
ABSTRACT
Techniques to isolate the small RNA fraction (<200nt) by column-based methods are commercially available. However, their use is limited because of the relatively high cost. We found that large RNA molecules, including mRNAs and rRNAs, are aggregated together in the presence of salts when RNA pellets are over-dried. Moreover, once RNA pellets are over-dried, large RNA molecules are barely soluble again during the elution process, whereas small RNA molecules (<100nt) can be eluted. We therefore modified the acid guanidinium thiocyanate-phenol-chloroform (AGPC)-based RNA extraction protocol by skipping the 70% ethanol washing step and over-drying the RNA pellet for 1 hour at room temperature. We named this novel small RNA isolation method “mirRICH.” The quality of the small RNA sequences was validated by electrophoresis, next-generation sequencing, and quantitative PCR, and the findings support that our newly developed column-free method can successfully and efficiently isolate small RNAs from over-dried RNA pellets.
Introduction
One of the method to extract total RNAs was originally developed by Piotr Chomczynski and Nicoletta Sacchi, which is based on acid guanidinium thiocyanate-phenol-chloroform (AGPC) extraction [Citation1]. The basic concept of this method is phase separation by centrifugation with phenol and chloroform. Guanidinium thiocyanate facilitates the denaturation of proteins and solubilizes them into the organic phase [Citation1]. The stability of DNA and RNA molecules is determined by the pH condition. Therefore, low pH conditions allow AGPC to selectively enrich RNA molecules in the aqueous phase by centrifugation [Citation2]. However, AGPC based method has several limitation to extract RNA. It is additionally required to remove the remaining salts in the sample by washing with 70% ethanol because RNA pellet precipitated by isopropanol contains high level of salt. Therefore, chaotropic reagents like ethanol are required to disrupt salt-nucleic acid complex thereby salts are selectively solubilized into water [Citation3]. In addition, it is difficult to isolate pure RNA, DNA and protein due to interphase contamination [Citation4].
Currently, a solid-phase based RNA extraction (SPE) method was developed to purify total RNA from cell lysates [Citation5,Citation6]. Solid-phase-based RNA extraction method has many advantages to purify by adsorbing onto glass or silica materials. This method provide quick and easy procedure to compare AGPC method under centrifugation [Citation7,Citation8]. Typical SPE methods comprise 4 distinct procedures: cell lysis, binding to the silica phase, washing with buffer, and elution with TE buffer or nuclease free water. Especially, adsorption processes make SPE method different from AGPC methods. Nucleic acid (RNA or DNA) can bind to the spin column under high pH and salt condition. The principle of hydrophilic and hydrophobic interaction is utilized in this selective elution of nucleic acid with chaotropic agents such as ethanol. Contents of solid phase is usually filled with glass particle, silica, diatomaceous and anion-exchange dependent of specific usage [Citation4]. Possibly, other components of cells such as protein also can bind to column. Thus, it is required to perform washing procedure to remove these contaminants by adding their competitive molecules [Citation4].
Recently, silica or cellulose coated magnetic beads are applied to capturing either DNA or RNA in acidic 4M guanidinium thiocyanate [Citation4,Citation9,Citation10]. This method is used for quick isolation of nucleic acid within 30min in the purpose of detection of viral infection [Citation9]. In addition, magnetic oligo (dT) bead is applied to purify RNA species having poly(A) tail of mRNAs. This method selectively purifies only mRNAs among total contents of RNAs because magnetic beads conjugated with poly(dT) oligo-nucleotide can interact with poly(A) tails of mRNAs [Citation4,Citation11]. However, cost of magnetic beads are relatively expensive than traditional RNA extraction technology.
Small RNAs (less than 200nt) have recently been highlighted to be crucial factors in regulating gene expression [Citation12,Citation13]. Numerous studies have shown that small non-coding RNAs are useful as biomarkers of human diseases such as cancer [Citation14–Citation16]. In order to study small RNAs, additional purification step are required because proportion of small RNAs are extremely low among total contents of RNAs extracted by conventional method either AGPC or SPE. Thus, after the isolation of total RNAs, the following approaches can be used: (1) separation of total RNAs by gel electrophoresis and elution of the miRNA fraction from the gel, (2) removal of large RNA molecules by polyethylene glycol to leave behind miRNA, (3) filtration of RNA molecules based on molecular weight, or (4) the use of a commercial kit (e.g., the mirVana miRNA isolation kit, Invitrogen, MA, USA). Most commercial kits use SPE, which binds to small RNAs, and this method is considered the most suitable for isolating small RNAs [Citation17]. However, column-based commercial miRNA extraction kits offer the advantage yielding high quantities of small RNAs, but the cost may be a burden [Citation18]. Therefore, it is necessary to develop a superior yet low-cost method to enrich small RNAs.
The AGPC method, meanwhile, allows the recovery of all types of RNA molecules, including small RNAs in relatively cheap price than column-based commercial small RNA extraction kits [Citation19,Citation20]. Nonetheless, in order to purify small RNA based on AGPC method, extra purification step is still necessary to separate only small RNA portions from the total RNAs and it require large amounts of total RNAs in the initial stage of the sample preparation. In this study, we developed a simple and economical method to isolate small RNA molecules using a modified AGPC method.
Over-dried RNA pellets are barely soluble in water so that and all known RNA isolation protocols highly recommend avoiding the over-drying of RNA pellets throughout the RNA preparation process. We accidently found that only small RNA fractions can still be released from over-dried RNA pellets. Here, we take advantage of our new finding of over-drying pellets and introduce an efficient procedure to enrich small RNA fractions without further extra small RNAs purification step in a AGPC method. We also validate that the quality of small RNAs from the over-dried pellets are comparable to a column-based commercial small RNA purification kit in many quality and quantity sensitive experiments including qPCR and next generation sequencing (NGS).
Results
Preparation of small RNAs by the mirRICH method
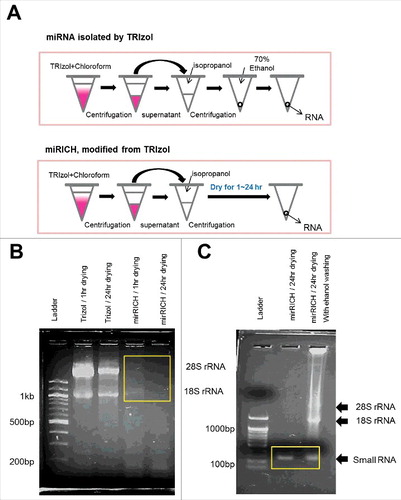
The typical AGPC-based RNA extraction method highly recommends a 70% ethanol washing step after isopropanol precipitation to remove extra salt molecules and to avoid the over-drying of RNA pellets. Once RNA pellets are over-dried, they can barely dissolve in distilled water during the elution step. Accidently, we observed that over-dried RNA pellets are still useful because they release at least small RNA molecules during the elution process. On the basis of this unexpected result, we hypothesize that over-drying RNA pellets might be advantageous in isolating small RNA molecules from total RNAs. Therefore, we modified the AGPC-based RNA extraction protocol by skipping the 70% ethanol washing step and adding a step of over-drying the RNA pellet for 1h∼24h at room temperature (). We named this newly developed small RNA isolation method “mirRICH.”
Figure 1. Over-drying based small RNA extraction method, mirRICH depletes large size of RNA molecules. (A) A schematic diagram describing differences in the isolation of RNAs between TRIzol and mirRICH methods (B) 1% agarose gel electrophoresis of RNA samples isolated from the breast cancer cell lines MDA-MB-231 by either TRIzol or mirRICH. Depleted 18S and 28S rRNA are boxed by yellow rectangle. M indicates DNA ladder (C) 2% agarose gel electrophoresis of RNA samples isolated from the breast cancer cell lines MDA-MB-231 by mirRICH with or without 70% ethanol washing. Small RNA fractions are boxed by yellow rectangle. Ladder indicates DNA ladder.
It is important to test whether the quantity and quality of small RNAs from mirRICH is good enough to use further sophisticated experiments such as qPCR or next generation sequencing (NGS). Since the column-free AGPC method using TRIzol and a column-based commercial method (mirVana kit) are currently and commonly used to isolate total RNAs, we tested the quantity and quality of small RNAs from mirRICH method with small RNAs from those two methods. In order to test the quality and quantity of small RNAs from mirRICH method, we used various experiments including Gel electrophoresis, PAGE, Denatured gel, bio-analyzer, next generation sequencing (NGS) and qPCR in this study.
To visualize the small RNA fractions, we isolated small RNAs from MDA-MB-231 and MCF7 breast cancer cells using the mirRICH method and performed agarose gel electrophoresis. As we expected, typical large RNA molecules such as 18S and 28S ribosomal RNAs were depleted the mirRICH method ( & S1A). It is important to find which factor causes the precipitation of RNA pellets when they are over-dried. Interestingly, if the RNA pellets were washed with 70% ethanol after TRIzol purification, significant amounts of rRNAs were still detected in both MDA-MB-231 and MCF7 cells even though the pellets were over-dried for 24hr (). These data indicates that skipping 70% ethanol washing step is critical to aggregate large RNA molecules with salts and isolate only small sizes of RNA fraction.
Since agarose gel does not sufficiently distinguish small nucleic acid molecules, we used 15% PAGE (Polyacrylamide gel electrophoresis) to separate small-sized RNAs obtained by TRIzol and mirRICH. Similar result was also shown in the PAGE gel that small RNAs from mirRICH was enriched better than that by the conventional TRIzol method ( & S1B). It is well known that RNA molecules are single-stranded nucleic acid and tend to form internal secondary or tertiary structure such as hair-pin. To avoid these internal secondary or tertiary structures, we performed 15% UREA denaturing gel electrophoresis. The result shows that mirRICH method is able to selectively enriched small RNA proportion compared to conventional TRIzol method (). These data together suggest that mirRICH method can isolate small sizes of RNA molecules comparable to the traditional RNA extraction methods.
Figure 2. Over-drying time does not affect quantity of small RNA in mirRICH method. (A) 15% PAGE (Polyacrylamide gel electrophoresis) and (B) 15% UREA denaturing gels electrophoresis of the RNA samples are prepared from two different breast cancer cells, MDA-MB-231 and MCF7 cells with either TRIzol with 1hr drying or 24hr drying, mirRICH with 1hr drying or 24hr drying. The 100-bp marker is indicated as ladder. (C) Peak images of experiment samples of MDA-MB-231 and MCF7 were obtained by Aglient RNA 6000 Nano kit respectively.
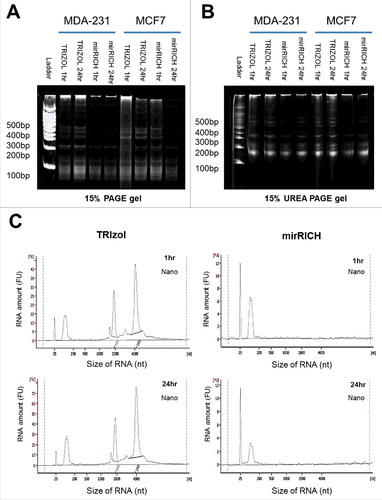
A bioanalyzer detects DNA or RNA of varying sizes with high sensitivity with low amounts of sample. We analyzed the quantity and quality of small RNA fractions from the mirRICH method using a bioanalyzer and compared the result with that of the AGPC-based RNA extraction protocols. We also investigated whether the over-drying time of RNA pellets affected the yields of small RNA molecules in the mirRICH method. We found that longer drying time does not increase the quality of small RNA isolated by mirRICH ().
Comparison to column-based small RNA extraction method
Next, we are questioned that the quantity and quality of small RNA fractions from the mirRICH method is comparable to the column-based small RNA extraction method such as mirVana small RNA extraction kit (Invitrogen, MA, USA). Bioanalyzer data showed that the size and peak patterns of the small RNA samples from both the mirVana kit and mirRICH method were almost identical. Both mirRICH method and mirVana kit can isolate only small RNA molecules and deplete most of the large RNA molecules such as 18S and 28S ribosomal RNAs in MDA-MB-231 and MCF7 cells ( & S2A). As expected, only total RNAs isolated using TRIzol showed both 18S and 28S ribosomal RNAs, as well as small RNAs ( & S2A). In addition, both mirRICH and mirVana kit method show dramatic reduced amount of RNA concentration and increase percentage of miRNA proportion comparing to TRIZOL method ().
Figure 3. Comparison of the quantity and the quality of small RNAs isolated by TRIzol, mirRICH and mirVana. Equal number of two different breast cancer cells, MDA-MB-231 and MCF7 cells were prepared and extracted with three different methods: TRIzol, mirRICH, and mirVana. Peak images of small RNA samples were obtained by Aglient RNA 6000 Nano kit (A) and small RNA analysis kit (B) respectively. (C) 15% PAGE (Polyacrylamide gel electrophoresis) and (D) 15% UREA denaturing gels electrophoresis of the RNA samples from two different breast cancer cells, MDA-MB-231 and MCF7 are prepared by three different methods, TRIzol (T), mirRICH (M) and mirVana (K). The 100-bp marker is indicated as ladder.
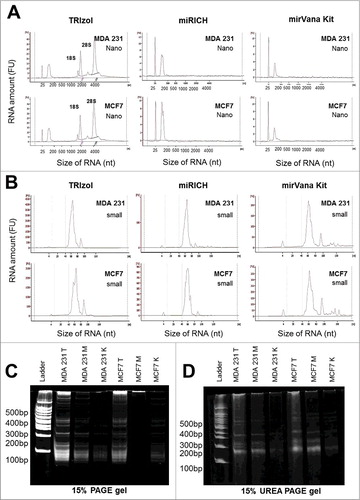
Although bioanalyzer nano-chip data show that both mirRICH method and mirVana kit deplete 18S and 28S rRNAs and enrich small RNAs, it is difficult to see the detail peaks of the small RNA fractions in the bioanalyzer nano-chip. Thus, we performed bioanalyzer small RNA chip analysis for the same samples. According to bioanalyzer small RNA chip data, there is no big different among TRIzol (no small RNA enriched), mirRICH and mirVana kit in MDA-MB-231 and MCF7 (). To further validate the data from the bioanalyzer small RNA chip, we also performed 15% PAGE and 15% UREA-denaturing gel electrophoresis and tested whether small RNA bands from mirRICH method is similar to mirVana kit in the PAGE gel (). In both 15% PAGE and 15% UREA-denaturing gel electrophoresis, we observed the similar result with the bioanalyzer small RNA chip. These data together indicate that mirRICH method isolated only small size of RNA and the quantity of small RNA fractions comparable to commercially available column-based small RNA isolation kit, mirVana Kit.
It is important to test whether mirRICH method is useful all types of cells instead of breast cancer cells. We isolated small RNAs using mirRICH method is well adapted to other types in various types of cells such as 293T, TPC-1 thyroid tumor cells (Fig. S4A-B). We observed that mirRICH methods properly isolate only small sizes of RNAs in all types of cells and it strongly support that mirRICH method is applicable to all types of cells.
Identifying the small RNA molecules isolated by the mirRICH method using NGS
Given that the sizes of the isolated RNAs were unknown, we performed Next generation sequencing (NGS) to sequence all the obtained small RNA molecules. However, to conduct NGS, the quality of DNA or RNA templates is critical [Citation21]. To perform small RNA NGS, it is generally recommended to exclude large RNAs (>100 nt) from the total RNAs because the contents of small RNAs are very low compared to the remaining mRNA and rRNA. Gel extraction after cutting out the undesirable sizes after PAGE is commonly used to remove large RNA molecules before NGS sequencing. Since the gel extraction process is inconvenient and inefficient because of the loss of considerable amounts of small RNA molecules, large amounts of small RNAs (more than 10μg) are typically needed for the NGS procedure.
Given that the mirRICH method could enrich small RNAs without gel extraction, we next investigated whether the quality and quantity of small RNAs isolated from the mirRICH method are applicable to NGS. First, we measured 260/280 and 260/230 ratio for all three samples, TRIzol, mirVana and mirRICH using nano-drop machine. Although 260/280 ratio for all three methods are similar, but 260/230 ratio from mirRICH method is slightly lower than TRIzol. However, 260/230 ratio from mirRICH method is comparable to mirVana kit (Table S1). As expected, the small RNAs from both mirRICH and mirVana enabled the successful construction of an NGS library (Fig. S5A–C and Table S2). Table S3 shows the raw data statistics for the NGS analysis of small RNAs isolated by the mirRICH method and commercial mirVana kit. The total read bases, total reads, GC content, and Q20 and Q30 statistics were nearly identical for both methods. Analysis of the trimmed data again showed similar GC content and Q20 and Q30, although the total read bases and total reads were slightly lower by mirRICH than by the mirVana kit (Table S3). After trimming out adapter dimers and adapter sequence end of small RNA, total read bases and total reads are similar between samples that extracted by mirRICH and mirVana kit respectively (Table S4). Although trimmed results of miRICH and mirVana kit are almost identical, the mirRICH method yielded slightly lower number of total reads than the commercial mirVana kit and it is because that the portion of short reads less than 18 bases are higher in mirRICH method than mirVana kit (Table S5).
Bioinformatic analysis was used to determine the proportion of each type of RNAs including rRNAs, tRNAs, snRNAs, snoRNAs and miRNAs. There was no significant difference in the RNA composition ratios between mirRICH and mirVana (). It is important to find out that the quality and quantity of miRNAs from both mirRICH method and mirVana kit are equivalent. Therefore, we compared the results of miRNA NGS between mirRICH and mirVana with total 2588 known human miRNAs (miRBase DB ver.21) and we found a strong correlation between both methods (). These data support that the quality and quantity of miRNAs obtained by the mirRICH method are comparable to mirVana kit and sufficiently high enough for use in NGS.
Figure 4. Comparison of next generation sequencing (NGS) and qPCR quantification between mirRICH and mirVana. Redrawing diagram with Excel program shows the composition patterns and ratios of various types of RNAs after next generation sequencing either in mirRICH (A) and mirVana (B) drawn by Rfam v9.1 [Citation38]. (C) The correlation of expression data of total 2588 known miRNAs obtained from two different small RNA preparation methods, mirRICH or mirVana were analyzed by using mirBase databases after NGS [Citation39]. (D) The relative expression of miRNA-23a, miR-218 and miR-708 during breast cancer metastasis. Samples were prepared by either mirRICH or mirVana kit and the quantification of each miRNAs were measured by qPCR analysis.
![Figure 4. Comparison of next generation sequencing (NGS) and qPCR quantification between mirRICH and mirVana. Redrawing diagram with Excel program shows the composition patterns and ratios of various types of RNAs after next generation sequencing either in mirRICH (A) and mirVana (B) drawn by Rfam v9.1 [Citation38]. (C) The correlation of expression data of total 2588 known miRNAs obtained from two different small RNA preparation methods, mirRICH or mirVana were analyzed by using mirBase databases after NGS [Citation39]. (D) The relative expression of miRNA-23a, miR-218 and miR-708 during breast cancer metastasis. Samples were prepared by either mirRICH or mirVana kit and the quantification of each miRNAs were measured by qPCR analysis.](/cms/asset/82b6ed90-9eac-479d-90f1-aaa5af9d74b2/krnb_a_1451723_f0004_oc.jpg)
qPCR validation of the NGS results
In general, to further validate the quantity of miRNAs from NGS, one of three methods are typically used, cloned small RNA library technologies [22], northern blot analysis [23], and qPCR [Citation24]. Among these, qPCR is widely used to validate the quantification of gene expression of mRNAs or miRNAs. In this study, we used the Taqman miRNA qPCR quantification technique to validate miRNA expression. The qPCR data showed a significant correlation between mirRICH and mirVana ( & S6A-B).
Next, we tested whether the quality of the small RNA fractions from mirRICH is sufficient to study differentially expressed miRNAs during metastasis in breast cancer. For this experiment, we selected 3 miRNAs, miR-23a, miR-218 and miR-708, and examined their expression in metastatic breast cancer cells (MDA-MB-231) and non-metastatic breast cancer cells (MCF7). According to our unpublished data of NGS results between MDA-MB-231 and MCF7, miR-218 is higher in MDA-MB-231 while miR-708 is lower in MDA-MB-231 comparing to MCF7. In addition, miR-708 expression was validated in our previous study [Citation25]. In consistence with NGS data and previous study, miR-218 had higher expression levels in the metastatic breast cancer cells (MDA-MB-231) than in the non-metastatic cells (MCF7). In the other hands, miR-708 had lower expression in MDA-MB-231 than MCF7 respectively. A similar pattern was observed in the small RNA samples isolated using mirVana (). This result indicates that small RNA samples isolated by using the mirRICH method can be used to study the differential expression of miRNAs. Interestingly, U6 expression was slightly higher with mirRICH than with mirVana, whereas miR-23a expression with mirRICH was slightly lower than with mirVana (Table S6).
Discussion
Recently, among the various types of RNAs, small RNAs, including miRNAs, have been extensively studied [Citation26]. To assess whether miRNAs are differentially expressed in certain conditions, it is necessary to isolate and analyze miRNAs, for which researchers often use a TRIzol-based approach or column-based commercial kits [Citation27]. However, both methods have certain limitations. With the TRIzol method, the fraction of miRNAs in total RNAs is extremely low, and therefore, large amounts of total RNAs need to be obtained to perform additional steps, such as removing rRNA [Citation28–Citation31]. Meanwhile, with a column-based commercial kit, such as mirVana, miRNAs can be enriched but at about five times higher cost.
During the past decade, next-generation sequencing (NGS) technology has enabled the sequencing of entire transcriptomes from cells. To apply the NGS platform for high-throughput sequencing of small RNA populations in cells, substantial amounts of total RNAs are required, owing to the low proportion of small RNAs compared to the remaining RNA types. In general, rRNA can account for up to 80% of the total RNA. Thus, additional steps are required to isolate small RNA fractions from total RNAs, such as the gel extraction procedure. However, considerable amounts of small RNAs can be lost during gel extraction.
The mirRICH method is a newly developed small RNA–enriching method, modified from the traditional TRIzol method. The major differences between the mirRICH and TRIzol methods are the absence of a 70% EtOH washing step and the inclusion of a pellet over-drying step for 1h-24h before the final elution process. The fundamental rule in RNA extraction is avoiding the over-drying of RNA pellets before the final elution because once RNA pellets are over-dried, pellets are precipitated and no longer solubilized in the water. However, we accidently discovered that small RNA molecules, <100 nt in size, can still be released from over-dried RNA pellets. We hypothesize that over-drying may cause trouble to purify mRNAs but it still useful to isolating small RNAs from the total RNAs. We named newly developed small RNA enriching method as “mirRICH” and successfully isolated small RNAs without contaminating 18S and 28S rRNA and mRNAs.
It is important to clarify the definition of “over-dry.” Therefore, we tested whether the over-drying time of 1 h or 24 h affected the enrichment of small RNAs. There is no significant difference between short (1hr) and long (24hr) drying time which implies that 1hr drying is enough to precipitate the RNA pellets perfectly (). This data support previous findings that drying RNA pellets will precipitate in a short time. Interestingly, in the TRIzol solution method, although most of large RNA molecules such as 18S and 28S rRNA are degraded but some of rRNAs still detected even though the RNA pellet is over-dried for 24hr, as long as the RNA pellet is washed with 70% ethanol solution (). These data suggest that the removal of salts with 70% ethanol is critical in causing the precipitation of the RNA pellet. We are concern that skipping the washing step with 70% ethanol may cause a contamination of phenol or salts trace in the sample and also cause a problem in further experiment. However, in our experiments, we did not found any problem to perform further experiments using small RNA extracted by mirRICH method such as RT-qPCR and next generation sequencing (NGS) in many independent replicated experiments ().
Most importantly, the quality of small RNAs obtained by mirRICH is suitable for sophisticated applications such as NGS. All the data showed that the quality and quantity of small RNAs isolated by using mirRICH is suitable for application to NGS or qPCR, and in fact equivalent to the small RNAs yielded by the column-based commercial mirVana kit ( & S7A-H). This result indicates that mirRICH method is suitable to isolate good quality of small RNA to perform downstream work. Moreover, NGS data showed that the composition of various types of RNAs (rRNAs, tRNAs, snoRNAs, and miRNAs) are almost the same between the mirRICH method and mirVana kit. This data is very important to evaluate mirRICH method because the small RNAs prepared by mirRICH do not arise from the degradation of larger RNA molecules during the over-drying process.
Only different between mirRICH and mirVana in NGS data is proportion of shorter reads less than 18 bases (Table S5). In bioinformatics pipeline to analyze miRNAs, shorter reads less than 18 bases is filtered out because the length of most miRNAs are ranged from 18 to 24 bases [Citation32]. It seems that mirVana kit tends to enrich certain range of small RNAs range like higher size than 18 bases. However, mirRICH does not show such biased enrichments for less than 18 or higher than 100 bases. We checked the strong tendency of enriching certain size of small RNAs in mirVana kit using qPCR technique.
Interestingly, we found that small RNAs prepared using mirRICH contain slightly more U6 and less miRNAs compared to small RNAs obtained by mirVana. We noticed that the size of U6 is relatively large (about 150nt), while most mature miRNAs are relatively smaller (22–23 nt). We assume that these size differences might affect the elution efficiency in a column-based small RNA extraction kit. This assumption suggests that column-based mirVana kit enriches more miRNA than other relatively larger-sized small RNAs like U6 (∼150 nt) which is generally used to normalize miRNAs. To prove our hypothesis, we compared quantity of U6 from mirVana and mirRICH with traditional TRIzol which is not enriching small RNA during the extraction process. The data showed that the value of U6 of mirRICH is similar to TRIzol (Table S6). This data suggest that U6 may not the best option for the internal normalization at least in the column-based small RNA extraction kit. Obviously, to prove this hypothesis, gathering more data is required.
Nevertheless, a strong correlation was observed between mirRICH and the mirVana kit when we compared the expressions of 2588 known miRNAs from NGS data (). It is important to assess whether mirRICH can be used to detect differentially expressed miRNAs in different types of cells. Therefore, we measured the differential expressions of two miRNAs, miR-23a and miR-218 during cancer development and metastasis [Citation33–Citation36]. The data clearly showed that the results were highly similar for both mirRICH method and mirVana kit. These data suggest that the mirRICH method is applicable to detect differentially expressed miRNAs in certain conditions. Although the fold change of miR-218 in mirRICH is slightly lower than mirVana, it may be due to the relatively low expression of U6 in mirVana compared to mirRICH.
In summary, our mirRICH approach enriches small RNAs of sufficient quality and quantity for applications in molecular biology studies. We believe that this technique will make a creditable contribution to research in the exciting and emerging field of small regulatory RNAs.
Materials and methods
Reagents and equipment
The reagents and instruments used were as follows: QIAzol® lysis reagent (Qiagen, cat. no. 79306), isopropanol (Sigma Aldrich, cat. no. I9516-500ML), ethanol (Sigma Aldrich), UREA (Sigma Aldrich), Tris-Borate-EDTA 10X solution (Thermo Scientific), chloroform (Sigma Aldrich, cat. no. 288306), mirVana small RNA extraction kit (Invitrogen, cat. no. AM1560), TaqMan universal master mix II, no UNG (Applied Biosystems, cat. no. 4440040), hsa-miR-218–5p (Applied Biosystems, cat. no. 000521), hsa-miR-23a-3p (Applied Biosystems, cat. no. 000399), High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, cat. no. 4368814), U6 primer (Applied Biosystems, cat. no. 001973). Concentration of RNA is measured by Nanodrop device (Thermo Fisher Scientific). A 2100 Bioanalyzer (Agilent, CA, USA) was used for analysis of contents of RNA. Agilent RNA 6000 Nano kit and small RNA analysis kit was purchased from Agilent. StepOnePlus-Real Time PCR system (Applied Biosystems) was used for real time qPCR analysis. In addition, the breast cancer cell lines MDA-MB-231, LM2, and MCF7 were purchased from the American Type Culture Collection (Manassas). The cells were cultured in Dulbecco's modified Eagle's medium (Mediatech Cellgro) with 10% fetal bovine serum (Equitech-Bio) containing 10,000 IU/mL penicillin, 10,000 μg/mL streptomycin (100 ×) (Mediatech Cellgro), and 200 mM l-glutamine (100 ×) (Gibco). All cell lines were maintained in an incubator at 37°C in 5% CO2.
Procedure
Small RNA extraction method
Culture the target cells (here, 5× 106 cells of the breast cancer cell line MDA-MB-231 and MCF7 for 3 days) and treat them with 1 mL QIAzol lysis reagent.
Scrape the contents with a scraper; and collect the samples in 1.5-mL tubes. Leave the samples for 5 min at room temperature.
Add 200 µL chloroform to each sample, mix thoroughly, vortex for 30 s, and leave for 3 min at room temperature.
Centrifuge the tubes at 12,000gfor 15 min at 4°C.
After centrifugation, carefully transfer the upper aqueous phase into fresh 1.5-mL tubes.
Add isopropanol (∼500 µL, 1.5 volumes of sample) and leave at room temperature for 10 min.
Centrifuge the tubes at 12,000gfor 15 min at 4°C.
Skip the washing step with 70% EtOH.*
After centrifugation, remove the supernatant and dry the pellet for 1 h or 24h.
Add nuclease-free water and resuspend the pellet for 5 min.
Quantify RNA concentration using a Nanodrop device.
*Different conditions were used to test the mirRICH method:
8) With 70% EtOH washing
A) Wash the pellet with 70% EtOH and dry it for 1 h.
B) Wash the pellet with 70% EtOH and dry it for 24 h.
9) Without 70% EtOH washing
A) Omit the 70% EtOH wash step and dry the pellet for 1 h.
B) Omit the 70% EtOH wash step and dry the pellet for 24 h.
Gel electrophoresis
To visualize the small RNA fractions isolated by the newly developed method, RNA samples were separated by both 2% agarose gel electrophoresis and 15% polyacrylamide gel electrophoresis (PAGE). Agarose gels display a wide size range of nucleic acids, whereas polyacrylamide gels show small-sized nucleic acids with high resolution. Making urea denaturing gels and running is followed the previous protocol [Citation37]. A 100-bp DNA ladder marker (ELPIS, Daejeon, South Korea) was used in both cases for determining nucleic acid size.
Validation of size and quality of obtained RNA
To analyze more precisely the size distributions of the small RNA fractions, we used an Agilent 2100 bioanalyzer with the Agilent RNA 6000 Nano kit or small RNA kit according to the manufacturer's protocol. To further validate the quality of RNA obtained by the newly developed protocol, we performed miRNA qPCR using the Taqman miRNA probe system (Applied Biosystems CA, USA). The U6 probe was used as the internal control.
Next generation sequencing (NGS)
To characterize the individual sequences of small RNAs and to determine the ratios of different types of small RNAs, NGS for small RNA fractions was performed using the Illumina sequencing platform. Detailed information is shown in supplemental data (Table S2).
[Construction of miRNA library]
To construct miRNA library, size selection from 18 ∼ 30 nt is necessary to obtain miRNAs in AGPC based method such as TRIzol reagent because proportion of miRNA is less than 1% among total amount of RNA. Agarose gel electrophoresis is recommended to select out the band corresponding to the size of miRNA. However, we skipped this procedure because mirRICH and mirVana only exclude large molecules of RNAs and enrich only small RNAs.
Specifically designed DNA oligomers called “adapter” are ligated to the size selected RNA molecules prepared by mirRICH and mirVana kit method respectively. This sequencing adapter allows reverse transcription and PCR amplification process like primer binding sites. T4 RNA ligase 2 is used for ligation step to ligate between adenylated single strand DNA 3' adapter followed by a 5' adapter and RNA templates together. This adapter is specifically designed to selectively capture small RNAs having 5′ phosphate group which is key feature of miRNAs while destructive fragments of RNA molecules have 5′ hydroxyl group.
miRNA fragments with ligated adapters are converted to cDNA fragments which are later used in the sequencing reaction. There are many commercial kits using some form of reverse transcriptase in order to carry out this step. PCR is then performed to amplify the pool of cDNA sequences.
Generally, the amplified cDNA library should be run on agarose gel and the band containing the molecules corresponding to the miRNA fragments with ligated adapters are cut out for subsequent sequencing between 140 ∼ 160 nt. However, in our protocols, we selected larger cDNA molecules than 140 nt to analysis whole spectrum of small RNA including long noncoding RNAs.
cDNA fragments are sequenced by HiSeq 2500 system from Illumina company. The detail analysis of NGS data are following,
[Quantification of miRNA expression]
After sequencing, the raw sequence reads are filtered out based on sequencing quality. And, the adapter sequences are also trimmed off from the raw sequence reads.
The trimmed reads are gathered forming a unique cluster. This cluster contains reads that are 100% match to the sequence identity as well as read length. The cluster is given its temporary cluster ID and the number of reads it holds.
To detect known and novel miRNAs, unique clustered reads are aligned to reference human genome database (UCSC hg19) and also aligned to precursor miRNAs obtained from miRBase v21. The miRDeep2 algorithm is used to predict potential hairpin structures and to assign scores that represent the probability that hairpins are true miRNA precursors.
Unique clustered reads are sequentially aligned to non-coding RNA database, Rfam9.1 to classify other type of RNA such as tRNA, snRNA, snoRNA etc.
The quantitation of each miRNA is obtained and normalized from the count of reads that mapped to miRNAs.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Suppl_mat_mirRICH__a_simple_method_to_enrich_the_small_RNA_fraction_from_over-dried_RNA.docx
Download MS Word (683 KB)Acknowledgment
C.C., Y.U, S.Y, S.S J.S and S.R. conceived and designed the experiments. C.C, Y.U., C.B., P.D. S.Y. T.V.N, S.B, J.W and S.R. performed the experiments. J.S, J.M. and S.S. analyzed the data. C.C., Y.U, S.S and S.R. wrote the manuscript. All authors contributed to critically revise the manuscript for important intellectual content, and gave final approval and agree to be accountable for all aspects of the work. This work was supported by the Soonchunhyang University Research Fund, the Ministry of Science and ICT and Business Belt Program (2017K000492), the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (2017R1A2B4010480) and a grant from the Marine Biotechnology Program (No. 20140428) funded by the Ministry of Oceans and Fisheries, South Korea.
Additional information
Funding
Related Research Data
References
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–9.
- Chomczynski P, Sacchi N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nat Protoc. 2006;1:581.
- Zeugin JA, Hartley JL. Ethanol precipitation of DNA. Focus. 1985;7:1–2.
- Tan SC, Yiap BC. DNA, RNA, and protein extraction: the past and the present. BioMed Res Int. 2009;574398:1–10.
- Hennion M-C. Solid-phase extraction: method development, sorbents, and coupling with liquid chromatography. J Chromatogr A. 1999;856:3–54.
- Augusto F, Hantao LW, Mogollon NG, et al. New materials and trends in sorbents for solid-phase extraction. TrAC Trends Anal Chem. 2013;43:14–23.
- Esser K-H, Marx WH, Lisowsky T. Nucleic acid-free matrix: regeneration of DNA binding columns. Biotechniques. 2005;39:270–1.
- Gjerde DT, Hoang L, Hornby D. RNA purification and analysis: sample preparation, extraction, chromatography. Hoboken, (NY): John Wiley & Sons; 2009.
- Sun N, Deng C, Liu Y, et al. Optimization of influencing factors of nucleic acid adsorption onto silica-coated magnetic particles: application to viral nucleic acid extraction from serum. J Chromatogr A. 2014;1325:31–9.
- Nargessi RD. CB, Inc. Magnetic isolation and purification of nucleic acids. United States patent US 6855499 B1. 2005.
- Cseke LJ, Kirakosyan A, Kaufman PB, et al. Handbook of molecular and cellular methods in biology and medicine. Boca Raton, (FL): CRC press; 2011.
- Ivey KN, Srivastava D. MicroRNAs as regulators of differentiation and cell fate decisions. Cell Stem Cell. 2010;7:36–41.
- Ivey KN, Srivastava D. microRNAs as developmental regulators. Cold Spring Harb Perspect Biol. 2015;7:a008144.
- Allegra A, Alonci A, Campo S, et al. Circulating microRNAs: new biomarkers in diagnosis, prognosis and treatment of cancer (review). Int J Oncol. 2012;41:1897–912.
- Lan H, Lu H, Wang X, et al. MicroRNAs as potential biomarkers in cancer: opportunities and challenges. BioMed Res Int. 2015;125094:1–17.
- Jansson MD, Lund AH. MicroRNA and cancer. Mol Oncol. 2012;6:590–610.
- Walleshauser III JG, Kessler T, Morse D, Tannous BA, Chiu NH. A simple approach for evaluating total MicroRNA extraction from mouse brain tissues. 2012.
- Zhang J, Li S, Li L, et al. Exosome and exosomal microRNA: trafficking, sorting, and function. Genomics, Proteomics & Bioinformatics. 2015;13:17–24.
- Castoldi M, Benes V, Hentze MW, et al. miChip: a microarray platform for expression profiling of microRNAs based on locked nucleic acid (LNA) oligonucleotide capture probes. Methods. 2007;43:146–52.
- Aravin A, Tuschl T. Identification and characterization of small RNAs involved in RNA silencing. FEBS Lett. 2005;579:5830–40.
- Raza K, Ahmad S. Principle, analysis, application and challenges of next-generation sequencing: a review. arXiv preprint arXiv:160605254. 2016.
- Berezikov E, Cuppen E, Plasterk RH. Approaches to microRNA discovery. Nat Genet. 2006;38:S2–S7.
- Pall GS, Hamilton AJ. Improved northern blot method for enhanced detection of small RNA. Nat Protoc. 2008;3:1077–84.
- Jiang J, Lee EJ, Gusev Y, et al. Real-time expression profiling of microRNA precursors in human cancer cell lines. Nucleic Acids Res. 2005;33:5394–403.
- Ryu S, McDonnell K, Choi H, et al. Suppression of miRNA-708 by polycomb group promotes metastases by calcium-induced cell migration. Cancer Cell. 2013;23:63–76.
- Cech TR, Steitz JA. The noncoding RNA revolution—trashing old rules to forge new ones. Cell. 2014;157:77–94.
- Tang F, Hajkova P, Barton SC, et al. MicroRNA expression profiling of single whole embryonic stem cells. Nucleic Acids Res. 2006;34:e9–e.
- Nilsen TW. Removing rRNA from deproteinized, phenol-extracted total RNA by enzymatic digestion. Cold Spring Harb Protoc. 2012;2012:pdb. prot072124.
- Rio DC, Ares M, Hannon GJ, et al. Purification of RNA by SDS solubilization and phenol extraction. Cold Spring Harb Protoc. 2010;2010:pdb. prot5438.
- Rio DC, Ares M, Hannon GJ, et al. Removal of ribosomal subunits (and rRNA) from cytoplasmic extracts before solubilization with SDS and deproteinization. Cold Spring Harb Protoc. 2010;2010:pdb. prot5442.
- Rio DC, Ares M, Hannon GJ, et al. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb Protoc. 2010;2010:pdb. prot5439.
- Ambros V. The functions of animal microRNAs. Nature. 2004;431:350.
- Ma F, Li W, Liu C, et al. MiR-23a promotes TGF-β1-induced EMT and tumor metastasis in breast cancer cells by directly targeting CDH1 and activating Wnt/β-catenin signaling. Oncotarget. 2017;8:69538–69550.
- Liu H, Pan Y, Han X, et al. MicroRNA-216a promotes the metastasis and epithelial–mesenchymal transition of ovarian cancer by suppressing the PTEN/AKT pathway. OncoTargets Ther. 2017;10:2701.
- Zhang D, Zhao L, Shen Q, et al. Down‐regulation of KIAA1199/CEMIP by miR‐216a suppresses tumor invasion and metastasis in colorectal cancer. Int J Cancer. 2017;140:2298–309.
- Xia H, Ooi LLP, Hui KM. MicroRNA‐216a/217‐induced epithelial‐mesenchymal transition targets PTEN and SMAD7 to promote drug resistance and recurrence of liver cancer. Hepatology. 2013;58:629–41.
- Summer H, Grämer R, Dröge P. Denaturing urea polyacrylamide gel electrophoresis (Urea PAGE). J Vis Exp: JoVE. 2009;32:1485.
- Gardner PP, Daub J, Tate JG, et al. Rfam: updates to the RNA families database. Nucleic Acids Res. 2008;37:D136–D40.
- Griffiths-Jones S. miRBase: the microRNA sequence database. MicroRNA Protoc. 2006;342:129–38.