
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Aminoacyl-tRNA synthetases (aaRSs) catalyze the aminoacylation of tRNAs to produce the aminoacyl-tRNAs (aa-tRNAs) required by ribosomes for translation of the genetic message into proteins. To ensure the accuracy of tRNA aminoacylation, and consequently the fidelity of protein synthesis, some aaRSs exhibit a proofreading (editing) site, distinct from the aa-tRNA synthetic site. The aaRS editing site hydrolyzes misacylated products formed when a non-cognate amino acid is used during tRNA charging. Because aaRSs play a central role in protein biosynthesis and cellular life, these proteins represent longstanding targets for therapeutic drug development to combat infectious diseases. Most existing aaRS inhibitors target the synthetic site, and it is only recently that drugs targeting the proofreading site have been considered. In the present study, we developed a robust assay for the high-throughput screening of libraries of inhibitors targeting both the synthetic and the proofreading sites of up to four aaRSs simultaneously. Thus, this assay allows for screening of eight distinct enzyme active sites in a single experiment. aaRSs from several prominent human pathogens (i.e., Mycobacterium tuberculosis, Plasmodium falciparum, and Escherichia coli) were used for development of this assay.
Introduction
Aminoacyl-tRNA synthetases (aaRSs) are essential components of the translation machinery. They are responsible for the specific pairing of each tRNA with its cognate amino acid (aa) to form the aminoacyl-tRNAs (aa-tRNA) used by ribosomes during translation of mRNA into proteins. Aa-tRNA synthesis consists of a two-step process. During the activation step, an aa is activated to form an aminoacyl-adenylate (aa-AMP), while ATP is consumed and inorganic pyrophosphate (PPi) is released. In the subsequent transfer step, the activated aa is transferred to the 3′-end of tRNA, while AMP and an aa-tRNA are released. tRNA aminoacylation must be accurate since errors would result in incorporation of incorrect aa during decoding of mRNA by the ribosome. However, certain aaRSs cannot sufficiently distinguish between cognate and non-cognate aa that differ minimally in their structures. Consequently, the active sites of certain aaRSs can sometimes produce improperly charged products (aa-AMP and aa-tRNA). To maintain the fidelity of tRNA aminoacylation, error prone aaRSs have evolved an editing activity, which specifically hydrolyses incorrectly acylated products, either prior to (pre-transfer editing) or following (post-transfer editing) the transfer step [Citation1]. Among the twenty-one existing aaRSs, ten enzymes possess an editing activity. In seven of these proteins (i.e., Ile, Val, Leu, Pro, Thr, Ala, and PheRS), editing is achieved during a post-transfer editing reaction, which hydrolyzes the aminoacylated tRNA. This reaction is carried out in an editing site that is distinct from the synthetic site. In the other three enzymes (i.e., Met, Ser, and LysRS), editing and synthesis occur within the same active site, and the editing reaction takes place prior to transfer of the amino acid (for review see [Citation1,Citation2]).
aaRSs are one of the leading targets for developing novel anti-infective agents. While these enzymes are ubiquitous and essential for cellular life, they exhibit structural differences between phyla that can be exploited to develop drugs targeting pathogenic species, but that don't interact with the human counterpart [Citation3]. During the past decade, natural and synthetic inhibitors targeting aaRSs from various human parasites and bacterial pathogens have been identified [Citation3-Citation5]. Most of these compounds bind to the synthetic sites of aaRSs and act as competitive inhibitors of the substrates for aminoacylation. Recently, inhibitors targeting the editing sites of aaRSs have also been discovered. Of the numerous inhibitors identified to date (for review see [Citation3,Citation5,Citation6]), only two compounds are currently available for clinical use. These two inhibitors exhibit different modes of action with one targeting the aaRS synthetic site and the other targeting the editing site. Mupirocin (pseudomonic acid), naturally produced by Pseudomonas fluorescens, is a broad-spectrum competitive inhibitor that blocks the synthetic site of various bacterial IleRSs, but not that of the human enzyme [Citation7,Citation8]. This compound is currently used as a topical ointment to treat various skin infections including impetigo and methicillin-resistant Staphylococcus aureus (MRSA) [Citation9-Citation11]. The compound AN2690, developed by Anacor for the topical treatment of onychomycosis, binds the proofreading site of fungal LeuRS. AN2690 inactivates the enzyme by reacting with the vicinal diol at the 3′ end of the tRNA to form a covalent adduct that is irreversibly bound to the enzyme [Citation12]. The success of this latter compound prompted development of similar drugs using a benzoxaborole core (containing an RNA-reactive boronic acid group) to target the LeuRS from M. tuberculosis and Pseudomonas aeruginosa, as well as other parasites such as Cryptosporidium and Toxoplasma [Citation13-Citation15].
Several colorimetric assays are available for measuring tRNA aminoacylation activity in vitro. Some have been adapted to a high-throughput format for screening large libraries of compounds for inhibitors of the aaRS synthetic site. These assays have the advantage of not requiring the use of radioactive compounds (i.e., radiolabelled ATP or aa), which are used for studying the biochemistry of aaRSs. Using an indirect readout of tRNA aminoacylation, these colorimetric assays typically monitor consumption of ATP, or formation of the reaction products AMP or PPi (). Several assays couple AMP formation during aminoacylation to reduction of NAD+, which can be monitored spectrophotometrically upon addition of the enzymes AMP deaminase and IMP dehydrogenase [Citation16]. In other methods, PPi accumulation during tRNA aminoacylation is monitored; the enzyme inorganic pyrophosphatase (PPase) is used to break down PPi to inorganic phosphate (Pi), which can be measured using a malachite green reagent [Citation17]. Alternatively, Pi can be detected enzymatically using purine nucleoside phosphorylase (PNPase), which uses free Pi to break down the nucleotide analog 2-amino-6-mercapto-7-methyl purine ribonucleoside (MESG), a product that is detectable by spectrophotometry [Citation18]. Except for the malachite green method described above, each of these enzymatically-coupled assays allows for continuous measurement of tRNA aminoacylation kinetics. Finally, it is worth mentioning a novel assay that measures the relative amount of ATP consumed during tRNA aminoacylation by monitoring the rate of light produced by the firefly luciferase (which also uses ATP as substrate [Citation19], ).
Figure 1. Colorimetric and luminescence-based assays to monitor tRNA aminoacylation in vitro. The sensitivity of these assays can be increased with aa-tRNA recycling systems, which utilize aa-tRNAs as substrates and regenerate the tRNAs needed for the aaRS reaction (e.g., tRNA-dependent pathways for synthesis of cyclodipeptide [Citation20] and modified lipids [Citation21], or aa-tRNA editing activities [Citation22], see text for details). FFluc: firefly luciferase, luc: luciferin, AMPD: adenosine monophosphate deaminase, IMPDH: inosine monophosphate dehydrogenase, PPase: pyrophosphatase, PNPase: purine nucleoside phosphorylase, AMMP: 2-amino-6-mercapto-7-methyl purine, MESG: AMMP ribonucleoside, Ribose-1P: ribose 1 phosphate. The compounds that can be detectable by spectrophotometry or by luminometry are indicated in gray.
![Figure 1. Colorimetric and luminescence-based assays to monitor tRNA aminoacylation in vitro. The sensitivity of these assays can be increased with aa-tRNA recycling systems, which utilize aa-tRNAs as substrates and regenerate the tRNAs needed for the aaRS reaction (e.g., tRNA-dependent pathways for synthesis of cyclodipeptide [Citation20] and modified lipids [Citation21], or aa-tRNA editing activities [Citation22], see text for details). FFluc: firefly luciferase, luc: luciferin, AMPD: adenosine monophosphate deaminase, IMPDH: inosine monophosphate dehydrogenase, PPase: pyrophosphatase, PNPase: purine nucleoside phosphorylase, AMMP: 2-amino-6-mercapto-7-methyl purine, MESG: AMMP ribonucleoside, Ribose-1P: ribose 1 phosphate. The compounds that can be detectable by spectrophotometry or by luminometry are indicated in gray.](/cms/asset/48379977-28ee-4f2d-9d46-352fc2b34752/krnb_a_1397262_f0001_b.gif)
The dynamic range of each of these assays is inherently weak because the concentration of tRNA in the reaction mixture is low (1–10 µM), making accurate determination of reagent consumption or formation difficult. However, addition of enzymes that utilize the aa-tRNA being formed can increase assay sensitivity. For instance, enzymes that recycle the tRNA for additional rounds of aminoacylation increase the amount of ATP consumed, or AMP and PPi produced (). This strategy was used to increase ATP consumption in an assay for TyrRS, by adding cyclodityrosine synthase or D-tyrosyl-tRNATyr deacylase, which utilize Tyr-tRNATyr to synthesize a cyclodipeptide or to hydrolyze the aa-tRNA, respectively [Citation20]. A similar strategy was developed for AlaRS using alanyl-diacylaglycerol synthase, which uses Ala-tRNAAla as an aa donor for synthesis of alanyl-diacylglycerol [Citation21]. Finally, the editing domain of PheRS, which hydrolyzes Tyr-tRNA (trans-editing), was recently used to recycle tRNA in an assay monitoring TyrRS activity [Citation22]. Each of these tRNA recycling strategies dramatically increases the sensitivity of colorimetric assays used to monitor tRNA aminoacylation. The AlaRS assay described above, in which alanyl-diacylglycerol synthesis is used to recycle tRNA, was recently adapted to a 384-well format for high-throughput screening (HTS) [Citation21], demonstrating the utility of this method.
Existing colorimetric assays test a single aaRS at a time, and are designed to probe the synthetic site of the protein. In this study, we report a continuous assay that uses the natural editing activity of aaRSs to recycle tRNAs in a sensitive HTS procedure targeting the synthetic and editing sites of multiple aaRSs, simultaneously. Because it tests multiple drug targets at a time this method cuts down on the screening time needed for discovery of novel inhibitors targeting these enzymes. This procedure can also be used for determining the steady state kinetic constants of aaRSs for their substrates and inhibitors. The sensitivity and reliability of this method enables us to assay up to four aaRSs at the same time, bringing the total number of enzyme active sites (i.e., synthetic and editing sites) included in a single run to eight.
Materials and methods
Cloning, protein expression and purification
Enzymes were cloned into the pet33b (Novagen) vector to generate constructs yielding N-terminally 6-His-tagged proteins. AlaRS, ValRS, and IleRS from M. tuberculosis (Mt-AlaRS, Mt-ValRS, and Mt-IleRS), and AlaRS from P. falciparum (Pf-AlaRS) were cloned using the fastcloning strategy [Citation23-Citation25] (see accession numbers of ORF and primers in Table S1). The PNPase from E. coli was cloned in the NcoI/XhoI sites of pet33b (see Table S1). Plasmid constructs (pCA24N) for expression of the N-terminal 6xHis tagged IleRS, ThrRS from E. coli (Ec-IleRS, Ec-ThrRS) were obtained from the ASKA clone collection [Citation26] and were expressed in the E. coli strains BL21 DE3 pLysS (Stratagen) or the strain AG126 respectively.
Table 1. Steady-state kinetic parameters for editing of non-cognate aa by various aaRSs.
Protein expression was achieved in Luria-Bertani (LB) medium at 37°C after 3 h of induction with 0.1 mM of isopropyl-D-thiogalactopyranoside (IPTG) under agitation. For the expression of Pf-AlaRS, cultures were grown at 37 °C in LB medium in the presence of 2% glucose, 50 mg/L kanamycin, and 30 mg/L chloramphenicol until an A600 of 2 (1 cm path length) was reached. The medium was then substituted with a solution lacking glucose, and aaRS expression was induced overnight at 18 °C with 0.25 mM IPTG. 6-His tagged proteins were purified using the TALON affinity chromatography resin (Clontech) according to the guideline of the manufacturer. Proteins were stored at −80 °C in a buffer containing 100 mM Tris•HCl (pH 8.0), 100 mM NaCl, 3 mM β-mercaptoethanol, and 50% (v/v) glycerol.
tRNA aminoacylation using [14C] radiolabeled amino acids
Aminoacylation was performed in 100 mM Hepes-NaOH (pH 7.6), 30 mM KCl, 10 mM MgCl2, 1 mM dithiothreitol (DTT), 2 mM adenosine triphosphate (ATP), 0.2 μCi L-[14C(U)]-amino acid mix (Perkin Elmer, this mixture contains all 20 aa except Met, Asn, and Gln), 1.5 mg/mL of E.coli total tRNA (Roche), and 2–10 nM of aaRS. After a 10 min incubation at 37 °C, reaction aliquots (10 μL) were spotted on 3MM filter discs (Whatman International Ltd, Maidstone, UK), washed 3 times with 5% trichloroacetic acid, and dried. The amount of L-[14C(U)] aminoacyl-tRNA retained on the discs was determined by liquid scintillation counting.
Purine nucleoside phosphorylase (PNPase) assay for monitoring tRNA aminoacylation and editing by aaRSs
Reactions were performed in a 50 μL reaction volume in a 384-well plate (Corning® Spheroid Microplate) in 100 mM Hepes-NaOH (pH 7.6), 30 mM KCl, 10 mM MgCl2, 1 mM dithiothreitol (DTT), 0.25 mM MESG (Seterah), 1 U/mL PPase (Hoffmann-La Roche Ltd, Basel,Switzerland), 1.07 μM PNPase from E. coli, 2 mM adenosine triphosphate (ATP), 3 mg/mL total tRNA (from E. coli MRE 600, Roche), and 0.03–500 mM of non-cognate amino acids. aaRSs were supplied in the reaction mixture as follows: Mt-ValRS (0.30 μM), Pf-AlaRS (0.40 μM), Mt-AlaRS (0.36 μM), Mt-IleRS (0.76 μM), Ec-IleRS (0.56 μM), Ec-ThrRS (0.40 μM), and Ec-LysRS (0.40 μM). The change in absorbance at 355 nm represents phosphate (PPi) accumulation over time, and each well was measured every 30 sec for the duration of the assay (10 min). The concentration of PPi at each time point was quantified using an NaK(PO4)2 standard curve. Except as otherwise noted, the reported rates of PPi accumulation were determined after subtracting the rate of spontaneous ATP hydrolysis (0.66 µM/min), which was determined using a reaction mixture lacking aaRS. The steady-state parameters of the editing reaction were determined by non-linear regression analysis of the kinetic data, and fitted with the Michaelis-Menten equation using GraphPad (Prism).
Ki determination of Ec-IleRS for mupirocin
50 μL editing reactions were performed in a 384-well plate (Corning® Spheroid Microplate) with 100 mM Hepes-NaOH (pH 7.6), 30 mM KCl, 10 mM MgCl2, 1 mM dithiothreitol (DTT), 0.25 mM MESG (Seterah), 1 U/mL PPase (Hoffmann-La Roche Ltd, Basel,Switzerland), 1.07 μM PNPase from E. coli, 2 mM adenosine triphosphate (ATP), 3 mg/mL total tRNA (from E. coli MRE 600, Roche), 300 nM Ec-IleRS, and L-Val (4 mM), with a range of concentration of mupirocin (1.56–160 nM) mupirocin (Gemini Bio-Products). The Ki was determined using non-linear regression analysis by fitting (GraphPad Prism) with the Morrison equation (1) modified for competitive inhibition [Citation27, Citation28].
vo and vi are the initial velocities determined in absence or in presence of a range of concentration of inhibitor (I). Substrate concentration (S) and K m determined without inhibitors are used as constants, and the enzyme concentration (E) and K i are the parameters of the equation.
Results
Continuous assay to monitor aaRS synthesis and editing of non-cognate products in vitro
Traditional in vitro assays for determining aaRS editing activity use radiolabelled compounds [Citation29]. These assays are labor intensive and are not practical for HTS. For our assay, we chose a method utilizing PPase and PNPase. PNPase has been used for continuous monitoring of various biochemical reactions that produce Pi [Citation30], and such an assay for monitoring tRNA aminoacylation was previously reported [Citation18]. We adapted this method to monitor the aa activation and cis-editing activities of aaRSs simultaneously. In this system, non-cognate aa are used to promote synthesis of misacylated products such as mischarged-AMP and tRNA. These products are hydrolyzed by the aaRS editing site, and the substrates (i.e., the liberated aa and tRNA) are recycled for additional rounds of aminoacylation (). Under these conditions, the limiting concentration of tRNA provided in the assay (5–10 µM) becomes non-rate-limiting, and larger amounts of PPi are generated. PPase added to the reaction hydrolyses the PPi into two Pi molecules, which are subsequently used by PNPase to break down the nucleoside MESG into a compound that is quantifiable at 355 nm. Non-cognate aa are poor substrates for aaRSs and can exhibit a rate of activation for formation of the aminoacyl-adenylate intermediate that is 3 to 5 orders of magnitude lower than that of the cognate aa [Citation31]. PPase in the coupled reaction helps to circumvent this problem because hydrolysis of PPi prevents reversal of the aa activation step and has been shown to drive aminoacylation forward for both cognate and non-cognate aa [Citation29,Citation32]. Thus, in this scheme, a drug targeting either of the aaRS's active sites (synthetic or editing) is predicted to decrease the kinetics of PPi synthesis. An inhibitor of the synthetic site would directly decrease the rate of formation of PPi and would also inhibit the pre-transfer editing reaction. Alternatively, an inhibitor targeting the editing site would prevent hydrolysis of the aminoacylated tRNA, which would subsequently decrease the rate of PPi synthesis.
Figure 2. Non-cognate aa used to assay the synthetic and editing sites of aaRSs simultaneously. A. Reaction scheme of the coupling assay, including PPase and PNPase for measuring PPi accumulation, to monitor the activities of the synthetic and proofreading sites of aaRSs. B. PPi accumulation catalyzed by Ec-ValRS in presence of total tRNA from Escherichia coli and L-Val (circle) or L-Thr (square).
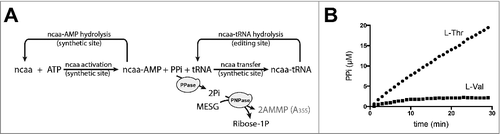
The assay described above () was used to determine the kinetics of PPi accumulation catalyzed by ValRS from E. coli (Ec-ValRS) in the presence of the cognate substrate L-Val, or the non-cognate substrate, L-Thr [Citation33] (). When L-Val was supplied in the reaction, PPi quickly reached a plateau corresponding to the concentration of tRNA added to the mixture (i.e., 2.5 µM as determined with [14C]-Val). In these conditions, tRNAAla was the limiting substrate, and no additional Pi was produced when tRNAAla was fully alanylated. In contrast, when L-Thr was supplied in the reaction mix, the concentration of Pi steadily increased over time to reach a plateau corresponding to the concentration of MESG (250 µM). In these conditions, MESG acted as the limiting substrate in the pathway. These results demonstrate the editing activity of ValRS and recycling of tRNA in the presence of L-Thr.
Determination of steady state kinetic parameters of the aaRSs editing reaction
To detect the effects of competitive inhibitors on the aaRS editing reaction, it is essential to use substrates at concentrations around their K m values. This is particularly important when high concentrations of a given inhibitor cannot be supplied in the reaction. Supplying substrates at levels near the K m values enables detection of weak inhibitory effects. For example, using a concentration of substrate equivalent to the K m, and a concentration of a competitive inhibitor that is 5-fold higher than the Ki , would result in a 71% decrease in enzymatic activity. In contrast, the activity would only be reduced by 31% if the substrate concentration were 10-fold higher than the K m value.
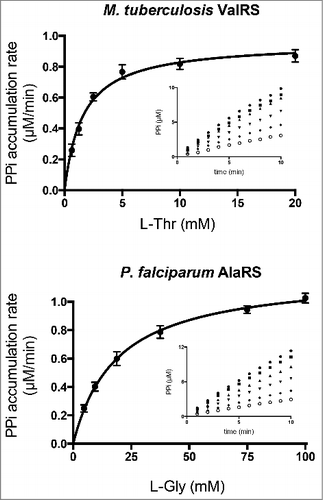
To determine the steady state kinetic parameters for non-cognate aa using the PPase/PNPase coupling system described above, it was necessary to ensure that editing by the aaRS was maintained as the rate-limiting step. To this end, non-rate-limiting amounts of PPase and PPi were determined empirically. 1 U/mL of PPase and 1.07 µM of PNPase were sufficient to maintain linearity between the rate of formation of PPi (between 0.25 and 6.5 µM/min) and the amount of AlaRS added to the reaction mixture (Fig. S1). To compensate for the low efficiency of the editing reaction, aaRSs were supplied at concentrations ranging from 0.1 to 0.5 µM. Because the K m for non-cognate aa was still several orders of magnitude higher than the concentration of enzyme, the Michaelis-Menten equation was applied to calculate the steady state parameters of the editing reaction (, ). K m values were in close agreement with values determined previously by other methods (i.e., reported K m values for Ec-IleRS and L-Val in ATP-PPi exchange reaction was 0.5 mM [Citation34] or Ec-ThrRS and L-Ser, 81.5 mM [Citation35]). These results demonstrate that the PPase/PNPase coupling reaction can be used to determine the steady-state parameters of aaRSs for non-cognate aa.
Figure 3. Michaelis-Menten kinetics of ValRS from M. tuberculosis and AlaRS from P. falciparum. Reaction kinetics of ValRS from M. tuberculosis (Mt-ValRS) and AlaRS from P. falciparum (Pf-AlaRS) with varying amounts of the non-cognate substrates L-Thr (0.6–20 mM) and L-Gly (4.6–100 mM), respectively. PPi synthesis kinetics are shown in insets (n = 4).
Multi-synthetase assay and validation for HTS
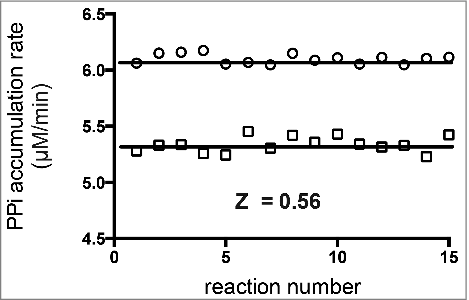
The Z'-factor is a widely accepted statistical parameter for evaluation and validation of HTS assays [Citation36,Citation37]. The Z'-factor describes an assay's ability to distinguish between the mean values for a positive and negative control, taking into account the data variability for both sample sets. A Z'-factor was calculated by comparing the velocities of formation of PPi in the presence and absence of a single aaRS. A determined value of Z' = 0.96 indicates that our assay is highly reliable and yields good separation between signal and background. Moreover, the standard deviations of the means of the observed initial velocities were low (<1.5%), indicating that the variability of the assay is small. Because of these parameters and the high dynamic range of the assay (velocities of up to at least 5.8 µM/min can be measured using high concentrations of a single aaRS), we hypothesized that 50% inhibition of a single aaRS could be detected in a mixture containing four aaRSs. This level of inhibition of a single enzyme would correspond to a 12% inhibition of the overall rate of PPi synthesis.
Four aaRSs and their non-cognate aa were assayed together as follows: Pf-AlaRS with L-Gly, Ec-LysRS with L-homocysteine, Ec-ThrRS with L-Ser, and Ec-IleRS with L-Val. The concentration of each aaRS was optimized so that each enzyme contributed to 25% of the overall rate of PPi synthesis (∼1.5 µM/min). It is important to point out that the non-cognate aa of any given aaRS may act as an editing substrate for other enzymes in the reaction mixture. Therefore, it is essential to optimize the amount of each enzyme using a mixture containing all of the non-cognate aa to be used in the final assay. For instance, L-Gly as well as L-Ser are editing substrates for AlaRS [Citation33]. shows that both of these amino acids are also edited by the Pf-AlaRS, with L-Gly being edited ten times more efficiently than L-Ser. The concentration of Pf-AlaRS was adjusted to account for 25% of the final rate of PPi synthesis in the presence of the four non-cognate aa used in the final reaction mixture. Predetermination of the K M and k cat values for each substrate of a given enzyme can be used to predict the effect of individual components on the overall rate of the reaction (i.e., using equations describing the kinetics of the enzymes in the presence of two competing substrates) [Citation38].
shows the results for determination of the Z'-factor of a reaction containing four aaRSs and their non-cognate aa (Pf-AlaRS/L-Gly, Ec-LysRS/L-homocysteine, Ec-ThrRS/L-Ser, and Ec-IleRS/L-Val). A positive control mimicking partial inhibition of one of the enzymes (by 50%) was included, along with a negative control where all four aaRSs were fully active. Each non-cognate aa was supplied at a concentration equal to the K M value (determined experimentally) for the respective aaRS (see ). The concentrations of individual aaRSs were optimized as described above. To simulate 50% inhibition of a single aaRS, the concentration of the enzyme was decreased by two-fold, resulting in a 12% decrease in the overall rate of PPi synthesis. Although a 12% difference in activity might be considered insufficient for some HTS assays, the data variability using this method is low, and a 12% loss in the rate of PPi synthesis is measurable with high accuracy. The standard deviations for the positive and negative control reactions were below 1.5%, and the calculated Z-factor was 0.56, a value indicating a robust HTS assay that can be used to report 50% inhibition of each enzyme with high confidence.
Figure 4. Scatter plot for determination of the Z'-factor for a reaction containing four aaRSs. The Z'-factor was calculated according to the formula described by Zhang et al. A negative control (representing no inhibition, circles) consisted of four aaRSs (Pf-AlaRS, Ec-LysRS, Ec-ThrRS, and Ec-IleRS) assayed simultaneously, with each contributing to the synthesis of PPi at an equal rate (1.5 µM/min). In the positive control (squares), the concentration of IleRS was cut in half in order to mimic 50 percent inhibition of a single enzyme. The Z'-factor calculated from fifteen replicate experiments was 0.56. Amino acids were supplied at concentrations near their K m values () (20 mM L-Gly, 45 mM homocysteine, 20 mM L-Ser, and 2 mM L-Val).
Determining the affinities of aaRS inhibitors
Determination of the inhibitory constant K i, is important for ranking the potency of inhibitors identified by HTS in the early stages of drug discovery. We selected the well-known IleRS inhibitor mupirocin as a model inhibitor to, i) demonstrate successful detection of an inhibitory compound targeting a single aaRS in the multi-enzyme assay described above, and ii) to validate the PPase/PNPase coupling assay, and determine the IC50 and K i of an aaRS inhibitor.
In an assay containing four aaRSs (Pf-AlaRS, Ec-LysRS, Ec-ThrRS, and Ec-IleRS), addition of 5 µM mupirocin decreased the overall rate of PPi accumulation by ∼14% compared to untreated samples (see ). This level of inhibition satisfies the primary hit threshold of ∼12% utilized for the Z'-factor calculation (). These results provide further validation of this method for detection of inhibitors of a single aaRS in a mixture containing several enzymes.
Figure 5. Inhibition of E. coli IleRS by mupirocin. A. Inhibition of a single enzyme (IleRS) in a mixture of four aaRSs (Pf-AlaRS, Ec-LysRS, Ec-ThrRS, and Ec-IleRS). Each enzyme was added in a sufficient amount to yield a rate of PPi synthesis of 1.5 µM/min. Bars and errors represent mean and standard deviation,*p<0.05, n = 3. B. Morrison plot for inhibition of Ec-IleRS by mupirocin. IleRS was assayed individually in presence of varying concentration of mupirocin and 4 mM of L-Val. The calculated K i for mupirocin was 0.8 nM, and the determined concentration of active IleRS was 33 nM. The fractional velocity (vi/v0) is the ratio of initial velocities measured in the presence of inhibitor (vi) and in the absence of inhibitor (v0). See Materials and Methods for details.
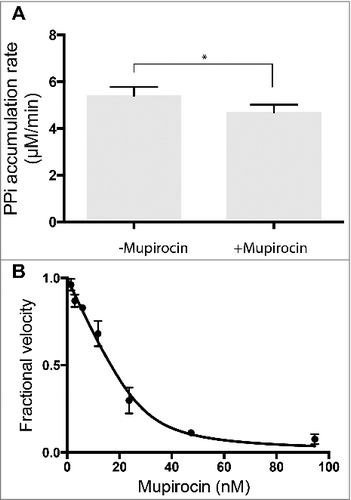
Previous work on mupirocin revealed a K i of 2.5 nM for inhibition of tRNA aminoacylation by Ec-IleRS [Citation39]. To determine the K i of this compound using the PPase/PNPase-coupled reaction, Ec-IleRS was tested in the absence of other aaRSs. The concentration of Ec-IleRS used in the assay was 0.2 µM, which is considerably higher than the reported K i for mupirocin (2.5 nM). These conditions (where K i << [Enzyme]) comply with the “tight-binding inhibition” conditions described by Morrison [Citation27] (see [Citation28] for review). In this scenario, the K i of a tight inhibitor cannot be determined using equations of the Michaelis-Menten type, but require the use of the Morrison equation for competitive inhibitors [Citation27,Citation28]. It has been suggested that Michaelis-Menten approximations should be abandoned whenever the K i is not more than 1000-fold greater than the concentration of the enzyme being tested [Citation28]. To determine the K i with this approach, various concentrations of inhibitor are tested in the presence of substrate, maintained at a fixed concentration around the K m value. It is worth mentioning that with the K m value for the substrate, the Morrison equation can be applied to determine the K i of the inhibitor, in addition to the concentration of active enzyme in the assay. Our determined K i value was 0.8 nM (), which is in close agreement with the previously reported value of 2.5 nM for mupirocin with Ec-IleRS [Citation39]. The calculated concentration of active Ec-IleRS in the assay was 33 nM. This strategy can be used to quickly determine K i values of various inhibitors in order to compare their relative potencies.
Discussion
The critical role of aaRSs in cellular viability has made them long-standing targets for development of drugs to combat bacterial pathogens. Most HTS endeavors have until now focused on discovery of inhibitors of the aaRS synthetic site, while inhibitors of the editing site of aaRS remain unexplored. The editing assay, coupled to the PPase/PNPase system presented here, fills a critical gap by reconstituting both the synthetic and editing activities of aaRSs. In this assay, several aaRSs can be targeted in a single experiment, allowing for simultaneous screening of compounds against the active sites of multiple enzymes. This method can also be used on aaRSs derived from several pathogenic species at a time, or for determination of the steady-state parameters of individual substrates and inhibitors.
Although the assay described here has multiple advantages over systems targeting a single enzyme, it also has some limitations. One obvious limitation is that an inhibitor identified by this method must be subjected to additional steps of validation to ensure that inhibition of an off-target component in the assay (e.g., PPase or PNPase) is not responsible for observed decreases in PPi accumulation. In addition, the assay described here is designed to work on a limited set of aaRSs. Of the twenty-two known aaRSs, only ten enzymes exhibit an editing activity and are therefore suitable for screening using this method. To circumvent this latter limitation, alternate factors for recycling tRNAs can be added in trans to study aaRSs lacking an editing activity [Citation16,Citation21].
The non-cognate aa of certain aaRSs serve as the cognate substrates for other aaRSs. Such pairings of aaRSs are therefore incompatible for simultaneous testing, since aminoacylation of a tRNA with the cognate aa would prevent its recycling by editing. For this reason, certain libraries of inhibitors, which inherently contain cognate aa (e.g., libraries of natural product extracts), cannot be used with this assay. provides a compatibility chart indicating which pairs of aaRSs can be assayed together, and also indicates incompatible non-cognate aa. This analysis shows that if care is taken with selection of non-cognate aa, most combinations of aaRSs are compatible. SerRS and ThrRS are the only two enzymes that are strictly incompatible, because the cognate substrate of SerRS, L-Ser, is the only non-cognate aa used by ThrRS. However, it is important to highlight that to overcome these incompatibility rules, four aaRS of identical specificity (e.g., four AlaRSs) from different organisms can be assayed simultaneously.
Table 2. aaRS compatibility chart for multi-enzyme HTS assay.
Finally, it is worth reiterating that due to the low efficiency of misacylated-tRNA editing, the concentration of aaRSs in this assay have to be maintained in the high nM range (100 −1000 nM). The K i determination of a tight-binding inhibitor (i.e., an inhibitor with a K i in the 1–100 nM) cannot be determined using Michaelis-Menten kinetics. In these conditions, the Morrison equation has to be applied [Citation27]. Methods for determination of inhibition modality of tight-binding inhibitors were reviewed by Copeland and these methods can be adapted to this assay to determine the mode of inhibition of aaRSs inhibitors [Citation28].
Supplementary_table_and_figure_Christopher.docx
Download MS Word (37.7 KB)Acknowledgments
This work was supported by National Institute of General Medical Sciences Grant R15-109404.
Related Research Data
References
- Perona JJ, Gruic-Sovulj I. Synthetic and editing mechanisms of aminoacyl-tRNA synthetases. Top Curr Chem. 2014;344:1–41.
- Jakubowski H. Quality control in tRNA charging. Wiley Interdiscip Rev RNA. 2012;3:295–310. doi:10.1002/wrna.122.
- Pham JS, Dawson KL, Jackson KE, et al. Aminoacyl-tRNA synthetases as drug targets in eukaryotic parasites. Int J Parasitol Drugs Drug Resist. 2014;4:1–13. doi:10.1016/j.ijpddr.2013.10.001.
- Hurdle JG, O'Neill AJ, Chopra I. Prospects for aminoacyl-tRNA synthetase inhibitors as new antimicrobial agents. Antimicrob Agents Chemother. 2005;49:4821–33. doi:10.1128/AAC.49.12.4821-4833.2005.
- Gadakh B, Van Aerschot A. Aminoacyl-tRNA synthetase inhibitors as antimicrobial agents: a patent review from 2006 till present. Expert Opin Ther Pat. 2012;22:1453–65. doi:10.1517/13543776.2012.732571.
- Vondenhoff GH, Van Aerschot A. Aminoacyl-tRNA synthetase inhibitors as potential antibiotics. Eur J Med Chem. 2011;46:5227–36. doi:10.1016/j.ejmech.2011.08.049.
- Nakama T, Nureki O, Yokoyama S. Structural basis for the recognition of isoleucyl-adenylate and an antibiotic, mupirocin, by isoleucyl-tRNA synthetase. J Biol Chem. 2001;276:47387–93. doi:10.1074/jbc.M109089200.
- Fuller AT, Mellows G, Woolford M, et al. Pseudomonic acid: an antibiotic produced by Pseudomonas fluorescens. Nature. 1971;234:416–7. doi:10.1038/234416a0.
- Hughes J, Mellows G. On the mode of action of pseudomonic acid: inhibition of protein synthesis in Staphylococcus aureus. J Antibiot (Tokyo). 1978;31:330–5. doi:10.7164/antibiotics.31.330.
- Pereira LB. Impetigo – review. An Bras Dermatol. 2014;89:293–9. doi:10.1590/abd1806-4841.20142283.
- Sutherland R, Boon RJ, Griffin KE, et al. Antibacterial activity of mupirocin (pseudomonic acid), a new antibiotic for topical use. Antimicrob Agents Chemother. 1985;27:495–8. doi:10.1128/AAC.27.4.495.
- Rock FL, Mao W, Yaremchuk A, et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316:1759–61. doi:10.1126/science.1142189.
- Palencia A, Li X, Bu W, et al. Discovery of Novel Oral Protein Synthesis Inhibitors of Mycobacterium tuberculosis That Target Leucyl-tRNA Synthetase. Antimicrob Agents Chemother. 2016;60:6271–80. doi:10.1128/AAC.01339-16.
- Palencia A, Liu RJ, Lukarska M, et al. Cryptosporidium and Toxoplasma Parasites Are Inhibited by a Benzoxaborole Targeting Leucyl-tRNA Synthetase. Antimicrob Agents Chemother. 2016;60:5817–27. doi:10.1128/AAC.00873-16.
- Hernandez V, Crepin T, Palencia A, et al. Discovery of a novel class of boron-based antibacterials with activity against gram-negative bacteria. Antimicrob Agents Chemother. 2013;57:1394–403. doi:10.1128/AAC.02058-12.
- First EA, Richardson CJ. Spectrophotometric assays for monitoring tRNA aminoacylation and aminoacyl-tRNA hydrolysis reactions. Methods. 2017;113:3–12. doi:10.1016/j.ymeth.2016.10.010.
- Cestari I, Stuart K. A spectrophotometric assay for quantitative measurement of aminoacyl-tRNA synthetase activity. J Biomol Screen. 2013;18:490–7. doi:10.1177/1087057112465980.
- Lloyd AJ, Thomann HU, Ibba M, et al. A broadly applicable continuous spectrophotometric assay for measuring aminoacyl-tRNA synthetase activity. Nucleic Acids Res. 1995;23:2886–92. doi:10.1093/nar/23.15.2886.
- Saint-Leger A, Ribas de Pouplana L. A new set of assays for the discovery of aminoacyl-tRNA synthetase inhibitors. Methods. 2017;113:34–45. doi:10.1016/j.ymeth.2016.10.011.
- Richardson CJ, First EA. A continuous tyrosyl-tRNA synthetase assay that regenerates the tRNA substrate. Anal Biochem. 2015;486:86–95. doi:10.1016/j.ab.2015.05.008.
- Grube CD, Roy H. A Quantitative Spectrophotometric Assay to Monitor the tRNA-Dependent Pathway for Lipid Aminoacylation In Vitro. J Biomol Screen. 2016;21:722–8. doi:10.1177/1087057116642987.
- Richardson CJ, First EA. Hyperactive Editing Domain Variants Switch the Stereospecificity of Tyrosyl-tRNA Synthetase. Biochemistry (Mosc). 2016;55:2526–37. doi:10.1021/acs.biochem.6b00157.
- Garcia-Nafria J, Watson JF, Greger IH. IVA cloning: A single-tube universal cloning system exploiting bacterial In Vivo Assembly. Sci Rep. 2016;6:27459. doi:10.1038/srep27459.
- Jacobus AP, Gross J. Optimal cloning of PCR fragments by homologous recombination in Escherichia coli. PLoS ONE. 2015;10:e0119221. doi:10.1371/journal.pone.0119221.
- Li C, Wen A, Shen B, et al. FastCloning: a highly simplified, purification-free, sequence- and ligation-independent PCR cloning method. BMC Biotechnol. 2011;11:92. doi:10.1186/1472-6750-11-92.
- Kitagawa M, Ara T, Arifuzzaman M, et al. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 2005;12:291–9. doi:10.1093/dnares/dsi012.
- Morrison JF. Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim Biophys Acta. 1969;185:269–86. doi:10.1016/0005-2744(69)90420-3.
- Copeland RA. Tight Binding Inhibitors. Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis, 2nd Edition. John Wiley & Sons, Inc. 2002:305–17.
- Splan KE, Musier-Forsyth K, Boniecki MT, et al. In vitro assays for the determination of aminoacyl-tRNA synthetase editing activity. Methods. 2008;44:119–28. doi:10.1016/j.ymeth.2007.10.009.
- Suarez AS, Stefan A, Lemma S, et al. Continuous enzyme-coupled assay of phosphate- or pyrophosphate-releasing enzymes. Biotechniques. 2012;53:99–103.
- Jakubowski H, Goldman E. Editing of errors in selection of amino acids for protein synthesis. Microbiol Rev. 1992;56:412–29.
- Wolfson AD, Uhlenbeck OC. Modulation of tRNAAla identity by inorganic pyrophosphatase. Proc Natl Acad Sci U S A. 2002;99:5965–70. doi:10.1073/pnas.092152799.
- Tsui WC, Fersht AR. Probing the principles of amino acid selection using the alanyl-tRNA synthetase from Escherichia coli. Nucleic Acids Res. 1981;9:4627–37. doi:10.1093/nar/9.18.4627.
- Fersht AR. Editing mechanisms in protein synthesis. Rejection of valine by the isoleucyl-tRNA synthetase. Biochemistry (Mosc). 1977;16:1025–30. doi:10.1021/bi00624a034.
- Sankaranarayanan R, Dock-Bregeon A-C, Rees B, et al. Zinc ion mediated amino acid discrimination by threonyl-tRNA synthetase. Nature Struct Biol. 2000;7:461–5. doi:10.1038/75856.
- Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi:10.1177/108705719900400206.
- Iversen PW, Eastwood BJ, Sittampalam GS, et al. A comparison of assay performance measures in screening assays: signal window, Z' factor, and assay variability ratio. J Biomol Screen. 2006;11:247–52. doi:10.1177/1087057105285610.
- Pocklington T, Jeffery J. Competition of two substrates for a single enzyme. A simple kinetic theorem exemplified by a hydroxy steroid dehydrogenase reaction. Biochem J. 1969;112:331–4. doi:10.1042/bj1120331.
- Hughes J, Mellows G. Interaction of pseudomonic acid A with Escherichia coli B isoleucyl-tRNA synthetase. Biochem J. 1980;191:209–19. doi:10.1042/bj1910209.
- Ling J, Reynolds N, Ibba M. Aminoacyl-tRNA synthesis and translational quality control. Annu Rev Microbiol. 2009;63:61–78. doi:10.1146/annurev.micro.091208.073210.