhibitor cocktail) by incubation at 100°C for 5 min. Insoluble matter was removed by centrifugation at 20,000 × g for 10 min and protein estimations were performed using a DC Protein Assay kit (Bio-Rad). Proteins were boiled in SDS-PAGE sample buffer (2% v/v mercaptoethanol, 2% w/v SDS, and 10% w/v sucrose in 0.1875 M Tris, pH 6.8, with bromophenol blue) and resolved by SDS-PAGE and transferred onto nitrocellulose membranes. Membranes were blocked with 3% w/v BSA or 5% skim milk w/v in TBS (pH 7.4) for 1 h before being probed with anti-PIWIL1 (1:500) primary antibodies in TBS containing 1% w/v BSA or skim milk and 0.1% v/v polyoxyethylenesorbitan monolaurate (Tween-20; TBST) overnight at 4°C. Blots were then washed three times in TBST followed by incubation with the appropriate horseradish peroxidase-conjugated secondary antibody (1:1000) in 1% w/v BSA or skim milk/TBS-T for 1 h. Following three washes in TBST, labeled proteins were detected using an enhanced chemiluminescence kit (GE Healthcare) and visualized on ImageQuant LAS 4000 (Fujifilm). All immunoblotting analyses were performed in triplicate and representative blots are presented. The negative controls used in this experiment included membranes probed under the same conditions except that the primary antibody was substituted with antibody buffer (secondary antibody only controls). An additional control was included in which anti-PIWIL1 antibodies were pre-absorbed with excess immunizing peptide prior to use for immunoblotting.
The identification of PIWIL1 in mouse spermatozoa was confirmed by mass spectrometry. Briefly, cauda epididymal spermatozoa were lysed by sonication in sodium carbonate containing buffer and tryptic peptides prepared as described.Citation71 Separation of tryptic peptides was achieved by nanoflow reversed phased liquid chromatography (Dionex Ultimate 3000RSLC, Dionex) using an in-house packed 20 cm column with pulled emitter and ReproSil-Pur 120 C18-AQ 3 μm material (Dr Maisch). Mass spectrometry was performed on a Q-Exactive Plus (Thermo Fisher Scientific) operating in positive ion mode and using data-dependent acquisition as described.Citation71 Multiply charged precursors were selected for higher-energy collision dissociation (HCD) fragmentation. Raw data was processed using Proteome Discoverer (Version 2, Thermo Fisher Scientific) and subjected to database searching using the Uniprot database using an in-house licensed MASCOT server (version 728 2.3.02, Matrix Science). Spectra meeting stringent selection criteriaCitation72 were validated by manual inspection to ensure accurate y- and b-ion detection with overlapping sequence coverage.
Statistical analysis
JMP Software (v12.2.0) was used to perform multivariate correlation analyses and Student T-tests to determine statistical significance with a significance threshold of P < 0.05. Unless otherwise specified, all experiments were performed in triplicate and all data are expressed as mean values ± SEM.
Disclosure of interest
The authors report no financial or non-financial conflict of interest.
Additional information
The datasets supporting the conclusions of this article are deposited in the NCBI Gene Expression Omnibus repository,Citation73 and are accessible through GEO Series accession numbers GSE70197 (epididymal epithelial cells; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE70197), GSE70198 (epididymal sperm cells; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE70198), and GSE79500 (epididymal epididymosomes; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE79500).
Funding details
This work was supported by the National Health and Medical Research Council of Australia under Project Grant number APP1062371.
Suppl_mat_tabl_Analysis_of_the_small_non-protein-coding_RNA_profile.pdf
Download PDF (126.1 KB)Suppl_mat_fig_Analysis_of_the_small_non-protein-coding_RNA_profile.pdf
Download PDF (98.3 KB)Related Research Data
References
- Hermo L, Pelletier RM, Cyr DG, Smith CE. Surfing the wave, cycle, life history, and genes/proteins expressed by testicular germ cells. Part 1: Background to spermatogenesis, spermatogonia, and spermatocytes. Microsc Res Tech. 2010;73:241–78. doi:10.1002/jemt.20783.
- Hermo L, Pelletier RM, Cyr DG, Smith CE. Surfing the wave, cycle, life history, and genes/proteins expressed by testicular germ cells. Part 2: Changes in spermatid organelles associated with development of spermatozoa. Microsc Res Tech. 2010;73:279–319. doi:10.1002/jemt.20783.
- Jodar M, Sendler E, Krawetz SA. The protein and transcript profiles of human semen. Cell Tissue Res. 2016;363:85–96. doi:10.1007/s00441-015-2237-1.
- Jodar M, Selvaraju S, Sendler E, Diamond MP, Krawetz SA, Reproductive Medicine N. The presence, role and clinical use of spermatozoal RNAs. Hum Reprod Update. 2013;19:604–24. doi:10.1093/humupd/dmt031.
- Krawetz SA, Kruger A, Lalancette C, Tagett R, Anton E, Draghici S, Diamond MP. A survey of small RNAs in human sperm. Hum Reprod. 2011;26:3401–12. doi:10.1093/humrep/der329.
- Miller D, Ostermeier GC, Krawetz SA. The controversy, potential and roles of spermatozoal RNA. Trends Mol Med. 2005;11:156–63. doi:10.1016/j.molmed.2005.02.006.
- Martins RP, Krawetz SA. RNA in human sperm. Asian J Androl. 2005;7:115–20. doi:10.1111/j.1745-7262.2005.00048.x.
- Kramer JA, Krawetz SA. RNA in spermatozoa: implications for the alternative haploid genome. Mol Hum Reprod. 1997;3:473–8. doi:10.1093/molehr/3.6.473.
- Yuan S, Schuster A, Tang C, Yu T, Ortogero N, Bao J, Zheng H, Yan W. Sperm-borne miRNAs and endo-siRNAs are important for fertilization and preimplantation embryonic development. Development. 2016;143:635–47. doi:10.1242/dev.131755.
- Miller D. Confrontation, Consolidation, and recognition: The oocyte's perspective on the incoming sperm. Cold Spring Harb Perspect Med. 2015;5:a023408. doi:10.1101/cshperspect.a023408.
- Sendler E, Johnson GD, Mao S, Goodrich RJ, Diamond MP, Hauser R, Krawetz SA. Stability, delivery and functions of human sperm RNAs at fertilization. Nucleic Acids Res. 2013;41:4104–17. doi:10.1093/nar/gkt132.
- Hosken DJ, Hodgson DJ. Why do sperm carry RNA? Relatedness, conflict, and control. Trends Ecol Evol. 2014;29:451–5. doi:10.1016/j.tree.2014.05.006.
- Holman L, Price TA. Even more functions of sperm RNA: A response to Hosken and Hodgson. Trends Ecol Evol. 2014;29:648–9. doi:10.1016/j.tree.2014.09.014.
- Hodgson DJ, Hosken DJ. Ultimate and proximate functions of sperm RNA: A reply to Holman and Price. Trends Ecol Evol. 2014;29:650. doi:10.1016/j.tree.2014.09.013.
- Lane M, Robker RL, Robertson SA. Parenting from before conception. Science. 2014;345:756–60. doi:10.1126/science.1254400.
- Wu H, Hauser R, Krawetz SA, Pilsner JR. Environmental susceptibility of the sperm epigenome during windows of male germ cell development. Curr Environ Health Rep. 2015;2:356–66. doi:10.1007/s40572-015-0067-7.
- Sharma U, Conine CC, Shea JM, Boskovic A, Derr AG, Bing XY, Belleannee C, Kucukural A, Serra RW, Sun F, et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science. 2016;351:391–6. doi:10.1126/science.aad6780.
- Fullston T, Ohlsson Teague EM, Palmer NO, DeBlasio MJ, Mitchell M, Corbett M, Print CG, Owens JA, Lane M. Paternal obesity initiates metabolic disturbances in two generations of mice with incomplete penetrance to the F2 generation and alters the transcriptional profile of testis and sperm microRNA content. FASEB Journal: official Publication Of the Federation of American Societies for Experimental Biology. 2013;27:4226–43. doi:10.1096/fj.12-224048.
- Chen Q, Yan M, Cao Z, Li X, Zhang Y, Shi J, Feng GH, Peng H, Zhang X, Zhang Y, et al. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science. 2016;351:397–400. doi:10.1126/science.aad7977.
- Short AK, Fennell KA, Perreau VM, Fox A, O'Bryan MK, Kim JH, Bredy TW, Pang TY, Hannan AJ. Elevated paternal glucocorticoid exposure alters the small noncoding RNA profile in sperm and modifies anxiety and depressive phenotypes in the offspring. Transl Psychiatry. 2016;6:e837. doi:10.1038/tp.2016.109.
- Dietz DM, Laplant Q, Watts EL, Hodes GE, Russo SJ, Feng J, Oosting RS, Vialou V, Nestler EJ. Paternal transmission of stress-induced pathologies. Biol Psychiatry. 2011;70:408–14. doi:10.1016/j.biopsych.2011.05.005.
- Rodgers AB, Morgan CP, Bronson SL, Revello S, Bale TL. Paternal stress exposure alters sperm microRNA content and reprograms offspring HPA stress axis regulation. J Neurosci. 2013;33:9003–12. doi:10.1523/JNEUROSCI.0914-13.2013.
- Rodgers AB, Morgan CP, Leu NA, Bale TL. Transgenerational epigenetic programming via sperm microRNA recapitulates effects of paternal stress. Proc Natl Acad Sci U S A. 2015;112:13699–704. doi:10.1073/pnas.1508347112.
- Marczylo EL, Amoako AA, Konje JC, Gant TW, Marczylo TH. Smoking induces differential miRNA expression in human spermatozoa: A potential transgenerational epigenetic concern? Epigenetics. 2012;7:432–9. doi:10.4161/epi.19794.
- Kim J, Kwon WS, Rahman MS, Lee JS, Yoon SJ, Park YJ, You YA, Pang MG. Effect of sodium fluoride on male mouse fertility. Andrology. 2015;3:544–51. doi:10.1111/andr.12006.
- Holt JE, Stanger SJ, Nixon B, McLaughlin EA. Non-coding RNA in spermatogenesis and epididymal maturation. Adv Exp Med Biol. 2016;886:95–120. doi:10.1007/978-94-017-7417-8_6.
- Miller D, Ostermeier GC. Spermatozoal RNA: Why is it there and what does it do? Gynecol Obstet Fertil. 2006;34:840–6. doi:10.1016/j.gyobfe.2006.07.013.
- Nixon B, Stanger SJ, Mihalas BP, Reilly JN, Anderson AL, Tyagi S, Holt JE, McLaughlin EA. The microRNA signature of mouse spermatozoa is substantially modified during epididymal maturation. Biol Reprod. 2015;93:91. doi:10.1095/biolreprod.115.132209.
- Holt JE, Stanger SJ, Nixon B, McLaughlin EA. Non-coding RNA in spermatogenesis and epididymal maturation. In: Wilhelm D, Bernard P, eds. Non-coding RNAs and the reproductive system. Heidelberg, Germany: Springer Publishing Company; 2015. p. 95–120.
- McIver SC, Roman SD, Nixon B, McLaughlin EA. miRNA and mammalian male germ cells. Hum Reprod Update. 2012;18:44–59. doi:10.1093/humupd/dmr041.
- McIver SC, Stanger SJ, Santarelli DM, Roman SD, Nixon B, McLaughlin EA. A unique combination of male germ cell miRNAs coordinates gonocyte differentiation. PLoS One. 2012;7:e35553. doi:10.1371/journal.pone.0035553.
- Eaton SA, Jayasooriah N, Buckland ME, Martin DI, Cropley JE, Suter CM. Roll over Weismann: Extracellular vesicles in the transgenerational transmission of environmental effects. Epigenomics. 2015;7:1165–71. doi:10.2217/epi.15.58.
- Belleannee C, Calvo E, Caballero J, Sullivan R. Epididymosomes convey different repertoires of microRNAs throughout the bovine epididymis. Biol Reprod. 2013;89:30. doi:10.1095/biolreprod.113.110486.
- Cornwall GA. New insights into epididymal biology and function. Hum Reprod Update. 2009;15:213–27. doi:10.1093/humupd/dmn055.
- Robaire B, Hinton BT, Orgebin-Crist MC. The Epididymis. In: Neill JD, ed. Knobil and Neill's physiology of reproduction, 3rd Edition. Elsevier; 2006. p. 1071–148.
- Bale TL. Epigenetic and transgenerational reprogramming of brain development. Nat Rev Neurosci. 2015;16:332–44. doi:10.1038/nrn3818.
- Reilly JN, McLaughlin EA, Stanger SJ, Anderson AL, Hutcheon K, Church K, Mihalas BP, Tyagi S, Holt JE, Eamens AL, et al. Characterisation of mouse epididymosomes reveals a complex profile of microRNAs and a potential mechanism for modification of the sperm epigenome. Sci Rep. 2016;6:31794. doi:10.1038/srep31794.
- Anderson AL, Stanger SJ, Mihalas BP, Tyagi S, Holt JE, McLaughlin EA, Nixon B. Assessment of microRNA expression in mouse epididymal epithelial cells and spermatozoa by next generation sequencing. Genom Data. 2015;6:208–11. doi:10.1016/j.gdata.2015.09.012.
- Nixon B, Stanger SJ, Mihalas BP, Reilly J, Anderson AL, Dun MD, Tyagi S, Holt JE, McLaughlin EA. Next generation sequencing analysis reveals segmental patterns of microRNA expression in mouse epididymal epithelial cells. PLoS One. 2015;10:e0135605. doi:10.1371/journal.pone.0135605.
- Weick EM, Miska EA. piRNAs: From biogenesis to function. Development. 2014;141:3458–71. doi:10.1242/dev.094037.
- Kim VN. Small RNAs just got bigger: Piwi-interacting RNAs (piRNAs) in mammalian testes. Genes & Development. 2006;20:1993–7. doi:10.1101/gad.1456106.
- Aravin AA, Sachidanandam R, Bourc'his D, Schaefer C, Pezic D, Toth KF, Bestor T, Hannon GJ. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell. 2008;31:785–99. doi:10.1016/j.molcel.2008.09.003.
- Rosenkranz D. piRNA cluster database: A web resource for piRNA producing loci. Nucleic Acids Res. 2016;44:D223–30. doi:10.1093/nar/gkv1265.
- Yamanaka S, Siomi MC, Siomi H. piRNA clusters and open chromatin structure. Mob DNA. 2014;5:22. doi:10.1186/1759-8753-5-22.
- Reuter M, Berninger P, Chuma S, Shah H, Hosokawa M, Funaya C, Antony C, Sachidanandam R, Pillai RS. Miwi catalysis is required for piRNA amplification-independent LINE1 transposon silencing. Nature. 2011;480:264–7. doi:10.1038/nature10672.
- Vourekas A, Zheng Q, Alexiou P, Maragkakis M, Kirino Y, Gregory BD, Mourelatos Z. Mili and Miwi target RNA repertoire reveals piRNA biogenesis and function of Miwi in spermiogenesis. Nat Struct Mol Biol. 2012;19:773–81. doi:10.1038/nsmb.2347.
- Robine N, Lau N, Balla S, Jin Z, Okamura K, Kuramochi-Miyagawa S, Blower MD, Lai EC. A broadly conserved pathway generates 3' UTR-directed primary piRNAs. Current Biology: CB. 2009;19:2066–76. doi:10.1016/j.cub.2009.11.064.
- Yamtich J, Heo SJ, Dhahbi J, Martin DIK, Boffelli D. piRNA-like small RNAs mark extended 3’UTRs present in germ and somatic cells. BMC Genomics. 2015;16:462. doi:10.1186/s12864-015-1662-6.
- Hirakata S, Siomi MC. piRNA biogenesis in the germline: From transcription of piRNA genomic sources to piRNA maturation. Biochim Biophys Acta. 2016;1859:82–92. doi:10.1016/j.bbagrm.2015.09.002.
- Beyret E, Liu N, Lin H. piRNA biogenesis during adult spermatogenesis in mice is independent of the ping-pong mechanism. Cell Res. 2012;22:1429–39. doi:10.1038/cr.2012.120.
- Aravin A, Gaidatzis D, Pfeffer S, Lagos-Quintana M, Landgraf P, Iovino N, Morris P, Brownstein MJ, Kuramochi-Miyagawa S, Nakano T, et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature. 2006;442:203–7.
- Girard A, Sachidanandam R, Hannon GJ, Carmell MA. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature. 2006;442:199–202.
- Goh WS, Falciatori I, Tam OH, Burgess R, Meikar O, Kotaja N, Hammell M, Hannon GJ. piRNA-directed cleavage of meiotic transcripts regulates spermatogenesis. Genes Dev. 2015;29:1032–44. doi:10.1101/gad.260455.115.
- Zhang P, Kang JY, Gou LT, Wang J, Xue Y, Skogerboe G, Dai P, Huang DW, Chen R, Fu XD, et al. MIWI and piRNA-mediated cleavage of messenger RNAs in mouse testes. Cell Res. 2015;25:193–207. doi:10.1038/cr.2015.4.
- Thomson T, Lin H. The biogenesis and function of PIWI proteins and piRNAs: Progress and prospect. Annu Rev Cell Dev Biol. 2009;25:355–76. doi:10.1146/annurev.cellbio.24.110707.175327.
- Grivna ST, Pyhtila B, Lin H. MIWI associates with translational machinery and PIWI-interacting RNAs (piRNAs) in regulating spermatogenesis. Proc Natl Acad Sci U S A. 2006;103:13415–20. doi:10.1073/pnas.0605506103.
- Eddy EM, Welch JE, O'Brien DA. Gene expression during spermatogenesis. In: de Kretser DM, ed. Molecular biology of the male reproductive system. New York: Academic Press; 1993.p. 181–232.
- Aitken RJ, Nixon B, Lin M, Koppers AJ, Lee YH, Baker MA. Proteomic changes in mammalian spermatozoa during epididymal maturation. Asian J Androl. 2007;9:554–64. doi:10.1111/j.1745-7262.2007.00280.x.
- Baker MA. Proteomics of post-translational modifications of mammalian spermatozoa. Cell Tissue Res. 2016;363:279–87. doi:10.1007/s00441-015-2249-x.
- Toth KF, Pezic D, Stuwe E, Webster A. The piRNA Pathway guards the germline genome against transposable elements. Adv Exp Med Biol. 2016;886:51–77. doi:10.1007/978-94-017-7417-8_4.
- Adelson DL, Buckley RM, Ivancevic AM, Qu Z, Zeng L. Retrotransposons: Genomic and trans-genomic agents of change. In: Pontarotti P, ed. Evolutionary biology: Biodiversification from genotype to phenotype. Switzerland: Springer International Publishing; 2015. p. 55–75.
- Ivancevic AM, Walsh AM, Kortschak RD, Adelson DL. Jumping the fine LINE between species: Horizontal transfer of transposable elements in animals catalyses genome evolution. Bioessays. 2013;35:1071–82. doi:10.1002/bies.201300072.
- Walsh AM, Kortschak RD, Gardner MG, Bertozzi T, Adelson DL. Widespread horizontal transfer of retrotransposons. Proc Natl Acad Sci U S A. 2013;110:1012–6. doi:10.1073/pnas.1205856110.
- Biggers JD, Whitten WK, Whittingham DG. The culture of mouse embryos in vitro. In: Daniel JC, ed. Methods in mammalian embryology. San Francisco, CA: Freeman Press; 1971. p. 86–116.
- Zuo WL, Li S, Huang JH, Yang DL, Zhang G, Chen SL, Ruan YC, Ye KN, Cheng CH, Zhou WL. Sodium coupled bicarbonate influx regulates intracellular and apical pH in cultured rat caput epididymal epithelium. PLoS One. 2011;6:e22283. doi:10.1371/journal.pone.0022283.
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet journal. 2011;17:10–12. doi:10.14806/ej.17.1.200.
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology. 2014;15:550. doi:10.1186/s13059-014-0550-8.
- Rosenkranz D, Zischler H. proTRAC–a software for probabilistic piRNA cluster detection, visualization and analysis. BMC Bioinformatics 2012;13:5. doi:10.1186/1471-2105-13-5.
- Quinlan AR, Hall IM. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2. doi:10.1093/bioinformatics/btq033.
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8. doi:10.1038/nprot.2008.73.
- Dun MD, Chalkley RJ, Faulkner S, Keene S, Avery-Kiejda KA, Scott RJ, Falkenby LG, Cairns MJ, Larsen MR, Bradshaw RA, et al. Proteotranscriptomic profiling of 231-BR breast cancer cells: Identification of potential biomarkers and therapeutic targets for brain metastasis. Molecular & Cellular proteomics: MCP. 2015;14:2316–30. doi:10.1074/mcp.M114.046110.
- Nixon B, Anderson AL, Smith ND, McLeod R, Johnston SD. The Australian saltwater crocodile (Crocodylus porosus) provides evidence that the capacitation of spermatozoa may extend beyond the mammalian lineage. Proc Biol Sci. 2016;283. doi:10.1098/rspb.2016.0495.
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–10. doi:10.1093/nar/30.1.207.