
ABSTRACT
The Poly-A Binding Protein (PABP) is a conserved eukaryotic polypeptide involved in many aspects of mRNA metabolism. During translation initiation, PABP interacts with the translation initiation complex eIF4F and enhances the translation of polyadenylated mRNAs. Schematically, most PABPs can be divided into an N-terminal RNA-binding region, a non-conserved linker segment and the C-terminal MLLE domain. In pathogenic Leishmania protozoans, three PABP homologues have been identified, with the first one (PABP1) targeted by phosphorylation and shown to co-immunoprecipitate with an eIF4F-like complex (EIF4E4/EIF4G3) implicated in translation initiation. Here, PABP1 phosphorylation was shown to be linked to logarithmic cell growth, reminiscent of EIF4E4 phosphorylation, and coincides with polysomal association. Phosphorylation targets multiple serine-proline (SP) or threonine-proline (TP) residues within the PABP1 linker region. This is an essential protein, but phosphorylation is not needed for its association with polysomes or cell viability. Mutations which do impair PABP1 polysomal association and are required for viability do not prevent phosphorylation, although further mutations lead to a presumed inactive protein largely lacking phosphorylated isoforms. Co-immunoprecipitation experiments were carried out to investigate PABP1 function further, identifying several novel protein partners and the EIF4E4/EIF4G3 complex, but no other eIF4F-like complex or subunit. A novel, direct interaction between PABP1 and EIF4E4 was also investigated and found to be mediated by the PABP1 MLLE binding to PABP Interacting Motifs (PAM2) within the EIF4E4 N-terminus. The results shown here are consistent with phosphorylation of PABP1 being part of a novel pathway controlling its function and possibly translation in Leishmania.
Introduction
The trypanosomatids constitute a group of parasitic flagellated protozoans which include major human pathogens belonging to the genera Leishmania and Trypanosoma. These organisms are characterized by a complex biology and singular molecular aspects rarely encountered in other eukaryotes. Unique features include polycistronic transcription and regulation of gene expression based mainly on post-transcriptional mechanisms, targeting events such as mRNA stability and translation [Citation1-Citation7]. A major target for regulation is possibly the initiation stage of protein synthesis where a number of eukaryotic translation initiation factors are known to act [Citation8]. Paramount among those is the translation initiation factor eIF4F, a complex of three distinct subunits including the RNA helicase eIF4A, the cap binding protein eIF4E and the scaffolding subunit eIF4G. During translation, eIF4F binds to the mRNA and facilitates the recruitment of the small ribosomal subunit to its 5′ end [Citation9-Citation12]. A key eIF4F partner is the cytoplasmic Poly-A Binding Protein (PABPC or simply PABP), a conserved eukaryotic protein involved in multiple cellular functions associated with the metabolism of messenger RNAs, including mRNA polyadenylation/deadenylation, export, surveillance, degradation, and translation [Citation13-Citation20]. At the initiation stage of protein synthesis, PABP is responsible for the enhanced translation of polyadenylated mRNAs, facilitating their recruitment by the translation machinery. In model eukaryotes, this is accomplished through a direct interaction between PABP and eIF4F. In mammals, yeasts and plants the interaction between eIF4F and PABP is mediated through the eIF4G subunit [Citation10,Citation13,Citation14,Citation16,Citation20].
Schematically, PABPs are formed by a N-terminal region, a non-conserved linker segment and the unique C-terminal MLLE domain, originally called PABC (for reviews see [Citation10,Citation13,Citation14,Citation16,Citation20]). The N-terminal region consists of roughly two-thirds of the protein and contains four conserved RNA binding domains (RRMs) positioned in tandem, each defined by the presence of two highly conserved RNP motifs (RNP1 and RNP2) [Citation21,Citation22]. The PABP RRMs 1 and 2 are responsible for the specific binding to poly-A and also mediate the protein's interaction with eIF4G [Citation23-Citation26]. RRMs 3 and 4 seem to bind to non-polymeric AU sequences with unknown functions and also mediate protein-protein interactions [Citation27-Citation29]. In mammals, the PABP linker region seems to be involved in the multimerization process where multiple PABP molecules associate with each other attached to the poly-A tail [Citation30,Citation31]. As for the C-terminal MLLE domain, it mediates the interaction of PABP with other polypeptides having a conserved peptide motif named PAM2 [Citation32-Citation34].
In Leishmania three PABP homologues were identified but only two of those (PABP1 and PABP2) are found conserved in most, if not all, trypanosomatids [Citation35,Citation36]. It has been shown that all three Leishmania PABPs and T. brucei PABP2 have affinity to poly-A, although the PABP2 orthologues share substitutions in conserved amino acids residues that are critical for poly-A recognition [Citation35,Citation37,Citation38]. All three Leishmania PABPs are abundant proteins which are constitutively expressed and localize to the cytoplasm although PABP2 and PABP3, but not PABP1, migrate to the nucleus upon inhibition of transcription. Co-precipitation studies have also shown a strong association between PABP2 and PABP3 which likely bind to the same set of target mRNAs [Citation35]. In T. brucei, through tethering assays, both PABP1 and PABP2 have been shown to stimulate translation of a reporter mRNA when tethered to its 3′ end [Citation39,Citation40] and have been found to be associated with polysomes [Citation36,Citation41]. RNAi depletion of the T. brucei PABP1 and PABP2 orthologues confirmed that both proteins are essential for cell viability [Citation35], however the two proteins localize to distinct sets of inducible RNP granules and PABP2, but not PABP1, can accumulate in the nucleus upon inhibition of mRNA maturation and heat shock treatment [Citation36].
Multiple homologues have also been identified in trypanosomatids for the eIF4E and eIF4G subunits of eIF4F. The six conserved eIF4Es (EIF4E1 through EIF4E6) and five conserved eIF4Gs (EIF4G1 through EIF4G5) differ substantially in sequence and in binding partners [Citation42-Citation46]. Two distinct eIF4F-like complexes, centered on the interactions between EIF4E4/EIF4G3 and EIF4E3/EIF4G4, have been characterized with properties which implicate them during translation initiation [Citation47-Citation51]. In both Leishmania and Trypanosoma species, the EIF4E4/EIF4G3 complex has been shown to be associated with PABP1 [Citation35,Citation36,Citation49] and the Leishmania PABP1 has been seen to bind directly to EIF4E4, through an interaction unknown from other eukaryotes [Citation49]. The novel PABP1/ EIF4E4 interaction seems to be stronger than the one between EIF4E4 and EIF4G3 and it was also shown to be more critical for EIF4E4 function in vivo [Citation52].
The Leishmania PABP1 was early on seen to be targeted by phosphorylation events, resulting in a clearly identifiable phosphorylated isoform which was seen to be reduced upon transcription inhibition [Citation53]. In contrast, no similar isoforms have been detected for PABP2 or PABP3 [Citation35], implying the existence of specific regulation directed to PABP1. In contrast, EIF4E4, the PABP1 partner, has also been found to be phosphorylated [Citation52], highlighting the two proteins as targets for regulatory mechanisms controlling their functions. Here, we have investigated PABP1 in more detail, assessing its phosphorylation as well as interactions with EIF4E4 and other protein partners, with the purpose of clarifying aspects of its function in Leishmania and other trypanosomatids and enhancing our knowledge on translation initiation in these organisms. Using wild-type and mutated PABP1 variants in L. infantum, protein-protein interaction assays and complementation studies after gene knockout, we defined a clear pattern of phosphorylation for PABP1, which is strikingly similar to the one previously reported for EIF4E4 [Citation52]. We further investigated requirements for phosphorylation as well as its role for PABP1 function and cell viability. Binding partners were identified as well as residues required for the protein to function properly. The novel EIF4E4/PABP1 interaction was further characterized in detail and shown to involve PAM2 like motifs in EIF4E4 and the MLLE domain in PABP1.
Results
PABP1 is constitutively expressed across the L. infantum life cycle but is hyperphosphorylated during growth conditions of active translation
PABP1 orthologues from both T. brucei and Leishmania species have been shown to be targeted by phosphorylation events leading to multiple isoforms which, nevertheless, were not clearly associated with any specific growth stage or biological process [Citation35,Citation36,Citation53]. In T. brucei, in a high throughput search for phosphoproteins, both PABP1 and PABP2 were found to be phosphorylated, with multiple residues targeted for phosphorylation in each protein [Citation54] but only PABP1 was represented by more than one isoform upon expression analysis using whole parasite lysates [Citation36]. Here, we first compared the expression of Leishmania PABP1 and its phosphorylated isoforms with PABP2 during standard promastigote growth conditions. In Leishmania, the original experiments focusing on the characterization of PABP1 were carried out in L. major [Citation35] but here we opted to use a second Leishmania species, L. infantum, which is more amenable to differentiation in culture. Growth curves were set up starting with stationary-phase promastigotes passaged to new media and evaluated for the expression of the two Leishmania PABP homologues, at selected time points, using rabbit polyclonal antiserum directed against the two proteins. As shown in , both proteins are constitutively expressed throughout all stages of the growth curve assayed, but only PABP1 is clearly represented by more than one isoform. In contrast to the constant pattern of expression seen for PABP2, the expression of the two detected PABP1 isoforms vary during the growth curve, with the top (phosphorylated) band being absent or nearly absent from stationary phase cells (days 5 to 6), but predominating soon after passaging (2 hours) and during the stages of fast, logarithmic, cell growth (up to 2 days).
Figure 1. Expression analysis of L. infantum PABP1 during both promastigote and amastigote life stages. (A) Western blotting comparing the PABP1 expression with that of PABP2 and alpha-tubulin during distinct promastigote growth phases. The results from a single growth curve are shown with aliquots taken immediately after passaging to start a new culture (0h), at 2, 4 and 6 hour time points and daily after that. For all lanes equal loads were run under denaturing conditions and blotted with whole rabbit polyclonal sera directed against Leishmania PABP1 and PABP2 or serum against alpha tubulin (protein loading control). The results shown are representative of multiple experiments carried out not only with L. infantum, but also with L. major and L. amazonensis. The graphic representation of the cell counts from every aliquot is shown below the blot. (B) Western blotting showing the PABP1 expression in L. infantum during two consecutive curves grown in amastigote media. The 1st curve, the differentiation curve, started with stationary phase promastigotes and the second curve started with fully differentiated, stationary phase amastigotes as previously described [Citation52] and, as previously shown, the differentiation into amastigotes was confirmed through the detection of the A2 amastigote-specific marker. STA – Stationary cells. LAG – Lag phase culture. LOG – Logarithmic phase culture. For the PABP blots relevant molecular weight markers are shown on the left.
![Figure 1. Expression analysis of L. infantum PABP1 during both promastigote and amastigote life stages. (A) Western blotting comparing the PABP1 expression with that of PABP2 and alpha-tubulin during distinct promastigote growth phases. The results from a single growth curve are shown with aliquots taken immediately after passaging to start a new culture (0h), at 2, 4 and 6 hour time points and daily after that. For all lanes equal loads were run under denaturing conditions and blotted with whole rabbit polyclonal sera directed against Leishmania PABP1 and PABP2 or serum against alpha tubulin (protein loading control). The results shown are representative of multiple experiments carried out not only with L. infantum, but also with L. major and L. amazonensis. The graphic representation of the cell counts from every aliquot is shown below the blot. (B) Western blotting showing the PABP1 expression in L. infantum during two consecutive curves grown in amastigote media. The 1st curve, the differentiation curve, started with stationary phase promastigotes and the second curve started with fully differentiated, stationary phase amastigotes as previously described [Citation52] and, as previously shown, the differentiation into amastigotes was confirmed through the detection of the A2 amastigote-specific marker. STA – Stationary cells. LAG – Lag phase culture. LOG – Logarithmic phase culture. For the PABP blots relevant molecular weight markers are shown on the left.](/cms/asset/dc22d961-7d99-4f1a-b128-10bcd80223f5/krnb_a_1445958_f0001_b.gif)
Next, we investigated the PABP1 expression during amastigote differentiation using two consecutive growth curves (). The differentiation curve started with stationary-phase promastigotes, which were allowed to differentiate in amastigote medium and it was followed by the second curve using fully differentiated cells passaged again into the new amastigote medium. As seen for promastigotes, the expression pattern of the PABP1 isoforms varied for both curves also according to the growth stage, with the top band being predominant soon after passaging and during logarithmic growth and the bottom band being more abundant in stationary cells. The changes in isoform profile for PABP1 is then clearly linked with the growth phase of the cells and is not influenced by its differentiation stage. This pattern of isoform expression observed for PABP1 and common to both promastigote and amastigote stages of the Leishmania life cycle is very similar to or identical to the one seen for EIF4E4, PABP1's binding partner. As previously reported, EIF4E4 is targeted by multiple phosphorylation events generating multiple isoforms which predominate in both promastigote and amastigote forms during logarithmic growth and presumably active protein synthesis [Citation52,Citation55].
PABP1 phosphorylation is linked to SP/TP motifs found within its linker region
L. infantum PABP1 has been shown to be phosphorylated in vitro by a p38 MAP kinase [Citation38]. The very fast changes in PABP1 isoform expression induced by the passaging to new media are also consistent with a MAP kinase response. In T. brucei the three PABP1 residues that were found to be targeted by phosphorylation are serine or threonine residues followed by a proline [Citation54], known targets of MAP kinases. To investigate possible phosphorylation sites within PABP1, and considering the similarity observed with the EIF4E4 phosphorylation targeting conserved serine-proline (SP) or threonine-proline (TP) motifs, we performed an alignment of multiple PABP1 orthologues from different trypanosomatid species and searched for conserved SP or TP motifs that are missing from equivalent regions from PABP2 and PABP3 (Fig. S1). Seven of these motifs were found in close proximity at the end of the linker region of PABP1 sequences from all Leishmania species investigated. These are indicated in the scheme from and highlighted in the alignment, comparing the distantly related L. infantum and L. braziliensis PABPs. Three of the motifs are also conserved in T. brucei and they coincide with the previously identified phosphorylation sites in PABP1 from this organism [Citation54]. The alignment also highlights the three motifs found within the T. brucei PABP1 (Fig. S1).
Figure 2. Identification of the PABP1 phosphorylation motifs. (A) Structural domain organization of the L. infantum PABP1 homologue. The protein's RRM region consisting of the first two thirds of PABP1 (encompassing all four RRMs) plus the “Linker” segment and the “MLLE” domain are shown. Seven putative phosphorylation sites are indicated, six of which were targeted by site-directed mutagenesis (circled). Also highlighted are the three sets of amino acid triplets chosen for mutagenesis and localized between RRMs 1 and 2 (LMW), within RRM2 (YGF) or within the MLLE domain (TGM) The N-terminus and C-terminus fragments used for the pull-down assays are also indicated. (B) Expression of HA-tagged, episomally encoded, PABP1 evaluated with a monoclonal commercial anti-HA antibody. The left panel compared the expression of wild-type PABP1 (HA-PABP1) during three representative stages of the parasite growth curve. The right panel evaluates the expression, under the same conditions of the PABP1 mutant (TP-SP Mut) where all six putative phosphorylation sites were targeted by site directed mutagenesis.
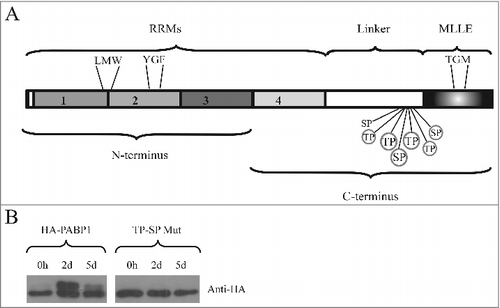
To investigate the identified motifs further, we opted here to use site directed mutagenesis in order to mutate, within the L. infantum PABP1 gene, the sequences encoding six out of the seven candidate SP/TP motifs found in the PABP1 linker region (circled in ). Each mutation resulted in the substitution of the serine/threonine by an alanine, effectively preventing the phosphorylation of the motif. The mutant protein (named TP-SP Mut) was then ectopically expressed after stable transfection of the mutated gene into L. infantum, generating a recombinant protein tagged with the HA epitope at its C-terminus. First, we compared its expression with that of an equivalent HA-tagged wild-type PABP1 (from now own named HA-PABP1). After blotting with an anti-HA monoclonal antibody, the HA-PABP1protein displayed the same isoform expression pattern seen for the endogenous protein (). A lower MW band was seen predominantly at the beginning of the growth curve and also at day 5 of the culture, when cells reach stationary phase. In contrast, during logarithmic growth at day 2, a higher MW band, corresponding to the phosphorylated PABP1 isoform, was detected (). The HA-tagged TP-SP mutant was also expressed and assayed under the same growth conditions as the wild-type protein (, TP-SP Mut). A single protein band was seen for the HA-tagged mutant, with no changes observed in size or expression between logarithmically grown or stationary cells, indicating a lack of phosphorylation throughout the growth curve. These results are consistent with multiple phosphorylation events targeting PABP1 and directed at the multiple conserved SP/TP motifs found in its linker region. These phosphorylation events lead to the high molecular weight isoform observed in the PABP1 expression analysis () and are possibly mediated by the same kinase(s) which target EIF4E4.
PABP1 is enriched in polysomal fractions during Leishmania logarithmic growth
We have previously attempted to correlate the presence of the phosphorylated EIF4E4 isoforms with a more active role in translation by investigating its association with mRNA bound polysomes. Unfortunately, in our hands at least, the very labile nature of this protein and its propensity to degradation during preparation of these extracts prevented a clear identification of EIF4E4 after polysome analysis (unpublished data). PABP1, however, is more abundant than EIF4E4 and is not as susceptible to degradation, so its association with polysomes could be investigated. Sucrose gradient fractionation of cellular extracts from cells grown logarithmically and after reaching stationary phase was then performed. Extracts from logarithmically grown cells yielded profiles with multiple peaks for the polysomes, compatible with robust protein synthesis. For the stationary cells, in contrast, the polysome peaks found in the sucrose-gradient profiles were minimal or non-existent, an indication of very limited protein synthesis (Fig. S2).
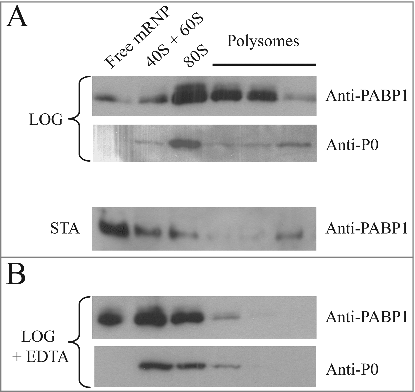
Fractions from the top of the gradients (“Free mRNP”) and from the 40S and 60S ribosomal subunits, 80S ribosomes and the polysomes were pooled and blotted with the PABP1 anti-serum. For the gradients derived from the logarithmically grown cells, PABP1 was found associated mostly with the 80S fraction and the polysomes (). However, in gradients from stationary cells, PABP1 was found mostly on the top of the gradient with only a very limited amount found in the fractions corresponding to the polysomes, compatible with the low levels of protein synthesis at this stage. In these experiments the top PABP1 isoform (phosphorylated) was not clearly visible, especially in the polysome fractions, and we believe this is likely due to dephosphorylation occurring during extract preparation and subsequent steps of gradient fractionation. Indeed when freshly prepared cytoplasmic extracts are compared with whole cell extracts, prepared by the resuspension of live cell directly in SDS-PAGE sample buffer, this isoform is already much reduced and this was seen even in the presence of phosphatase inhibitors (not shown). This contrasts to what has been described from T. brucei, where phosphorylated PABP1 can be detected both in polysomal and non-polysomal fractions [Citation36]. In , a control blot was also carried out using a rabbit polyclonal antiserum directed against the ribosomal protein P0, a constituent of the 60S ribosomal subunit. As expected, P0 was detected on the polysome fractions from logarithmically grown cells as well as in the 40S/60S and 80S fractions (). Another control was performed by treating extracts from logarithmically grown cells with EDTA prior to loading on the sucrose gradient, followed by blotting to assay for both PABP1 and P0 (). Polysome dissociation was confirmed by the detection of P0 mainly in the 40S/60S and 80S fractions. PABP1, although more abundant, displayed a profile similar to P0 with most of it shifting to the top of the gradient upon EDTA treatment. Altogether, these results indicate that the Leishmania PABP1 protein is strongly associated with the polysomes during logarithmic growth.
Figure 3. Polysome profile analysis of PABP1 in logarithmically growing and stationary cells. (A) Sucrose gradients (15-45%) for polysome profile analysis of L. infantum promastigotes were carried out using extracts from both logarithmic (LOG) or stationary (STA) phase stages of representative growth curves. Samples from 260 nm peaks derived from the free mRNPs, 40/60S and 80S ribosomal fractions, as well as the polysomes, were pooled and blotted with the anti-PABP1 antibodies and antibodies directed against the Leishmania ribosomal protein P0. (B) EDTA was used with the samples from logarithmic growth (LOG + EDTA) to demonstrate the dissociation of polysomes and corresponding effect on the migration of PABP1 and P0 on the sucrose gradients. The results shown are representative of a minimum of two different experiments carried out with different batches of cell growth.
Identification of motifs relevant for PABP1 phosphorylation and polysome association
In order to better define the mechanism(s) associated with PABP1 phosphorylation and its role in translation, we opted to search for motifs that would be required for specific aspects of PABP1 function and that could be identified by mutagenesis. This also follows a similar approach carried out with EIF4E4 where, for instance, the elimination of its interaction with its EIF4G3 partner did not prevent its phosphorylation [Citation52], contrasting to mammalian systems where eIF4E phosphorylation requires binding to eIF4G, which in turn recruits the kinase Mnk [Citation56]. Here, three sets of three consecutive amino acid residues were chosen for mutagenesis and which are indicated in . All three are strictly conserved in different PABP1 orthologues and are also mostly conserved between PABP homologues from different organisms, including metazoans, plants and yeast, but the first two are specifically modified in trypanosomatid PABP2 orthologues. The first triplet, leucine-methionine-tryptophan (LMW) maps to a segment between the PABP RRMs 1 and 2 which has been shown to mediate PABP binding to eIF4G and eIF4B homologues [Citation10]. The second triplet, tyrosine-glycine-phenylalanine (YGF), is part of the RNP2 motif from RRM2 and includes residues which have been implicated in poly-A binding and recognition [Citation22]. A third triplet, chosen within the conserved structural core of the MLLE domain [Citation57], consists of threonine-glycine-methionine (TGM), and is the only one also found in PABP2. All mutations replaced the selected residues for alanines, generating the LMW/AAA, YGF/AAA and TGM/AAA mutants which were also expressed as HA-tagged proteins in transfected L. infantum cells. The expression of the respective recombinant proteins was then evaluated under conditions of logarithmic growth where the phosphorylated isoform of PABP1 predominates (). All three mutants were represented by the two isoforms seen with wild-type PABP1, with the top phosphorylated isoform being in general predominant and no indication of an effect induced by the mutations on phosphorylation (). A fourth mutant, containing the first two triplets mutated (LMW/AAA + YGF/AAA) was then generated and evaluated likewise. This double mutant seemed to be expressed at lower levels than the previous proteins, although no proper quantitation was carried out to evaluate this, and very little expression of the top isoform was seen, indicating a significant impact on PABP1 phosphorylation when both LMW and YGF motifs were mutated.
Figure 4. Identification of relevant motifs involved in PABP1 phosphorylation and polysome binding. (A) Expression analysis of HA-tagged PABP1 mutants containing selected triplets of amino acid residues replaced by alanines. The expression of the resulting proteins was evaluated in logarithmically grown cells, as shown at the 2d time point in . The effect on PABP1 phosphorylation of mutating the triplets LMW, YGF or TGM, individually, or both sets of LMW and YGF was evaluated. The results shown were reproduced using a minimum of two growth curves for each mutant and, as much as possible, the cultures were grown in parallel and assayed at the same stage of the growth, using the same number of passages following transfection. (B) Polysome profile analysis of L. infantum promastigotes expressing wild-type or mutant HA-tagged PABP1 proteins, under logarithmic growth, were carried out to investigate the association of each individual mutant to polysomes. Samples were pooled as shown in and blotted with the anti-HA antibody. For the LMW/AAA + YGF/AAA mutant, as control, the same samples were also blotted using the anti-P0 and anti-PABP1 antisera.
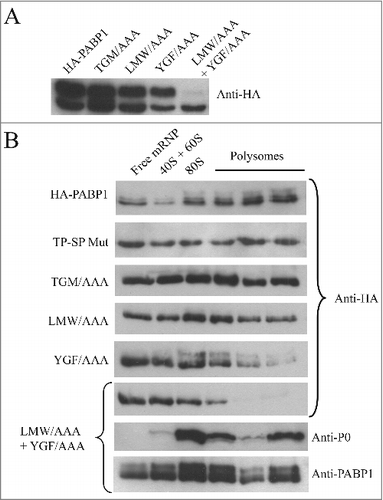
To investigate if PABP1 phosphorylation was a requirement for it to bind to polysomes, wild-type and mutant HA-tagged proteins were evaluated through polysome profiling using sucrose gradients carried out as described in with promastigote cells grown logarithmically. As expected, wild-type HA-tagged PABP1 associated efficiently with the polysomes, with most of the ectopically expressed protein seen in the polysomal fractions (). A similar profile was seen for the TP-SP mutant, where phosphorylation was abolished, indicating that phosphorylation of PABP1 is not a requirement for its association with polysomes. The various mutants assayed in were also investigated using the same approach. Both LMW/AAA and TGM/AAA mutants were also found with the polysomes although, when compared to the wild-type protein, these seemed to be found in greater quantities in the lighter non-polysomal fractions of the gradients, indicating a possible reduction in efficiency in polysome association (). The YGF/AAA mutant, in contrast, was clearly impaired on its ability to associate with mRNA bound to polysomes, presumably a consequence of the mutations targeting residues required for mRNA binding. This effect was further enhanced in the LMW/AAA + YGF/AAA double mutant, which showed no indication of association with polysomes with most of the protein found on the top fractions of the gradients (). For this last mutant, the same sucrose gradient fractions were also blotted with anti-sera directed against the P0 ribosomal protein and the native PABP1 (this anti-sera recognizes both the HA-tagged mutant as well as the endogenous PABP1 present in the extracts). Both P0 and native PABP1 were mainly found with the polysome fractions, confirming the quality of the sucrose gradient fractionation and that the lack of association of the double mutant with the polysomes was due to a disruption of poly(A) tract binding. In comparison to the isoforms observed in the whole cell extracts from , phosphorylated isoforms in the gradient fractions from different HA tagged proteins are much diminished in intensity and, when present, are reduced in apparent molecular weight. As for the native PABP1 shown in we believe this is due to dephosphorylation and their presence/absence from different fractions does not necessarily have any functional implications. Nonetheless, the sucrose gradient results with the mutant proteins are consistent with phosphorylation not being required for PABP1 to be recruited by the translation apparatus. These experiments also highlight the functional relevance of the LMW and YGF motifs within the PABP1 RRM region.
The Leishmania PABP1 gene is essential for parasite growth
In T. brucei, RNAi mediated knockdown of PABP1 prevented cell viability and growth in procyclic cells confirming it is essential for survival in these cells [Citation35]. However, the existence of a third PABP homologue in Leishmania species (PABP3), lost from the Trypanosoma lineage [Citation36], means that the results from T. brucei cannot readily be extrapolated to all trypanosomatids. Here, attempts were made to generate haploid or diploid knockouts for the PABP1 gene and evaluate its effect on L. infantum survival. The gene replacement strategy used for generating the knockouts is the same applied previously to study EIF4E4 [Citation52], (see also ). The first PABP1 copy gene replacement was done using the hygromycin resistance gene (HYG) expression cassette for the generation of the haploid PABP1/PABP1::HYG cell line (HYG KO). Transfected cells were recovered with no difficulties followed by cloning in order to have multiple lineages having the single knockout (SKO) of the PABP1 gene. Integration into the endogenous PABP1 locus was confirmed through a PCR based analysis using a primer annealing to the PABP1 gene intergenic region, external to the fragment used for the integration procedure, and an internal primer annealing to the hygromycin coding sequence (). The presence of the remaining PABP1 allele was also confirmed using a similar PCR procedure, as illustrated in . This second wild type allele was then targeted with a cassette expressing the puromycin resistance gene (PUR KO). Multiple transfection events were carried out using independent clones generated with the haploid genotype. These transfections did not always yield viable transfected cells but sometimes cell lines were recovered which had the PUR gene integrated into the PABP1 locus (PABP1::PUR /PABP1::HYG), suggesting a double knockout of the PABP1 genes (DKO). When investigated through PCR, however, an intact copy of the endogenous PABP1 gene was still detected (not shown). Thus, our conclusion is that the PABP1 gene is essential for cell viability and susceptible to gene duplication events, leading to the generation of a trisomic gene copy upon targeted gene replacement of the two PABP1 alleles.
Figure 5. Generation of haploid and diploid PABP1 gene knockout mutants in L. infantum. (A) Schematic representation of the endogenous PABP1 gene as well as gene replacements following the single integration of the hygromycin (HYG) and/or puromycin (PUR) drug selectable markers. The 500 bp intergenic regions upstream and downstream of the PABP1 coding region (named 5′IR and 3′IR in the figure), and that were used to generate the knockout cell lines through homologous recombination, are indicated by the small rectangles in dark gray. The position of the primers added to the PCR reactions used to confirm the integration events are indicated by arrows, while the position of the restriction sites used for the Southern blots are also shown. (B) Agarose gel showing the PCR fragments amplified from the wild type L. infantum as well the single knockout (SKO) lineages and the double knockout (DKO) lineages generated in the presence of a plasmid encoded PABP1 gene. The oligonucleotides used for the PCR reactions are listed in Table S5. (C) Southern blots confirming the efficiency of the knockout procedures. Total genomic DNA from wild-type L. infantum as well as the SKO and DKO cells lines was digested with either ClaI or PvuII restriction enzymes and probed with the gene fragment corresponding to the PABP1 gene 5′ intergenic region (5′ IR). For (B) and (C), fragments corresponding to the wild-type PABP1 gene as well as the hygromycin (HYG KO) and puromycin (PUR KO) integration events are shown. Size markers in bp are shown on the left of the panels, while the sizes of the bands of interest are also indicated on the right. All transfection experiments were performed at least twice, generally more, with the DKOs performed using a minimum of two sets of SKO cell lines. Selected samples were used for the PCR or Southern blots.
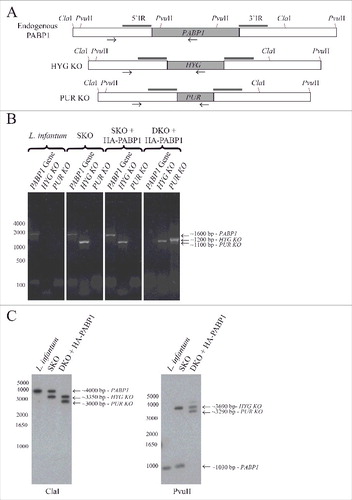
To evaluate if both copies of the PABP1 gene could be targeted without inducing the gene duplication event, and to eliminate the possibility of the PABP1 gene being originally trisomic, an alternative complementation strategy, also previously applied to the EIF4E4 gene [Citation52], was tested. This strategy requires transfecting in the HYG SKO cell lines a plasmid encoding the PABP1 gene (conferring resistance to neomycin) prior to the transfection with the puromycin expression cassette (PUR KO). Again, multiples HYG SKO cell lines were used for the transfection with the plasmid encoded PABP1 gene, followed by neomycin selection and subsequent transfection using the PUR KO cassette and selection with puromycin. Using this strategy, the second PABP1 allele was successfully replaced by the puromycin cassette, as confirmed by the lack of PCR amplification of the endogenous PABP1 gene fragment in the presence of the PUR KO gene fragment. For these gene replacement experiments, in order to independently confirm the deletion of the two endogenous copies of the PABP1 gene, we also opted to use Southern-blot, as previously applied for EIF4E4. DNA from the wild-type L. infantum as well as the single (SKO) and double (DKO) knockout cells lines was probed with the gene fragment corresponding to the PABP1 gene 5′ intergenic region, a fragment missing from the plasmid encoded gene. As shown in , the band corresponding to the endogenous PABP1 gene is missing from the DKO cell lines, confirming the deletion of both gene copies by the knockout procedures. Altogether, the results from and confirm the essential nature of the PABP1 gene, at least for the promastigote stage of the Leishmania cell cycle.
Complementation studies reveal motifs strictly required for PABP1 function
Next, attempts were made to recover double knockout (DKO) cells from the HYG SKO lines expressing the different recombinant proteins in order to see which of the mutants could complement the lack of endogenous PABP1. Successful recovery of cell lines in the presence of both hygromycin and puromycin selection was achieved for cells expressing all mutant proteins, although, as seen for HYG SKO derived lines (discussed below), the recovery was significantly delayed in comparison with cells expressing the wild-type PABP1 (not shown). DNA recovered from all putative DKO lines was assayed, by PCR only, for the presence of the endogenous PABP1 gene and representative results are shown in . Successful depletion of the endogenous gene was reproducibly achieved for the cells expressing the TP-SP phosphorylation mutant as well as the one having the TGM/AAA mutation. In contrast, for those cells complemented with the LMW/AAA and YGF/AAA mutants, the endogenous PABP1 gene was still present after hygromycin and puromycin dependent integration events, indicating a gene duplication event similar to the one observed during our attempts to generate the PABP1::PUR /PABP1::HYG cell lines.
Figure 6. Evaluation of the diploid PABP1 knockout in L. infantum cell lineages complemented with various HA-tagged PABP1 mutants. (A) PCR results carried out as shown in evaluating the presence of the endogenous PABP1 gene in recombinant cell lines complemented with different PABP1 mutants. Size markers in bp are shown on the left and the sizes of the amplified bands are also indicated. (B) Western-blot using the serum directed against native PABP1 to assess expression of the non-phosphorylated and phosphorylated PABP isoforms in the cell lines complemented with the wild-type PABP1 gene (HA-PABP1) or the phosphorylation mutant (TP-SP Mut). Notice the extra phosphorylated band in the presence of the wild-type HA-tagged PABP1, which is slightly bigger in comparison with the native PABP1 (L. infantum lane) due to the presence of the HA-tag (phosphorylated and non-phosphorylated isoforms indicated by arrows). The experiments shown in (A) and (B) are representative of results generated with a minimum of two sets of independently transfected cells.
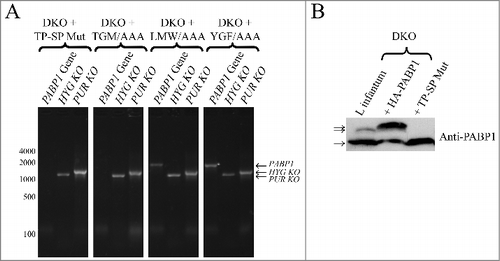
The absence of the endogenous PABP1 in the DKO lines complemented with the phosphorylation mutant was also assessed using the anti-PABP1 serum. As shown in the western blot of , only the non-phosphorylated PABP1 isoform was detected in cells complemented with the TP-SP mutant in contrast to the cell line overexpressing the HA-tagged wild-type PABP1 which produces both phosphorylated and non-phosphorylated isoforms. Thus, a conclusion from the complementation experiments is that phosphorylation of PABP1 is not strictly required for cell viability, since the non-phosphorylatable mutant can complement the loss of both copies of the otherwise essential PABP1 gene. Likewise, the protein having the TGM/AAA mutation can also function sufficiently to maintain cell viability in the absence of the endogenous protein. The mutant impaired in its ability to bind to mRNAs and associate with polysomes (YGF/AAA), however, cannot complement the lack of the endogenous PABP1. The same applies for the LMW/AAA mutant, indicating that the LMW motif is involved in critical interactions required for the PABP1 function.
Effects on L. infantum growth induced by the overexpression of PABP1 mutants
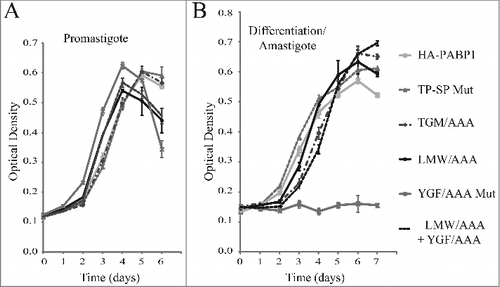
The essential nature of the PABP1 function in Leishmania might be linked to a role during translation or to other processes associated with the metabolism of mRNAs. Since a PABP1 double knockout cell line cannot be generated, an indirect way to study its function is to evaluate the effects induced by the overexpression of the native protein or selected mutants in transgenic cells through the transfection with the plasmid encoded genes. Here, to investigate more specifically the effect of the various PABP1 mutations on promastigote and amastigote growth, haploid PABP1/PABP1::HYG (HYG SKO) cell lines were used. The SKO cell lines were chosen since the 50% reduction in copy number for the endogenous PABP1 gene might increase the cells’ sensitivity to any effects induced by the overexpressed recombinant proteins. Indeed, during the selection phase of the transfection procedure with the plasmids encoding the PABP1 wild-type and mutant proteins, the cells transfected with several of the mutants did take a longer time to recover, when compared with wild-type PABP1 (data not shown). Once recovered, growth of the different cell lines was then compared using standard promastigote growth curves. As described in , these curves were also started with stationary phase promastigotes passaged to new media and allowed to grow for several days, with growth monitored daily. For these experiments cell lines generated from independent transfection events of at least two distinct SKO cell lines were used and the results described were reproduced with a minimum of two independent cell. No major differences in growth were observed, however, when the cells expressing either the wild-type PABP1 or any of the mutants were compared (). The same cell lines from stationary cultures were then transferred to amastigote media, and allowed to differentiate. Similarly, no significant differences were observed in differentiation and growth as amastigotes for the phosphorylation mutant (TP-SP Mut) as well as the LMW/AAA and TGM/AAA mutants in comparison with the wild-type protein (). In contrast, cells overexpressing the YGF/AAA mutant did not grow as amastigotes and this phenotype suggests a dominant negative effect, since the overexpression of the mutant PABP1 might be interfering with the activity of the endogenous protein and inhibiting growth as amastigotes. This effect was not seen with the double mutant protein (LMW/AAA + YGF/AAA), confirming that the growth inhibitory phenotype requires an intact LMW motif.
Figure 7. Cell growth phenotype of the haploid PABP1 knockout cell lineages complemented with various HA-tagged PABP1 mutants. (A) and (B) L. infantum cells lacking one of the PABP1 gene copies (SKO) were transfected with episomal vectors expressing either the wild-type HA-PABP1 or the various mutant proteins. Cells were grown as promastigotes (A) or as axenic amastigotes (B) and their growth monitored daily. Standard deviations were derived from a single experiment done with three replicates grown in parallel. Two sets of experiments were done for two sets of independently transfected cells.
Identification of known and novel PABP1 partners
PABP1 likely executes its functions by binding to poly-A as well as to proteins which are involved in different processes associated to the metabolism of mRNAs, including translation. Leishmania PABP1 has been shown to co-immunoprecipitate with both partners of the EIF4E4/EIF4G3 complex and direct interactions with these two proteins have also been demonstrated [Citation35,Citation49], providing a possible explanation as to how PABP1 would function in translation initiation. Nevertheless, it is not clear what other protein factors or even homologs to eIF4F subunits might also be interacting with PABP1 and which could be affected by the identified phosphorylation events. Here, in order to investigate the association of PABP1 with known or novel protein binding partners, cytoplasmic extracts from the cells expressing the HA-tagged wild-type PABP1 were used in immunoprecipitation (IP) assays with an anti-HA monoclonal antibody bound to magnetic beads. Control assays were also carried out with cytoplasmic extracts from non-transfected L. infantum promastigotes incubated with the anti-HA bound magnetic beads and the precipitated samples from both reactions were then submitted to mass-spectrometry analysis. Two sets of independent experiments were carried out and, following polypeptide identification, only those which were substantially enriched in the PABP1 sample in comparison with the control, in both experiments, were considered and are listed on . As expected, the top hit was the HA-tagged PABP1 and both EIF4G3 and EIF4E4 were also found among the polypeptides bound to PABP1, highlighting the strong association between the Leishmania PABP1 and the EIF4E4/EIF4G3 complex. In contrast, none of the other eIF4E or eIF4G homologues were found. The remaining bound polypeptides include the RRM-containing protein RBP23 (LinJ.17.0610), an uncharacterized protein having both a Nuclear Transport Factor 2 (NTF2) and RRM domains (LinJ.18.0300), a Zinc-finger protein having both KH and Helicase domains (ZC3H41 – LinJ.27.1220) and yet another uncharacterized protein (LinJ.05.0450). The two T. brucei PABP homologues as well as both EIF4E4 and EIF4G3 have been shown to stimulate translation of a reporter mRNA when tethered to its 3′UTR [Citation39,Citation40] and the same applies for the T. brucei orthologues of RBP23 and LinJ.18.0300.
Table 1. Proteins co-purifying with HA-tagged PABP1.
The association of several known translational activators with PABP1 suggests a common function involving ribosome recruitment during translation initiation. Parallel IP assays were also carried out using the HA-tagged PABP1 phosphorylation TP-SP mutant, however no clear differences in co-immunoprecipitated proteins were observed, when compared with the wild-type protein, which were reproducible in the two experiments performed (data not shown). Since the mutant protein co-exists with the wild-type native PABP1 in the transgenic cells, and multiple PABP1 molecules may bind to the same mRNA poly-A tail, it is possible that the effects induced by the lack of phosphorylation of the mutant protein may be masked by its association with the native PABP1. Overall, the mass-spectrometry data confirm a strict association between PABP1 and EIF4E4/EIF4G3, but no other eIF4F-like complex or subunits, and identifies novel proteins, which might be required for PABP1 to function properly during translation or other processes.
Defining the EIF4E4 binding region within PABP1
The motifs within EIF4E4 that are involved in the interaction with PABP1 have been recently identified and seen to be localized within the unique N-terminal segments found only in the trypanosomatid EIF4E3 and EIF4E4 orthologues [Citation52]. However, the binding motifs within PABP1 have not been identified yet and here an attempt was made to better define this interaction. First, truncated PABP1 proteins were expressed as GST-fusions in E. coli and tested for their ability to bind to 35S-labeled EIF4E4 through co-precipitation/pull-down assays. EIF4E4 was seen to bind to the C-terminal half of PABP1 in a segment including the 4th RRM, its linker region and the MLLE domain (C-terminus in ). The PABP1 mutants having the LMW/AAA + YGF/AAA or the TGM/AAA mutations were expressed also as GST-fusions and tested for their ability to interact with labelled EIF4E4. Both wild-type and the LMW/AAA + YGF/AAA full-length proteins bound efficiently to EIF4E4, but no binding was seen with the TGM/AAA mutation, directed to the protein's MLLE domain. These results define then the MLLE domain of PABP1 as the binding site for EIF4E4 () with a role for the TGM residues in this interaction.
Figure 8. Mapping the EIF4E4 binding region within PABP1. Co-immunoprecipitation assay to map the localization of the EIF4E4 binding motif within PABP1. (A) Recombinant L. major PABP1 expressed in Escherichia coli fusioned at its N-terminus with Glutathione S-transferase (GST), was assessed for its ability to bind to the [35S]-labeled wild-type EIF4E4. Full-length PABP1 or truncated mutants lacking part of its N- or C-terminal regions (as shown in ) were evaluated for their ability to bind to the labeled EIF4E4. (B) Full-length L. infantum GST-PABP1 was also compared with the “TGM” or the double “LMW”/”YGF” mutants as to their ability to bind to EIF4E4. The upper panels shows the Coomassie-blue stained gels indicating the recombinant GST (negative control), the wild-type GST-PABP1 fusion or the truncated proteins and mutants assayed (the recombinant proteins are indicated by arrows and the sizes of molecular weight markers are shown on the left). The panels below shows the result from the assays carried out evaluating their binding to the wild-type EIF4E4.
![Figure 8. Mapping the EIF4E4 binding region within PABP1. Co-immunoprecipitation assay to map the localization of the EIF4E4 binding motif within PABP1. (A) Recombinant L. major PABP1 expressed in Escherichia coli fusioned at its N-terminus with Glutathione S-transferase (GST), was assessed for its ability to bind to the [35S]-labeled wild-type EIF4E4. Full-length PABP1 or truncated mutants lacking part of its N- or C-terminal regions (as shown in Fig. 2A) were evaluated for their ability to bind to the labeled EIF4E4. (B) Full-length L. infantum GST-PABP1 was also compared with the “TGM” or the double “LMW”/”YGF” mutants as to their ability to bind to EIF4E4. The upper panels shows the Coomassie-blue stained gels indicating the recombinant GST (negative control), the wild-type GST-PABP1 fusion or the truncated proteins and mutants assayed (the recombinant proteins are indicated by arrows and the sizes of molecular weight markers are shown on the left). The panels below shows the result from the assays carried out evaluating their binding to the wild-type EIF4E4.](/cms/asset/422d7525-e230-4eab-b4ea-82066782dbb8/krnb_a_1445958_f0008_b.gif)
Molecular modeling of the PABP1/EIF4E4 interaction
It is well known that the MLLE domain found in PABP homologues binds to PAM2 motifs which have been identified in proteins with distinct functions in different organisms, including plants and mammals [Citation33]. The interaction between the MLLE domain and a PAM2 motif has been more recently characterized through X-ray crystallography and the latter has been defined as a region of 12 residues, with five of those (at positions 3, 5, 7, 10 and 12) being directly implicated in the interaction. Binding to PABP requires a critical stacking interaction between the conserved Phe/Tyr within PAM2 at position 10 and another conserved Phe/Tyr localized to the MLLE's second alpha helix [Citation57]. The TGM motif lies within a conserved stretch localized in the middle of the MLLE (see Fig. S1), within its central third helix, and both the threonine and methionine residues within this motif have been implicated in the interaction with PAM2. With the identification of the MLLE domain from PABP1 as the site for its interaction to EIF4E4, and considering that three motifs (boxes A, B and C) have been found in EIF4E4 to be implicated in the binding to PABP1 [Citation52], this interaction was further analyzed. Indeed, when the sequences from the three EIF4E4 boxes were compared with PAM2 motifs found in PABP interacting proteins from various eukaryotes, all three aligned with the consensus motif derived from these sequences (). Based on this alignment all three boxes can be confidently defined as PAM2 motifs, with box B more closely resembling the consensus motif while boxes A and C are characterized by some divergent residues in positions which are generally conserved in most other PAM2 sequences.
Figure 9. Evaluating the EIF4E4 PAM2 motifs and the EIF4E4/PABP1 interaction. (A) Alignment of multiple PAM2 motifs identified in various putative PABP interacting proteins isolated from different organisms with the three box (A, B and C) motifs reported for L. infantum EIF4E4 [Citation52]. Proteins from human (Homo sapiens – HS), plant (Arabidopsis thaliana – AT), fungi (Schizosaccharomyces pombe – SP) and other unicellular organisms (Dyctiostelium discoideum – DD; Plasmodium falciparum – PF) are shown, as previously described [Citation33]. The positions of the five hydrophobic residues which characterize the PAM2 motif are shown (3, 5, 7, 10 and 12). (B) and (C) Modeling of the interaction between the L. infantum EIF4E4 PAM2 motif from box B and the PABP1 MLLE domain. A segment starting at proline 484 and ending in glutamine 588 from the full-length PABP1 was used for the modeling. The alpha-helical segments of the MLLE are in blue with relevant aminoacid residues colored in yellow while the peptide backbone from the PAM2 motif is in red with selected residues colored in green. The image in (C) is horizontally rotated 180 degrees in comparison with (B).
![Figure 9. Evaluating the EIF4E4 PAM2 motifs and the EIF4E4/PABP1 interaction. (A) Alignment of multiple PAM2 motifs identified in various putative PABP interacting proteins isolated from different organisms with the three box (A, B and C) motifs reported for L. infantum EIF4E4 [Citation52]. Proteins from human (Homo sapiens – HS), plant (Arabidopsis thaliana – AT), fungi (Schizosaccharomyces pombe – SP) and other unicellular organisms (Dyctiostelium discoideum – DD; Plasmodium falciparum – PF) are shown, as previously described [Citation33]. The positions of the five hydrophobic residues which characterize the PAM2 motif are shown (3, 5, 7, 10 and 12). (B) and (C) Modeling of the interaction between the L. infantum EIF4E4 PAM2 motif from box B and the PABP1 MLLE domain. A segment starting at proline 484 and ending in glutamine 588 from the full-length PABP1 was used for the modeling. The alpha-helical segments of the MLLE are in blue with relevant aminoacid residues colored in yellow while the peptide backbone from the PAM2 motif is in red with selected residues colored in green. The image in (C) is horizontally rotated 180 degrees in comparison with (B).](/cms/asset/7a1f9c29-aea3-4845-b655-69859bba7ebd/krnb_a_1445958_f0009_oc.jpg)
Next, we attempted to model the interaction between the MLLE domain from Leishmania PABP1 with the PAM2 motif found in the EIF4E4 box B. The interaction between the human PABP MLLE and PAM2 motifs from binding partners has been solved by NMR and X-ray crystallography and the MLLE has been characterized as a peptide-binding domain capable of specifically recognizing the PAM2 motif [Citation57,Citation58]. The molecular modeling of the interaction between the Leishmania PABP1 MLLE and the EIF4E4 PAM2 motif is shown in and , with both the MLLE domain and the PAM2 motif conforming to the published structure. The critical PAM2 residues at positions 3, 4, 7 and 10 (M143, N144, A147 and F150, respectively) are indicated as well as several residues from the MLLE domain involved in the binding between the two polypeptides (also highlighted in Fig. S1). Noteworthy is the stacking interaction between the phenylalanine at position 10 in PAM2 with another phenylalanine residue in MLLE (F507), the single most important event required for the binding to occur [Citation59]. Overall, the modeling analysis strongly supports that the individual residues found in the EIF4E4 box B perform all the expected interactions with the conserved residues found within the PABP1 MLLE domain, compatible with a role as a PAM2 motif, and that the motif on its own would be sufficient to mediate the EIF4E4 interaction with PABP1.
Discussion
The data presented in this work are consistent with phosphorylation of PABP1 being part of a novel pathway in Leishmania species controlling the protein's function and possibly translation. PABP1 is phosphorylated simultaneously with its partner EIF4E4 (this work and [Citation52]) and the similarities in the targeted residues are compatible with both proteins being targeted by the same kinase(s). Indeed, both T. brucei orthologues of PABP1 and EIF4E4, as well as a few unrelated proteins, were found to be targeted by the cell cycle dependent kinase CRK1, in complex with the cyclin CYC2 [Citation60]. CRK1 is conserved in Leishmania and is involved in controlling the G1/S transition [Citation61], so phosphorylation of both PABP1 and EIF4E4 might be required for the cells to proceed through the cell cycle. Nevertheless, the phosphorylated pattern seen by both proteins soon after passaging of stationary Leishmania promastigotes to new media also indicates a phosphorylation mediated by MAP-kinase like enzymes, which yet need to be identified. Phosphorylation of PABP1 is not needed for its association to polysomes and is not required for the protein to function and maintain cell viability. In vitro the strong association between PABP1 and EIF4E4 does not require phosphorylation and the efficient phosphorylation of the TGM/AAA mutant implies that binding of the two proteins might not be required for phosphorylation to occur. Likewise, the efficient phosphorylation of the YGF/AAA mutant, impaired in its association to polysomes and presumably to mRNAs, indicate that PABP1 does not necessarily need to be tightly bound to mRNAs to be phosphorylated, although the lack of phosphorylation by the double mutant (LMW/AAA + YGF/AAA) does indicate that the protein needs to retain some functionality in order to be properly phosphorylated. The fact that both binding partners, EIF4E4 and PABP1, are simultaneously phosphorylated is a strong indication of an impact on translation that needs to be further resolved.
In different organisms, PABP phosphorylation has been seen to affect many of its interactions and functions. In plants, for example, PABP hypophosphorylation lowers its binding activity to the poly(A) and eIF4B while its interaction with eIF4G is enhanced [Citation62]. In Xenopus oocytes, hyperphosphorylation of an embryo homologue of PABP, ePABP, also targets the protein's linker region but the phosphorylated serine or threonine residues do not require a neighboring proline, as seen for the Leishmania PABP1. The ePABP phosphorylation is dispensable for translational activation, but it is required for the cytoplasmic polyadenylation of maternal mRNAs [Citation63]. In HeLa cells, PABP phosphorylation has also been confirmed [Citation64], although subsequently PABP1 was shown to be targeted by multiple post-translational modifications events, which include aspartate and glutamate methylation, lysine and arginine methylation and lysine acetylation in a report where no PABP phosphorylation was seen [Citation65]. It is likely then that different organisms evolved alternative mechanisms of post-translationally modifying PABP homologues in order to regulate its function.
The solution structure of the C-terminal domain from the T. cruzi PABP2 confirms the conservation of the MLLE domain from trypanosomatid PABP homologues and its ability to bind PAM2-like motifs [Citation66]. PAM2 motifs have been shown to localize to disordered regions mapped near to protein phosphorylation sites [Citation67] and some PABP partners have been found to have two PAMs, one of which is responsible for specific PABP recognition, while the other might be required to stabilize the interaction [Citation68,Citation69]. All these features seem to be conserved within the motifs found in the Leishmania EIF4E4 N-terminus and support the in vitro binding assays and bioinformatic analysis, which have defined the PABP1/EIF4E4 interaction. The characterization of this interaction highlights even further the diversity observed in PABP interacting proteins which bind through the PAM2/MLLE interaction. This has been more recently discussed in plants, where the large number of PABP homologues is probably associated with multiple processes involved in different aspects of mRNA metabolism and which might have major impacts on regulation of gene expression and translation [Citation34,Citation70].
It is still not clear what role phosphorylation might have in regulating both PABP1 and EIF4E4 functions and presumably leading to enhanced translation of the bound mRNAs under conditions of logarithmic cell growth. Human proteins have been identified having PAM2 motifs similar to those observed for EIF4E4 and localized next to phosphorylation sites within disordered regions. Phosphorylation of these proteins inhibits the interaction with PABP [Citation16,Citation67], therefore it seems likely that simultaneous phosphorylation of both PABP1 and EIF4E4 would inhibit or remodel their interaction, something that needs to be confirmed. Evaluating this possibility, however, has so far been prevented by the strong susceptibility of both proteins to dephosphorylation, and degradation in the case of EIF4E4, during cytoplasmic extract preparation. The lack of a clear phenotype by either the PABP1 or EIF4E4 phosphorylation mutants in the complementation assays (this work and [Citation52]) also prevented a proper evaluation of the role of phosphorylation in translation. This might be due to the fact that when one protein is mutated the other is still phosphorylated and both might be required to be in a dephosphorylated state in order to induce differences in growth profile or protein synthesis, so both proteins would have to be targeted simultaneously. Further work then will be required in order to investigate these issues properly.
The identification of protein partners which specifically co-immunoprecipitated with PABP1 also helped clarify its functional role. PABP1 was found to interact with EIF4E4/EIF4G3, constituting the eIF4F complex most likely involved in translation initiation in Leishmania [ Citation44], and none of the other eIF4E or eIF4G homologues identified in this organism. Likewise, neither PABP2 or PABP3 seem to co-precipitate with the EIF4E4/EIF4G3 complex [Citation35], highlighting the strong association between PABP1 and EIF4E4/EIF4G3 with likely common roles in translation initiation. Both RBP23 and the uncharacterized LinJ.18.0300 polypeptide are likely RNA binding proteins, which may help recruiting PABP1 and the EIF4E4/EIF4G3 complex to selected mRNAs in order to promote their translation. The other two putative PABP1 partners, the CCCH zinc finger protein ZC3H41 and the uncharacterized LinJ.05.0450, might be involved in regulating PABP1 function either in translation or otherwise. In mammals PABP interacting proteins (Paip1 and Paip2) are well known translation regulators which function by directly binding to PABP, either enhancing or repressing translation [Citation71]. More recently, in T. brucei, another CCCH zinc finger protein, ZC3H11, has been shown to act as a post-transcriptional regulator required to stabilize stress response mRNAs following heat shock [Citation72]. It is possible that ZC3H41 also regulates translation acting upon selected messages or otherwise functions in other processes associated with the metabolism of mRNAs. So far however, and in view of the fact that PABP2, but not PABP1, can shuttle to the nucleus upon inhibition of mRNA synthesis or processing [Citation35,Citation36], it is more likely that PABP1 and binding partners would not be involved in processes such as mRNA processing and transport, which would require a nuclear localization.
Materials and methods
Parasite growth and transfections
Leishmania infantum MHOM/MA/67/ITMAP-263 promastigotes were cultured in SDM-79 medium supplemented with 10% heat-inactivated FCS (Multicell Wisent Inc.) and 5 µg/ml hemin and axenic amastigotes in MAA/20 medium supplemented with 20% FCS in 5% CO2 atmosphere as described previously [Citation52]. Growth curves were set up using late-stationary phase cells passaged to fresh medium at a pre-established cell density of 106 cells/ml. Samples were taken at selected time points for processing, SDS-PAGE and blotting. Transfection procedures used for both circular plasmids (episomal expression) and linear DNA fragments (for integration and gene deletion) were carried out by electroporation as described [Citation73]. Cells transfected with the PABP1 wild-type and mutant constructs were selected with neomycin (G418, 20 µg/ml, Sigma) and those transfected with the linear DNA cassettes for PABP1 gene deletion events with hygromycin B (40 µg/ml, Sigma) or puromycin (70 µg/ml, Sigma).
Antibodies and western-blotting
Western-blotting was performed using standard procedures. Native Leishmania PABP1 and PABP2 were detected using rabbit polyclonal sera raised against the two proteins as described previously [Citation35]. The anti-mouse alpha-tubulin antibody (1:10000 dilution; Sigma) was used for normalizing protein loading. For the detection of the different HA-tagged proteins, an anti-HA monoclonal antibody (100 ηg/ml) from Applied Biological Materials was used. For the Leishmania large ribosomal P0 subunit (TriTrypDb accession: LmjF.27.1380), antiserum was raised by immunizing adult New Zealand White rabbits with a His-tagged recombinant protein. Recombinant P0 was produced after amplification of the L. major gene flanked by restriction sites for BamHI and XhoI immediately before and after the AUG and stop codons, respectively (oligonucleotides listed in Table S1). The amplified fragment was cloned into the same sites of the pET21a vector (Novagen), followed by transformation into Escherichia coli BL21 cells, protein expression and affinity purification using Ni-NTA Agarose (Qiagen).
Sucrose gradient methods
Cytoplasmic cellular extracts were produced from late-logarithmically grown L. infantum promastigotes incubated for 10 minutes with 100 µg/ml cycloheximide (Sigma). For extract preparation, the cells were first harvested and washed once in PBS and once again in Hepes-lysis buffer (20 mM Hepes-KOH, pH7.4, 75 mM potassium acetate, 4 mM magnesium acetate, 2 mM DTT), both supplemented with 1x protease inhibitors (EDTA-free EASYpack Roche) and 100 µg/ml cycloheximide. The weight of the final pelleted cells was measured prior to their resuspension to a concentration of 2–4 × 109 cells/ml in Hepes-lysis buffer plus protease inhibitors and cycloheximide. Twice the weight of the pelleted cells in glass beads, 150–200 µm in diameter (from Sigma) was then added. Lysis was carried through vortexing for 15 minutes at 4°C, followed by passaging five times through a 30 gauge needle and centrifugation at 13,000g for 15 minutes to remove cellular debris. Roughly 15 to 20 OD260 nm units of the supernatants, the cytoplasmic extracts, were then layered on top of 15% to 45% linear sucrose gradients. For the EDTA-treated control gradients, 0.1 M EDTA was added to the cytoplasmic extracts prior to loading on the gradients. Sucrose gradient preparations and fractionation were carried out as described previously [Citation74,Citation75] and aliquots from each fraction were prepared for SDS-PAGE and western-blotting through 10% TCA precipitation followed by acetone washes and resuspension in SDS-PAGE sample buffer.
Sequence analysis and molecular modeling
Sequence analysis and alignment of the various PABP1 homologues were carried out as previously described [Citation52] using selected trypanosomatid sequences recovered from TriTrypDB and sequences from other organisms from GenBank. TriTrypDB accessions: Leishmania infantum (Li) PABP1 – LinJ.35.5360; LiPABP2 – LinJ.35.4200; LiPABP3 – LinJ.25.0080; L. braziliensis (Lb) PABP1 – LbrM.34.4980; LbPABP2 – LbrM.34.4130; LbPABP3 – LbrM.25.0080; Trypanosoma brucei (Tb) PABP1 – Tb927.9.9290; TbPABP2 – Tb927.9.10770. Further GeneBank accessions: Homo sapiens (Hs) PABP1 – NP_002559.2; Saccharomyces cerevisiae (Sc) PABP – NP_011092; Arabidopsis thaliana (At) PABP2 – NP_195137 (the most conserved of the various A. thaliana PABPs). The alignment of the various PAM2 motifs was carried out using similar procedures but using colored shadings when more than 60% of the sequences were identical or similar. All PAM2 sequences used in the alignment have been previously described [Citation33,Citation52].
Modeling of the MLLE domain from L. infantum PABP1 was performed using the Modeller program [Citation76], version 9.14, with the crystal structure from the human PABP MLLE domain bound to a PAM2 motif from the PABP partner PAIP2 [Citation57] used as template and downloaded from PDB (http://www.rcsb.org/pdb/home/home.do, PDB ID: 3KUS). The PyMOL software, version 1.7 (PyMOL Molecular Graphics System, Version 1.7 Schrödinger, LLC), was then applied to overlap the best model produced with the original crystallized structure of the human PABP MLLE bound to the PAIP2 PAM2 motif. The next step was the manual removal of the crystallized MLLE domain, resulting in a model consisting of the L. infantum PABP1 MLLE domain bound to the PAM2 motif from human PAIP2. To replace the original PAM2 motif with the one mapped to the N-terminus from L. infantum EIF4E4, tools from the Rosetta package [Citation77] were used. First, fixbb was used to replace residues non-conserved between the crystallized PAM2 motif and the selected motif from EIF4E4. This replacement does not allow movement for the backbone of the PAM2 motif, which may produce unstable final conformations. The flexpepdock tool, also from the Rosetta package, was thus applied with 300 solutions in order to make the PAM2 backbone flexible and better accommodate the EIF4E4 motif on the MLLE domain model from the L. infantum PABP1. The final models were assessed by PROCHECK [Citation78], version 3.5, which validated them to be used to infer the interaction between the PABP1 MLLE domain and the EIF4E4 PAM2 motif.
Plasmid constructs, DNA manipulations
L. infantum genomic DNA extraction and PCR amplification were carried out as described previously [Citation52]. The full length wild-type PABP1 gene was amplified together with flanked BamHI and HindIII restriction sites and a 27 nucleotide extension encoding a single copy of the HA epitope (YPYDVPDYA) added immediately prior to the PABP1 translation stop codon. Site-directed mutagenesis was also carried out as described [Citation52]. All PCR fragments were first cloned into the pGEM-T Easy vector (Promega) prior to sequencing and subcloning into the BamHI-HindIII sites of the Leishmania expression vector pSPBT1YNEOα [Citation79] with the 3′ HA tag. The PABP1 gene deletion and complementation strategies followed the same procedures used previously with the L. infantum EIF4E4 gene [Citation52]. All oligonucleotides used for the various amplification and mutagenesis reactions are listed in Tables S2 through S4. The genes encoding selected L. infantum PABP1 mutants were subsequently subcloned into the BamHI-HindIII sites of a modified pGEX4T3 vector (GE Healthcare) for the expression of GST-tagged recombinant proteins. The two truncated versions of the L. major GST-tagged PABP1 (only five amino acid differences between the L. infantum and L. major orthologues) were generated from the wild-type PABP1 cloned into the pGEX4T3 vector [Citation35]. For the N-terminus, the plasmid was digested with BamHI/NotI and the 897 pb fragment encoding for the first three PABP1 RRMs (residues 1 to 313 from the wild type protein) was gel-purified and subcloned into the same sites of pGEX4T3. For the C-terminus, a gene fragment encoding residues 302 to 560 was amplified flanked by sites for NcoI/HindIII, purified and cloned likewise into the same sites of a modified pGEX4T3 vector. Southern blots were carried out using standard procedures after total DNA digestion with ClaI or PvuII. The probe used was derived from the 500 bp PCR fragment corresponding to the 5′ intergenic region immediately before the PABP1 gene start codon and generated using the primers listed in Tables S3.
Extract preparation for immunoprecipitation
Whole cytoplasmic extracts from wild-type L. infantum or recombinant strains expressing selected HA-tagged PABP1 variants were generated after lysing the cells through cavitation, aiming to reduce undesired debris and rupture of the nuclei and/or organelles. First, late-logarithmically grown Leishmania infantum promastigotes were harvested and washed once in ice cold PBS, followed by resuspension in HEPES-lysis buffer (20 mM HEPES-KOH, pH 7.4, 75 mM potassium acetate, 4 mM magnesium acetate, 2 mM DTT, supplemented with and 1x EDTA-free protease inhibitors from Roche) to a concentration of 1–2 × 109 cells/ml. Lysis was carried out using nitrogen cavitation as described [Citation80], with modifications. Briefly, the resuspended cells were transferred into the cavitation chamber of the cell disruption vessel (Parr Instruments) and incubated at 4ºC under 70 bar pressure for 40 min, followed by rapid decompression and lysis. The lysates were submitted to centrifugation at 17,000g for 10 min to remove cellular debris and the supernatants, the cytoplasmic extracts, aliquoted and stored.
For the immunoprecipitation assays (IPs), the cytoplasmic extracts were mixed with Pierce™ Anti-HA Magnetic Beads as per manufacturer's protocol. Briefly, 0.2 mg of these anti-HA magnetic beads were washed three times with PBS followed by the incubation with the cytoplasmic extract for 1 h at 4ºC. After, the depleted supernatant was removed and the beads were washed three times with PBS, the resulting, specifically bound, immunoprecipitated antigen-antibody complexes were eluted in SDS-PAGE sample buffer. They were then assayed by SDS-PAGE and Western-blotting using antibodies against PABP1 and the HA-tag to confirm the efficiency of the precipitation reaction.
Mass spectrometry analysis
For mass spectrometry (MS), the sets of eluted proteins were loaded onto 15% SDS-PAGE gels and allowed to migrate into the resolving gel. Gel slices containing the whole IP products were then excised and submitted to an in-gel tryptic digestion and mass spectrometry analysis as previously described [Citation81]. Protein identification was based on the L. infantum protein sequence database downloaded from UniProt. For validation of the identified proteins a minimum of six amino acids for peptide length and two peptides per protein were required. In addition, a false discovery rate (FDR) threshold of 0.01 (using the decoy database approach) was applied at both peptide and protein levels. To confirm the specificity of the IP assays for each polypeptide, the ratio between the intensity generated from the IPs using the extracts expressing the HA-tagged PABP1 and the intensity from the control IP using an extract from non-transfected cells was first determined. The base 2 logarithms of the values produced were then calculated for two independent experiments carried out with different cytoplasmic extracts and only the values >3.0 were considered. Identified polypeptides were then ranked and listed in with the highest ranking values obtained for the first experiment listed on top.
In vitro pull-down assays
Co-precipitation/pull-down assays were essentially performed as described previously [Citation42,Citation52] using Glutathione-Sepharose 4B beads (GE Healthcare) and affinity purified GST-tagged recombinant proteins. GST alone and the different GST-tagged PABP1 variants (wild-type, truncations and mutants) were expressed in Escherichia coli, immobilized on the beads and assayed for their ability to bind to 35S-labeled EIF4E4. The labeled protein was obtained through the linearization of the corresponding pET21a derived plasmids with NotI, followed by transcription with T7 RNA polymerase in the presence of the cap analogue and translation in rabbit reticulocyte lysates (Promega or Ambion) supplemented with 35S-methionine (Perkin Elmer).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
suppl_mat_Osvaldo_P._de_Melo_Neto.docx
Download MS Word (90.7 KB)Acknowledgments
We thank Carole Dumas for technical assistance and other members of Dr. Papadopoulou's and Dr. de Melo Neto's laboratories for helpful discussions. We are also thankful to the Proteomics platform of the CHU de Quebec Research Center for preliminary mass-spectrometry analysis of PABP1 bound complexes. The work in Dr. Papadopoulou's lab was supported by the Canadian Institutes of Health Research (CIHR) operating grant MOP-12182, the Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant GC100741 (418444), and the Ministère du Développement Économique de l'Innovation et de l'Exportation (MDEIE) PSR-SIIRI-439 grant awarded to BP. The Leishmania work in Dr. de Melo Neto's lab was more recently funded with grants provided by the Brazilian funding agencies FACEPE (APQ-0239-2.02/12 and APQ-1662-2.02/15), CNPq (480899/2013-4, 313934/2013-4 and 401282/2014-7) and CAPES (23038.007656/2011-92). Dr. de Melo Neto was the recipient of a Visiting Scholar grant from CAPES for a medium term stay at the University of Laval (BEX 3743/10-1). To cover their stays at the same institution, TPR, TDCL and LMN received funding from CNPq, the Agence Universitaire de la Francophonie and CAPES, respectively. Studentships for the graduate students in Brazil (TPR, KCM, POR, LAA, LMN and CCX) were provided by CNPq, FACEPE or CAPES. The authors thank the Nucleo de Plataformas Tecnológicas (NPT) at the Instituto Aggeu Magalhães for the use of its automatic sequencing facility, the Instituto Carlos Chagas for the use of its proteomics facility and FIOCRUZ for the funding support.
Additional information
Funding
Related Research Data
References
- Ouellette M, Papadopoulou B. Coordinated gene expression by post-transcriptional regulons in African trypanosomes. J Biol. 2009;8:100. doi:10.1186/jbiol203
- Fernandez-Moya SM, Estevez AM. Posttranscriptional control and the role of RNA-binding proteins in gene regulation in trypanosomatid protozoan parasites. Wiley Interdiscip Rev RNA. 2010;1:34–46. doi:10.1002/wrna.6
- De Gaudenzi JG, Noe G, Campo VA, Frasch AC, et al. Gene expression regulation in trypanosomatids. Essays Biochem. 2011;51:31–46. doi:10.1042/bse0510031
- Schwede A, Kramer S, Carrington M. How do trypanosomes change gene expression in response to the environment? Protoplasma. 2012;249:223–38. doi:10.1007/s00709-011-0282-5
- Kramer S. Developmental regulation of gene expression in the absence of transcriptional control: the case of kinetoplastids. Mol Biochem Parasitol. 2012;181:61–72. doi:10.1016/j.molbiopara.2011.10.002
- Clayton CE. Networks of gene expression regulation in Trypanosoma brucei. Mol Biochem Parasitol. 2014;195:96–106. doi:10.1016/j.molbiopara.2014.06.005
- Clayton CE. Gene expression in Kinetoplastids. Curr Opin Microbiol. 2016;32:46–51. doi:10.1016/j.mib.2016.04.018
- van der Kelen K, Beyaert R, Inzé D, et al. Translational control of eukaryotic gene expression Crit Rev Biochem Mol Biol. 2009;44:143–68. doi:10.1080/10409230902882090
- Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem. 1999;68:913–63. doi:10.1146/annurev.biochem.68.1.913
- Gallie DR. The role of the poly (A) binding protein in the assembly of the cap-binding complex during translation initiation in plants. Translation. 2014;731:1–14
- Merrick WC. eIF4F: A Retrospective. J Biol Chem. 2015;290:24091–9. doi:10.1074/jbc.R115.675280
- Aylett CHS, Ban N. Eukaryotic aspects of translation initiation brought into focus. Philos Trans R Soc London B Biol Sci. 2017;372: doi:10.1098/rstb.2016.0186
- Kühn U, Wahle E. Structure and function of poly(A) binding proteins. Biochim Biophys Acta. 2004;1678:67–84. doi:10.1016/j.bbaexp.2004.03.008
- Gorgoni B, Gray NK. The roles of cytoplasmic poly(A)-binding proteins in regulating gene expression: a developmental perspective. Brief Funct Genomic Proteomic. 2004;3:125–41. doi:10.1093/bfgp/3.2.125
- Lemay J-F, Lemieux C, St-André O, et al. Crossing the borders: Poly(A)-binding proteins working on both sides of the fence. RNA Biol. 2010;7:291–5. doi:10.4161/rna.7.3.11649
- Eliseeva IA, Lyabin DN, Ovchinnikov LP. Poly(A)-binding proteins: structure, domain organization, and activity regulation. Biochem. 2013;78:1377–91
- Smith RWP, Blee TKP, Gray NK. Poly(A)-binding proteins are required for diverse biological processes in metazoans. Biochem Soc Trans. 2014;42:1229–37. doi:10.1042/BST20140111
- Wigington CP, Williams KR, Meers MP, et al. Poly(A) RNA-binding proteins and polyadenosine RNA: new members and novel functions. Wiley Interdiscip Rev RNA. 2014;13:601–22. doi:10.1002/wrna.1233
- Gray NK, Hrabálková L, Scanlon JP, et al. Poly(A)-binding proteins and mRNA localization: who rules the roost? Biochem Soc Trans. 2015;43:1277–84. doi:10.1042/BST20150171
- Goss DJ, Kleiman FE. Poly(A) binding proteins: are they all created equal? Wiley Interdiscip Rev RNA. 2013;4:167–79. doi:10.1002/wrna.1151
- Burd CG, Dreyfuss G. Conserved structures and diversity of functions of RNA-binding proteins. Science. 1994;265:615–21. doi:10.1126/science.8036511
- Deo RC, Bonanno JB, Sonenberg N, et al. Recognition of polyadenylate RNA by the poly(A)-binding protein. Cell. 1999;98:835–45. doi:10.1016/S0092-8674(00)81517-2
- Kuhn U, Pieler T, Kühn U, et al. Xenopus poly(A) binding protein: functional domains in RNA binding and protein-protein interaction. J Mol Biol. 1996;256:20–30. doi:10.1006/jmbi.1996.0065
- Kessler SH, Sachs AB. RNA recognition motif 2 of yeast Pab1p is required for its functional interaction with eukaryotic translation initiation factor 4G. Mol Cell Biol. 1998;18:51–7. doi:10.1128/MCB.18.1.51
- Imataka H, Gradi A, Sonenberg N. A newly identified N-terminal amino acid sequence of human eIF4G binds poly(A)-binding protein and functions in poly(A)-dependent translation. EMBO J. 1998;17:7480–9. doi:10.1093/emboj/17.24.7480
- Cheng S, Gallie DR. eIF4G, eIFiso4G, and eIF4B bind the poly(A)-binding protein through overlapping sites within the RNA recognition motif domains. J Biol Chem. 2007;282:25247–58. doi:10.1074/jbc.M702193200
- Khanam T, Muddashetty RS, Kahvejian A, et al. Poly(A)-binding protein binds to A-rich sequences via RNA-binding domains 1+2 and 3+4. RNA Biol. 2006;3:170–7. doi:10.4161/rna.3.4.4075
- Sladic RT, Lagnado CA, Bagley CJ, et al. Human PABP binds AU-rich RNA via RNA-binding domains 3 and 4. Eur J Biochem. 2004;271:450–7. doi:10.1046/j.1432-1033.2003.03945.x
- Khacho M, Mekhail K, Pilon-Larose K, et al. eEF1A Is a Novel Component of the Mammalian Nuclear Protein Export Machinery. Mol Biol Cell. 2008;19:5296–308. doi:10.1091/mbc.E08-06-0562
- Melo EO, Dhalia R, Martins de SC, et al. Identification of a C-terminal poly(A)-binding protein (PABP)-PABP interaction domain: role in cooperative binding to poly (A) and efficient cap distal translational repression. J Biol Chem. 2003;278:46357–68. doi:10.1074/jbc.M307624200
- Lin J, Fabian M, Sonenberg N, et al. Nanopore detachment kinetics of poly(A) binding proteins from RNA molecules reveals the critical role of C-terminus interactions. Biophys J. 2012;102:1427–34. doi:10.1016/j.bpj.2012.02.025
- Khaleghpour K, Kahvejian A, De CG, Roy G, et al. Dual interactions of the translational repressor Paip2 with poly(A) binding protein. Mol Cell Biol. 2001;21:5200–13. doi:10.1128/MCB.21.15.5200-5213.2001
- Albrecht M, Lengauer T. Survey on the PABC recognition motif PAM2. Biochem Biophys Res Commun. 2004;316:129–38. doi:10.1016/j.bbrc.2004.02.024
- Jiménez-López D, Bravo J, Guzmán P. Evolutionary history exposes radical diversification among classes of interaction partners of the MLLE domain of plant poly(A)-binding proteins. BMC Evol Biol. 2015;15:195. doi:10.1186/s12862-015-0475-1
- da Costa Lima TD, Moura DMN, Reis CRS, et al. Functional characterization of three leishmania poly(a) binding protein homologues with distinct binding properties to RNA and protein partners. Eukaryot Cell. 2010;9:1484–94. doi:10.1128/EC.00148-10
- Kramer S, Bannerman-Chukualim B, Ellis L, et al. Differential localization of the two T. brucei poly(A) binding proteins to the nucleus and RNP granules suggests binding to distinct mRNA pools. PLoS One. 2013;8:e54004. doi:10.1371/journal.pone.0054004
- Pitula J, Ruyechan WT, Williams N. Trypanosoma brucei: identification and purification of a poly(A)-binding protein. Exp Parasitol. 1998;88:157–60. doi:10.1006/expr.1998.4211
- Guerra N, Vega-Sendino M, Pérez-Morgado MI, et al. Identification and functional characterization of a poly(A)-binding protein from Leishmania infantum (LiPABP). FEBS Lett. 2011;585:193–8. doi:10.1016/j.febslet.2010.11.042
- Erben ED, Fadda A, Lueong S, et al. A genome-wide tethering screen reveals novel potential post-transcriptional regulators in Trypanosoma brucei. PLoS Pathog. 2014;10:e1004178. doi:10.1371/journal.ppat.1004178
- Lueong S, Merce C, Fischer B, et al. Gene expression regulatory networks in Trypanosoma brucei: Insights into the role of the mRNA-binding proteome. Mol Microbiol. 2016;100:457–71. doi:10.1111/mmi.13328
- Klein C, Terrao M, Inchaustegui GD, et al. Polysomes of Trypanosoma brucei: Association with Initiation Factors and RNA-Binding Proteins. PLoS One. 2015;10:e0135973. doi:10.1371/journal.pone.0135973
- Dhalia R, Reis CR, Freire ER, et al. Translation initiation in Leishmania major: characterisation of multiple eIF4F subunit homologues. Mol Biochem Parasitol. 2005;140:23–41. doi:10.1016/j.molbiopara.2004.12.001
- Jagus R, Bachvaroff TR, Joshi B, et al. Diversity of eukaryotic translational initiation factor eIF4E in protists. Comp Funct Genomics. 2012;2012:134839. doi:10.1155/2012/134839
- de Melo Neto OP, Reis CR, Moura DM, Freire ER, et al. Unique and conserved features of the protein synthesis apparatus in trypanosomatid (Trypanosoma and Leishmania) species. In: Hernandez G, Jagus R, editors. Evolution of the protein synthesis machinery and its regulation. Springer International Publishing; 2016. p. page 435–75
- Zinoviev A, Shapira M. Evolutionary conservation and diversification of the translation initiation apparatus in trypanosomatids. Comp Funct Genomics. 2012;2012:813718. doi:10.1155/2012/813718
- Freire E, Sturm N, Campbell D, et al. The role of cytoplasmic mRNA cap-binding protein complexes in Trypanosoma brucei and other trypanosomatids. Pathogens. 2017;6:55. doi:10.3390/pathogens6040055
- Yoffe Y, Leger M, Zinoviev A, et al. Evolutionary changes in the Leishmania eIF4F complex involve variations in the eIF4E-eIF4G interactions. Nucleic Acids Res. 2009;37:3243–53. doi:10.1093/nar/gkp190
- Freire ER, Dhalia R, Moura DMN, et al. The four trypanosomatid eIF4E homologues fall into two separate groups, with distinct features in primary sequence and biological properties. Mol Biochem Parasitol. 2011;176:25–36. doi:10.1016/j.molbiopara.2010.11.011
- Zinoviev A, Leger M, Wagner G, et al. A novel 4E-interacting protein in Leishmania is involved in stage-specific translation pathways. Nucleic Acids Res. 2011;39:8404–15. doi:10.1093/nar/gkr555
- Zinoviev A, Manor S, Shapira M. Nutritional stress affects an atypical cap-binding protein in Leishmania. RNA Biol. 2012;9:1450–60. doi:10.4161/rna.22709
- Moura DMN, Reis CRS, Xavier CC, et al. Two related trypanosomatid eIF4G homologues have functional differences compatible with distinct roles during translation initiation. RNA Biol. 2015;12:305–19. doi:10.1080/15476286.2015.1017233
- de Melo Neto OP, da Costa Lima TDC, Xavier CC, et al. The unique Leishmania EIF4E4 N-terminus is a target for multiple phosphorylation events and participates in critical interactions required for translation initiation. RNA Biol. 2015;12:1209–21. doi:10.1080/15476286.2015.1086865
- Bates EJ, Knuepfer E, Smith DF. Poly(A)-binding protein I of Leishmania: functional analysis and localisation in trypanosomatid parasites. Nucleic Acids Res. 2000;28:1211–20. doi:10.1093/nar/28.5.1211
- Urbaniak MD, Martin DM, Ferguson MA. Global quantitative SILAC phosphoproteomics reveals differential phosphorylation is widespread between the procyclic and bloodstream form lifecycle stages of Trypanosoma brucei. J Proteome Res. 2013;12:2233–44. doi:10.1021/pr400086y
- Pereira MMC, Malvezzi AM, Nascimento LM, et al. The eIF4E subunits of two distinct trypanosomatid eIF4F complexes are subjected to differential post-translational modifications associated to distinct growth phases in culture. Mol Biochem Parasitol. 2013;190:82–6. doi:10.1016/j.molbiopara.2013.06.008
- Pyronnet S, Imataka H, Gingras AC, et al. Human eukaryotic translation initiation factor 4G (eIF4G) recruits mnk1 to phosphorylate eIF4E. EMBO J. 1999;18:270–9. doi:10.1093/emboj/18.1.270
- Kozlov G, Ménade M, Rosenauer A, et al. Molecular determinants of PAM2 recognition by the MLLE domain of poly(A)-binding protein. J Mol Biol. 2010;397:397–407. doi:10.1016/j.jmb.2010.01.032
- Kozlov G, De CG, Lim NS, Siddiqui N, et al. Structural basis of ligand recognition by PABC, a highly specific peptide-binding domain found in poly(A)-binding protein and a HECT ubiquitin ligase. EMBO J. 2004;23:272–81. doi:10.1038/sj.emboj.7600048
- Kozlov G, Gehring K. Molecular basis of eRF3 recognition by the MLLE domain of poly(A)-binding protein. PLoS One. 2010;5:e10169. doi:10.1371/journal.pone.0010169
- Hu H, Gourguechon S, Wang CC, et al. The G1 cyclin-dependent kinase CRK1 in Trypanosoma brucei regulates anterograde protein transport by phosphorylating the COPII subunit Sec31. J Biol Chem. 2016;291:15527–39. doi:10.1074/jbc.M116.715185
- Naula C, Parsons M, Mottram JC. Protein kinases as drug targets in trypanosomes and Leishmania. Biochim Biophys Acta. 2005;1754:151–9. doi:10.1016/j.bbapap.2005.08.018
- Le H, Browning KS, Gallie DR. The phosphorylation state of poly(A)-binding protein specifies its binding to poly(A) RNA and its interaction with eukaryotic initiation factor (eIF) 4F, eIFiso4F, and eIF4B. J Biol Chem. 2000;275:17452–62. doi:10.1074/jbc.M001186200
- Friend K, Brook M, Bezirci FB, et al. Embryonic poly(A)-binding protein (ePAB) phosphorylation is required for Xenopus oocyte maturation. Biochem J. 2012;445:93–100. doi:10.1042/BJ20120304
- Ma S, Musa T, Bag J. Reduced stability of mitogen-activated protein kinase kinase-2 mRNA and phosphorylation of Poly(A)-binding Protein (PABP) in cells overexpressing PABP. J Biol Chem. 2006;281:3145–56. doi:10.1074/jbc.M508937200
- Brook M, McCracken L, Reddington JP, et al. The multifunctional poly(A)-binding protein (PABP) 1 is subject to extensive dynamic post-translational modification, which molecular modelling suggests plays an important role in co-ordinating its activities. Biochem J. 2012;441:803–12. doi:10.1042/BJ20111474
- Siddiqui N, Kozlov G, D'Orso I, et al. Solution structure of the C-terminal domain from poly(A)-binding protein in Trypanosoma cruzi: a vegetal PABC domain. Protein Sci. 2003;12:1925–33. doi:10.1110/ps.0390103
- Huang K-L, Chadee AB, Chen C-Y a, et al. Phosphorylation at intrinsically disordered regions of PAM2 motif-containing proteins modulates their interactions with PABPC1 and influences mRNA fate. RNA. 2013;19:295–305. doi:10.1261/rna.037317.112
- Huntzinger E, Braun JE, Heimstädt S, et al. Two PABPC1-binding sites in GW182 proteins promote miRNA-mediated gene silencing. EMBO J. 2010;29:4146–60. doi:10.1038/emboj.2010.274
- Kononenko A V., Mitkevich VA, Atkinson GC, et al. GTP-dependent structural rearrangement of the eRF1:eRF3 complex and eRF3 sequence motifs essential for PABP binding. Nucleic Acids Res. 2010;38:548–58. doi:10.1093/nar/gkp908
- Gallie DR, Liu R. Phylogenetic analysis reveals dynamic evolution of the poly(A)-binding protein gene family in plants. BMC Evol Biol. 2014;14:238. doi:10.1186/s12862-014-0238-4
- Derry MC, Yanagiya A, Martineau Y, et al. Regulation of poly(A)-binding protein through PABP-interacting proteins. Cold Spring Harb Symp Quant Biol. 2006;71:537–43. doi:10.1101/sqb.2006.71.061
- Minia I, Clayton C. Regulating a post-transcriptional regulator: protein phosphorylation, degradation and translational blockage in control of the trypanosome stress-response RNA-Binding Protein ZC3H11. PLoS Pathog. 2016;12:1–31. doi:10.1371/journal.ppat.1005514
- Papadopoulou B, Roy G, Ouellette M. A novel antifolate resistance gene on the amplified H circle of Leishmania. EMBO J. 1992;11:3601–8
- McNicoll F, Muller M, Cloutier S, et al. Distinct 3′-untranslated region elements regulate stage-specific mRNA accumulation and translation in Leishmania. J Biol Chem. 2005;280:35238–46. doi:10.1074/jbc.M507511200
- Cloutier S, Laverdière M, Chou MN, et al. Translational control through eIF2alpha phosphorylation during the Leishmania differentiation process. PLoS One. 2012;7:e35085. doi:10.1371/journal.pone.0035085
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi:10.1006/jmbi.1993.1626
- Leaver-Fay A, Tyka M, Lewis SM, et al. ROSETTA3: An object-oriented software suite for the simulation and design of macromolecules. Methods Enzimol. 2011;487:545–74. doi:10.1016/B978-0-12-381270-4.00019-6
- Laskowski RA, MacArthur MW, Moss DS, et al. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–91. doi:10.1107/S0021889892009944
- Padmanabhan PK, Samant M, Cloutier S, et al. Apoptosis-like programmed cell death induces antisense ribosomal RNA (rRNA) fragmentation and rRNA degradation in Leishmania. Cell Death Differ. 2012;19:1972–82. doi:10.1038/cdd.2012.85
- Mureev S, Kovtun O, Nguyen UTT, et al. Species-independent translational leaders facilitate cell-free expression. Nat Biotechnol. 2009;27:747–52. doi:10.1038/nbt.1556
- Rezende AM, Assis LA, Nunes EC, et al. The translation initiation complex eIF3 in trypanosomatids and other pathogenic excavates–identification of conserved and divergent features based on orthologue analysis. BMC Genomics. 2014;15:1175. doi:10.1186/1471-2164-15-1175