
ABSTRACT
Therapeutics that directly target RNAs are promising for a broad spectrum of disorders, including the neurodegenerative diseases. This is exemplified by the FDA approval of Nusinersen, an antisense oligonucleotide (ASO) therapeutic for spinal muscular atrophy (SMA). RNA targeting therapeutics are currently under development for amyotrophic lateral sclerosis (ALS), Huntington's disease (HD), and spinocerebellar ataxias. We have used an ASO approach toward developing a treatment for spinocerebellar ataxia type 2 (SCA2), for targeting the causative gene ATXN2. We demonstrated that reduction of ATXN2 expression in SCA2 mice treated by intracerebroventicular injection (ICV) of ATXN2 ASO delayed motor phenotype onset, improved the expression of several genes demonstrated abnormally reduced by transcriptomic profiling of SCA2 mice, and restored abnormal Purkinje cell firing frequency in acute cerebellar sections. Here we discuss RNA abnormalities in disease and the prospects of targeting neurodegenerative diseases at the level of RNA control using ASOs and other RNA-targeted therapeutics.
Therapeutic modalities and oligonucleotide therapeutics
Development of a treatment strategy for disease is largely determined by the disease mechanism. In monogenic, mendelian diseases where a likely therapeutic target is known, modifying the target directly may be possible using an RNA-targeted therapeutic approach, and oligonucleotide-based therapeutics are well matched for these diseases. Antisense oligonucleotides (ASOs) provide multiple ways to alter gene expression: they can either block translation, alter stability, modify splicing, or elicit degradation pathways altering the target mRNA's expression [Citation1]. Other methods of targeting gene expression that are RNA based or RNA targeted include RNA interference (RNAi), and ribozymes & deoxyribozimes with intrinsic catalytic activities [Citation2], and RNA aptamers such as pegaptanib, which binds and deactivates VEGF for age-related macular degeneration (AMD) [Citation3] and an RNA decoy aptamer sequestering NFκB [Citation4]. Depending on the application, RNAi therapeutics can be in the form of shRNAs that are plasmid based [Citation5], siRNAs complexed with carriers [Citation6], or naked and unmodified siRNAs [Citation7]. The delivery of shRNAs by adeno-associated viruses (AAVs) is very promising for treating neurodegenerative diseases [Citation5]. AAVs carrying shRNAs targeting a human ATXN1 transgene delayed the progression of SCA1 mouse motor and anatomical phenotypes, when infused into the deep cerebellar nucleus (DCN) [Citation8]. This is exciting proof-of-concept that the technique might be effective for treating multiple polyglutamine and other CNS disorders [Citation9,Citation10]. Also, AAVs of various serotypes delivered to the CSF were able to reach CNS targets in non-human primates [Citation11]. The benefit of the AAV technique is permanent episomal shRNA expression, thus patients might require only a single treatment. However, like most therapeutics, the approach is not without risk. For one, there is the possibility that the AAVs delivered by intraparenchymal infusion may not be widely taken up. Thus, multiple AAV infusions might be necessary for effective therapeutic benefit. The integration of AAVs into the genome is thought to be very rare [Citation12]. Oligonucleotide-based approaches are particularly attractive for monogenic diseases that are characterized by a gain-of-function mutation such as for the spinocerebellar ataxias and Huntington's disease, as are siRNAs and microRNAs (miRNAs) that function by RNAi. Oligonucleotide-based approaches are also well suited for some loss-of-function diseases such as spinal muscular atrophy where the oligonucleotide-based therapeutic nursinersen restores gene expression by altering splicing.
Most of our understanding of gene expression mechanisms involving RNAi, miRNAs, long noncoding RNAs (lncRNAs) and ASOs was developed within the past two decades. Small double-stranded RNAs able to regulate gene expression were discovered in 1998, with the finding of RNA interference by Andrew Fire and Craig Mello [Citation13] who were awarded a Nobel Prize for the discovery eight years later. Only five years earlier in 1993, the first discovery of naturally occurring microRNAs was made [Citation14]. Long non-coding RNAs have a more drawn-out history with hints of their presence appearing in the 1970s, but it was almost 40 years later that it was understood that lncRNAs could up- or down-regulate other genes in numerous ways involving protein interactions [Citation15]. Understanding on how antisense oligonucleotides could be used as tools for manipulating gene expression developed in the early to mid ‘90s. During this time, Stanly Crooke, now the founder of Ionis Pharmaceuticals, had begun publishing his research describing the mechanism. While in the early 90's it was first revealed that nucleotides with a phosphorothioated backbone were resistant to RNase H1 cleavage [Citation16], Crooke's team demonstrated that RNase H1 was recruited to RNA-DNA duplexes, and that RNAs adjacent to phosphorothioated DNA oligonucleotides in these duplexes were cleaved by the nuclease [Citation17]. Crooke and colleagues also predicted the clinical utility of ASOs [Citation17].
Enormous effort continues to be made developing oligonucleotide therapeutics because of their potential for treating human disease. The benefit of ASOs is broad distribution and long-lasting effects (in the order of months) that is not permanent and thus can be ended by terminating treatment. Unlike ASO therapeutics, RNAi-based therapeutics when delivered as an shRNA by AAV can develop non-reversible efficacy, which may be beneficial but also comes with risk since a therapeutic commitment is made. RNAi therapeutics may also be challenging to deliver to the relevant tissues or cell types. However, very few ASO therapeutics have been approved for use in humans. The first approval of an antisense oligonucleotide drug came in 1998 for fomivirsen an RNase H1-stimulating ASO injected into the vitreous of the eye for CMV retinitis [Citation18,Citation19]. Although formivirsen is seen as an effective drug, it has been discontinued for favor of newer more effective therapeutics for CMV retinitis. Other ASO drugs have now been approved for use in the clinic, and these are discussed in some detail below with emphasis on developing ASOs for neurodegenerative disease.
RNA-targeted vs. RNA-based therapeutics
There is considerable overlap between RNA-targeted therapeutics and RNA-based therapeutics; some examples are provided here. An RNA-targeted therapeutic might be composed of an RNA, such as an siRNA or miRNA, and is therefore RNA-based. But not all RNA-targeted therapeutics are RNA in composition and not all RNA-based therapeutics target RNAs [Citation20]. RNA-based therapeutics are RNAs that can function therapeutically in multiple ways, either by targeting other RNAs or proteins. Antisense oligonucleotides and double-stranded oligonucleotides (siRNAs and shRNAs) that function via the RNAi pathway are usually RNA-targeted. ASOs used clinically are often chimeric with various 2′-sugar modifications on flanking bases, and thus are mixmers, or have 2′-sugar modifications on all base positions (2′-O-methoxyethyl (MOE)), as for nusinersen that does not function by recruiting RNase H1 but instead blocks activity of the spliceosome. There are also other RNA-targeted approaches for altering splicing therapeutically [Citation21]. Other types of RNA-based therapeutics include catalytically active ribozymes [Citation2], suppresser tRNAs for premature stop codon read-through [Citation22], long non-coding RNAs [Citation23], and RNA aptamers [Citation3,Citation4]. summarizes RNA-targeted approaches to therapy.
Figure 1. Common types of RNA-targeted therapeutics.
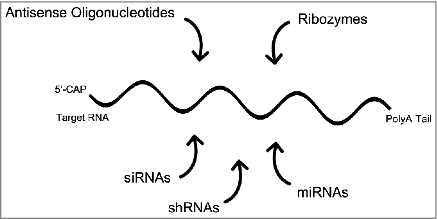
Delivery
Oligonucleotide and other RNA targeted therapeutics are in part determined by how therapeutics are taken up by the target tissues, which is dependent also on the therapeutic composition. Here we provide a brief discussion on delivery approaches. Naked delivery of siRNAs has been demonstrated to be effective for localized treatments, including in the eye for age-related macular degeneration [Citation24,Citation25], by inhalation for pulmonary disease [Citation7], and transdermally for various skin conditions [Citation26]. Antisense oligonucleotides are also employed without carriers. The mechanism of ASO uptake is thought to be mediated by endocytosis, and recently the main proteins involved in ASO uptake and cellular distribution have been identified [Citation27,Citation28]. Naked delivery of siRNA can also be enhanced by co-delivery of a phosphorothiorated oligonucleotide to stimulate the endocytosis pathway [Citation29]. Plasmids encoding siRNAs, miRNAs, or shRNA can be combined with to carriers to increase the stability, efficacy, and cellular uptake of the therapeutic cargo. There are multiple types of carriers for siRNAs and shRNAs. Nanoparticles are promising as carriers for siRNA and shRNA therapeutics for neurodegenerative disorders [Citation30], including shRNAs targeting a α-synuclein in a mouse model of Parkinson's disease [Citation31], siRNAs targeting BACE1 and APP in mouse CNS toward treating Alzheimer disease [Citation32]. Nanopartical systems can also be used to deliver siRNAs to CNS targets intranasally [Citation33]. A promising approach for siRNA delivery is next-generation ionizable cationic lipid nanoparticles (LNPs) [Citation34]. LNPs have promising toxicity and immunogenicity profiles and are used for ALN-TTR02, an siRNA-based therapeutic targeting transthyretin for treating familial amyloid polyneuropathy (FAP) [Citation34]. Delivery and distribution of antisense oligonucleotides can also be modified by various carrier approaches including conjugating ASOs to either of N-acetylgalactosamine, octaguanidine dendrimer, or cell-penetrating peptides, or by employing liposomes or nanoparticles [Citation35]. An engineered non-viral gene delivery system made from adenovirus serotype 5 (Ad5) penton capsid proteins can also deliver siRNA cargoes [Citation36]. Ad5 capsids are self-assembling and can be constructed to incorporate ligand properties for cell-specific uptake, demonstrated by the specific uptake of cargoes carried by pentons fused to EGF-like peptides by HER2-positive tumor cells [Citation37].
Antisense oligonucleotides
ASOs are highly versatile therapeutics that can be designed for modifying gene expression in multiple ways, depending on sequence and chemistry [Citation38]. Strategies for lowering target RNA expression typically employ gapmer ASOs that are phosphorothioates (PS) supporting RNase H1 activity, with further modifications at the 2′-sugar positions enhancing binding affinity to the target mRNA (see for examples of some common ASO modifications). Cleavage of targeted mRNAs by RNase H1 or by RNAi activity supported by ASOs usually results in mRNA elimination by nonsense mediated decay () [Citation39]. In addition, nonsense mediated decay as a mechanism for target reduction has also been observed for ASOs that induce exon skipping to alter the target mRNA reading frame [Citation39]. Not all RNA-targeted therapeutics elicit target degradation as for SMN2 splicing correction mediated by the nusinersen ASO, or other approaches that alter mRNA translation. This is the case for the SMN2 ASO because it is fully 2′-sugar modified thus it does not support RNAse-H1 function, and the splicing that it elicits does not result in an out-of-frame mRNA. presents a selection of ASO therapies for neurodegenerative diseases that have been approved by the FDA or are in development.
Figure 2. ASO modifications and action of ASOs supporting RNase H1 activity. a) Commonly used ASO modifications. b) Typical action of an ASO supporting RNase H1 activity. The structure of the gapmer ASO is shown, where all positions are phosphorothioate (PS) modified and 5 bp on each flank are also 2′-MOE modified. Following cellular uptake the ASO engages the target and RNase H1 is recruited and cleaves the RNA target opposite of the PS ASO bps. The cleaved RNAs are then eliminated by nonsense mediated decay (NMD).
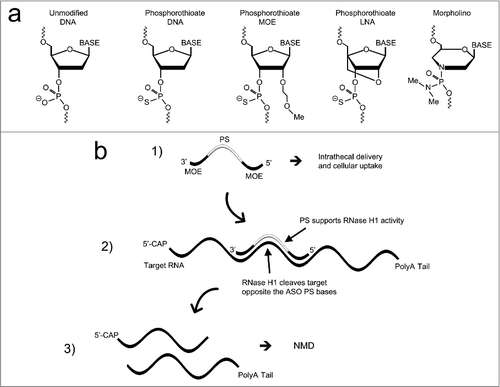
Table 1. ASO therapeutics for neurodegenerative disease.
ASOs and allelic specificity
Ideally, an ASO therapeutic for treating dominantly inherited disease would specifically target the mutant allele leaving expression of the normal allele intact. However, this can be exceptionally challenging. A large scale ASO screen of the scale needed for identifying highly potent and non-toxic ASOs would be limited by constraining the ASO target sites to those overlapping the specific mutation. The development of an ASO that specifically targets a missense or other type of mutation might lead to partial wildtype allele targeting. However, partial reduction of wildtype allele may be acceptable.
To address this problem in developing an ASO for treating Huntington's disease, ASOs were produced that target HTT pre-mRNA at SNPs in linkage disequilibrium with the mutant allele [Citation40]. Theoretically, this strategy would allow for the treatment of >75% of patients with HD [Citation41]. Despite this promising strategy, ASOs targeting both the wildtype and mutant HTT have also been developed and are being prepared for testing in HD patients [Citation42]. This “non-specific” approach is the strategy that we used for developing ASOs targeting the ATXN2 gene toward producing a treatment for SCA2 [Citation43]. While huntingin is essential for normal cell function since homozygous knockout of HTT is embryonic lethal [Citation44–Citation46], mice null for the Atxn2 gene are healthy, fertile, and display no neurodegeneration, but develop obesity [Citation47,Citation48]. Therefore, ATXN2 knockdown in the CNS may be well tolerated in patients with SCA2. The same reasoning was invoked in the development of an ATXN3 ASO toward developing a treatment for SCA3 [Citation49]. Another approach for treating polyglutamine diseases with ASOs is to target the expanded CAG repeat allele with chemically modified CTG ASOs antisense to the CAG repeats of HTT or ATXN3 [Citation50,Citation51].
Off-target effects and cytotoxicity
Assessment of off target effects and cytotoxicity is important in developing ASO therapeutics. These side-effects of ASOs may be sequence-dependent and can also depend on the ASO backbone chemistry. There are multiple ways of assessing ASO off-target effects, summarized here.
Gliosis: ASOs altering off-target expression may be associated with stimulation of gliosis in the CNS as an indication of cytotoxicity or inflammation. Therefore, in vivo screens for identifying ASOs that do not activate gliosis are useful. This is typically done by assessing for microgliosis or astrogliosis indicated by activation of Aif1 or Gfap, respectively, by qPCR, western blotting, and immunohistochemial staining in the brain and other tissues that may uptake the ASO.
RNA expression analysis: Assessment of off-target RNA expression could be a particularly effective method for assessing ASO specificity. Sequences with close complementarity to the target ASO can be evaluated by qPCR to demonstrate lack of engagement by the therapeutic ASO. For example, in the development of an ASO targeting ATXN2, we confirmed by qPCR that the lead ASO did not alter the expression of the closely homologous ATXN2L mRNA [Citation43]. Transcriptome analysis can also be employed for identifying off-targets. One effective approach is to treat mice null for the target gene to detect off-target hybridization in the complete absence of the target mRNA.
Protein binding: Exogenous ASOs can interact with various RNA or DNA binding proteins stimulating a host defense response. Reviewed by Rigo et al. (2014) [Citation52], ASOs can interact with defense-response proteins resulting activation of innate immunity and complement systems, and ASOs can interact and deactivate coagulation factors associated with clearance of infectious agents.
ASO chemistry and off-targets. The spectrum of off-targets can also be related to ASO chemistry. For an ASO gapmer with homologies to unwanted RNA target sites there is the possibility for RNase H1 recruitment and degradation of the off-target mRNAs. However, an ASO that is modified at all 2′ sugar positions such as nusinersen is not expected to support RNase H1 activation, while such an ASO may alter the off-target expression in other ways including RNA translation and stability. Therefore, in vivo screening for off target effects might be more challenging for gapmer ASOs vs. those that are modified at all 2′ sugar positions.
ASO treatment strategy for neurodegenerative disease
Overall there are relatively few ASO-based therapeutics that are FDA approved. Ionis Pharmaceuticals presently has three that are approved for use by the FDA. These include two that treat non-neurodegenerative diseases, mipomersen targeting APOB for treating homozygous familial hypercholesterolemia and alicaforsen targeting ICAM-1 for pouchitis, and nusinersen for spinal muscular atrophy which represents one of the first ever disease modifying therapeutics for a neurodegenerative disorder.
One challenge for treatment of neurodegenerative diseases is that ASOs do not cross the blood brain barrier. However, they are able to reach targets throughout the CNS when delivered intrathecally [Citation53]. While the chemical modifications made to ASOs used therapeutically protect them from nucleases, the modified ASOs do remain susceptible to reduced biological efficacy over time. The biological half-life of ASOs is in part dependent on the chemical modification. When delivered to CSF, phosphorothioates that are 2′-MOE modified at all positions have been observed with biological half-lives up to 6 months and may be detected in CNS after 1 year of treatment [Citation53,Citation54], while gapmer ASOs typically have shorter half-lives [Citation42].
Current state of ASO development for neurodegenerative disease
Spinal muscular atrophy: Nusinersen for spinal muscular atrophy is likely the most intriguing of all the ASO therapeutics targeting neurodegenerative diseases since it is among the first ever disease modifying therapeutics for a neurodegenerative disease that has been approved by the FDA. SMA a familiarly-inherited recessive early-onset neurodegenerative disorder characterized by progressive loss of motor neurons, caused by loss of function mutation in the SMN1 gene and resultant loss of the survival motor neuron protein. The SMN2 gene is nearly identical to SMN1, with the difference resulting in exclusion of exon 7. Nusinersen is a splice switching oligonucleotide (SSO) targeted to SMN2 that blocks an intronic splicing silencer (ISS) element in SMN2 preventing exon 7 exclusion, resulting in the expression of the functional, full-length survival motor neuron protein from SMN2. In preclinical work, the therapeutic strategy proved remarkable for restoring the tail and ear necrosis phenotypes of Smn1−/−; SMN2+/+ mice [Citation54]. Pharmacokinetic analysis verified the candidate ASO with widespread CNS distribution after intrathecal administration in rodents and nonhuman primates without activation of gliosis markers [Citation53]. Phase I clinical trial showed that the therapeutic strategy was well tolerated in patients with type II and III SMA [Citation55]. In Phase II an open-label study of patients with type I SMA demonstrated improved Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND) scores for drug treated children compared to placebo [Citation56]. On December 23, 2016, nusinersen (Spinraza) became the first FDA approved drug for the treatment of SMA [Citation57].
SOD1 Amyotrophic Lateral Sclerosis: An ASO strategy for treating SOD1 ALS is presently under development. Mutations in SOD1 account for approximately 10% of all familial ALS cases and reducing the abundance of the toxic SOD1 protein is hypothesized to be beneficial for these patients with SOD1 ALS and the strategy may be effective for treating sporadic ALS cases as well [Citation58]. Preclinincal testing demonstrated widespread uptake in transgenic SOD1G93A rats for SOD1 ASO (ISIS 333611) delivered to CSF, and target engagement [Citation59]. Phase I clinical testing included a randomized placebo control study of SOD1 ALS patients demonstrated no toxicities and no serious adverse events for up to 3 mg ISIS 333611 infused to CSF [Citation60]. However, the Phase I trial of ISIS 333611 did not reduce the abundance of the mutant SOD1 protein, and phase I clinical trials are now ongoing using a reformulated version of the SOD1 ASO (BIIB067 or IONIS-SOD1Rx) [Citation61].
C9ORF72 Amyotrophic Lateral Sclerosis/frontotemporal dementia (FTD): C9ORF72 ALS is caused by a GGGGCC repeat expansion in intron 1 of the C9ORF72 gene [Citation62,Citation63]. Repeat expansion in C9ORF72 results in repeat associated non-AUG (RAN) translation in six reading frames, and aggregation of C9ORF72 mRNA in nuclear foci [Citation64-Citation66]. Targeting of expanded C9ORF72 in neurons differentiated from patient-derived induced pluripotent stem cells (iPSCs) using ASOs targeting the expanded repeat reduced toxic RNA aggregations, rescued the expression of five genes associated with C9ORF72 mutation, and rescued glutamate excitotoxicity, without lowering the abundance of the C9ORF72 mRNA [Citation67]. Another study that used ASOs to target expanded C9ORF72 in ALS/FTD patient derived fibroblasts was unable to demonstrate rescue of gene expression characteristic of disease determined by transcriptome analysis, and concluded that the result was likely due to targeting of only the sense C9ORF72 strand [Citation68]. However, the strategy of targeting C9ORF72 in mice proved to be effective for improving ALS phenotypes and well tolerated. Lowering nonmutant mouse C9orf72 using an ASO strategy did not result in ALS-related phenotypes (locomotion, anxiety, coordination, grip strength) [Citation68], while targeting expanded human C9ORF72 expressing 450 repeats in a BAC C9ORF72 mouse model with an ASO improved anxiety and cognitive function/learning and memory-related phenotypes of ALS (marble burying, and three maze tests) [Citation69]. This established proof-of-concept for targeting C9ORF72 in ALS in future clinical research.
Alzheimer's disease/tauopathies: Elevated tau expression is characteristic of multiple neurodegenerative disease including Alzheimer's disease (AD), FTD, and others, resulting in pathological aggregates known as neurofibiliary tangles. When delivered by ICV injection, an ASO targeting the mouse microtubule associated protein tau gene Mapt was effective for lowering tau abundance throughout the CNS without altering multiple behavioral readouts [Citation70]. The study also demonstrated that reduction of tau expression by the ASO could protect against picrotoxin-induced seizures using EEG recordings to determine hyperexcitability-related spike frequencies, and pentylenetetrazole-induced seizures recorded using a videotape system [Citation70]. In a second study the same group of investigators evaluated a tau ASO in a mouse model expressing human tau with a P301S mutation (PS19) [Citation71]. Treatment of 9-month-old PS19 mice resulted in reduced total and phosphorylated tau, reduced neuronal tau aggregations in the piriform cortex, and reduced loss of hippocampal neurons. Tau ASO also significantly lengthened survival of PS19 mice from 312 to 348 days (median survival) [Citation71]. Finally, a single intrathecal bolus of tau ASO in cynomolgus monkeys resulted in significant tau reduction in multiple CNS tissues evaluated at 6 wks post treatment [Citation71]. ASOs for Alzheimer's disease are also in development that target the apolipoprotein E (APOE) gene, and have promise for improving learning and memory readouts in AD mice (TgCRND8 mice harboring mutant human APP695) [Citation72].
Huntington Disease: HD is caused by a CAG repeat expansion in an encoded region of the HTT gene beginning at the 18th codon, resulting in a gain of toxic function with a loss-of-function playing a potential role. Discussion on the preclinical development of an ASO targeting HTT expression was provided above in the section entitled ASOs and Allelic Specificity. A phase I/II double-blind placebo controlled clinical trial for testing of IONIS-HTTRx (ISIS 443139), a second generation gapmer HTT ASO, is now ongoing (clinicaltrials.gov).
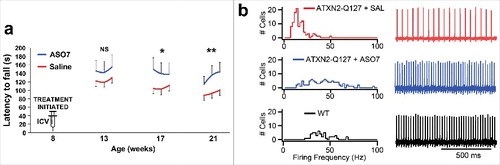
Spinocerebellar ataxia type 2: Spinocerebellar ataxia type 2 is caused by a CAG repeat expansion in an encoded region located in exon 1 of the ATXN2 gene. Beginning with 152 ASOs targeting ATXN2, we performed in vitro screening followed by in vivo by treating SCA2 mice transgenic for ATXN2 by ICV injections of ASOs, leading to the identification of ASO7. ASO7 lowered ATXN2 expression in SCA2 mouse cerebella by >60 percent for up to 13 wks treatment, and did not activate markers of gliosis. We then tested ASO7 in SCA2 mouse trials using two models, including Pcp2-ATXN2-Q127 transgenic mice with ATXN2 expression driven by the Purkinje cell protein 2 (Pcp2) promoter for strong expression in Purkinje cells, and BAC-ATXN2-Q72 mice that have ATXN2 driven by its own promoter which may be more “SCA2-like”. We demonstrated that the expression of six genes that we had previously shown were reduced in the cerebella of SCA2 mice [Citation73] were restored after treatment with ASO7 [Citation43]. We also showed that the rotarod motor phenotypes of SCA2 mice were delayed, and that the intrinsic Purkinje cell firing frequency was normalized by ASO7 treatment [Citation43]. Example data from our study for Pcp2-ATXN2-Q127 treated with ASO7 are presented in . These findings were replicated in both SCA2 mouse models. The robust modification of SCA2 mouse molecular, motor, and neurophysiological phenotypes by ASO7 provided robust proof-of-concept supporting ongoing additional preclinical testing that we expect will result in a reformulated lead ASO suitable for clinical trial for SCA2.
Figure 3. ATXN2 ASO7 improved SCA2 mouse phenotypes. a) SCA2 mice (Pcp2-ATXN2-Q127) were treated ICV with ASO7 at 8 weeks of age then motor behavior was tested on the accelerating rotarod. Values shown are means and standard deviations for three daily trials per week of testing (NS = non-significant; * = p < 0.05; ** = p < 0.01). b) At the endpoint of the experiment in A, Purkinje cell firing frequencies were determined in using extracellular recordings from acute cerebellar slices. Normal firing frequencies were recorded from ASO7 treated mice. Firing frequency distributions are shown on the left and representative firing traces for 1s of recording on the right.
Targeting the ATXN2 gene may also be an effective therapeutic strategy for treating TDP-43 ALS. By performing a screen for genes modifying Tdp-43 toxicity in yeast, Aron Gitler's group discovered yeast null for Atxn2 had improved survival. Sequencing of the ATXN2 gene in ALS patients revealed that intermediate CAG repeat expansions in ATXN2 increased the risk for ALS [Citation74]. This suggested that targeting normal ATXN2 might be therapeutic for ALS. As proof-of-concept, Gitler's group then performed a genetic interaction study by crossing TDP-43 transgenic mice with our Atxn2 knockout mice demonstrating mice null for Atxn2 had significantly improved survival in a dose dependent manner: mice null for both Atxn2 alleles had improved survival by tens of days, while those null for both Atxn2 alleles had survival extended by hundreds of days [Citation75]. Next the group showed that TDP-43 mice treated with an ASO targeting Atxn2 also had significantly improved survival much like TDP-43 mice null for Atxn2 [Citation75]. Reduction of Atxn2 expression also improved gait scores and reduced TDP-43 inclusions [Citation75].
Duchenne muscular dystrophy: DMD is an X-linked recessive disorder caused by mutation in the DMD gene encoding dystrophin, the largest known gene in the genome. A subset of DMD mutations cause dystrophin truncations. For some of these, the exclusion of exon 51 predicts dystrophin proteins with long open reading frames, but with partially impaired function related to the excluded exon. Eteplirsen is a morpholino-based ASO that interacts with the DMD pre-mRNA at exon 51 causing exon 51 exclusion [Citation76]. It was approved by the FDA for DMD in 2016. Approximately 14% of DMD cases may be treatable by this strategy [Citation77].
Concluding remarks
Few RNA-targeting therapeutics have been approved by the FDA, and there are only a handful for use in human neurodegenerative disease, including nusinersen and eteplirsen. These successes show that we are at a new horizon in the development of well-tolerated drugs effective for treating neurological disease. A strong outlook is also supported by promising new preclinical and clinical trial outcome data for RNA-targeted therapeutics with CNS activities.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
A portion of this work was supported by grants R21NS081182 and R37NS033123 from the National Institutes of Neurological Disorders and Stroke (NINDS).
Additional information
Funding
References
- Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. 2010;50:259–293.
- Dallas A, Vlassov AV. RNAi: a novel antisense technology and its therapeutic potential. Med Sci Monit. 2006;12(4):RA67–74.
- Gragoudas ES, Adamis AP, Cunningham ET, Jr., et al. Pegaptanib for neovascular age-related macular degeneration. N Eng J Med. 2004;351(27):2805–2816.
- Lebruska LL, Maher LJ, 3rd. Selection and characterization of an RNA decoy for transcription factor NF-kappa B. Biochemistry. 1999;38(10):3168–3174.
- Boudreau RL, Davidson BL. Generation of hairpin-based RNAi vectors for biological and therapeutic application. Methods Enzymol. 2012;507:275–296.
- Yin H, Kanasty RL, Eltoukhy AA, et al. Non-viral vectors for gene-based therapy. Nat Rev Genet. 2014;15(8):541–555.
- Tahara K, Hashimoto W, Takeuchi H. Inhalation properties and stability of nebulized naked siRNA solution for pulmonary therapy. Chem Pharm Bull (Tokyo). 2016;64(1):63–67.
- Keiser MS, Boudreau RL, Davidson BL. Broad therapeutic benefit after RNAi expression vector delivery to deep cerebellar nuclei: implications for spinocerebellar ataxia type 1 therapy. Mol Ther. 2014;22(3):588–595.
- Hocquemiller M, Giersch L, Audrain M, et al. Adeno-associated virus-based gene therapy for CNS diseases. Hum Gene Ther. 2016;27(7):478–496.
- Ramachandran PS, Keiser MS, Davidson BL. Recent advances in RNA interference therapeutics for CNS diseases. Neurotherapeutics. 2013;10(3):473–485.
- Gray SJ, Nagabhushan Kalburgi S, McCown TJ, et al. Global CNS gene delivery and evasion of anti-AAV-neutralizing antibodies by intrathecal AAV administration in non-human primates. Gene Ther. 2013;20(4):450–459.
- Deyle DR, Russell DW. Adeno-associated virus vector integration. Curr Opin Mol Ther. 2009;11(4):442–447.
- Fire A, Xu S, Montgomery MK, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391(6669):806–811.
- Almeida MI, Reis RM, Calin GA. MicroRNA history: discovery, recent applications, and next frontiers. Mutat Res. 2011;717(1-2):1–8.
- Kung JT, Colognori D, Lee JT. Long noncoding RNAs: past, present, and future. Genetics. 2013;193(3):651–669.
- Gao WY, Han FS, Storm C, et al. Phosphorothioate oligonucleotides are inhibitors of human DNA polymerases and RNase H: implications for antisense technology. Mol Pharmacol. 1992;41(2):223–229.
- Crooke ST, Lemonidis KM, Neilson L, et al. Kinetic characteristics of Escherichia coli RNase H1: cleavage of various antisense oligonucleotide-RNA duplexes. The Biochemical journal. 1995;312(Pt 2):599–608.
- Marwick C. First “antisense” drug will treat CMV retinitis. JAMA. 1998;280(10):871.
- Crooke ST. Vitravene–another piece in the mosaic. Antisense Nucleic Acid Drug Dev. 1998;8(4):vii–viii.
- Burnett JC, Rossi JJ. RNA-based therapeutics: current progress and future prospects. Chem Biol. 2012;19(1):60–71.
- Sune-Pou M, Prieto-Sanchez S, Boyero-Corral S, et al. Targeting splicing in the treatment of human disease. Genes (Basel). 2017;8(3):E87.
- Keeling KM, Xue X, Gunn G, et al. Therapeutics based on stop codon readthrough. Annu Rev Genomics Hum Genet. 2014;15:371–394.
- Adams BD, Parsons C, Walker L, et al. Targeting noncoding RNAs in disease. J Clin Invest. 2017;127(3):761–771.
- Soutschek J, Akinc A, Bramlage B, et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432(7014):173–178.
- Kleinman ME, Yamada K, Takeda A, et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008;452(7187):591–597.
- Deng Y, Chen J, Zhao Y, et al. Transdermal delivery of siRNA through Microneedle Array. Sci Rep. 2016;6:21422.
- Crooke ST, Wang S, Vickers TA, et al. Cellular uptake and trafficking of antisense oligonucleotides. Nat Biotechnol. 2017;35(3):230–237.
- Geary RS, Norris D, Yu R, et al. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliv Rev. 2015;87:46–51.
- Detzer A, Overhoff M, Mescalchin A, et al. Phosphorothioate-stimulated cellular uptake of siRNA: a cell culture model for mechanistic studies. Curr Pharm Des. 2008;14(34):3666–3673.
- Prakash S, Malhotra M, Rengaswamy V. Nonviral siRNA delivery for gene silencing in neurodegenerative diseases. Methods Mol Biol. 2010;623:211–229.
- Niu S, Zhang LK, Zhang L, et al. Inhibition by multifunctional magnetic nanoparticles loaded with Alpha-synuclein RNAi plasmid in a Parkinson's disease model. Theranostics. 2017;7(2):344–356.
- Shyam R, Ren Y, Lee J, et al. Intraventricular delivery of siRNA nanoparticles to the central nervous system. Mol Ther Nucleic Acids. 2015;4:e242.
- Malhotra M, Tomaro-Duchesneau C, Saha S, et al. Intranasal delivery of chitosan-siRNA nanoparticle formulation to the brain. Methods Mol Biol. 2014;1141:233–247.
- Rietwyk S, Peer D. Next-generation lipids in RNA interference therapeutics. ACS Nano. 2017;11(8):7572–7586.
- Godfrey C, Desviat LR, Smedsrod B, et al. Delivery is key: lessons learnt from developing splice-switching antisense therapies. EMBO Mol Med. 2017;9(5):545–557.
- Medina-Kauwe LK. Development of adenovirus capsid proteins for targeted therapeutic delivery. Ther Deliv. 2013;4(2):267–277.
- Medina-Kauwe LK, Maguire M, Kasahara N, et al. Nonviral gene delivery to human breast cancer cells by targeted Ad5 penton proteins. Gene Ther. 2001;8(23):1753–1761.
- DeVos SL, Miller TM. Antisense oligonucleotides: treating neurodegeneration at the level of RNA. Neurotherapeutics. 2013;10(3):486–497.
- Ward AJ, Norrbom M, Chun S, et al. Nonsense-mediated decay as a terminating mechanism for antisense oligonucleotides. Nucleic Acids Res. 2014;42(9):5871–5879.
- Carroll JB, Warby SC, Southwell AL, et al. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene / allele-specific silencing of mutant huntingtin. Mol Ther. 2011;19(12):2178–2185.
- Southwell AL, Skotte NH, Bennett CF, et al. Antisense oligonucleotide therapeutics for inherited neurodegenerative diseases. Trends Mol Med. 2012;18(11):634–643.
- Kordasiewicz HB, Stanek LM, Wancewicz EV, et al. Sustained therapeutic reversal of Huntington's disease by transient repression of huntingtin synthesis. Neuroendocrinology. 2012;74(6):1031–1044.
- Scoles DR, Meera P, Schneider MD, et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature. 2017;544:362–366.
- Duyao MP, Auerbach AB, Ryan A, et al. Inactivation of the mouse Huntington's disease gene homolog Hdh. Science. 1995;269(5222):407–410.
- Nasir J, Floresco SB, O'Kusky JR, et al. Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81(5):811–823.
- Zeitlin S, Liu JP, Chapman DL, et al. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat Genet. 1995;11(2):155–163.
- Kiehl TR, Nechiporuk A, Figueroa KP, et al. Generation and characterization of Sca2 (ataxin-2) knockout mice. Biochem Biophys Res Commun. 2006;339(1):17–24.
- Huynh DP, Maalouf M, Silva AJ, et al. Dissociated fear and spatial learning in mice with deficiency of ataxin-2. PLoS One. 2009;4(7):e6235.
- Moore LR, Rajpal G, Dillingham IT, et al. Evaluation of Antisense Oligonucleotides Targeting ATXN3 in SCA3 Mouse Models. Mol Ther Nucleic Acids. 2017;7:200–210.
- Gagnon KT, Pendergraff HM, Deleavey GF, et al. Allele-selective inhibition of mutant huntingtin expression with antisense oligonucleotides targeting the expanded CAG repeat. Biochemistry. 2010;49(47):10166–10178.
- Hu J, Matsui M, Gagnon KT, et al. Allele-specific silencing of mutant huntingtin and ataxin-3 genes by targeting expanded CAG repeats in mRNAs. Nat Biotechnol. 2009;27(5):478–484.
- Rigo F, Seth PP, Bennett CF. Antisense oligonucleotide-based therapies for diseases caused by pre-mRNA processing defects. Adv Exp Med Biol. 2014;825:303–352.
- Rigo F, Chun SJ, Norris DA, et al. Pharmacology of a central nervous system delivered 2'-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J Pharmacol Exp Ther. 2014;350(1):46–55.
- Hua Y, Sahashi K, Hung G, et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010;24(15):1634–1644.
- Chiriboga CA, Swoboda KJ, Darras BT, et al. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology. 2016;86(10):890–897.
- Finkel RS, Chiriboga CA, Vajsar J, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. 2016;388(10063):3017–3026.
- Aartsma-Rus A. FDA approval of nusinersen for spinal muscular atrophy makes 2016 the year of splice modulating oligonucleotides. Nucleic Acid Ther. 2017;27(2):67–69.
- Rotunno MS, Bosco DA. An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesis. Front Cell Neurosci. 2013;7:253.
- Winer L, Srinivasan D, Chun S, et al. SOD1 in cerebral spinal fluid as a pharmacodynamic marker for antisense oligonucleotide therapy. JAMA Neurol. 2013;70(2):201–207.
- Miller TM, Pestronk A, David W, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 2013;12(5):435–442.
- Schoch KM, Miller TM. Antisense oligonucleotides: translation from mouse models to human neurodegenerative diseases. Neuroendocrinology. 2017;94(6):1056–1070.
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuroendocrinology. 2011;72(2):245–256.
- Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuroendocrinology. 2011;72(2):257–268.
- Taylor JP, Brown RH, Jr., Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539(7628):197–206.
- Mori K, Arzberger T, Grasser FA, et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013;126(6):881–893.
- Zu T, Liu Y, Banez-Coronel M, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A. 2013;110(51):E4968–4977.
- Donnelly CJ, Zhang PW, Pham JT, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuroendocrinology. 2013;80(2):415–428.
- Lagier-Tourenne C, Baughn M, Rigo F, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A. 2013;110(47):E4530–4539.
- Jiang J, Zhu Q, Gendron TF, et al. Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuroendocrinology. 2016;90(3):535–550.
- DeVos SL, Goncharoff DK, Chen G, et al. Antisense reduction of tau in adult mice protects against seizures. J Neurosci. 2013;33(31):12887–12897.
- DeVos SL, Miller RL, Schoch KM, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med. 2017;9(374):eaag0481.
- Hinrich AJ, Jodelka FM, Chang JL, et al. Therapeutic correction of ApoER2 splicing in Alzheimer's disease mice using antisense oligonucleotides. EMBO Mol Med. 2016;8(4):328–345.
- Dansithong W, Paul S, Figueroa KP, et al. Ataxin-2 regulates RGS8 translation in a new BAC-SCA2 transgenic mouse model. PLoS Genet. 2015;11(4):e1005182.
- Elden AC, Kim HJ, Hart MP, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466(7310):1069–1075.
- Becker LA, Huang B, Bieri G, et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature. 2017;544:367–371.
- Nelson SF, Crosbie RH, Miceli MC, et al. Emerging genetic therapies to treat Duchenne muscular dystrophy. Curr Opin Neurol. 2009;22(5):532–538.
- Lim KR, Maruyama R, Yokota T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des Devel Ther. 2017;11:533–545.