
ABSTRACT
Cancer metastasis is defined as the dissemination of malignant cells from the primary tumor site, leading to colonization of distant organs and the establishment of a secondary tumor. Metastasis is frequently associated with chemoresistance and is the major cause of cancer-related mortality. Metastatic cells need to acquire the ability to resist to stresses provided by different environments, such as reactive oxygen species, shear stress, hemodynamic forces, stromal composition, and immune responses, to colonize other tissues. Hence, only a small population of cells has a metastasis-initiating potential. Several studies have revealed the misregulation of transcriptional variants during cancer progression, and many splice events can be used to distinguish between normal and tumoral tissue. These variants, which are abnormally expressed in malignant cells, contribute to an adaptive response of tumor cells and the success of the metastatic cascade, promoting an anomalous cell cycle, cellular adhesion, resistance to death, cell survival, migration and invasion. Understanding the different aspects of splicing regulation and the influence of transcriptional variants that control metastatic cells is critical for the development of therapeutic strategies. In this review, we describe how transcriptional variants contribute to metastatic competence and discuss how targeting specific isoforms may be a promising therapeutic strategy.
1. Introduction
In recent years, intensive studies have contributed to advances in early cancer detection and in the treatment of surgically resected primary tumors and adjuvant therapy [Citation1,Citation2]. These efforts increased survival rates for the majority of patients diagnosed during the initial stages of cancer [Citation1,Citation2]. However, metastatic cancer treatment faces resistance to chemotherapeutic compounds, making it necessary to understand the underlying mechanisms of the metastatic cascade [Citation1,Citation2]. In this sense, metastasis is responsible for 90% of cancer-related mortality and morbidity in patients [Citation2].
Metastasis is a multistage process in which malignant cells disseminate from the primary site and grow on distant organs, leading to new tumors [Citation3]. Nonetheless, malignant cell transformation is not sufficient for the acquisition of metastatic competence [Citation3]. Metastasis mainly occurs due to genome instability, the loss of effective DNA repair, checkpoint control failure and gene expression regulation [Citation4]. Hence, genome comparisons between primary tumor and distant metastasis show distinct mutations in metastatic cells compared with the primary tumors [Citation4].
Recently, several studies have established the importance of alternative transcription and alternative splicing in promoting the initiation and maintenance of cancer cells [Citation5–Citation10]. The use of alternative promoters and/or alternative termination sites can result in multiples pre-mRNA that undergoes splicing, generating a huge diversity of transcripts from a single gene [Citation6]. During splicing, premature mRNA (pre-mRNA) is processed and exons are assembled together to compose a mature mRNA, while the remaining introns are subsequently degraded [Citation11]. Additionally, some sequences can be alternatively included (or excluded) and differentially combined to give rise to different mature mRNA isoforms via a process named alternative splicing [Citation11]. This event may have different consequences in the final product: (i) the alternative splicing does not alter the reading frame, and the resulting in-frame protein may have (or not) a different function from canonical protein, or (ii) alternative splicing causes a frameshift, which leads to the generation of a premature termination codon (PTC) that is directed to nonsense-mediated decay (NMD), a different protein arrangement or a truncated protein production [Citation12,Citation13]. It is estimated that reading frame alteration occurs approximately in 50% of all alternative splicing events [Citation14].
In this sense, alternative splicing provides proteomic diversity through the production of multiple mRNA transcripts from protein-coding genes [Citation11]. More than 95% of human genes are differentially spliced depending on the tissue/cell type or signal transduction in response to developmental stages and environmental stimuli [Citation11,Citation15,Citation16]. Currently, the role of alternative splicing has been associated with different diseases, and disturbances in alternative splicing regulation are considered a hallmark of cancer [Citation10,Citation15–Citation17].
This review is focused on describing the influence of alternative splicing during the metastatic cascade, focusing on current knowledge about alternative transcripts that may be intrinsically related to metastatic competence and how targeting specific isoforms that contribute to metastatic progression may be a promising therapeutic strategy.
2. General view of splicing
Alternative RNA isoforms are regulated by genetic and epigenetic mechanisms and can be formed by means of alternative promoters or by undergoing alternative splicing [Citation6]. In this section, we will briefly summarize these mechanisms, focusing on alternative splicing. For more information, please refer to [Citation6,Citation11,Citation18–Citation20].
The alternative transcription mechanism is defined by the use of alternative promoters and transcriptional termination sites, resulting in multiple pre-mRNA [Citation6]. Its regulation involves the presence of cis-regulatory elements, DNA methylation, histone modifications and chromatin remodeling [Citation6]. Transcription and RNA processing are highly interdependent events and occur simultaneously [Citation6,Citation11]. During transcription, RNA polymerase II (RNA Pol II) elongation produces pre-mRNA, which undergoes cotranscriptional splicing, resulting in mature mRNA, during which pre-mRNA processing introns are removed and exons are joined together [Citation6,Citation19]. However, some exons and introns can be alternatively spliced, generating different isoforms with distinct activities, and such transcriptome variability is observed at the protein level [Citation10]. This process is catalyzed by the spliceosome, a large complex of RNA composed of five small nuclear ribonucleoproteins (snRNPs; U1, U2, U4, U5 and U6) and several associated proteins cofactors, resulting in a highly dynamic conformation [Citation5,Citation21].
The spliceosome recognizes short consensus sequences, called splice sites, in nascent transcripts located at the intron-exon junctions, which are 5ʹ splice sites, 3’splice sites, and a branch point region near the 3’splice site [Citation5,Citation21]. These splice sites can be classified as ‘strong’ when they are similar to consensus sequences and, thus, more efficiently recognized [Citation21]. In general, cognate splicing factors tend to more efficiently recognize ‘strong’ splice sites, which leads to constitutive splicing [Citation21]. However, neighboring ‘strong’ and ‘weak’ splice sites compete with themselves for affinity with the splicing factors, and different positions of these splice sites can cause different alternative splicing outcomes [Citation21]. Thus, additional splicing regulation is performed by trans-acting factors and cis-regulatory sequences [Citation21]. Cis-regulatory sequences include exonic and intronic splicing silencers (ESSs and ISSs) and enhancers (ESEs and ISEs) ()) [Citation21]. It is common for alternative exons to have weaker splice sites and ESE, indicating an evolutionary advantage of the use of alternative exons [Citation22].
Figure 1. (a) Sequences located at the intron-exon junctions, termed 5ʹ and 3ʹ splice sites, together with branch point positioned near the 3ʹ splice site, are responsible for defining the intron and exon limits. These sequences are recognized by the spliceosome machinery and associated factors. Additional regulation of the site splice choice can be mediated by cis-regulatory sequences, including exonic and intronic splicing silencers (ESSs and ISSs) and enhancers (ESEs and ISEs), and trans-acting factors, such as serine- and arginine-rich (SR) proteins and heterogeneous nuclear ribonucleoprotein (hnRNP). These splicing factors are able to inhibit or promote the recognition of nearby splice sites affecting the activity of the spliceosome components, such as the snRNPs U1 and U2. (b) Representation of the five main alternative splicing events.
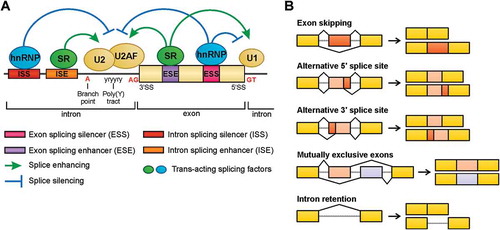
Trans-acting factors bind to cis-regulatory sequences to either promote or prevent spliceosome assemblement at an adjacent splice site [Citation5,Citation11,Citation21]. Most of these factors are serine- and arginine-rich (SR) proteins and heterogeneous nuclear ribonucleoprotein (hnRNP), and numerous tissue-specific factors and additional proteins have been described to stimulate or inhibit splicing ()) [Citation5,Citation11,Citation21]. SR proteins and hnRNPs are described to act as proto-oncogenes and tumor suppressors. For example, the RNA-binding protein hnRNP I is a pre-mRNA splicing regulator in alternative splicing and is related to biological processes, such as cell structure and motility, cell cycle, immunity and others [Citation23]. Its overexpression is observed in colorectal, brain, ovarian and breast cancers, and it has been related to tumor aggressiveness, especially in glioma and ovarian tumors [Citation23]. By contrast, hnRNP I inhibition decreases cell proliferation, migration and invasion in colorectal cancer cells [Citation23].
Another example is the SR splicing factor SRSF1, which is related to malignant transformation and is overexpressed in lung, breast and colon cancers [Citation24]. This protein leads to activation of the mTOR pathway, inducing the missplicing of MNK2 and RPS6KB1 and the alternative splicing of BCL2L11 and BIN1 to produce isoforms without pro-apoptotic activity [Citation24]. In addition, overexpression of SRSF3, SRSF6 and SRSF10 is also related to tumorigenesis [Citation24]. Although SR proteins generally act as oncogenes, hnRNPs can exert both roles, suppressing and promoting tumorigenesis [Citation24].
Alternative splicing regulation is typically influenced by three different aspects of co-transcriptional RNA processing: (i) CTD (RNA Pol II carboxy-terminal domain)-mediated recruitment, (ii) kinetic competition and (iii) chromatin structure [Citation21,Citation25]. In CTD-mediated recruitment, splicing factors are recruited to transcription sites by associating splicing factors with the CTD [Citation21,Citation25]. In kinetic competition, the RNA Pol II elongation rate influences the output timing of splice sites and regulatory sequences from the nascent pre-mRNA [Citation21,Citation25]. In this sense, a fast RNA Pol II elongation rate limits kinetic competition and favors the inclusion of exons flanked by strong splice sites, while weaker splice sites are favored by a slow elongation rate [Citation25]. The coupling of transcription and splicing processes is suggested to be associated with the effects of histone modification and nucleosome positioning [Citation25]. In fact, histone post-translational modifications are related to active transcription, such as histone H3K36me3, which is enriched in exonic compared with intronic regions and is implicated in the alternative splice site choice [Citation21,Citation25]. In human kidney cancer, mutations in SETD2, a histone methyltransferase that specifically catalyzes H3K36me3, promote increased chromatin accessibility and lead to widespread changes in RNA processing and aberrant splicing [Citation26]. In this tumor, chromatin accessibility is pronounced in the proximal region of misspliced exons, where the deficiency of H3K36me3 is related to decreased nucleosome occupancy of these exons [Citation26]. In fact, evidence provides a key role for nucleosomes in splice site recognition and exon definition [Citation25]. Moreover, the roles of nucleosome are enriched in exons, preferentially exons flanked by weaker splice sites [Citation25]. Thus, nucleosome positioning contributes not only to exon definition but also to splicing regulation [Citation25].
Currently, five main types of alternative splicing are recognizable:(i) exon skipping; (ii) alternative 5ʹ splice site; (iii) alternative 3ʹ splice site; (iv) mutually exclusive exons; and (v) intronic retention ()) [Citation11]. In vertebrates and invertebrates, the most prevalent alternative splicing type is exon skipping, which represents approximately 30% of all alternative splicing events [Citation11]. However, mutations in splice sites and regulatory sequences may result in aberrant intron retention with pathological consequences [Citation13]. Actually, intron retention is enriched in tumor-suppressor genes when somatic single-nucleotide variants (SNVs) are present [Citation27]. Consequently, these transcripts undergo proteins truncation or NMD by the generation of the PTC [Citation27].
A subset of retained introns has been investigated recently, which was called exitrons because they have features of both introns and protein-coding exons [Citation28]. Exitrons are a class of alternatively spliced introns defined as a protein-coding intron sequence within protein-coding exons, whose regulation is made by internal splice site motifs in the exon [Citation12,Citation28,Citation29]. They have different characteristics from others introns, such as the absence of stop codons, high GC content, a preferential size that corresponds to multiples of three, and prevalence of synonymous substitutions, also frequently observed in exons [Citation29]. Unlikely of retained conventional introns transcripts that are inclined to stay in the nucleus, retained exitrons transcripts are transported to the cytoplasm and translated, being enriched in disordered regions of the codified proteins [Citation12,Citation28,Citation29]. Marquez et al. [Citation28] investigated the role of exitrons in ERBB2-positive breast cancer and identified 29 cases with differential exitron splicing between breast cancer and normal tissue, including the suggested tumor suppressors CIZ1, ZNF238 and EXT1.
Alternative splicing precision, efficiency and plasticity generate the vast transcriptomic and proteomic diversity required for eukaryotic cell biology. However, disturbances in this regulation can lead to incorrect decisions and, consequently, disease development.
3. Metastasis and implications of alternative splicing for the metastatic cascade
Cancer metastasis is described as a dissemination of malignant cells from a primary tumor site to different organs through the blood and lymphatic circulation. In 1889, Stephen Paget described metastasis not as a random process, but rather, he proposed that cells of certain tumors have a preferential affinity to specific organs [Citation2,Citation30]. Paget hypothesized the ‘seed-and-soil’ theory of metastatic outgrowth, where the establishment and growth of malignant cell (‘seeds’) are dependent on particular organs (‘soil’) to survive and proliferate [Citation2,Citation30].
Successful metastasis consists of achieving the five basic steps of the invasion-metastasis cascade: local invasion, intravasation, circulation survival, extravasation and colonization (). In epithelium-derived cancers, the cells are maintained together by cell-to-cell adherence and by adherence to the extracellular matrix (ECM) [Citation2].
Figure 2. Cell spreading from the primary tumor involves the acquisition of capabilities to disrupt the basement membrane, invade the surrounding tissue (stroma) and enter the blood circulation. After that, the spreading cells in the circulation must survive in the hostile environment until they can reach distant organs and establish a niche to grow and form a new lesion (secondary tumor).
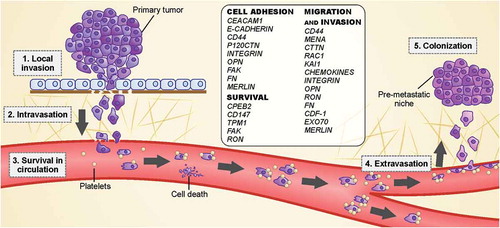
Once normal cells have lost their adhesion to other cells or the basement membrane, the detached cells undergo anoikis, a mechanism of apoptosis induced by cell detachment [Citation2]. However, metastatic cells are able to overcome this phenomenon by altering the signaling pathways responsible for anoikis regulation, including NF-κB, small GTPases and its effectors, some receptor tyrosine kinases and cytoplasmic kinases, and epithelial-to-mesenchymal transition (EMT) factors [Citation31].
Overcoming anoikis is followed by the invasion of surrounding tissues of the primary tumor, which is initiated by metastatic cells migrating through the basement membrane and ECM [Citation32]. In this sense, malignant cells need to acquire invasive features that are frequently associated with the suppression of epithelial properties and the acquisition of mesenchymal markers to disrupt cell-cell and cell-ECM adhesion [Citation33]. This plastic program, called EMT, is reversible and may provide the ability to the tumor cell to travel to metastatic sites and establish the growth of a secondary tumor [Citation33]. During EMT, epithelial cells acquire a mesenchymal cell phenotype that consists of a (i) loss of adhesion structures, (ii) increased cell death resistance, (iii) altered polarity, (iv) cytoskeleton reorganization, and (v) enhanced migratory capacity [Citation33,Citation34]. However, the reversion of disseminated tumor cells into an epithelial phenotype is required for seeding in distant organs and the formation of a secondary tumor through a process called mesenchymal-epithelial transition (MET) [Citation35]. Some studies using breast and pancreatic cancer cell lines have shown that EMT is not indispensable for metastasis establishment but contributes largely to its aggressiveness by increasing resistance to anticancer therapy [Citation36].
Invasive cells undergo actin cytoskeleton reorganization and the formation of membrane protrusions enriched in filamentous actin (F-actin) that, along with adhesive interactions, are crucial for invasion and migration through the stroma [Citation2,Citation36]. Malignant cells exit the tumor as single cells or as clusters and enter the vasculature, spreading throughout the whole body () [Citation36]. Additionally, platelets and granulocytes are recruited to form niches that contribute to metastatic progression by forming a shield around tumor cells, promoting adhesion to endothelial cells and increasing endothelial permeability [Citation37].
These tumor cells halt their circulation in capillaries at distant sites and start to extravasate into the target organ parenchyma, where these cells adopt strategies to survive and initiate colonization () [Citation3,Citation36].
Although metastasis itself is believed to be an inefficient process, the dissemination of cells from a one-centimeter-sized primary tumor corresponds to an infiltration rate of one million cells/day in the circulatory system [Citation32]. However, due to the limited compatibility of distal sites (‘seed-and-soil’ theory), only < 0.1% of tumor cells reach an appropriate site and are able to survive in the new ‘soil’ and avoid immune surveillance [Citation32]. This low metastatic competence can be explained by the death of tumor cells due to their inability to resist the many types of stresses, such as reactive oxygen species, shear stress, hemodynamic forces, stromal composition, detachment, and immune response [Citation38,Citation39]. For these reasons, only a small population of tumor cells are able to exert an adaptive response to the environmental stress, thus carrying a metastasis-initiating potential [Citation38].
Many splice events can be used to distinguish normal and tumor tissue and contribute to an adaptive response of tumor cells and the success of the metastatic cascade. These contributions frequently alter normal biological processes, such as cell cycle control, cytoskeleton organization, migration, cell-cell adhesion and insulin signaling [Citation40]. Splicing regulatory proteins have been the main investigated targets because the abnormal activity of these effectors could be the key related to pathological splicing alterations. Many articles have provided excellent reviews of master changes affecting the splicing program [Citation24,Citation41,Citation42]. Moreover, cellular features acquired by tumor cells drive a splice program shift that could be exerted even beyond the sole aberrant function of splicing factors. In the next section, we will address the most relevant genes related to metastasis with reported transcriptional variants that contribute to tumor cell adaptation. A brief summary is represented in .
Table 1. Summary of the main mentioned isoforms and representative examples of therapeutic approaches to targeting them. M.C., metastasis contribution (↑ high contribution; ↓ low contribution).
3.1. E-cadherin
E-cadherin is an adhesion regulator that is also known as an important tumor suppressor, which, along with catenins, maintains cell-cell adhesion and cell-matrix interactions [Citation30]. During EMT, cadherin type switching occurs, in which epithelial E-cadherin is downregulated and the mesenchymal marker N-cadherin is upregulated [Citation43]. Nonetheless, a study has demonstrated that E-cadherin downregulation can result from missplicing [Citation44]. Sharma et al. [Citation44] showed that E-cadherin exon 11 is skipped, resulting in a frameshift and PTC that will direct the transcript to NMD. The splicing factor SRSF2 has been suggested to be responsible for the aberrant splicing of E-cadherin, since in cancer cells with skipped exon 11, SRSF2 is upregulated, and its silencing increases wild-type E-cadherin RNA levels [Citation44].
In gastric cancer, the lack of the final 83 base pairs of exon 8 (1054del83) results in a nonfunctional transcript [Citation45]. Although it does not result from abnormal activity of SRSF2, this phenomenon is related to decreased acetylation of histones H3 and H4K16Ac on the E-cadherin gene surrounding the alternative exon 8 [Citation45]. H3K36me3 surrounding the exon 8 region is correlated with abnormal splicing of this same region, which indicates that epigenetic modifications patterns can be critical in gastric cancer [Citation45].
3.2. CEACAM1
Carcinoembryonic antigen-related cell adhesion molecule-1 (CEACAM1) is a transmembrane glycoprotein involved in cell-cell adhesion, and its abnormal expression is associated with a variety of human malignancies [Citation46,Citation47]. CEACAM1 alternative splicing generates 12 isoforms, resulting in three C2-like domains and forming isoforms that differ in the length of the extracellular region [Citation48]. Furthermore, CEACAM1 splicing is able to encode two major cytoplasmic domains that differ by the inclusion (termed long (-L) tail) or exclusion (termed short (-S) tail) of exon 7, respectively, forming CEACAM1-L and CEACAM1-S [Citation47]. Dery et al. [Citation47] observed that stress-induced cytoplasmic accumulation of hnRNP A1 leads to the formation of CEACAM1-L in breast cancer and, consequently, leads to an inappropriate deregulation of CEACAM1-S expression. The prevalence of CEACAM1-L isoform expression is associated with accelerated metastasis progression and a lower survival rate in patients with colon tumors [Citation49]. By contrast, transfection of CEACAM1-S has been described to mediate the reversion of mammary tumor cells to a normal phenotype, as noted by acini structures with the central lumen formation via the apoptosis mechanism [Citation50].
3.3. CD44
The cell surface glycoprotein CD44 also plays an important role in cell adhesion and EMT, in addition to being involved in proliferation, migration and invasion during metastasis, in an extracellular matrix ligand-dependent manner [Citation51,Citation52]. CD44 pre-mRNA has 20 exons and shows several isoforms encoded by standard exons 1–5 (c1-c5) and 16–20 (c6-c10), whereas exons 6–15 (v1-v10) are considered variant exons that are regulated by alternative splicing [Citation52]. In this sense, the inclusion and exclusion of a variant exon represent tissue and environment-specific-dependent factors, for which a single or combined exons can be selected to compose the final transcript [Citation52]. Through these variable combinations, CD44 can interact with different cognate receptors and cooperate in the activation of several signaling pathways that contribute to tumor progression and dissemination [Citation53]
In mesenchymal cells, variant exons are skipped, and only standard exons are included (termed standard isoform or CD44s) [Citation52]. Nonetheless, proliferating and epithelial cells predominantly show variant-exon-encoded sequences (termed the variant isoform or CD44v) [Citation52]. Both CD44s and CD44v may be associated with cellular malignancy depending on their partners, type and tumor stage. For example, CD44v is enriched in normal breast tissue and in lower-grade breast tumors; however, a switch in the expression of CD44v rather than CD44s accelerates the EMT process and contributes to cancer progression [Citation34,Citation54]. Furthermore, CD44s has been reported to be an integral component of invadopodia, which contribute to extracellular matrix degradation and the dissemination of metastatic cells from the primary site to distant organs [Citation55]. In pancreatic cancer, CD44s is related to the EMT phenotype, collaborating to participate in invasiveness and chemoresistance in vivo [Citation56].
In addition, the switch from CD44v to CD44s has been correlated to the mesenchymal phenotype and is prevented by the splicing factors ESRP1 and ESRP2 [Citation41,Citation54,Citation57]. ESRP1 and ESRP2 are involved in alternative splicing events of epithelial cell phenotypes, and their targets are frequently involved in cell-cell adhesion, cell motility, actin cytoskeletal organization and EMT [Citation41,Citation58]. These splicing factors mediate the variable exon inclusion and, consequently, the generation of CD44v transcripts [Citation41,Citation54,Citation57]. Nonetheless, ESRP1 and ESRP2 downregulation by TGF-β regulates CD44 alternative splicing and also leads the phosphorylation of the T179 residue of SMAD3, mediating an interaction of this latter gene product with PCBP1, an RNA-binding protein [Citation59]. This SMAD3-PCBP1 complex interacts with the variable exon region of CD44 pre-mRNA, inhibiting spliceosome assembly and favoring the expression of the mesenchymal isoform CD44s [Citation59]. Finally, another splicing factor, hnRNP M, competes with ESRP1 to bind to the cis-regulatory element in the CD44 pre-mRNA, leading to the generation of CD44s by skipping the variable exons and, thus, promoting EMT and contributing to breast cancer metastasis [Citation60].
Although CD44s is more frequently associated with cellular malignancy, CD44v has also been described in tumorigenesis. A study revealed that a subpopulation of breast cancer stem cells with increased expression of ESRP1 and CD44v (mainly CD44v3-10) had enhanced metastatic capacity to invade lung tissue [Citation61]. Furthermore, in the lung microenvironment, OPN interacts with CD44v through the V3-V7 exon region, but not CD44s, promoting cancer cell invasion in an ESRP1-dependent manner and lung colonization [Citation61]. In fact, CD44-dependent chemotaxis is induced by OPN, while CD44-dependent cell aggregation can be stimulated by hyaluronan, a glycosaminoglycan that belongs to the extracellular membrane [Citation51,Citation62].
CD44v has been correlated to malignancy in colorectal cancer stem cells, where CD44v6 expression and its cooperation with c-MET drive a highly tumorigenic population with metastatic potential and have been correlated with a poor prognosis [Citation63]. This expression is regulated by HGF, OPN and SDF-1, which induce the expression of EMT-related genes [Citation63]. Furthermore, CD44v6 acts as a coreceptor and activates c-MET and VEGFR-2 in response to HGF and VEGF-A, inducing endothelial cell migration, sprouting and tubule formation [Citation64]. Some SR proteins (SRSF1, SRSF6 and SRSF9) also regulate CD44 splicing, inhibiting formation of the CD44v6 isoform, while SRSF2 reduces the expression of V6 but increases the expression of isoforms containing exons V6-10 and V6, V8-V10 [Citation65]. CD44v6 has been suggested to be important for drug resistance since its knockdown prevents colony formation in response to several antitumoral drugs [Citation66]. In addition, CD44v6 knockdown reduces the expression of mesenchymal markers, such as vimentin, Snail1/2 and Twist [Citation66]. Another study showed that hnRNP LL is involved in the regulation of CD44 splicing in colorectal cancer since the knockdown of this splicing factor leads to increased expression of CD44v6 [Citation67]. In colon cancer cell lines and clinical samples, the downregulation of hnRNP LL is associated with EMT and contributes to the increased cell invasion ability in these samples [Citation67].
3.4. Mena
Human enabled homolog (Mena), a relative of enable/vasodilator-stimulated phosphoprotein (ENA/VASP), is an actin regulatory protein that influences cell adhesion, protrusion, motility and invasion [Citation68]. The upregulation of this protein has been linked to cell malignancy in breast, pancreas, colon and cervical cancers [Citation68]. In agreement with this statement, Mena deficiency is related to reduced invasion, intravasation and metastatic dissemination [Citation68]. The variant isoforms of Mena that are produced during tumorigenesis are Mena11a, MenaΔv6 and MenaINV [Citation68–Citation71]. The first one is an epithelial-associated isoform that has 21 additional amino acids in the EVH2 domain, which is encoded by exon 11a and is described in highly cohesive and well differentiated tumor phenotypes, whereas is downregulated in invasive cells and ones that have undergone EMT [Citation68,Citation69]. In fact, the epithelial regulatory splicing factors ESRP1 and ESRP2 regulate exon 11a inclusion, which contributes to epithelial phenotype and the formation of tumors with a differentiated morphology [Citation68]. Furthermore, Mena11a contributes to survival and resistance to phosphatidylinositol-3-kinase (PI3K) inhibition therapies [Citation72]. MenaΔv6 lacks exon 6 and is related to increased invasiveness and drug resistance of breast tumors in the absence of Mena11a [Citation71]. In pancreatic cancer, TGF-β1 differentially regulates MenaΔv6 upregulation along with the downregulation of Mena11a, promoting EMT [Citation70].
MenaINV has an additional 19 amino acids codified by the ‘INV’ exon. Its expression is not enriched in highly proliferative cells and reveals an inverse correlation of E-cadherin expression in breast cancer [Citation69]. In primary tumor from breast cancer, this isoform contributes to a discohesive morphology and the loss of epithelial cell-cell contact, as well as increased cell motility [Citation73]. The expression of MenaINV is responsible for sensitizing tumor cells to EGF, eliciting tumor cells to a more invasive phenotype in low EGF microenvironments [Citation68,Citation69]. Additionally, a subpopulation of highly motile cancer cells can respond to the chemotactic stimuli provided by macrophages through EGF and promotes CSF1 production by tumor cells [Citation74,Citation75]. This motile tumor cells show increased expression of both Menaclassic and MenaINV and decreased expression of the Mena11a isoform [Citation75,Citation76]. Mena-expressing tumor cells and macrophages mutually exert chemotaxis to each other so tumor cells can co-migrate with macrophages towards blood vessels and, consequently, intravasate at sites enriched with perivascular macrophages [Citation74,Citation75]. These intravasation sites are called tumor microenvironment of metastasis, and the number of these regions are positively correlated with the MenaINV abundance [Citation75,Citation76]. Furthermore, macrophages also can facilitate cancer cell intravasation by physically interacting with perivascular macrophages and tumor cells, increasing the formation of invadopodia by inducing RhoA activity and leading to macrophage-dependent transendothelial migration [Citation76]. In this case, MenaINV was observed to stimulate the maturation of invadopodia by promoting the phosphorylation state of CTTN at the tyrosine 421 residue and inhibit normal dephosphorylation mediated by phosphatase PTP1B [Citation77].
3.5. Rac1
Rac1 belongs to the Rho family of small GTPase and acts by cycling between an inactive GDP-bound state to an active GTP-bound state in response to different stimuli [Citation78]. Rac1 is crucial in the regulation of the cell cycle, cytoskeletal dynamics and formation of filopodia, lamellipodia and stress fibers [Citation79]. Rac1 mRNA is alternatively spliced by the inclusion of exon 3b, generating Rac1b, which shows the insertion of 19 amino acids at the end of the switch II region, leading to a predominantly GTP-bound state, preventing the inactive state [Citation78]. This mRNA isoform is overexpressed in colorectal, lung, pancreatic, thyroid and breast cancers [Citation79,Citation80].
In breast cancer, hnRNP A1 prevents Rac1b expression, whereas the activation of SRSF1 (through phosphorylation mediated by SR kinases (SRPK1)) in colorectal cancer is related to increased expression of Rac1b [Citation81,Citation82]. A study has shown that upon EGF treatment, hnRNP A1 is ubiquitylated and retained in the cytoplasm, preventing its repressive splicing activity [Citation83]. Furthermore, EGF signaling stimulates the action of SR kinases, which in turn leads to the activation of SR proteins that mediate exon 3b inclusion, generating Rac1b and promoting cell migration [Citation83]. Conversely, Rac1b promotes growth transformation, causing a loss of density and anchorage-dependent growth regulation in the NIH3T3 cell line in comparison to Rac1 [Citation79].
However, Rac1b lacks some Rac1 functions and is incapable of forming lamellipodia or interacting with PAK1 and Rho-GDI [Citation78]. Rac1b also induces NF-κB activation, which stimulates proliferation and apoptosis resistance in neoplastic thyroid cells [Citation78,Citation84].
3.6. CTTN
Cortactin (CTTN) is a cytoplasmic F-actin binding protein and acts as a Src substrate, participating in the assembly and cross-linkage of F-actin, both of which are controlled Rac1 activity [Citation85,Citation86]. Branched actin offers structural support for the plasma membrane and, in response to a specific stimulus, provides lamellipodial protrusions by branching of existing filaments, which requires the activity of CTTN to promote Arp2/3 complex activation leading to actin nucleation [Citation85]. CTTN has three transcriptional isoforms: CTTN-A, CTTN-B and CTTN-C [Citation23]. CTTN-A is the dominant isoform and has a region that consists of six motifs containing tandem amino acids repeats, where the 3rd to 6th units are codified by exons 8, 9, 10 and 11, respectively [Citation87]. These six units represent the full functional domain responsible for filamentous actin-binding, cross-linking and cell migration [Citation23,Citation87]. CTTN-B and CTTN-C have five and four CTTN repeats, respectively, and bind to F-actin less robustly [Citation87]. In this sense, cells overexpressing CTTN-B and CTTN-C exhibit reduced migration in comparison to cells overexpressing CTTN-A due to the absence of the full F-actin binding domain [Citation23]. In colorectal cancer, hnRNP I promotes the inclusion of exon 11, leading to the overexpression of CTTN-A in comparison to normal tissue and increasing cell migration and invasion [Citation23]. However, when hnRNP I was knocked down, the total mRNA of CTTN remains the same, although CTTN-A levels decline, indicating the preference of hnRNP I in the generation of CTTN-A in colorectal cancer [Citation23]. This isoform is also overexpressed in B cells of patients with chronic lymphocytic leukemia, which suggests that it is related to disease progression [Citation88].
3.7. CPEB2
In triple-negative breast cancer (TNBC), a subpopulation with anoikis resistance has been observed to exhibit altered pre-mRNA splicing of cytoplasmic polyadenylation element binding 2 (CPEB2), a translation regulator [Citation89]. CPEB2 is associated with TNBC resistance by regulating cellular stress response [Citation89]. CPEB2 is localized to RNA stress granules, where it binds to specific transcripts, maintaining them in an inactive state [Citation89]. During the stress response, dissociation of the CPEB2 target transcript occurs, and the transcript is translated [Citation89]. However, altered alternative splicing of CPEB2 leads to CPEB2B isoform expression in a subpopulation of TNBC cells [Citation89]. This alternative isoform includes exon 4, is sufficient to promote anoikis resistance and is associated with higher metastasis and mortality rates [Citation89].
3.8. KAI1
The transmembrane glycoprotein Kangai1 (KAI1, also known as CD82) has been described as a metastasis suppressor, and its loss or downregulation is correlated with a poor prognosis in prostate, lung, gastric, breast, bladder, colon, skin, cervix, endometrium and ovary tumors [Citation90,Citation91]. The expression of KAI1 reduces cell motility and invasiveness through the crosstalk with integrin signaling, adherens and tight junctions components [Citation92]. A splice variant of KAI1 (KAI1-SP) has been detected in gastric cancer and associated with a poor prognosis and increased invasive potential in vivo [Citation90,Citation91]. KAI1-SP lacks exon 7 encoding 28 amino acids from the distal part of the second extracellular loop (ECL2) to the proximal part of the fourth transmembrane region [Citation90,Citation91]. Colon carcinoma cell lines transfected with KAI1-SP display a greater binding capacity to ECM components, such as type IV collagen and fibronectin, compared to those transfected with wild-type KAI1 [Citation91]. In this study, KAI1-SP transfected cells showed increased motility mediated by cell-ECM adhesion components (especially fibronectin) and a weaker interaction with integrin α3β1, the major receptor for laminin [Citation91]. Successful cell migration depends on an intermediate level of cell adhesiveness to the underlying matrix, but stronger cellular attachment may be related to cell immobilization [Citation90]. In this sense, fibronectin and its receptors have been reported to act not only in attachment and cellular adhesion but also to provide signals that regulate cellular locomotion [Citation91]. Therefore, in vitro ovarian cancer cells expressing KAI1-SP display decreased cell adhesiveness to vitronectin and type I collagen and increased cell motility mediated by αvβ3 integrin and vitronectin [Citation90]. Finally, KAI1-SP fails to promote E-cadherin-mediated cell-to-cell contacts, which may prevent cell spreading [Citation90].
3.9. CD147
CD147, called basigin, is a highly enriched glycosylated transmembrane protein found on the plasma membrane of various human cancer cells and is related to the increased tumorigenicity and invasive potential of such cells [Citation93]. The most known activity is related to the expression, secretion, and activity regulation of matrix metalloproteinases (MMPs) [Citation94]. A study has shown that CD147 expression increases cancer cell viability by modulating intercellular contacts through fibronectin assembly and preventing anoikis by downregulating Bim via the MAP kinase pathway [Citation93]. CD147 has four isoforms, termed CD147/Bsg-1 to 4. Among them, the most predominant is CD147/Bsg-2, while CD147/Bsg-1 is retina-isoform specific and CD147/Bsg-3 and −4 were first identified in human endometrial cells and cervical carcinoma cell lines [Citation94]. In hepatocellular carcinoma, the expression levels of CD147/Bsg-2 and −3 were higher compared to adjacent normal tissue [Citation94]. However, these isoforms have contrasting effects, while CD147/Bsg-2 overexpression promotes cell proliferation and invasiveness, CD147/Bsg-3 is responsible for inhibiting the aforementioned processes, possibly by hetero-oligomerization of CD147/Bsg-3 with CD147/Bsg-2, thus preventing the invasive role of the latter [Citation94].
3.10. RON
The recepteur d’origine nantais (RON) proto-oncogene encodes a transmembrane tyrosine kinase receptor and is activated by interacting with MSP, leading to RON kinase activity upregulation [Citation95]. RON downstream signaling involves the regulation of many intracellular processes, such as EMT stimulation, cell proliferation, survival, migration, invasion, chemoresistance and angiogenesis [Citation95]. In normal and malignant cells, two transcripts encoded by different RON gene promoters are observed. These transcripts are the short-form RON (SF-RON) and the full-length RON, although different isoforms can be generated through post-transcriptional modifications (mostly alternative mRNA splicing as well as protein truncation) [Citation95].
SF-RON is generated by an alternative transcription start site controlled by a second promoter located between introns 8 and 10 [Citation95]. This isoform is constitutively active with ligand-independent activity [Citation96]. In ovarian cancer, SF-RON expression is related to increased tumor growth and spreading to the abdominal cavity [Citation96]. Additionally, the RONΔ165 variant is formed by an in-frame exon 11 and results in the accumulation of the single-chain pro-RONΔ165 [Citation97]. Furthermore, RONΔ165 is constitutively phosphorylated by oligomerization due to the deletion of exon 11 and is involved in mobile and invasive phenotypes [Citation97,Citation98].
RONΔ170 is a defective variant, the alternative splicing of which generates the loss of exon 19, which leads to the absence of kinase activity, inhibiting the oncogenic activity mediated by another oncogenic variant, RONΔ160 [Citation97,Citation98]. RONΔ160 results from an in-frame deletion of 109 amino acids encoded by exons 5 and 6 [Citation95,Citation97]. RONΔ160 and RONΔ155 are associated with tumor growth and metastasis, and RONΔ160 leads to the accumulation of β-catenin and the increased gene expression of β-catenin targets [Citation97,Citation98]. RONΔ155 carries deletions of exons 5, 6 and 11, and due to the loss of exon 11, the translated protein is retained in the cytoplasm [Citation95,Citation97,Citation98].
Moreover, RONΔ110 is a constitutively active proteolytic truncated isoform that is present on the cell surface of cancer cells overexpressing RON, but its role in tumorigenicity it is not completely understood [Citation95,Citation97,Citation98]. Among the splicing factors, SRSF1 is responsible for inhibiting the inclusion of exon 11, while hnRNP A2/B1 and hnRNP H are also described to mediate glioma cell transformation through exclusion of exon 11 from the proto-oncogene RON [Citation99–Citation101].
3.11. TPM1
Tropomyosin-1 (TPM1) belongs to a family of actin-binding proteins that provide stability to actin filaments in non-muscle cell cytoskeleton and the contractility of skeletal and smooth muscle cells, as well as affecting integrin expression and activity [Citation102–Citation104]. Previous studies have shown that TPM1 is downregulated in bladder, breast, colorectal, esophageal, oral cavity and oropharynx, ovarian and renal cancers [Citation102,Citation105]. Apoptosis evasion and cell invasiveness may be associated with disrupted cytoskeleton structures and components. Hence, TPM1 seems to have a crucial role as a potential tumor suppressor by mediating cytoskeletal reorganization and preventing metastasis [Citation102,Citation105,Citation106]. In this sense, the restoration of TPM1 expression in breast cancer cells promotes cytoskeletal reorganization and recovery of cell-ECM interactions, reestablishing cell adhesion and promoting anoikis of detached cells [Citation102].
The TPM1 isoforms are classified according to the number of amino acids in length as high molecular weight (HMW) or low molecular weight (LMW) and result from alternative promoter usage [Citation103]. The use of exon 1a and 2a (or 2b) generates HMW, whereas exon 1b is used for the LMW isoform [Citation103]. Furthermore, alternative splicing of exon 6 may occur via mutually exclusive splicing of 6a (or 6b) and exon 9, and 14 isoforms have been identified to date [Citation103,Citation106]. Among them, TPM1δ and TPM1γ are HMW expressed in normal human breast epithelial cells (but downmodulated in human breast cancer), the product of which is incorporated into stress fibers [Citation106]. In fact, the downregulation of HMW isoforms is related to increased anoikis resistance, and tumorigenesis and malignant cells often undergo to a switch from HMV to LMW [Citation103,Citation104]. By contrast, the HMW isoform TPM1λ is upregulated in malignant breast cell lines and shows an inverse correlation with stress fiber formation [Citation106].
Other tropomyosin isoforms, such as the LMW TM5NM1 and TM4, have been related to metastasis progression in melanoma and breast cancer, respectively [Citation103]. This information indicates that their regulation is crucial for the fate of malignant cells that can potentially undergo anoikis when specific isoforms are expressed or possess increased invasiveness through isoform switching.
3.12. IL-7
Interleukin 7 (IL-7) is a cytokine related to tumor development and progression and, together with its receptor IL-7R, has been identified in several cancers, such as colorectal, renal, breast, lung and central nervous system cancers [Citation107]. An IL-7 variant lacking exon 5 (IL-7δ5) has been related to promoting cellular proliferation and cell cycle progression in breast cancer via PI3K/Akt pathway activation [Citation107]. Furthermore, IL-7δ5 is able to induce EMT through the PI3K/Akt pathway, in addition to exogenous IL-7δ5, that promotes downregulation of E-cadherin and upregulation of N-cadherin expression [Citation107].
3.13. Chemokines
Chemokines are chemotactic cytokines that are subdivided into four main classes (CC, CXC, C and CX3C) according to the position of the first two cysteine residues [Citation108]. Considering the tumor microenvironment, chemokines can be secreted by tumor cells or stromal and immune cells [Citation108]. Specific chemokines drive immune cell homing to the tumor microenvironment, influencing immune-cell infiltration and affecting tumor progression [Citation108]. Furthermore, chemokines can target tumor and stromal cells when these cells express chemokine receptors and are able to respond to different chemokine gradients [Citation108]. In this sense, chemokines affect cell proliferation, invasiveness, cancer stem-like cell properties and metastasis [Citation108]. Several chemokine receptors have been described to undergo aberrant splicing, which may contribute to tumor progression and metastasis.
CXCR3 is a receptor for CXC chemokines and is activated by the ligands CXCL4/PF4, CXCL9/MIG, CXCL10/IP10 and CXCL11/IP9/I-TAC [Citation109]. The activity observed for this receptor is related to its two major splice isoforms: CXCR3-A, formed by exon 1 and exon 3 and considered the classical isoform, and CXCR3-B, which is assembled by exon 2 and exon 3 [Citation110]. Interestingly, these isoforms have opposing functions, CXCR3-A is a pro-proliferative isoform that is related to invasion and migration promotion in several tumors, while CXCR3-B has no chemotactic functions and inhibits migration [Citation109,Citation110]. CXCR3-A, activated by its ligand CXCL9, has been shown to change the levels of ERK1/2 phosphorylation, a member of MAPK signaling, leading to the overexpression of MMP2 and MMP9 and, consequently, contributing to the invasion and metastasis of CD133+ liver cancer cells [Citation111]. CXCR3-A is expressed at higher levels than CXCR3-B in malignant cells and supports cell proliferation and migration, invasiveness and metastasis, whereas CXCR3-B reduces the metastatic potential and colonizing capacity [Citation109,Citation110]. However, in bulk populations, CXCR3-B inhibits cell proliferation and shows a supportive role for cells with cancer stem-like properties [Citation110]. In this sense, CXCR3-B may contribute to resistance in therapies that target highly proliferative cells after those stem cells have achieved a low proliferation rate, contrasting with general populations of malignant cells [Citation110].
SDF-1, also known as CXCL12, is a chemokine member of the CXC class and the ligand of the chemokine receptors CXCR4 and CXCR7 [Citation112]. Serum SDF-1 concentration is positively correlated with the occurrence of distant metastasis in patients with gastric cancer [Citation113]. This chemokine has six alternative spliced isoforms, comprising SDF-1α, SDF-1β, SDF-1γ, SDF-1δ, SDF-1ε and SDF-1θ [Citation112]. In tumors, there is an increase in the expression of the SDF-1α G801A polymorphism associated with lymph node metastasis in patients with colorectal cancer, whereas SDF-1β is involved in angiogenesis and, together with SDF-1γ, is related to tumor size in patients with metastasis [Citation112].
3.14. Integrin
Integrins are the major cell surface receptors and are responsible for mediating cell adhesion to the ECM [Citation114]. They are crucial for the maintenance of homeostatic balance in the cell, but are also involved in inflammation, leukocyte trafficking, tumor metastasis and wound repair [Citation114]. Integrins can transmit an ‘outside-in’ signal through the plasma membrane, as well as an ‘inside-out’ signal, functioning as an important intermediate a bidirectional communication between cells and their microenvironment [Citation114]. In tumors, integrins regulate different processes through the tumor cell-microenvironment interface, influencing the cancer response and progression [Citation114].
The α6 integrin subunit (CD49f) can heterodimerize with either β1 or β4, forming α6β1 or α6β4 integrins and then acting as the main laminin receptor [Citation115,Citation116]. The α6 subunit undergoes alternative splicing, forming two cytoplasmic isoforms: α6A, which is enriched in epithelial populations, and α6B, which is enriched in mesenchymal populations, where the latter has an important role in slow-cycling stem cell functions and is directly involved in the stem cell phenotype; it is related to a poor outcome in TNBC patients [Citation116]. In this sense, VEGF signaling can affect the alternative splicing of the α6 subunit, promoting the repression of α6A and favoring the expression of α6B via induction of the oncogene GLI1 and transcriptional inhibition of the splicing factor ESRP1 [Citation116]. However, isoform α6A knockdown reduces tumor growth, although it does not affect α6B levels [Citation115]. In fact, α6A has been described to have a positive proliferative effect in colorectal cancer, which is involved in the Wnt/β-catenin pathway, where the knockdown of α6A leads to enhanced phosphorylation of β-catenin by GSK3β [Citation115].
Integrin β1 alternative splicing leads to the formation of four isoforms, called β1A, β1B, β1C and β1D [Citation117]. The expression levels of isoforms β1B, β1C and β1D are downregulated in several tumors, whereas β1A is related to focal adhesion formation, cell spreading and motility [Citation117]. A study has shown that β1A expression is related to metastasis in pancreatic cancer, while Fyn inhibition and hnRNP E1 overexpression promote the splicing of β1C, which reduces the migration and invasiveness of tumor cells [Citation117].
3.15. FAK
Focal adhesion kinase (FAK) mediates cell communication with the extracellular environment, triggering intracellular signaling in response to ECM remodeling, nutrient availability and growth factors [Citation118]. Furthermore, its action is involved in cell adhesion, motility, survival, vasculogenesis and proliferation [Citation118]. During tumorigenesis, FAK overexpression is frequently observed in breast, colon, cervical, ovary and prostate cancers, and it is associated with metastasis [Citation118,Citation119]. When detached cells undergo anoikis, FAK is proteolyzed by caspase-3 and −7, and anoikis evasion is recurrently associated with increased FAK levels or mutations [Citation119]. FAK shows four traditional alternatively spliced exons: 13, 14, 16 and 31 [Citation119,Citation120]. However, in human breast cancers, an aberrant alternative isoform that lacks exon 26, a region that contains the caspase-3/caspase-7-like cleavage motif, was observed [Citation119,Citation120]. Thus, although this isoform has the same kinase activity as wild-type FAK, the missing segment causes aberrant FAK to become resistant to caspase-mediated proteolysis, promoting tumor cell survival [Citation119].
3.16. OPN
Osteopontin (OPN) is a glycophosphoprotein that is secreted by several cells and has been broadly overexpressed in plasma and tumor tissues, as well as described to promote tumor progression and metastasis [Citation121]. OPN interacts with different integrins and CD44, activating signal transduction pathways involved in cell proliferation, adhesion, migration and invasion [Citation121].
The primary transcript of OPN can be alternatively spliced in the upstream region of the central integrin binding domain and the C-terminal CD44 binding domain to form (i) the full-length OPN-a, which consists of 7 exons; (ii) the isoform OPN-b, which lacks exon 5; (iii) and OPN-c, which is generated by the lack of exon 4 [Citation121].
A study has shown that OPN-a and OPN-b both act through integrin signaling, while OPN-c has an integrin-independent function [Citation122]. Furthermore, OPN-b augments cell migration in vitro compared with OPN-c [Citation122]. In the same study, stable cells transfected with OPN-b showed increased cell adhesion, proliferation and invasion, while OPN-c transfected cells exhibited decreased cell invasion and migration but enhanced cell detachment [Citation122]. OPN-c is present in 75% of breast cancers, and it is directly related to anchorage-independent growth and metastasis [Citation123]. In non-small cell lung cancer, OPN-a is highly expressed compared to OPN-b and OPN-c [Citation124]. However, both OPN-b and OPN-c are overexpressed in gastric cancer and showed anti-apoptotic activity when induced by H2O2, but not OPN-a [Citation125]. This pro-survival effect is promoted by OPN-b, possibly through the interaction with CD44v6 and CD44v7, which triggers downstream anti-apoptotic signaling [Citation125].
In addition, OPN-a overexpression has been related to the promotion of proliferation, migration, adhesion and invasion in A549 cells through the interaction with ανβ3 integrin [Citation124]. This interaction allows lung cancer cells to invade the bone matrix during metastasis, and once OPN-a is silenced, significant inhibition of bone matrix attachment occurs [Citation124]. In thyroid cancer cell lines, increased levels of OPN-a have been related to cell growth and enhancement of cell migration and motility, possibly through the induction of MMP2 and MMP9 activity, leading to extracellular matrix degradation [Citation126]. In papillary thyroid carcinoma, OPN-a is correlated with extrathyroid extension, vascular invasion and BRAFV600E mutation, which are significantly related to a poor prognosis [Citation126].
Finally, in breast cancers, all tumor-derived OPN transcriptional isoforms increase the tumor expression of the immunosuppressive cytokines TGF-β1 and MCP-1 [Citation127]. Additionally, these tumor-derived OPN transcripts lead to the inhibition of pro-inflammatory cytokines TNF-α and increase anti-inflammatory IL-10 production by monocytes [Citation127]. This mechanism may be related to alternative immune cell activation, which is associated with tumor-associated macrophages (TAMs) and possibly contributes to immunological evasion mediated by OPN [Citation127].
3.17. FN
Fibronectin (FN) is a glycoprotein present in the plasma and ECM that has cell adhesiveness and migration activities [Citation128]. Fibronectin is formed by the combination of three repeating domains, called Types I, II and III [Citation128]. The Type III domain is alternatively spliced by the inclusion of extra domain-A (EDA) or extra domain-B (EDB) and a variable type III connecting segment (IIICS), which can be excluded (or included) completely or partially [Citation128,Citation129]. FN can also be found either as a plasmatic FN (pFN) or as a cellular FN (cFN), and only the latter comprises variable proportions of EDA or EDB [Citation128].
In normal tissues, these isoforms are not detected by antibodies, but they were highly representative in ECM components of aggressive tumors and other diseases that exhibit considerable tissue remodeling, serving as markers for tumor angiogenesis [Citation128,Citation130]. EDA-FN has been related to the promotion of neo-lymphangiogenesis in colorectal carcinoma cells through integrin α9 activation, in addition to the stimulation of angiogenesis, cell adhesion, migration and proliferation in malignant tumors [Citation131]. EDB-FN can be regulated by ECM stiffness, which mediates splicing through PI3K/Akt signaling activation, promoting the regulation of SR protein phosphorylation, such as SRp40 [Citation132]. The high level of proteolytic degradation and remodeling of the stroma surrounding the tumors alters the stiffness, enabling the invasion of migrating cells [Citation133]. This remodeling leads to the release of activated growth factors stored in the ECM, influencing cell signaling [Citation133]. Finally, in urinary bladder cancer, IIICS shows superexpression by 21.5-fold in invasive cells compared with noninvasive cells, as well as increased de novo O-linked glycosylation [Citation129,Citation134].
Another alternatively spliced FN isoform known as migration stimulating factor (MSF) is overexpressed by TAMs [Citation133,Citation135]. The MSF isoform consists of intron 12 retention followed by a two-step series of intra-intronic cleavage events, which results in a 70-kDa truncated protein [Citation133,Citation136]. Furthermore, MSF is highly chemotactic for monocytes and tumor cells, stimulating cell migration and possibly contributing to metastasis, which it is not observed for full-length FN [Citation135]. In established tumors, M2-like macrophages become TAMs and show poor cytotoxicity against tumor cells, acting in the production and secretion of chemotactic molecules that stimulate tumor cell migration to distant sites and metastatic niche preparation [Citation135]. MSF has been reported to be upregulated in several tumors, and in addition to TAMs, MSF is also expressed by neoplastic cells and vascular endothelial cells [Citation133,Citation136].
3.18. Tp53
The tumor suppressor TP53 is well known for its multiple roles in cell cycle arrest, genomic stability, DNA repair, apoptosis, autophagy, differentiation, stemness, migration, drug resistance, metabolism and angiogenesis [Citation137]. The relationship of TP53 with cancer is frequently associated with mutations that are located in the TP53 DNA binding domain (DBD) or its conformational mutants [Citation137]. TP53 encodes twelve isoforms through different mechanisms, which involve the usage of the alternative promoter and splicing: TAp53α, TAp53β, TAp53γ, Δ40p53α, Δ40p53β, Δ40p53γ, Δ133p53α, Δ133p53β, Δ133p53γ, Δ160p53α, Δ160p53β and Δ160p53γ [Citation137,Citation138]. TAp53 and Δ40p53 are long TP53 isoforms that contain the transactivation domain, which it is not present in either of the short isoforms Δ133p53 or Δ160p53 [Citation137]. Furthermore, β and γ have an additional domain with an unknown function and do not contain a canonical C-terminal oligomerization domain [Citation137,Citation138]. The isoform TAp53α is the canonical isoform expressed and found in normal tissues, but Δ133p53β has been shown to increase the risk of recurrence by eight times and the risk of death by three times in breast cancer [Citation139]. High levels of Δ133p53 positively regulate the cancer stem cell potential in the MCF-7 cell line, stimulating mammosphere formation and the expression of pluripotency and stemness regulators, such as SOX2, OCT3/4, NANOG and CD24/CD44 [Citation140]. Concomitantly, the metastatic capacity, invasiveness and chemoresistance are associated with increased Δ133p53 expression in different cancer cell lines, which is not associated with TP53 mutations; the depletion of Δ133p53 in wild-type TP53 eliminates the scattering and invasive activities, as well cancer stem cell formation [Citation139,Citation140].
3.19. CSF-1
Colony stimulating factor 1 (CSF-1) is a hematopoietic growth factor found in the bloodstream and has been related to the survival, differentiation and proliferation of mononuclear phagocytic lineages, as well as to macrophage spreading and motility [Citation141,Citation142]. CSF-1 is composed of at least 10 exons, and its mRNA undergoes alternative splicing, which it involved in the alternative use of a 3ʹ non-coding region represented by exon 9 and 10 [Citation142]. Exon 6 is also alternatively spliced, during which a large portion of the 5ʹ end is removed in one of the alternative transcripts [Citation142]. In addition, exon 6 has a variable length, depending on which of the two spliced acceptor sites is used, forming different transcripts with different roles [Citation142,Citation143]. After alternative splicing and differential post-translational proteolytic processing, CSF-1 generates three mature homodimeric proteins: a cell surface-associated glycoprotein (csCSF-1), a secreted extracellular matrix-anchored proteoglycan (spCSF-1) and a secreted soluble glycoprotein (sgCSF-1) [Citation141,Citation142]. spCSF-1 and sgCSF-1 are secreted isoforms and regulate cells nearby or at a distance, while cell surface isoforms act on local regulation [Citation141,Citation142].
CSF-1 has an important role in the chemotaxis and activation of macrophages, which are important for innate immunity and for tissue morphogenesis and/or remodeling [Citation141]. CSF-1 is related to cancer, and its activity has been associated with the recruitment and re-education of macrophages, which are also known as TAMs [Citation133,Citation144]. In fact, blood circulating monocytes are recruited locally and differentiate into TAMs in response to signals from the tumor microenvironment, such as chemokines and growth factors secreted by stromal and tumor cells [Citation133,Citation144]. TAMs are regarded to have an M2-like phenotype and are suggested to have pro-tumor effects due to their higher levels of anti-inflammatory cytokines, scavenging receptors, angiogenic factors and proteases, promoting an immunosuppressive microenvironment and contributing to tumor progression [Citation144,Citation145]. This ability results from to secretion of molecules that directly affect cell growth, motility, migration, angiogenesis, intravasation, invasion, inflammatory response, ECM remodeling and proliferation [Citation135,Citation145].
Different CSF-1 isoforms have been described to be associated with TAM activation [Citation145]. Elevated levels of CSF-1 have been detected in breast, ovarian, endometrial, cervical, and colorectal cancers, leukemia, lymphoid malignancies and myelodysplastic syndromes [Citation145]. Among the isoforms, sgCSF-1 is related to poor survival outcomes in patients with colorectal and breast cancer [Citation145]. One study detected more infiltrating TAMs in the microenvironment containing csCSF-1 and sgCSF-1 isoforms [Citation145]. Both isoforms were able to promote macrophage re-education, but they induced different spectra of M2 phenotype-related genes [Citation145]. In this sense, csCSF-1 is more related to the stimulation of TAM proliferation in the tumor microenvironment, whereas sgCSF-1 is more prone to the recruitment and induction of TAM differentiation [Citation145].
3.20. Prrx1
Alternative splicing of the paired-related homeobox transcription factor (Prrx1) results in two isoforms, termed Prrx1a and Prrx1b, which differ from each other at the C-terminus but are identical in the 199 N-terminal amino acids [Citation35,Citation146]. Both isoforms maintain the homeobox domain, but only Prrx1a has a conserved OAR domain with potentially important functions in transactivation [Citation35,Citation146].
In pancreatic carcinogenesis, Prrx1 isoform switching regulates epithelial and mesenchymal phenotypes in primary and metastatic cancer: Prrx1b is upregulated in primary tumor cells, where it promotes EMT, tumor invasion and differentiation [Citation35]. In addition, Prrx1b facilitates tumor cell invasion by upregulating HGF through signaling mediated by its receptor (c-MET), driving metastasis by promoting the invasion of tumor cells and EMT [Citation35].
However, higher levels of Prrx1a expression have been found in metastatic cells, contributing to MET, which is crucial to the colonization of distant organs after extravasation, as well as an increased capacity for anchorage-independent growth and self-renewal [Citation35]. Furthermore, knockdown of both isoforms in the KPC2 cell line decreased the invasiveness and self-renewal capacity, and in vivo transplantation generated a smaller primary tumor volume and a reduced number of circulating tumor cells [Citation35].
3.21. EGFR
Epidermal growth factor receptor (EGFR) is a transmembrane glycoprotein and a member of the ErbB receptor tyrosine kinase family of growth factor receptors [Citation147]. It is described as providing a substantial contribution to cell survival and proliferation motility [Citation147]. Its overexpression indicates a poor prognosis in breast, lung, liver, prostate and ovarian cancers [Citation147].
EGFR is composed of 30 exons that can be spliced to form EGFRv1, EGFRv2, EGFRv3, EGFRv4, EGFRvIII and de4 EGFR [Citation148–Citation150]. The EGFRvIII isoform results in a gain-of-function mutation that leads to genomic deletion of exons 2–7, and its expression is restricted to tumors, such as lung carcinoma, glioma, glioblastoma and head and neck squamous cell carcinoma [Citation150]. de4 EGFR forms a truncated EGF variant due to a deletion of exon 4, and it has been detected in glioma, prostate and ovarian cancers, and like EGFRvIII, de4 EGFR is also found exclusively in tumoral tissues [Citation150,Citation151]. Both de4 EGFR and EGFRvIII have minimal EGF-binding activity and ligand-independent autophosphorylation, exhibiting similarities in their biological activities and extracellular structures [Citation151]. Some de4 EGFR tyrosine residues are constitutively phosphorylated even without ligand activation, which leads to downstream signaling pathway activation [Citation147,Citation150]. In this sense, de4 EGFR promotes the upregulation of FAK, Src, ERK, Akt and MMP9, whereas E-cadherin is downregulated in ovarian cancer cells [Citation147]. Furthermore, de4 EGFR contributes to the increase in cell proliferation and malignant transformation, stimulating metastasis-promoting activities and higher cisplatin tolerance [Citation147].
3.22. Id1b
The inhibitor of differentiation (Id1) belongs to the bHLH family consisting of three other members, Id2 to Id4 [Citation152]. Its role in tumorigenesis, angiogenesis and metastasis has been described, and it is associated with a poor prognosis in patients with glioblastoma and breast cancer [Citation152]. Id1 has an important role in maintaining self-renewal and pluripotency in embryonic stem cells, and its role extends to cancer stem cells [Citation152]. The alternative splicing of Id1 forms two isoforms: the classical isoform, known as Id1a, containing 155 amino acids; and the alternative isoform, referred as Id1b, containing 149 amino acids [Citation152]. The difference between the two isoforms is found in the C terminal region, which alters their capacity to interact with specific transcription factors [Citation152]. Id1a is related to increased cell proliferation, whereas overexpression of Id1b reduces proliferation, promotes self-renewal, and leads to a stem cell-like phenotype, increasing the expression of cancer stem cell markers, such as ALDH1A1, Notch-1, Sca-1, Tert, Sox-2 and Oct-4 [Citation152]. Thus, Id1b may contribute to cell quiescence, a strategy adopted by many primary tumor or metastatic cells [Citation152,Citation153]. These quiescent cells are maintained in a non-proliferative state for long periods, and the awakening of these dormant cells leads to tumor progression and metastatic relapse [Citation153,Citation154].
3.23. XAF1
XIAP-associated factor 1 (XAF1) is a nuclear protein that when overexpressed leads to apoptosis and tumor growth inhibition [Citation155]. XAF1 splice variants have been detected in several cancer cell lines and are regulated by aberrant epigenetic modifications [Citation155]. The aberrant variants frequently contain a PTC, which leads to NMD [Citation155]. However, suppression of the NMD pathway may occur due to cellular stresses influenced by the tumor microenvironment, stabilizing the NMD-target mRNA [Citation155]. In this sense, the upregulated XAF1 variant was found to lack exon 5 and contain an extended region from exon 3, termed XAF1F [Citation155]. XAF1F harbors a PTC and was found in highly metastatic cell lines compared to parental cells and in peripheral blood containing circulating tumor cells from patients with gastric cancer, indicating a suppression of the NMD pathway and, consequently, an accumulation of XAF1F [Citation155]. Furthermore, although the function of XAF1F has not yet been reported, its mRNA expression levels in circulating tumor cells is correlated with venous invasion, lymph node metastasis, and the depth of tumor invasion [Citation155].
3.24. FAM3B
Family with sequence similarity 3 member B (FAM3B) is a member of the FAM3 family of cytokine-like genes and has been described to have at least seven alternatively spliced isoforms [Citation156]. Among them, FAM3B-258 is a non-secretory variant with 258 amino acids that is highly expressed in colon adenocarcinomas. It is related to poor cancer cell differentiation and has been shown to promote invasion and migration in vitro and metastasis in nude mice [Citation156]. FAM3B-258 promotion of invasion is regulated by Slug-dependent activation, which inhibits the expression of JAM and E-cadherin, leading to the breakdown of cell-cell interactions by downregulating cellular adhesion proteins [Citation156].
3.25. KLF6
The transcription factor Krüppel-like factor 6 (KLF6) encodes a zinc finger protein that regulates the cell cycle, differentiation, and apoptosis [Citation157]. KLF6 acts as a tumor suppressor and is inactivated in many cancers [Citation157]. KLF6 alternative splicing generates three cytoplasmic isoforms: KLF6-SV1, KLF6-SV2 and KLF6-SV3 [Citation158]. In prostate cancer, a germline single nucleotide polymorphism located in an intronic region of KLF6 is responsible for disruption of their own splicing regulation by creating a novel functional SRp40 DNA binding site and removing two overlapping SR protein sites [Citation158]. This process leads to increased expression of three alternatively spliced isoforms from KLF6 and an augmented risk of prostate cancer [Citation158].
KLF6-SV1 and KLF6-SV2 are described to antagonize the activity of wild-type KLF6 during p21 upregulation and to suppress cell proliferation [Citation158,Citation159]. Furthermore, KLF6-SV1 promotes metastasis, and its upregulation is considered to be a poor prognostic factor for patients with prostate, lung, ovarian and breast cancers [Citation159,Citation160]. Its expression is detected in both healthy and tumor tissues, but it is frequently upregulated in the latter [Citation159].
In breast cancer, KLF6-SV1 expression in primary tumors is correlated with an increased metastatic potential and EMT-like phenotype [Citation159]. Furthermore, overexpression of KLF6-SV1 leads to increased colony formation in tumorigenic and non-tumorigenic cell lines, promoting cell survival, migration and invasion [Citation159]. KLF6-SV1 knockdown decreases the metastatic capacity via diminished invasion and reduced expression of N-cadherin with concomitant E-cadherin overexpression, reverting the mesenchymal-like phenotype [Citation159].
3.26. Exo70
Exocyst is an octameric protein complex composed of Sec3, Sec5, Sec6, Sec8, Sec10, Sec15, Exo70 and Exo84 [Citation161]. This complex is crucial for exocytosis and is related to plasma membrane expansion during migration [Citation161,Citation162]. Furthermore, Exo70 is crucial for cell invasion by mediating MMP secretion at focal degrading sites [Citation162]. However, during the transition of the EMT state, the epithelial Exo70 isoform (Exo70-E) is switched to a mesenchymal Exo70 isoform (Exo70-M) by the splicing factor ESRP1 [Citation161]. Exo70-E differs from Exo70-M by an insertion of a portion of exon 8 generated by ESRP1-mediating alternative splicing [Citation161]. This switch does not affect exocyst complex function, but it is implicated to have different functions according to the assembled Exo70 isoform [Citation161].
When Exo70 knockdown cell lines are transfected with Exo70-M, these cells recover their invasive ability, while Exo70-E transfection fails to do the same [Citation161]. These observations were explained by the high invadopodial activity levels in cells expressing Exo70-M, whereas Exo70-E cells had lower invadopodial activity [Citation161]. Additionally, Exo70-M interacts with the Arp2/3 complex and stimulates actin branching, which is crucial for membrane protrusion formation during migration and invasion [Citation161].
3.27. Merlin
Moesin-ezrin-radixin-like protein (Merlin) is a tumor suppressor that participates in the regulation of cell proliferation, motility and survival [Citation163]. Merlin is codified by the Nf2 gene, which is heterozygous in mice and is related to the development of highly metastatic malignant tumors [Citation163]. Merlin is important for the prevention of tumorigenesis by promoting adherens junctions stabilization and controlling cell-cell contact mediated by cadherin and activation of Rac [Citation163]. Splicing variants of Merlin lacking exon 2 (MerlinΔ2), exon 3 (MerlinΔ3), or both of them (MerlinΔ2/3) are related to their inactivity as a tumor suppressor [Citation163]. In addition, another alternative splicing variant lacking exons 2, 3 and 4 (MerlinΔ2−4) was identified in hepatocellular carcinoma and in portal vein tumor thrombus [Citation163]. MerlinΔ2−4 overexpression promotes migration and invasion by upregulating Twist1, and it impairs the ability of wild-type Merlin to bind to β-catenin and Ezrin/Radixin/Moesin proteins [Citation163]. This isoform also leads to stemness activity by increasing spheroid formation and inducing the expression of stem-related genes, including Sox2, OCT4, Klf4, c-MYC and Nanog [Citation163].
3.28. CD99
CD99 is a transmembrane glycoprotein that is involved in the cell adhesion, migration, apoptosis, differentiation, activation and proliferation of lymphocytes, monocyte migration, transport of several transmembrane proteins, and intercellular adhesion between lymphocytes and endothelial cells [Citation164,Citation165].
CD99 produces two distinct transcripts: the full-length CD99 isoform (CD99wt) and a truncated shorter isoform (CD99sh) resulting from alternative splicing [Citation164,Citation165]. CD99sh carries a deletion of the intracytoplasmic fragment and an 18-bp insertion at the boundary of exons 8 and 9, which introduces an in-frame stop codon [Citation164]. Studies in breast cancer cell lines have revealed that CD99sh stimulates MMP9 activity and has an FN-augmented binding potential, in addition to increasing cell motility and invasiveness [Citation165]. The higher invasion capacity can result in diminished cell-cell adhesion in cells expressing CD99sh and an augmented interaction between FN-CD99sh [Citation165]. Furthermore, CD99sh upregulates the AP-1 signaling pathway, which mediates cell motility and MMP9 expression [Citation165]. The higher activity of proteolytic enzymes (such as MMP) allows ECM degradation, contributing to cell movement and invasion [Citation165].
In osteosarcoma and prostate cancer cells, CD99 isoforms have opposing effects: CD99wt-expressing cells show a less migrating phenotype, with organized actin filaments and greater collagen type I adhesion, laminin and fibronectin and sensitivity to anoikis [Citation164]; CD99sh activates c-Src kinase activity, which can act as a mediator of CD99 functions in stimulating cell motility [Citation164].
4. Therapy
The differences observed between the tumor and normal transcriptome are vast as a consequence of several changes in genomic and transcriptomic regulation. In summary, the major genomic and transcriptomic changes are as follows: (i) the presence of mutations in transcription factor binding and splice sites; (ii) mutations that generates aberrant proteins; (iii) different DNA arrangements and histone modifications; (iv) changes in chromatin accessibility; and (v) altered expression of splicing factors and transcription factors [Citation6]. In the past, drugs that act on specific cellular targets were used without regard for the aberrant isoforms generated during oncogenesis, mostly due to missing information regarding the isoform variants in a gene [Citation6,Citation166]. The differential expression of several isoforms encoded by the same gene might be a contributing factor to the observed variation in patient responses during drug treatment [Citation6,Citation166].
Studies have shown that the analysis of isoform-level expression offers an effective discrimination of cancer and non-cancer cells, providing a better identification of different subgroups within a cancer-type and allowing a robust signature to classify patients in molecular subgroups [Citation6,Citation167]. Furthermore, during tumor progression, alternative splicing occurs in a disease-specific manner and contributes to the disease outcome [Citation168]. The observation that some isoforms are detected only in tumor cells suggests that cancer-specific isoforms may be good diagnostic markers and therapeutic targets.
Validation of the isoform-specific expression and role in tumor cells highlights a therapeutic potential and an opportunity to develop drugs that target specific isoforms. However, it is important to maintain deep large-scale studies that provide information concerning whether an isoform is really cancer-specific or has a pro- or anti-oncogenic role, before developing a therapeutic design to ensure the correct drug-to-isoform (or drug-to-isoform product) assignment.
Some studies addressing cancer-specific isoforms as targets have shown promising results and are listed in . A successful example is EGFRvIII, which is the target of several immunotherapies for patients with glioblastoma, such as peptide vaccines, dendritic cell vaccination therapy, monoclonal antibodies and modified T cells [Citation169]. Among them, the peptide vaccine rindopepimut was approved by the US FDA based on phase I and II clinical trials showing a significantly higher survival rate in patients with glioblastoma expressing EGFRvIII [Citation169]. Advances have also been realized with cell therapy using the chimeric antigen receptor T (CAR-T) as an antitumor treatment, and CAR-T targeting the EGFRvIII isoform efficiently eliminates glioma cells [Citation169].
The modulation of alternative splicing may reduce the severity of disease and prevent metastasis and its incidence. In this sense, different approaches to perform this manipulation have been studied, including targeting protein isoforms, isoform expression, alternative splicing through trans-acting elements, oligonucleotides specific to specific cis-acting elements in the transcript and inhibition of the specific isoform by RNAi [Citation168]. One example is the use of compounds that bind to SF3B (a component of the U2 snRNP), leading to disruption of the early stages of spliceosome assembly [Citation5]. These compounds are E7107, spliceostatin A and sudemycins, which promote unspliced (or incompletely spliced) pre-mRNA accumulation in the nucleus and show anti-proliferative activity in vitro [Citation5]. E7107 was used in phase 1 clinical trials, but its use was suspended due to its high toxicity in patients [Citation5,Citation170]. Moreover, the spliceosome modulation in MYC-driven cancers has been suggested as an alternative to therapeutic intervention. MYC is a transcription factor that is frequently amplified in human cancers. It is highly related to cell transformation, and studies have revealed that the role for MYC as an oncogene is dependent on spliceosome activity [Citation171,Citation172].
5. Conclusive remarks and future directions
Although great advances have been achieved, understanding all of the mechanisms that contribute to tumor formation and progression is a great challenge. The presence or absence of specific isoforms may affect the metastatic potential of malignant cells, and variations in isoform expression from different genes are observed in cancer compared to normal tissues. Currently, the growth of large-scale studies has allowed us to integrate information and fill knowledge gaps in all areas of molecular biology – including information regarding splicing mechanisms and isoform variety. Certainly, further investigations are still necessary to understand the links between the splicing mechanisms, isoform diversity and the context of the disease. Finally, comprehension of the functional characteristics of abnormal splicing and their relationship with genomic instability is crucial for the development of novel therapeutic approaches.
Acknowledgments
This work was supported by CNPq under Grant 302969/2016–0 and CAPES.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Valastyan S, Weinberg RA. Tumor Metastasis : molecular Insights and Evolving Paradigms. Cell. 2011;147(2):275–292.
- Guan X. Cancer metastases: challenges and opportunities. Acta Pharm Sin B. 2015;5(5):402–418.
- Nguyen DX, Bos PD, Massagué J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9(4):274–284.
- Steeg PS. Targeting metastasis. Nat Rev Cancer. 2016;16(4):201–218.
- Lee SC-W, Abdel-Wahab O. Therapeutic targeting of splicing in cancer. Nat Med. 2016;22(9):976–986.
- Pal S, Gupta R, Davuluri RV. Alternative transcription and alternative splicing in cancer. Pharmacol Ther. 2012;136(3):283–294.
- Corkery DP, Holly AC, Lahsaee S, et al. Connecting the speckles: splicing kinases and their role in tumorigenesis and treatment response. Nucleus. 2015;6(4):279–288.
- Silipo M, Gautrey H, Tyson-Capper A. Deregulation of splicing factors and breast cancer development. J Mol Cell Biol. 2015;7(5):388–401.
- Lu Z, Huang Q, Park JW, et al. Transcriptome-wide Landscape of Pre-mRNA Alternative Splicing Associated with Metastatic Colonization. Mol Cancer Res. 2015;13(2):305–318.
- Oltean S, Bates DO. Hallmarks of alternative splicing in cancer. Oncogene. 2013;33(46):5311–5318.
- Wang Y, Liu J, Huang BO, et al. Mechanism of alternative splicing and its regulation. Biomed reports. 2015;3(2):152–158.
- Sibley CR, Blazquez L, Ule J. Lessons from non-canonical splicing. Nat Rev Genet. 2016;17(7):407–421.
- Jacob AG, Smith CWJ. Intron retention as a component of regulated gene expression programs. Hum Genet. 2017;136(9):1043–1057.
- Kovacs E, Tompa P, Liliom K, et al. Dual coding in alternative reading frames correlates with intrinsic protein disorder. Proc Natl Acad Sci USA. 2010;107(12):5429–5434.
- Sveen A, Kilpinen S, Ruusulehto A, et al. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene. 2015;35(19):2413–2427.
- Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat Rev Genet. 2016;17(1):19–32.
- Paronetto MP, Passacantilli I, Sette C. Alternative splicing and cell survival: from tissue homeostasis to disease. Cell Death Differ. 2016;23(12):1919–1929.
- Matera AG, Wang Z. A day in the life of the spliceosome. Nat Rev Mol Cell Biol. 2014;15(2):108–121.
- Merkhofer EC, Hu P, Johnson TL. Introduction to Cotranscriptional RNA Splicing. Methods Mol Biol. 2014;1126:83–96.
- Papasaikas P, Valcárcel J. The Spliceosome: the Ultimate RNA Chaperone and Sculptor. Trends Biochem Sci. 2016;41(1):33–45.
- Kornblihtt AR, Schor IE, Alló M, et al. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol. 2013;14(3):153–165.
- Kelemen O, Convertini P, Zhang Z, et al. Function of alternative splicing. Gene. 2013;514(1):1–30.
- Wang Z, Liu D, Yin B, et al. High expression of PTBP1 promote invasion of colorectal cancer by alternative splicing of cortactin. Oncotarget. 2017;8(22):36185–36202.
- Dvinge H, Kim E, Abdel-Wahab O, et al. RNA splicing factors as oncoproteins and tumour suppressors. Nat Rev Cancer. 2016;16(7):413–430.
- Iannone C, Valcárcel J. Chromatin’s thread to alternative splicing regulation. Chromosoma. 2013;122(6):465–474.
- Simon JM, Hacker KE, Singh D, et al. Variation in chromatin accessibility in human kidney cancer links H3K36 methyltransferase loss with widespread RNA processing defects. Genome Res. 2014;24(2):241–250.
- Jung H, Lee D, Lee J, et al. Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat Genet. 2015;47(11):1242–1248.
- Marquez Y, Höpfler M, Ayatollahi Z, et al. Unmasking alternative splicing inside protein-coding exons defines exitrons and their role in proteome plasticity. Genome Res. 2015;25(7):995–1007.
- Staiger D, Simpson GG. Enter exitrons. Genome Biol. 2015;16(1):136.
- Jiang WG, Sanders AJ, Katoh M, et al. Tissue invasion and metastasis: molecular, biological and clinical perspectives. Semin Cancer Biol. 2015;35:S244–75.
- Buchheit CL, Weigel KJ, Schafer ZT. Cancer cell survival during detachment from the ECM: multiple barriers to tumour progression. Nat Rev Cancer. 2014;14(9):632–641.
- Van Zijl F, Krupitza G, Mikulits W. Initial steps of metastasis: cell invasion and endothelial transmigration. Mutat Res - Rev Mutat Res. 2011;728(1–2):23–34.
- Chaffer CL, San Juan BP, Lim E, et al. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016;35(4):645–654.
- Warzecha CC, Carstens RP. Complex changes in alternative pre-mRNA splicing play a central role in the epithelial-to-mesenchymal transition (EMT). Semin Cancer Biol. 2012;22(5–6):417–427.
- Takano S, Reichert M, Bakir B, et al. Prrx1 isoform switching regulates pancreatic cancer invasion and metastatic colonization. Genes Dev. 2016;30(2):233–247.
- Massagué J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016;529(7586):298–306.
- Labelle M, Begum S, Hynes RO. Platelets guide the formation of early metastatic niches. Proc Natl Acad Sci. 2014;111(30):E3053–61.
- Senft D, Ronai ZA. Adaptive Stress Responses During Tumor Metastasis and Dormancy. Trends in Cancer. 2016;2(8):429–442.
- Lorusso G, Rüegg C. New insights into the mechanisms of organ-specific breast cancer metastasis. Semin Cancer Biol. 2012;22(3):226–233.
- Abou Faycal C, Gazzeri S, Eymin B. RNA splicing, cell signaling, and response to therapies. Curr Opin Oncol. 2016;28(1):58–64.
- Anczuków O, Krainer AR. Splicing-factor alterations in cancers. RNA. 2016;22(9):1285–1301.
- Luz FAC, Brígido PC, Moraes AS, et al. Aberrant Splicing in Cancer: mediators of Malignant Progression through an Imperfect Splice Program Shift. Oncology. 2016.
- Seidel B, Braeg S, Adler G, et al. E- and N-cadherin differ with respect to their associated p120ctn isoforms and their ability to suppress invasive growth in pancreatic cancer cells. Oncogene. 2004;23(32):5532–5542.
- Sharma S, Liao W, Zhou X, et al. Exon 11 skipping of E-cadherin RNA downregulates its expression in head and neck cancer cells. Mol Cancer Ther. 2011;10(9):1751–1759.
- Li X-W, Shi B-Y, Yang Q-L, et al. Epigenetic regulation of CDH1 exon 8 alternative splicing in gastric cancer. BMC Cancer. 2015;15(1):954.
- Yang CC, He P, Liu Y, et al. Down-regulation of CEACAM1 in breast cancer. Acta Biochim Biophys Sin (Shanghai). 2015;47(10):788–794.
- Dery KJ, Gaur S, Gencheva M, et al. Mechanistic control of carcinoembryonic antigen-related cell adhesion molecule-1 (CEACAM1) splice isoforms by the heterogeneous nuclear ribonuclear proteins hnRNP L, hnRNP A1, and hnRNP M. J Biol Chem. 2011;286(18):16039–16051.
- Dankner M, Gray-Owen SD, Huang YH, et al. CEACAM1 as a multi-purpose target for cancer immunotherapy. Oncoimmunology. 2017;6(7):1–16.
- Ieda J, Yokoyama S, Tamura K, et al. Re-expression of CEACAM1 long cytoplasmic domain isoform is associated with invasion and migration of colorectal cancer. Int J Cancer. 2011;129(6):1351–1361.
- Kirshner J, Chen C-J, Liu P, et al. CEACAM1-4S, a cell-cell adhesion molecule, mediates apoptosis and reverts mammary carcinoma cells to a normal morphogenic phenotype in a 3D culture. Proc Natl Acad Sci U S A. 2003;100(2):521–526.
- Senbanjo LT, Chellaiah MA. CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells. Front cell Dev Biol. 2017;5:18.
- Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol. 2003;4(1):33–45.
- Birzele F, Voss E, Nopora A, et al. CD44 Isoform status predicts response to treatment with Anti-CD44 antibody in cancer patients. Clin Cancer Res. 2015;21(12):2753–2762.
- Brown RL, Reinke LM, Damerow MS, et al. CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J Clin Invest. 2011;121(3):1064–1074.
- Zhao P, Xu Y, Wei Y, et al. The CD44s splice isoform is a central mediator for invadopodia activity. J Cell Sci. 2016;129(7):1355–1365.
- Zhao S, Chen C, Chang K, et al. CD44 expression level and isoform contributes to pancreatic cancer cell plasticity, invasiveness, and response to therapy. Clin Cancer Res. 2016;22(22):5592–5604.
- Prochazka L, Tesarik R, Turanek J. Regulation of alternative splicing of CD44 in cancer. Cell Signal. 2014;26(10):2234–2239.
- Warzecha CC, Shen S, Xing Y, et al. The epithelial splicing factors ESRP1 and ESRP2 positively and negatively negulate diverse types of alternative splicing events. RNA Biol. 2009;6(5):546.
- Tripathi V, Sixt KM, Gao S, et al. Direct Regulation of Alternative Splicing by SMAD3 through PCBP1 Is Essential to the Tumor-Promoting Role of TGF-β. Mol Cell. 2016;64(5):1010.
- Xu Y, Gao XD, Lee JH, et al. Cell type-restricted activity of hnRNPM promotes breast cancer metastasis via regulating alternative splicing. Genes Dev. 2014;28(11):1191–1203.
- Hu J, Li G, Zhang P, et al. A CD44v+ subpopulation subpopulation of breast cancer stem-like cells with enhanced lung metastasis capacity. Cell Death Dis. 2017;8(3):e2679.
- Weber GF, Ashkar S, Glimcher MJ, et al. Receptor-Ligand Interaction Between CD44 and Osteopontin (Eta-1). Science (80-). 1996;271(5248):509–512.
- Todaro M, Gaggianesi M, Catalano V, et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell. 2014;14(3):342–356.
- Tremmel M, Matzke A, Albrecht I, et al. A CD44v6 peptide reveals a role of CD44 in VEGFR-2 signaling and angiogenesis. Blood. 2009;114(25):5236–5244.
- Loh TJ, Moon H, Jang HN, et al. SR proteins regulate V 6 exon splicing of CD44 pre-mRNA. BMB Rep. 2016;49(11):612–616.
- Hernandez JR, Kim JJ, Verdone JE, et al. Alternative CD44 splicing identifies epithelial prostate cancer cells from the mesenchymal counterparts. Med Oncol. 2015;32(5):159.
- Sakuma K, Sasaki E, Kimura K, et al. HNRNPLL, a newly identified colorectal cancer metastasis suppressor, modulates alternative splicing of CD44 during epithelial-mesenchymal transition. Gut. 2018;67(6):1103–1111.
- Balsamo M, Mondal C, Carmona G, et al. The alternatively-included 11a sequence modifies the effects of Mena on actin cytoskeletal organization and cell behavior. Sci Rep. 2016;6:35298.
- Oudin MJ, Hughes SK, Rohani N, et al. Characterization of the expression of the pro-metastatic MenaINV isoform during breast tumor progression. Clin Exp Metastasis. 2016;33(3):249–261.
- Melchionnaa R, Iapiccaa P, Di MF, et al. The pattern of hMENA isoforms is regulated by TGF-β1 in pancreatic cancer and may predict patient outcome. Oncoimmunology. 2016;5(12):e1221556.
- Bonomi S, Gallo S, Catillo M, et al. Oncogenic alternative splicing switches: role in cancer progression and prospects for therapy. Int J Cell Biol. 2013;2013:962038.
- Trono P, Di Modugno F, Nisticò P. hMENA 11a, a hMENA isoform sending survival signals. Mol Cell Oncol. 2016;3(2):e1083648.
- Roussos ET, Goswami S, Balsamo M, et al. Mena invasive (MenaINV) and Mena11a isoforms play distinct roles in breast cancer cell cohesion and association with TMEM. Clin Exp Metastasis. 2011;28(6):515–527.
- Roussos ET, Condeelis JS, Patsialou A. Chemotaxis in cancer. Nat Rev Cancer. 2011;11(8):573–587.
- Oktay MH, Jones JG. TMEM: A novel breast cancer dissemination marker for the assessment of metastatic risk. Biomark Med. 2015;9(2):81–84.
- Pignatelli J, Goswami S, Jones JG, et al. Invasive breast carcinoma cells from patients exhibit MenaINV-and macrophage-dependent transendothelial migration. Sci Signal. 2014;7(353):ra112.
- Weidmann MD, Surve CR, Eddy RJ, et al. MenaINV dysregulates cortactin phosphorylation to promote invadopodium maturation. Sci Rep. 2016;6(1):36142.
- Fiegen D, Haeusler L-C, Blumenstein L, et al. Alternative Splicing of Rac1 Generates Rac1b, a Self-activating GTPase. J Biol Chem. 2004;279(6):4743–4749.
- Singh A, Karnoub AE, Palmby TR, et al. Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene. 2004;23(58):9369–9380.
- Faria M, Capinha L, Simões-Pereira J, et al. Extending the Impact of RAC1b Overexpression to Follicular Thyroid Carcinomas. Int J Endocrinol. 2016;2016:1–7.
- Gonçalves V, Henriques A, Pereira J, et al. Phosphorylation of SRSF1 by SRPK1 regulates alternative splicing of tumor-related Rac1b in colorectal cells. RNA. 2014;20(4):474–482.
- Pelisch F, Khauv D, Risso G, et al. Involvement of hnRNP A1 in the matrix metalloprotease-3-dependent regulation of Rac1 pre-mRNA splicing. J Cell Biochem. 2012;113(7):2319–2329.
- Wang F, Fu X, Chen P, et al. SPSB1-mediated HnRNP A1 ubiquitylation regulates alternative splicing and cell migration in EGF signaling. Cell Res. 2017;27(4):540–558.
- Faria M, Matos P, Pereira T, et al. RAC1b overexpression stimulates proliferation and NF-kB-mediated anti-apoptotic signaling in thyroid cancer cells. PLoS One. 2017;12(2):e0172689.
- Weaver AM. Cortactin in tumor invasiveness. Cancer Lett. 2008;265(2):157–166.
- Head JA, Jiang D, Li M, et al. Cortactin Tyrosine Phosphorylation Requires Rac1 Activity and Association with the Cortical Actin Cytoskeleton. Mol Biol Cel. 2003;14(8):3216–3229.
- Katsube T, Togashi S, Hashimoto N, et al. Filamentous actin binding ability of cortactin isoforms is responsible for their cell-cell junctional localization in epithelial cells. Arch Biochem Biophys. 2004;427(1):79–90.
- Gattazzo C, Martini V, Frezzato F, et al. Cortactin, another player in the Lyn signaling pathway, is over-expressed and alternatively spliced in leukemic cells from patients with B-cell chronic lymphocytic leukemia. Haematologica. 2014;99(6):1069–1077.
- Johnson RM, Vu NT, Griffin BP, et al. The alternative splicing of cytoplasmic polyadenylation element binding protein 2 drives anoikis resistance and the metastasis of triple negative breast cancer. J Biol Chem. 2015;290(42):25717–25727.
- Upheber S, Karle A, Miller J, et al. Alternative splicing of KAI1 abrogates its tumor-suppressive effects on integrin αvβ3-mediated ovarian cancer biology. Cell Signal. 2015;27(3):652–662.
- Lee JH, Seo Y, Park SR, et al. Expression of a Splice Variant of KAI1, a Tumor Metastasis Suppressor Gene, Influences Tumor Invasion and Progression Expression of a Splice Variant of KAI1, a Tumor Metastasis Suppressor Gene, Influences Tumor Invasion and Progression. Group. 2003;63(21):7247–7255.
- Miranti CK. Controlling cell surface dynamics and signaling: how CD82/KAI1 suppresses metastasis. Cell Signal. 2009;21(2):196–211.
- Yang JM, O’Neill P, Jin W, et al. Extracellular matrix metalloproteinase inducer (CD147) confers resistance of breast cancer cells to anoikis through inhibition of Bim. J Biol Chem. 2006;281(14):9719–9727.
- Liao C-G, Kong L-M, Song F, et al. Characterization of basigin isoforms and the inhibitory function of basigin-3 in human hepatocellular carcinoma proliferation and invasion. Mol Cell Biol. 2011;31(13):2591–2604.
- Yao H-P, Zhou Y-Q, Zhang R, et al. MSP–RON signalling in cancer: pathogenesis and therapeutic potential. Nat Rev Cancer. 2013;13(7):466–481.
- Moxley KM, Wang L, Welm AL, et al. Short-form Ron is a novel determinant of ovarian cancer initiation and progression. Genes Cancer. 2016;7(5–6):169–181.
- Lu Y, Yao HP, Wang MH. Multiple variants of the RON receptor tyrosine kinase: biochemical properties, tumorigenic activities, and potential drug targets. Cancer Lett. 2007;257(2):157–164.
- Mayer S, Hirschfeld M, Jaeger M, et al. RON alternative splicing regulation in primary ovarian cancer. Oncol Rep. 2015;34(1):423–430.
- LeFave CV, Squatrito M, Vorlova S, et al. Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. EMBO J. 2011;30(19):4084–4097.
- Golan-Gerstl R, Cohen M, Shilo A, et al. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res. 2011;71(13):4464–4472.
- Pagliarini V, Naro C, Sette C. Splicing Regulation: A Molecular Device to Enhance Cancer Cell Adaptation. Biomed Res Int. 2015;2015:543067.
- Bharadwaj S, Thanawala R, Bon G, et al. Resensitization of breast cancer cells to anoikis by tropomyosin-1: role of Rho kinase-dependent cytoskeleton and adhesion. Oncogene. 2005;24(56):8291–8303.
- Gunning P, Neill GO, Hardeman E. Tropomyosin-Based Regulation of the Actin Cytoskeleton in Time and Space. Physiol Rev. 2008;88(1):1–35.
- Desouza M, Gunning PW, Stehn JR. The actin cytoskeleton as a sensor and mediator of apoptosis. Bioarchitecture. 2012;2(3):75–87.
- Pan H, Gu L, Liu B, et al. Tropomyosin-1 acts as a potential tumor suppressor in human oral squamous cell carcinoma. PLoS One. 2017;12(2):e0168900.
- Dube S, Yalamanchili S, Lachant J, et al. Expression of Tropomyosin 1 Gene Isoforms in Human Breast Cancer Cell Lines. Int J Breast Cancer. 2015;2015:859427.
- Yang J, Zeng Z, Peng Y, et al. IL-7 splicing variant IL-7δ5 induces EMT and metastasis of human breast cancer cell lines MCF-7 and BT-20 through activation of PI3K/Akt pathway. Histochem Cell Biol. 2014;142(4):401–410.
- Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17(9):559–572.
- Wu Q, Dhir R, Wells A. Altered CXCR3 isoform expression regulates prostate cancer cell migration and invasion. Mol Cancer. 2012;11:3.
- Li Y, Reader JC, Ma X, et al. Divergent roles of CXCR3 isoforms in promoting cancer stem-like cell survival and metastasis. Breast Cancer Res Treat. 2015;149(2):403–415.
- Ding Q, Xia Y, Ding S, et al. An alternatively spliced variant of CXCR3 mediates the metastasis of CD133 + liver cancer cells induced by CXCL9. Oncotarget. 2016;7(12):14405–14414.
- Allami RH, Graf C, Martchenko K, et al. Analysis of the expression of SDF-1 splicing variants in human colorectal cancer and normal mucosa tissues. Oncol Lett. 2016;11(3):1873–1878.
- Woo IS, Hong SH, Byun JH, et al. Circulating stromal cell derived factor-1α (SDF-1α) is predictive of distant metastasis in gastric carcinoma. Cancer Invest. 2008;26(3):256–261.
- Seguin L, Desgrosellier JS, Weis SM, et al. Integrins and cancer: regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015;25(4):234–240.
- Groulx J-F, Giroux V, Beauséjour M, et al. Integrin α6A splice variant regulates proliferation and the Wnt/β-catenin pathway in human colorectal cancer cells. Carcinogenesis. 2014;35(6):1217–1227.
- Goel HL, Gritsko T, Pursell B, et al. Regulated splicing of the α6 integrin cytoplasmic domain determines the fate of breast cancer stem cells. CellReports. 2014;7(3):747–761.
- Jiang P, Li Z, Tian F, et al. Fyn/heterogeneous nuclear ribonucleoprotein E1 signaling regulates pancreatic cancer metastasis by affecting the alternative splicing of integrin β1. Int J Oncol. 2017;51(1):169–183.
- Yoon H, Dehart JP, Murphy JM, et al. Understanding the Roles of FAK in Cancer: inhibitors, Genetic Models, and New Insights. J Histochem Cytochem. 2015;63(2):114–128.
- Yao L, Li K, Peng W, et al. An aberrant spliced transcript of focal adhesion kinase is exclusively expressed in human breast cancer. J Transl Med. 2014;12(1):136.
- Despeaux M, Chicanne G, Rouer E, et al. Focal adhesion kinase splice variants maintain primitive acute myeloid leukemia cells through altered Wnt signaling. Stem Cells. 2012;30(8):1597–1610.
- Hao C, Cui Y, Owen S, et al. Human osteopontin: potential clinical applications in cancer (Review). Int J Mol Med. 2017;39(6):1327–1337.
- Lin J, Myers AL, Wang Z, et al. Osteopontin (OPN/SPP1) isoforms collectively enhance tumor cell invasion and dissemination in esophageal adenocarcinoma. Oncotarget. 2015;6(26):22239–22257.
- Shen H, Weber GF. The osteopontin-c splice junction is important for anchorage-independent growth. Mol Carcinog. 2014;53(6):480–487.
- Hao C, Cui Y, Hu M, et al. OPN-a Splicing Variant Expression in Non-small Cell Lung Cancer and its Effects on the Bone Metastatic Abilities of Lung Cancer Cells In Vitro. Anticancer Res. 2017;37(5):2245–2254.
- Tang X, Li J, Yu B, et al. Osteopontin splice variants differentially exert clinicopathological features and biological functions in gastric cancer. Int J Biol Sci. 2013;9(1):55–66.
- Bueno Ferreira L, Tavares C, Pestana A, et al. Osteopontin-a splice variant is overexpressed in papillary thyroid carcinoma and modulates invasive behavior. Oncotarget. 2016;7(32):52003–52016.
- Sun J, Feng A, Chen S, et al. Osteopontin splice variants expressed by breast tumors regulate monocyte activation via MCP-1 and TGF-β1. Cell Mol Immunol. 2013;10(2):176–182.
- White ES, Muro AF. Fibronectin splice variants: understanding their multiple roles in health and disease using engineered mouse models. IUBMB Life. 2011;63(7):538–546.
- White E, Baralle F, Muro A. New insights into form and function of fibronectin splice variants. J Pathol. 2008;216(1):1–14.
- Frey K, Fiechter M, Schwager K, et al. Different patterns of fibronectin and tenascin-C splice variants expression in primary and metastatic melanoma lesions. Exp Dermatol. 2011;20(8):685–688.
- Ou JJ, Wu F, Liang HJ. Colorectal tumor derived fibronectin alternatively spliced EDA domain exserts lymphangiogenic effect on human lymphatic endothelial cells. Cancer Biol Ther. 2010;9(3):186–191.
- Bordeleau F, Califano JP, Abril YLN, et al. Tissue stiffness regulates serine/arginine-rich protein-mediated splicing of the extra domain B-fibronectin isoform in tumors. Proc Natl Acad Sci. 2015;112(27):8314–8319.
- Liguori M, Solinas G, Germano G, et al. Tumor-associated macrophages as incessant builders and destroyers of the cancer stroma. Cancers (Basel). 2011;3(4):3740–3761.
- Richter P, Junker K, Franz M, et al. IIICS de novo glycosylated fibronectin as a marker for invasiveness in urothelial carcinoma of the urinary bladder (UBC). J Cancer Res Clin Oncol. 2008;134(10):1059–1065.
- Solinas G, Schiarea S, Liguori M, et al. Tumor-Conditioned Macrophages Secrete Migration-Stimulating Factor: A New Marker for M2-Polarization, Influencing Tumor Cell Motility. J Immunol. 2010;185(1):642–652.
- Perrier S, Woolston A-M, Purdie CA, Kazmi S, Preece PE, Davey KJ, Schor SL, Schor AM. Migration Stimulating Factor (MSF). A Novel Biomarker of Breast Cancer Progression. Transl Med. 2012;S1:003.
- Olivos DJ, Mayo LD. Emerging Non-Canonical Functions and Regulation by p53: p53 and Stemness. Int J Mol Sci. 2016;17(12):E1982.
- Pflaum J, Schlosser S, Müller M. p53 Family and Cellular Stress Responses in Cancer. Front Oncol. 2014;4:285.
- Gadea G, Arsic N, Fernandes K, et al. TP53 drives invasion through expression of its Δ133p53β variant. Elife. 2016;5:e14734.
- Arsic N, Gadea G, Lagerqvist EL, et al. The p53 isoform Δ133p53β promotes cancer stem cell potential. Stem Cell Reports. 2015;4(4):531–540.
- Pixley FJ, Stanley ER. CSF-1 regulation of the wandering macrophage : complexity in action. Trends Cell Biol. 2004;14(11):628–638.
- Stanley ER, Berg KL, Einstein DB, et al. Biology and Action of Colony – stimulating Factor-1. Mol Reprod Dev. 1997;46(1):4–10.
- Ladner MB, Martin GA, Noble JA, et al. Human CSF-1: gene structure and alternative splicing of mRNA precursors. EMBO J. 1987;6(9):2693–2698.
- Yang L, Zhang Y. Tumor-associated macrophages : from basic research to clinical application. J Hematol Oncol. 2017;10(1):58.
- Liao J, Feng W, Wang R, et al. Diverse in vivo effects of soluble and membrane-bound M-CSF on tumor-associated macrophages in lymphoma xenograft model. Oncotarget. 2016;7(2):1354–1366.
- Norris RA, Kern MJ. The Identification of Prx1 Transcription Regulatory Domains Provides a Mechanism for Unequal Compensation by the Prx1 and Prx2 Loci. J Biol Chem. 2001;276(29):26829–26837.
- Zhang P, Zhang P, Zhou M, et al. 4 decletion variant of epidermal growth factor receptor enhances invasiveness and cisplatin resistance in epithelial ovarian cancer. Carcinogenesis. 2013;34(11):2639–2646.
- Greenall SA, Johns TG. EGFRvIII: the promiscuous mutation. Cell Death Discov. 2016;2:16049.
- Guillaudeau A, Durand K, Bessette B, et al. Egfr soluble isoforms and their transcripts are expressed in meningiomas. PLoS One. 2012;7(5):e37204.
- Wang H, Zhou M, Shi B, et al. Identification of an Exon 4-Deletion Variant of Epidermal Growth Factor Receptor with Increased Metastasis-Promoting Capacity. Neoplasia. 2011;13(5):461–471.
- Wang H, Shi B, Zhang Q, et al. Growth and metastasis suppression of glioma xenografts expressing exon 4-deletion variant of epidermal growth factor receptor by monoclonal antibody CH12-mediated receptor degradation. FASEB J. 2012;26(1):73–80.
- Manrique I, Nguewa P, Bleau AM, et al. The inhibitor of differentiation isoform Id1b, generated by alternative splicing, maintains cell quiescence and confers self-renewal and cancer stem cell-like properties. Cancer Lett. 2015;356(2 Pt B):899–909.
- Giancotti FG. Mechanisms Governing Metastatic Dormancy and Reactivation. Cell. 2013;155(4):750–764.
- Chen W, Dong J, Haiech J, et al. Cancer stem cell quiescence and plasticity as major challenges in cancer therapy. Stem Cells Int. 2016;2016:1740936.
- Hatakeyama K, Yamakawa Y, Fukuda Y, et al. A novel splice variant of XIAP-associated factor 1 (XAF1) is expressed in peripheral blood containing gastric cancer-derived circulating tumor cells. Gastric Cancer. 2015;18(4):751–761.
- Li Z, Mou H, Wang T, et al. A non-secretory form of FAM3B promotes invasion and metastasis of human colon cancer cells by upregulating Slug expression. Cancer Lett. 2013;328(2):278–284.
- Kong L, Yao L, Lu N, et al. Interaction of KLF6 and Sp1 regulates basigin-2 expression mediated proliferation, invasion and metastasis in hepatocellular carcinoma. Oncotarget. 2016;7(19):27975–27987.
- Narla G, Difeo A, Yao S, et al. Targeted inhibition of the KLF6 splice variant, KLF6 SV1, suppresses prostate cancer cell growth and spread. Cancer Res. 2005;65(13):5761–5768.
- Hatami R, Sieuwerts AM, Izadmehr S, et al. KLF6-SV1 Drives Breast Cancer Metastasis and Is Associated with Poor Survival. Sci Transl Med. 2013;5(169):169ra12.
- DiFeo A, Martignetti JA, Narla G. The role of KLF6 and its splice variants in cancer therapy. Drug Resist Updat. 2009;12(1–2):1–7.
- Lu H, Liu J, Liu S, et al. Exo70 isoform switching upon epithelial-mesenchymal transition mediates cancer cell invasion. Dev Cell. 2013;27(5):560–573.
- Xiao L, Zheng K, Lv X, et al. Exo70 is an independent prognostic factor in colon cancer. Sci Rep. 2017;7(1):5039.
- Luo Z, Cheng S, Shi J, et al. A splicing variant of Merlin promotes metastasis in hepatocellular carcinoma. Nat Commun. 2015;6:8457.
- Scotlandi K, Zuntini M, Manara MC, et al. CD99 isoforms dictate opposite functions in tumour malignancy and metastases by activating or repressing c-Src kinase activity. Oncogene. 2007;26(46):6604–6618.
- Byun HJ, Hong IK, Kim E, et al. A splice variant of CD99 increases motility and MMP-9 expression of human breast cancer cells through the AKT-, ERK-, and JNK-dependent AP-1 activation signaling pathways. J Biol Chem. 2006;281(46):34833–34847.
- Da Silva MR, Moreira GA, Gonçalves Da Silva RA, et al. Splicing regulators and their roles in cancer biology and therapy. Biomed Res Int. 2015;2015:150514.
- Zhang ZF, Pal S, Bi Y, et al. Isoform level expression profiles provide better cancer signatures than gene level expression profiles. Genome Med. 2013;5(4):33.
- Le K-Q, Prabhakar BS, Hong W-J, et al. Alternative splicing as a biomarker and potential target for drug discovery. Acta Pharmacol Sin. 2015;36(10):1212–1218.
- Ren P, Li M, Li T, et al. Anti-EGFRvIII Chimeric Antigen Receptor-Modified T Cells for Adoptive Cell Therapy of Glioblastoma. Curr Pharm Des. 2017;23(14):2113–2116.
- Hong DS, Kurzrock R, Naing A, et al. A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Invest New Drugs. 2014;32(3):436–444.
- Koh CM, Bezzi M, Low DHP, et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature. 2015;523(7558):96–100.
- Hsu TY, Simon LM, Neill NJ, et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature. 2015;525(7569):384–388.
- Jordaan G, Liao W, Sharma S. E-cadherin gene re-expression in chronic lymphocytic leukemia cells by HDAC inhibitors. BMC Cancer. 2013;13(1):88.
- Yan Y, Zuo X, Wei D. Concise Review: emerging Role of CD44 in Cancer Stem Cells: A Promising Biomarker and Therapeutic Target. Stem Cells Transl Med. 2015;4(9):1033–1043.
- Zöller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer. 2011;11(4):254–267.
- Misra S, Hascall VC, De Giovanni C, et al. Delivery of CD44 shRNA/nanoparticles within cancer cells. Perturbation of hyaluronan/CD44v6 interactions and reduction in adenoma growth in Apc Min/+mice. J Biol Chem. 2009;284(18):12432–12446.
- Orian-Rousseau V, Ponta H. Perspectives of CD44 targeting therapies. Arch Toxicol. 2015;89(1):3–14.
- Ying L, Li G, Wei -S-S, et al. Sanguinarine inhibits Rac1b-rendered cell survival enhancement by promoting apoptosis and blocking proliferation. Nat Publ Gr. 2014;36(10):229–240.
- Matos P, Kotelevets L, Jordan P, et al. Ibuprofen Inhibits Colitis-Induced Overexpression of Tumor Related Rac1b. Neoplasia. 2013;15(1):102–111.
- Bieniasz M, Radhakrishnan P, Faham N, et al. Pre-clinical efficacy of Ron kinase inhibitors alone and in combination with PI3K inhibitors for treatment of sfRon- expressing breast cancer patient-derived xenografts. Clin Cancer Res. 2015;21(24):5588–5600.
- Femel J, Huijbers EJM, Saupe F, et al. Therapeutic vaccination against fibronectin ED-A attenuates progression of metastatic breast cancer. Oncotarget. 2014;5(23):12418–12427.
- Han Z, Zhou Z, Shi X, et al. EDB fibronectin specific peptide for prostate cancer targeting. Bioconjug Chem. 2015;26(5):830–838.
- Menrad A, Menssen HD. ED-B fibronectin as a target for antibody-based cancer treatments. Expert Opin Ther Targets. 2005;9(3):491–500.
- Chen H, Chen L, Sun L, et al. A small interfering RNA targeting the KLF6 splice variant, KLF6-SV1, as gene therapy for gastric cancer. Gastric Cancer. 2011;14(4):339–352.