
ABSTRACT
Horizontal gene transfer is crucial for the adaptation of microorganisms to environmental cues. The acidophilic, bioleaching bacterium Acidithiobacillus ferrooxidans encodes an integrative-conjugative genetic element (ICEAfe1) inserted in the gene encoding a tRNAAla. This genetic element is actively excised from the chromosome upon induction of DNA damage. A similar genetic element (ICEAcaTY.2) is also found in an equivalent position in the genome of Acidithiobacillus caldus. The local genomic context of both mobile genetic elements is highly syntenous and the cognate integrases are well conserved. By means of site directed mutagenesis, target site deletions and in vivo integrations assays in the heterologous model Escherichia coli, we assessed the target sequence requirements for site-specific recombination to be catalyzed by these integrases. We determined that each enzyme recognizes a specific small DNA segment encoding the anticodon stem/loop of the tRNA as target site and that specific positions in these regions are well conserved in the target attB sites of orthologous integrases. Also, we demonstrate that the local genetic context of the target sequence is not relevant for the integration to take place. These findings shed new light on the mechanism of site-specific integration of integrative-conjugative elements in members of Acidithiobacillus genus.
Introduction
Gene acquisition by horizontal gene transfer (HGT) is crucial in the evolution of bacteria, allowing microbial survival and adaptation to environmental change Citation[1-Citation3]. One of the most relevant mechanisms of HGT is conjugation, which occurs through the physical contact between donor and recipient cells Citation[4,Citation5]. In the last decades many different mobile genetic elements (MGEs) have been identified and characterized Citation[6,Citation7]. Integrative-conjugative elements (ICEs) are MGEs capable of excision from the bacterial genome and conjugative transfer Citation[8]. These elements carry a variable number of genes and integrate in the recipient bacterial genome by site specific recombination, preferentially within tRNA coding genes Citation[9]. These genetic elements have a modular organization. A typical ICE contains modules for integration, conjugation and its regulation Citation[10]. ICEs can also carry accessory modules encoding genes that confer the microorganisms with adaptive advantages to particular environmental conditions (e.g. antibiotic resistance, heavy metal resistance, etc.) Citation[6,Citation11,Citation12]. Under cellular stress conditions the mechanism of ICE transfer is activated. It starts with the excision of the element from the genome and its circularization, and it is followed by the mobilization of the element to a recipient cell using the ICE's conjugation machinery, which entails a Type IV Secretion System (T4SS) specialized in DNA transfer. This step also involves the relaxase-mediated recognition and nicking of the DNA at the oriT, the establishment of a cell-to-cell connecting bridge (or pilus), and the replicative transfer of the ICE circular ssDNA complexed with the coupling protein. Once in the recipient cell, and if the recipient organism has the appropriate target sequence, an ICE-encoded integrase catalyzes the element integration in the host genome by a site-specific recombination process Citation[13]. The integration module is generally composed of the integrase Citation[14] and a recombination directionality factor (RDF)[15], commonly named excisionase, which participates in the excision of the ICE out of the bacterial genome.
Site-specific recombination involves the strand exchange between two DNA segments catalyzed by an integrase, an enzyme capable of recognition and breaking/joining of the recombining DNA segments by an energy conservative mechanism Citation[16]. There are two families of enzymes that allow site-specific recombination; serine recombinases (that catalyze the resolution of co-integrates from transposition, integration-excision and inversion of DNA segments) and tyrosine recombinases (that catalyze the integration and excision of phages and other MGEs into and out of the host genome, the resolution of replicon dimers into monomers, the mobilization of gene cassettes of integrons, among others) Citation[17]. Both enzyme families share some features and generate similar final products, but the specific mechanisms for each one is different Citation[14]. The tyrosine recombinase family is named after the nucleophile tyrosine residue in the catalytic site of these enzymes, which allows the formation of a transient phospho-tyrosine covalent bond between the recombinase and the 3´-end of the DNA substrate as an intermediate in the recombination process Citation[18]. Serine recombinases present a different mechanism compared to tyrosine recombinases, and the products in general are solved by the resolvase/invertase activities of these enzymes Citation[19]. There is a third family of integrases called DD[E/D]-transposases for Asp-Asp-Glu/Asp-transposases (named after their catalytic amino acids) that possess a common structural RNase H-like motif, shared with members of polynucleotidyl transferases superfamily Citation[20]. Transposase enzymes bind to the inverted terminal repeats (ITRs) of a transposon and catalyze the mobilization of the transposon to new location. Some DD[E/D] transposases are able to cut one DNA strand at the end of the transposon generating a replicative transposition, other transposases cut both DNA strands generating a cut-and-paste transposition Citation[21].
The best-studied model for site-specific recombination mediated by tyrosine recombinases is the insertion of the lambda (λ) bacteriophage in the Escherichia coli chromosome. Briefly, the process starts with the binding of a tetrameric form of the integrase to DNA specific regions named attachment or att regions and the establishment of a synaptic complex with the two recombining DNA molecules Citation[22]. Next, the nucleophilic attack of the catalytic tyrosine residue over a phosphodiester bond generates a transient 3’-end phospho-tyrosine linkage. Subsequent DNA rearrangements take place in two rounds of pairwise cleavage, strand exchange and ligation in a concerted way, forming the characteristic Holliday junction intermediate. After the second strand exchange, the nucleoprotein synaptic complex and ligation is resolved and the recombination products are released Citation[23-Citation25].
Acidithiobacillus ferrooxidans is part of a consortium of microorganisms that participate in the bioleaching of minerals Citation[26]. It is a Gram negative, chemolithotrophic, mesophilic acidophile that belongs to the Acidithiobacillia class Citation[27]. It obtains its energy from the oxidation of iron and sulfur compounds Citation[28]. Complete genomic sequences of a number of strains are nowadays available Citation[29] and their comparative analyses have contributed to the identification of genes related to mineral dissolution and adaptation to the extreme environments, providing insights into bioleaching processes Citation[30]. Bioinformatic analysis of the genome sequence from A. ferrooxidans strain ATCC 23270 (NC011761) revealed the presence of a large MGE (∼300 Kbp) that encodes around 300 ORFs, including all the gene modules characteristic of ICE-type elements. This genetic element, coined ICEAfe1, is found in the type strain of the species (ATCC 23270), but not in strain ATCC 53993 (NC011206) Citation[12,Citation31]. ICEAfe1 is actively excised from the genome under DNA damaging conditions that trigger the SOS response Citation[32]. A tRNAAla (GGC) gene is predicted to be the integration target site of ICEAfe1. When integrated in the genome the ICE is flanked by 48 nucleotides long direct repeats, attR and attL, corresponding in sequence to the 3´end half of the tRNAAla gene. Other four tRNAAla genes are present in the A. ferrooxidans type strains´ genome, with nucleotide sequence identities ranging between 72% and 75% with respect to tRNAAla (GGC). Other predicted ICE using different tRNA genes as target sites have been mapped in the genome of A. ferrooxidans ATCC 23270, as well as in other members of the Acidithiobacillus genus Citation[33,Citation34].
The tRNAAla (GGC) gene is in a single copy in the genome from A. ferrooxidans, and its immediate genetic context is highly conserved among strains of the species and partially conserved in other species of the Acidithiobacillus genus. For instance, in Acidithiobacillus caldus strains ATCC 51756 this region of the genome contains a predicted ICE element Citation[34] encoding a similar tyrosine recombinase and integrated within an adjacent tRNAAsn gene. These relatively conserved genetic segments seem to be hot spots for the integration of ICEs in the acidithiobacilli. The aim of this work was to determine whether the tRNA gene is enough to guide the macromolecular machinery for integration to occur, or whether the local genetic context of the target site is required for integrative recombination. To this end we analyzed the target specificity of the predicted recombination systems of ICEAfe1 and ICEAca TY.2 from A. ferrooxidans ATCC 23270 and A. caldus ATCC 51756, respectively. Our results led to the identification of the minimal DNA sequence that guides the site-specific integration of the cognate ICE elements in this taxon.
Results
Integrases from integrative-conjugative elements from acidithiobacillus species
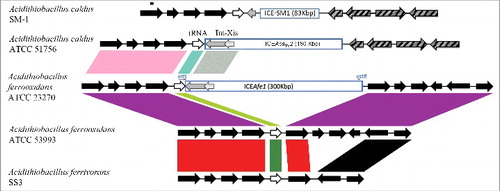
In the acidithiobacilli three ICE elements have been reported, named ICEAfe1, Citation[12] ICEAfe2 Citation[33] and ICEAca TY.2 Citation[34]. The local context of the integration site for the ICEAfe1 from A. ferrooxidans ATCC 23270 is partially conserved among members of the Acidithiobacillus genus (). The predicted integration site of ICEAfe1 is a tRNAAla gene, which is the most frequent tRNA in the region downstream of the phnP gene in the sequenced representatives of the Acidithiobacillus genus. In A. caldus ATCC 51756 and other related strains a tRNAAsn gene is found in this genomic location instead, and is followed by a major genomic rearrangement that disrupts synteny downstream ( ). Genes upstream the integration target-site are conserved at varying degrees among Acidithiobacillus species at both the DNA and protein levels (, and Fig. S1).
Figure 1. Comparison of the ICEAfe1 integration site context from Acidithiobacillus ferrooxidans with other members of the Acidithiobacillus genus. ICEs are also integrated in the same genetic context in Acidithiobacillus caldus ATCC 51756 and SM-1. White arrows show the predicted target tRNA genes. Light and dark green bars symbolizes 100% and 97,4% identity of the tRNAAla genes respectively and light blue bar represents 65.8% percent identity between the tRNA genes from Acidithiobacillus ferrooxidans 23270 and Acidithiobacillus caldus ATCC 51756. Gray and light gray arrows indicate the predicted integrase-excisionase genes respectively. Black arrows represent conserved predicted genes. Dark grey dashed arrows are non-related ORFs with the other species in the same context. Amino acid similarity was represented by purple bars (98%), red bars (90%), and pink bar (77%). Occurrence of attachment sites attL and attR in the genomic sequences are indicated in ICEAfe1.
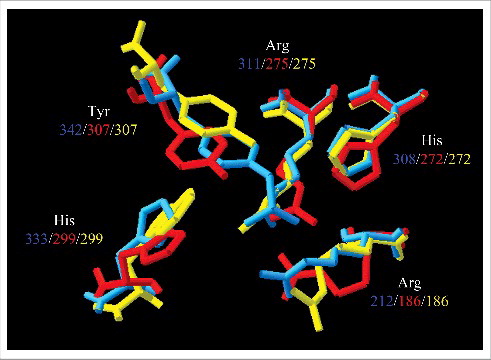
Comparative analysis of the integrases from ICEAfe1 and ICEAca TY.2 reveal their intrinsic sequence conservation (76% similarity), which extends to other orthologs of the recombinase encoded in sequenced Acidithiobacillus (Fig. S2A). The λ phage integrase, available on the RCSB PDB database, was chosen as structural template to model the A. ferrooxidans and A. caldus integrases by threading using Phyre2 and I-TASSER. Further refinement of the models was performed using Modeller. Structure prediction revealed the presence of the three domains described for the λ integrase Citation[35]: an N-terminal, a core-binding and a catalytic C-terminal domains (Fig. S2B, C). According to the structural model, amino acids at the active site are arranged to fit the structural orientation of λ integrase, in particular the tyrosine at the catalytic site that forms the phospho-tyrosine intermediate in the recombination reaction ().
Figure 2. Overlapping of amino acids at the catalytic sites. The predicted orientation of the amino acid at the catalytic site of integrases from λ phage (blue), A. ferrooxidans ICEAfe1 (red) and A. caldus ICEAcaTY.2 (yellow) are shown. Below the amino acid name, the position in each integrase is indicated.
Interaction of the integrases with attL and attR regions
To determine whether ICEAfe1 integrase binds to the predicted cognate attR and attL regions, EMSA analyses were carried out using the corresponding DNA segments and the purified ICEAfe1 integrase. Band shifts were observed in the presence of the purified protein, when both the attR (Fig. S3A) and the attL sequences (Fig. S3B) were used as probes. Unlabeled probes competed for the binding. To explore the contribution of the local genomic context, binding assays with the purified integrase from A. ferrooxidans ICEAfe1 and the predicted attR region from the A. caldus ICEAca TY.2 were also carried out. Band shifts were observed at all the integrase concentrations tested (Fig. S3C), suggesting that the formation of the integrase-DNA complex occurs regardless of the local genomic sequence context, provided that a suitable integration target site is present. Two different non-related sequences, NRS 1 (120 bp region inside the mazF toxin coding sequence from A. ferrooxidans ATCC 23270) and NRS 2 (180 bp inside the hypothetical protein excisionase coding region from A. ferrooxidans ATCC 23270) showed no band shift upon incubation with the ICEAfe1 integrase (Fig. S3D).
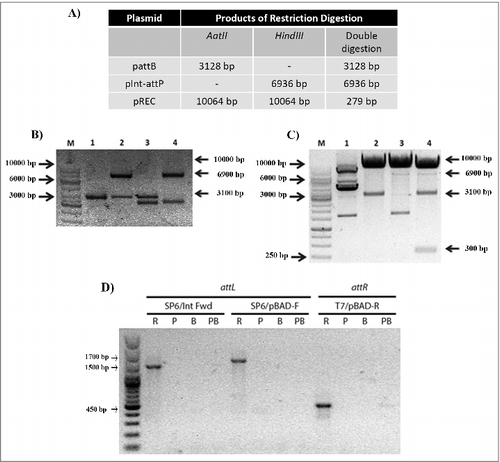
Figure 3. Analysis of plasmidial co-integrates in E. coli JM109. A. Predicted size of the plasmids restriction digestion products using AatII or HindIII individually, or in a double digestion. B. Restriction digestion products of the plasmids extracted from a selected control colony grown in the absence of arabinose (not induced). M, 1 Kb ladder; lane 1, undigested total plasmidial DNA; lane 2 digestion with HindIII (pInt-attP linearized); lane 3 restriction pattern with AatII (pattB linearized); lane 4 double digestion with AatII and HindIII. C. Restriction digestion products of the plasmids extracted from a selected experimental colony grown with arabinose (induced). M, 1 Kb ladder; lane 1, undigested total plasmidial DNA; lane 2 restriction pattern with AatII (pattB and pREC linearized); lane 3 digestion with HindIII (pInt-attP and pREC linearized); lane 4 double digestion with AatII and HindIII. In all digestion experiments an approximately 10 kbp band that corresponds to the linearized pREC is observed (C). D. PCR products from att regions of pREC. The DNA templates were: R, total plasmidial DNA from a liquid culture induced with arabinose; P, purified pInt-attP; B, purified pattB; PB, both purified plasmids pInt-attP and pattB mixed together (see Fig S4 for details on PCR primers).
λ phage integrase core-binding domain (CBD) and the N-terminal domain (ND) are well described Citation[14]. Both domains were identified in the ICEAfe1 integrase. The individual domains were obtained by PCR amplification and cloned and expressed in E. coli. These domains were also tested for their capacity to bind to the predicted ICEAfe1 att regions. Band shifts were observed in the presence of both individual domains of the enzyme, the N terminal domain (ND) and the core binding domain (CDB), when tested with the cognate attR or the attL sites derived from the tRNAAla (GGC) (Fig. S3E).
Altogether, these results showed that the predicted att regions contain the information to serve as binding sites for the ICEAfe1 integrase. The formation of a number of additional band shifts upon incubation with the different enzymatic forms tested (entire enzyme or fragments) suggests that different complexes might be forming which accommodate an increasing number of subunits. Similar multiple binding patterns have been reported in gel shift assays in other microbial models, e.g. Bacteroides thetaiotaomicron Citation[36,Citation37].
Acidithiobacillus integrases are active in vivo in E. coli.
To test the function of the acidithiobacilli integrases, the specificity of predicted attB sites as well as the contribution of the genetic context in the recombination process, an in vivo integration assay in E. coli JM109 was developed. Two types of plasmids were constructed: 1) the plasmid pattB (ApR), containing the target attB region (Fig. S4A) and 2) the plasmid pInt-attP (CmR), containing the gene encoding the integrase and the cognate attP (Fig. S4B). Plasmid pInt-attP emulates the circular intermediate in the recombination process. In order to modulate the expression of the integrase gene in E. coli, its transcription was placed under the control of BAD promoter, which is inducible by arabinose and repressed with glucose Citation[38]. The expected product of this recombination assay is a circular cointegrate (plasmid pREC) of ∼10 Kbp, carrying the double antibiotic resistance selection markers (CmR, ApR), two compatible origins of replication and the attL and attR products of the recombination (Fig. S4C).
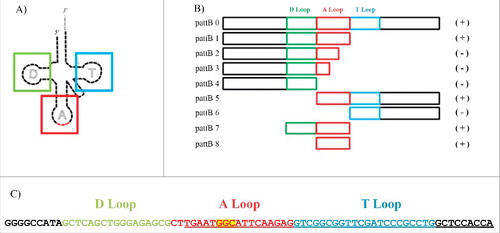
Figure 4. Sequence of tRNAAla gen and schematic representation of deletions tested in the integration assays. A. Two-dimensional representation of a tRNA, where the colored boxes represent the segments in the deletion constructs corresponding to the D (green), anticodon (red) and T (blue) stem/loops. B. Deletion mutant constructs pattB 0 to pattB 8. Green, red and blue boxes represent the length and location of the tRNAAla gene D, A and T loops fragments included in the different constructions. Signs at the right of each construct indicate whether they are functional (+) or not (−) in integration. C. Nucleotide sequence of the tRNAAla gene adjacent to the ICEAfe1 integrase orthologs. Yellow highlighted nucleotides corresponding to the anticodon region. Underlined nucleotides represent direct repeats present in the attL, attR, attB and attP regions. Red nucleotides correspond to the anticodon stem/loop.
Clones resistant to both selection antibiotics were obtained and the 10,064 bp cointegrate recovered, upon induction of the expression of the integrase with arabinose and in the presence of cognate attachment sites. The sizes of the digestion products are summarized in . and the expected single and double digestion products were also obtained (, ). Alternative, the presence of attL and attR in pREC was evidence by PCR with primers designed to identify the integration products ().
Based on the structure of λ phage integrase, the predicted catalytic tyrosine from the ICEAfe1 integrase corresponds to amino acid Tyr307 () Citation[35]. To test whether this amino acid residue is indeed responsible of the catalysis, two mutant integrase genes were constructed. In one mutant the tyrosine 307 was replaced by phenylalanine (Y307F) and in the other by alanine (Y307A). The in vivo experiments using these mutant genes did not render the recombination product pREC compared with the wild type control assay. These results confirm that the ICEAfe1 integrase is an active tyrosine recombinase and that the active catalytic residue is Tyr307 ().
Table 1. Effect of mutations in tyrosine 307 on integrase activity.
DNA encoding the anticodon loop spans the minimal recombination site
To identify the minimal DNA sequence recognized by the ICEAfe1 integrase that allows the integration reaction to occur, deletion mutant versions of the tRNAAla gene (attB) were constructed (). Inverted and complementary sequences that form the stems and loops in the mature tRNA (, ) were considered in the design of the deletions, since tyrosine integrases are speculated to use these structural sequence elements during site-specific recombination Citation[9]. The deletions constructed are schematized in the . New pattB plasmids were constructed using these deleted attB sequence variants and tested in E. coli JM109 as indicated before. The in vivo assays using the constructions pattB-0 to pattB-8 demonstrated that all fragments that contain the intact anticodon loop (A loop) sequence (pattB 0, 1, 5, 7 and 8) could recombine to produce the cointegrate plasmid pREC () and generated the expected attL and attR products (data not shown). The minimal segment tested that allowed the integration reaction between pInt-attP and pattB to form pREC contained 19 nucleotides from the anticodon stem/ loop of the tRNAAla (pattB 8). Whether an even shorter segment is functional as minimal target site merits further analysis.
Target sites recognition specificity by the ICE-encoded tyrosine integrases
Diverse lines of evidence suggested that the ICEAfe1 and the ICEAca TY.2 attB sequences could be cross-recognized as target sites by the integrases from A. caldus and A. ferrooxidans, respectively. Namely: 1) the amino acidic sequence similarity between the two ICE integrases (76% similarity; Fig. S2A, C), 2) the conserved genomic context of the integration sites between the two bacterial species (, Fig. S1A), 3) binding of the ICEAfe1 integrase to the extended attB (110 bp) region from A. caldus (Fig. S3C), and 4) the sequence identity between the tRNA genes at the attB site (79% identity) (Fig. S1). To test this hypothesis, we constructed genetic chimeras that contained the integrase gene from A. ferrooxidans ICEAfe1 immediately adjacent to the attP sequence from A. caldus ICEAca TY.2. This chimera was co-transformed with the pattB plasmid that contains A. caldus attB sequence. Conversely, a second chimera that contained the integrase gene from A. caldus ICEAca TY.2 adjacent to A. ferrooxidans ICEAfe1attP was constructed and co-transformed with the plasmid containing the attB region from A. ferrooxidans. Both the full-size predicted attB (110 bp) and the minimal attB encompassing the anticodon stem/loop (19 bp) from each bacterium were used in the assay. The components of A. ferrooxidans ICEAfe2, where a tRNAVal is predicted as the target site, were also tested Citation[33]. shows that integration takes place when cognate integration systems are assayed, i.e. when the integrase, attB and attP are derived from the same ICE, but not otherwise. These results demonstrate that the ICE integrases are highly specific. These assays also reinforced the notion that 19 nucleotides from the target site are sufficient for site-specific integration to take place and that the local genetic context is not required for the specific recombination.
Table 2. Summary of the in vivo cross-integration assay.
Molecular docking of the DNA-protein complex
To visualize the position the DNA adopts in the catalytic domain of the integrase, a molecular docking approach was used. For simplicity, during simulation the 19 nucleotides representing the minimal attB regions required for site-specific recombination were used together with the tridimensional model of each integrase. The following DNA-protein combinations were tested: 1) the ICEAfe1 integrase with its cognate attB sequence; 2) the ICEAcaTY .2 integrase with its cognate attBC sequence; 3) a combination between the ICEAfe1 integrase and the heterologous attB from A. caldus (attBC); or 4) the integrase from ICEAcaTY .2 and the heterologous attB from ICEAfe1. The catalytic tyrosine (Tyr307) of each integrase was selected as reference point and the interaction between the enzyme and its target attB was determined. Using the webserver HADDOCK 2.2 Citation[39,Citation40] several possible complexes can be retrieved (e.g. on the basis of variations in the free energy, or by an iterative search between the surfaces and molecules disposition). The models that presented the lower HADDOCK score (which correlates with the intermolecular Van der Waals energy) were selected for further analysis. The lower this value is, the more energetically favored is the model Citation[41]. Cognate recombination systems (e.g. ICEAfe1 integrase and ICEAfe attB) score lower HADDOCK values than non-cognate or reciprocate systems (e.g. ICEAfe1 integrase and ICEAcaTY .2 attBC), suggesting that these combinations are less energetically favored (). The interaction between the integrases and the att regions coincide in the four cases analyzed; the binding pattern is conserved and varies slightly between the nucleotide pair T-G, adjacent to the anticodon sequence (, underlined nucleotides).
Table 3. Summary of molecular docking energy values for the homologous and heterologous systems.
Discussion
Integrases of the tyrosine recombinase family are highly specific in the selection of their integration target sites Citation[4,Citation42]. These enzymes preferentially use tRNA genes as target for integration. A survey of 43 integrases recognized as tyrosine recombinases encoded in ICEs from a variety of bacterial and archaeal species revealed that 53% use tRNA genes as target sites Citation[4]. Based on the location of the recognized segment within the tRNA gene, these enzymes have been classified in type Ia and Ib, type II and type III. Type I and II recognize a symmetric sequence element (anticodon or T stem/loop in tRNA) as the target for integration Citation[9]. Despite this fact, the molecular basis of the enzymes capacity to recognize a specific target site is still unclear. Several hypotheses have been proposed to address this knowledge gap. Accessory cellular factors, specific DNA context or specific recognition domains in proteins have been proposed to underlie the minimal recognition requirements Citation[43]. Although all these factors might contribute to target site selection, there is still no consensus on the global mechanism for the recognition of the recombination site by these enzymes. In this work, we hypothesized that the local genetic context might be an important factor that contributes to the recognition of the target site by the integrases from Acidithiobacillus genus members. This idea was based on the existence of ICE-type elements in A. ferrooxidans and in A. caldus sharing orthologous integrases, both inserted in tRNA genes with different amino acid identity but localized in the genomes in a conserved genetic context (). The work performed demonstrated that 19 nucleotides encoding the anticodon stem/loop of the corresponding tRNA were sufficient to determine the specific integration catalyzed by each integrase. Yet, the genetic context where these segments are integrated has no influence on the specific recognition of the integration site by the integrases. These small DNA segments concentrate several differences in the nucleotide sequences between the target tRNA genes since 8 out of 19 nucleotides (42%) are not conserved among them, including the anticodon itself being determinant for the specificity of integration.
Since only 19 nucleotides (whether fewer than 19 nucleotides are still active is not yet known) are required for specific recognition by the integrases, we further assessed the enzymes determinants for specific recognition of the target site. Known tyrosine recombinases have highly conserved carboxyl-terminal catalytic domains, and fully conserved arginine, lysine, histidine and tyrosine residues that form part of the catalytic site. Although this is also the case for the ICEAfe1 and ICEAcaTY .2 integrases from A. ferrooxidans and A. caldus ( and Fig. S2C), docking of the 19 nucleotides segments to the catalytic domain of each integrase predicts that cognate interactions are favored over the reciprocate combinations. This evidence, and the higher divergence found in the amino-terminal and central domains of the integrases, which are described as the DNA binding motifs, might contribute to the specificity of the interaction between the enzymes and the target sites Citation[35]. A high similarity in the primary structure between the two integrases analyzed in this work, together with the similarity between the target sites in the tRNA genes, led us to think, as proposed by Williams Citation[9], that mutational events at the attP site of an ancestral common ICE element might have contributed to evolve the specificity of integration of ICEAfe1 and ICEAcaTY.2 from each organism. Altogether, these findings shed new light on the mechanism of site-specific integration of integrative-conjugative elements in members of Acidithiobacillus genus.
Materials and methods
Bacteria, media and antibiotics
Escherichia coli strains Rosetta BL21 (DE3) (donated by Jeff Gardner's Lab, University of Illinois) and DH5α (Promega) were grown in Luria-Bertani (LB) medium (Bacto) and the antibiotics (Sigma) were used at the following concentrations: ampicillin (Ap) 100 μg/ml, chloramphenicol (Cm) 25 μg/ml, and kanamycin (Kn) 50 μg/ml.
Plasmidial constructions
For protein overexpression, the integrase gene from Acidithiobacillus ferrooxidans ICEAfe1 was amplified by PCR using genomic DNA as template and specific primers baring NcoI and XhoI restriction sites. The amplicon was cloned into the pET-33b expression vector to generate the plasmid p33int. The same procedure was performed to clone the separate domains of the ICEAfe1 integrase; the N-terminal domain (ND) and the core-binding domain (CBD) to generate the p33N and p33CBD plasmids, respectively. The different integration vectors pInt-attP were generated in pBAD33, which carries the pACYC184/p15A origin of replication and a chloramphenicol resistance gene, by separately cloning into the plasmid two PCR products in tandem; the integrase gene and the predicted attP region. This construct emulates the ICEAfe1 circular intermediary generated through excision. The integration vector pattB was created cloning a predicted integration region attB for the ICEAfe1 into the pGEM-T Easy vector (Promega), carrying the phage f1 origin of replication and the gene encoding the resistance to ampicillin. To ensure the successful recombination of the plasmids pInt-attP and pattB to form the cointegrate pREC and avoid the plasmids integration in the heterologous model chromosome, the Escherichia coli JM109 genome sequence was inspected for conserved attB-like sequences. The deletion mutants were constructed by cloning different attB constructions (attB1 – attB8) in the pGEM-T Easy vector. The same procedure was performed to generate the cognate plasmids with the Acidithiobacillus caldus recombination modules. The complete list of the plasmids used in this work is summarized in the Table S1.
In vivo integration assay
An E. coli JM109 double transformation was performed using the plasmids pInt-attP and pattB. The positive colonies were selected from LB agar plates with chloramphenicol and ampicillin (35 and 100 μg/mL respectively) and then grown in LB liquid culture media with the same two antibiotics. To induce the integrase gene transcription 0.2% w/v arabinose was added to the medium, or glucose 0.2% w/v to repress the transcription Citation[38]. The cultures were incubated for 16 hours at 37°C with shaking (180 r.p.m.). Finally, a total plasmidial DNA extraction was performed to evaluate the formation of cointegrates. Plasmids were digested with the restriction enzymes AatII or HindIII that linearize the plasmid pattB (∼3,100 bp) and pInt-attP (∼6,900 bp), respectively. The two enzymes together digest the co-integrate plasmid pREC producing a 10,000 bp and 300 bp lineal products. Integration generated regions attL and attR were detected by PCR analysis using the primers pairs T7/pBAD R to detect attR and SP6/Int Fwd or SP6/pBAD F for attL.
Protein overexpression and purification
Rosetta BL21 (DE3) cells were transformed with p33int and grown in LB medium with kanamycin at 37°C to an OD600 of 0.3 – 0.4. Overexpression of the integrase and its individual domains was achieved by induction with 0.025 mM IPTG for 5 hours at 22°C. Cells were collected by centrifugation and the pellet was suspended in a solubilization buffer containing 50mM Tris-HCl pH 7.5, 300 mM NaCl, 10mM imidazole, 1mM DTT and protease inhibitors cocktail tablet Complete (Roche). The cells were lysed by sonication. The extract passed through a Ni2+-sepharose column (GE) and eluted with 300 mM imidazole. The eluted fractions were analyzed by SDS-PAGE 10% and the fractions where the protein was visualized were dialyzed into a storage buffer (50 mM Tris-HCl, 300 mM NaCl, 1 mM DTT, 1 mM EDTA pH 8.0 and 20% glycerol) and stored at −20 °C until further use.
Electrophoretic mobility shift assays
Electrophoretic mobility shift assays (EMSA) were performed as described previously Citation[36], in a 50 mM Tris-HCl pH 8.0, 1 mM EDTA, 50 mM NaCl, 10% glycerol and 75 μg/mL DNA from salmon testis buffer. Substrates attB, attR and attL (110 bp, 237 bp and 417 bp) were generated by PCR and the double-stranded products were then labeled with the modified nucleotide dideoxyuridine phosphate digoxigenin conjugate (ddUTP-DIG) using the reagents provided in the DIG Oligonucleotide 3´-End Labeling Kit (Roche). Probes were incubated with different concentrations of protein for 30 min at 23 °C and then loaded in a prerun 8% acrylamide gel. DNA was transferred to a Hybond N+ nylon membrane (GE) by electrotransference and the products were detected using Anti-Digoxigenin AP conjugate antibody (Roche) and revealed with the NBT/BCIP substrate (Thermo).
Protein modeling and molecular docking
Homology modeling based in the threading approach was performed using the web servers Phyre2 Citation[44,Citation45] and I-TASSER Citation[46]. Structure depuration was done with Modeller Citation[47]. The λ phage integrase model (PDB number 1Z1B)[35] was selected as template for the threading analysis. Protein – DNA docking was performed with the web server HADDOCK version 2.2 Citation[39,Citation40].
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Suppl_Fig_A_DNA_segment_encoding_the_anticodon_stem.pdf
Download PDF (1.7 MB)Funding
Fondecyt Chile to OO, 1150834 and 111203, Conicyt Chile PhD fellowship to AC, 21120316, USACH Chile to MT, USA 1555, Fondecyt Chile to RQ, 1140048.
Additional information
Funding
Related Research Data
References
- Cheetham BF, Katz ME. A role for bacteriophages in the evolution and transfer of bacterial virulence determinants. Mol Microbiol. 1995;18:201–8. doi:10.1111/j.1365-2958.1995.mmi_18020201.x
- Koonin E V., Makarova KS, Aravind L. Horizontal gene transfer in prokaryotes: quantification and classification. Annu Rev Microbiol. 2001;55:709–42. doi:10.1146/annurev.micro.55.1.709
- Jain R, Rivera MC, Moore JE, Lake JA. Horizontal gene transfer accelerates genome innovation and evolution. Mol Biol Evol. 2003;20:1598–602. doi:10.1093/molbev/msg154
- Bellanger X, Payot S, Leblond-Bourget N, Guédon G. Conjugative and mobilizable genomic islands in bacteria: Evolution and diversity. FEMS Microbiol Rev. 2014;38:720–60. doi:10.1111/1574-6976.12058
- Soucy SM, Huang J, Gogarten JP. Horizontal gene transfer: building the web of life. Nat Rev Genet. 2015;16:472–82. doi:10.1038/nrg3962
- Boyd EF, Almagro-Moreno S, Parent MA. Genomic islands are dynamic, ancient integrative elements in bacterial evolution. Trends Microbiol. 2009;17:47–53. doi:10.1016/j.tim.2008.11.003
- Menouni R, Hutinet G, Petit MA, Ansaldi M. Bacterial genome remodeling through bacteriophage recombination. FEMS Microbiol Lett. 2015;362:1–10. doi:10.1093/femsle/fnu022
- Burrus V, Pavlovic G, Decaris B, Guédon G. Conjugative transposons: The tip of the iceberg. Mol Microbiol. 2002;46:601–10. doi:10.1046/j.1365-2958.2002.03191.x
- Williams KP. Integration sites for genetic elements in prokaryotic tRNA and tmRNA genes: sublocation preference of integrase subfamilies. Nucleic Acids Res [Internet]. 2002;30:866–75. doi:10.1093/nar/30.4.866
- Burrus V, Waldor MK. Shaping bacterial genomes with integrative and conjugative elements. Res Microbiol. 2004;155:376–86. doi:10.1016/j.resmic.2004.01.012
- Frost LS, Leplae R, Summers AO, Toussaint A. Mobile genetic elements: the agents of open source evolution. Nat Rev Microbiol. 2005;3:722–32. doi:10.1038/nrmicro1235
- Levicán G, Katz A, Valdés JH, Quatrini R, Holmes DS, Orellana O. A 300 kpb Genome Segment, Including a Complete Set of tRNA Genes, is Dispensable for Acidithiobacillus Ferrooxidans. Adv Mater Res. 2009;71–73:187–90. doi:10.4028/www.scientific.net/AMR.71-73.187
- Wozniak RAF, Waldor MK. Integrative and conjugative elements: mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat Rev Microbiol. 2010;8:552–63. doi:10.1038/nrmicro2382
- Hirano N, Muroi T, Takahashi H, Haruki M. Site-specific recombinases as tools for heterologous gene integration. Appl Microbiol Biotechnol. 2011;92:227–39. doi:10.1007/s00253-011-3519-5
- Lewis JA, Hatfull GF. Control of directionality in integrase-mediated recombination: examination of recombination directionality factors (RDFs) including Xis and Cox proteins. Nucleic Acids Res. 2001;29:2205–16. doi:10.1093/nar/29.11.2205
- Craig NL. The mechanism of conservative site-specific recombination. Annu Rev Genet 1988;22:77–105. doi:10.1146/annurev.ge.22.120188.000453
- Grindley NDF, Whiteson KL, Rice PA. Mechanisms of Site-Specific Recombination. Annu Rev Biochem. 2006;75:567–605. doi:10.1146/annurev.biochem.73.011303.073908
- Esposito D, Scocca JJ. The integrase family of tyrosine recombinases: Evolution of a conserved active site domain. Nucleic Acids Res. 1997;25:3605–14. doi:10.1093/nar/25.18.3605
- Smith MCM, Brown WR a, McEwan AR, Rowley P a. Site-specific recombination by phiC31 integrase and other large serine recombinases. Biochem Soc Trans. 2010;38:388–94. doi:10.1042/BST0380388
- Nesmelova I V., Hackett PB. DDE transposases: Structural similarity and diversity. Adv Drug Deliv Rev. 2010;62:1187–95. doi:10.1016/j.addr.2010.06.006
- Hickman AB, Dyda F. Mechanisms of DNA Transposition. Microbiol Spectr. 2015;3: 1–22. doi:10.1128/microbiolspec.MDNA3-0034-2014.
- Van Duyne GD. Lambda Integrase: Armed for Recombination. Curr Biol. 2005;15:658–60. doi:10.1016/j.cub.2005.08.031
- Grainge I, Jayaram M. The integrase family of recombinase: organization and function of the active site. MolMicrobiol 1999;33:449–56. doi:10.1046/j.1365-2958.1999.01493.x
- Mumm JP, Landy A, Gelles J. Viewing single lambda site-specific recombination events from start to finish. EMBO J. 2006;25:4586–95. doi:10.1038/sj.emboj.7601325
- Landy A. The λ Integrase Site-specific Recombination Pathway. Microbiol Spectr. 2015;3: 1–27. doi:10.1128/microbiolspec.MDNA3-0051-2014.
- Rawlings DE, Kusano T. Molecular genetics of Thiobacillus ferrooxidans. Microbiol Rev 1994;58:39–55.
- Williams KP, Kelly DP. Proposal for a new class within the phylum Proteobacteria, Acidithiobacillia classis nov., with the type order Acidithiobacillales, and emended description of the class Gammaproteobacteria. Int J Syst Evol Microbiol. 2013;63:2901–6. doi:10.1099/ijs.0.049270-0
- Rawlings DE. Heavy metal mining using microbes. Annu Rev Microbiol. 2002;56:65–91. doi:10.1146/annurev.micro.56.012302.161052
- Cárdenas JP, Quatrini R, Holmes DS. Genomic and metagenomic challenges and opportunities for bioleaching: a mini-review. Res Microbiol. 2016;167:529–38. doi:10.1016/j.resmic.2016.06.007
- Valdés J, Pedroso I, Quatrini R, Dodson RJ, Tettelin H, Blake R, Eisen J a, Holmes DS. Acidithiobacillus ferrooxidans metabolism: from genome sequence to industrial applications. BMC Genomics. 2008;9:597. doi:10.1186/1471-2164-9-597
- Holmes DS, Cárdenas JP, Valdés JH, Quatrini R, Esparza M, Osorio H, Duarte F, Lefimil C, Jedlicki E. Comparative Genomics Begins to Unravel the Ecophysiology of Bioleaching. Adv Mater Res. 2009;71–73:143–50. doi:10.4028/www.scientific.net/AMR.71-73.143
- Bustamante P, Covarrubias PC, Levicán G, Katz A, Tapia P, Holmes D, Quatrini R, Orellana O. ICEAfe1, an actively excising genetic element from the biomining bacterium Acidithiobacillus ferrooxidans. J Mol Microbiol Biotechnol. 2013;22:399–407. doi:10.1159/000346669
- Bustamante P, Tello M, Orellana O. Toxin-antitoxin systems in the mobile genome of Acidithiobacillus ferrooxidans. PLoS One. 2014;9. doi:10.1371/journal.pone.0112226
- Acuña LG, Cárdenas JP, Covarrubias PC, Haristoy JJ, Flores R, Nuñez H, Riadi G, Shmaryahu A, Valdés J, Dopson M, et al. Architecture and gene repertoire of the flexible genome of the extreme acidophile Acidithiobacillus caldus. PLoS One. 2013;8. doi:10.1371/journal.pone.0078237
- Biswas T, Aihara H, Radman-Livaja M, Filman D, Landy A, Ellenberger T. A structural basis for allosteric control of DNA recombination by lambda integrase. Nature. 2005;435:1059–66. doi:10.1038/nature03657
- Keeton CM, Hopp CM, Yoneji S, Gardner JF. Interactions of the excision proteins of CTnDOT in the attR intasome. Plasmid. 2013;70:190–200. doi:10.1016/j.plasmid.2013.03.009
- Keeton CM, Park J, Wang GR, Hopp CM, Shoemaker NB, Gardner JF, Salyers AA. The excision proteins of CTnDOT positively regulate the transfer operon. Plasmid. 2013;69:172–9. doi:10.1016/j.plasmid.2012.12.001
- Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 1995;177:4121–30. doi:10.1128/JB.177.14.4121-4130.1995
- Dominguez C, Boelens R, Bonvin AMJJ. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125:1731–7. doi:10.1021/ja026939x
- Van Zundert GCP, Rodrigues JPGLM, Trellet M, Schmitz C, Kastritis PL, Karaca E, Melquiond ASJ, Van Dijk M, De Vries SJ, Bonvin AMJJ. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J Mol Biol. 2016;428:720–5. doi:10.1016/j.jmb.2015.09.014
- Wassenaar TA, van Dijk M, Loureiro-Ferreira N, van der Schot G, de Vries SJ, Schmitz C, van der Zwan J, Boelens R, Giachetti A, Ferella L, et al. WeNMR: Structural Biology on the Grid. J Grid Comput. 2012;10:743–67. doi:10.1007/s10723-012-9246-z
- Jayaram M, Ma C, Kachroo AH, Rowley PA, Guga P, Fan H, Voziyanov Y. An Overview of Tyrosine Site-specific Recombination: From an Flp Perspective. Microbiol Spectr. 2015;3:1–28. doi:10.1128/microbiolspec.MDNA3-0021
- Cheng Q, Swalla BM, Beck M, Alcaraz R, Gumport RI, Gardner JF. Specificity determinants for bacteriophage Hong Kong 022 integrase: Analysis of mutants with relaxed core-binding specificities. Mol Microbiol. 2000;36:424–36. doi:10.1046/j.1365-2958.2000.01860.x
- Kelley LA, Sternberg MJE. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–71. doi:10.1038/nprot.2009.2
- Kelley LA, Mezulis S, Yates C, Wass M, Sternberg M. The Phyre2 web portal for protein modelling, prediction, and analysis. Nat Protoc. 2015;10:845–58. doi:10.1038/nprot.2015-053
- Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2011;5:725–38. doi:10.1038/nprot.2010.5
- Šali A, Blundell TL. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993;234:779–815. doi:10.1006/jmbi.1993.1626