
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Recently, increasing studies have shown that miRNAs are involved in the development and progression of various complex diseases. Consequently, predicting potential miRNA-disease associations makes an important contribution to understanding the pathogenesis of diseases, developing new drugs as well as designing individualized diagnostic and therapeutic approaches for different human diseases. Nonetheless, the inherent noise and incompleteness in the existing biological datasets have limited the prediction accuracy of current computational models. To solve this issue, in this paper, we propose a novel method for miRNA-disease association prediction based on global linear neighborhoods (GLNMDA). Specifically, our method obtains a new miRNA/disease similarity matrix by linearly reconstructing each miRNA/disease according to the known experimentally verified miRNA-disease associations. We then adopt label propagation to infer the potential associations between miRNAs and diseases. As a result, GLNMDA achieved reliable performance in the frameworks of both local and global LOOCV (AUCs of 0.867 and 0.929, respectively) and 5-fold cross validation (average AUC of 0.926). Case studies on five common human diseases further confirmed the utility of our method in discovering latent miRNA-disease pairs. Taken together, GLNMDA could serve as a reliable computational tool for miRNA-disease association prediction.
Introduction
MicroRNAs(miRNAs) are highly enriched small non-coding RNAs of approximately 22 nucleotides that normally regulate gene expression at the post-transcriptional level by targeting mRNA for cleavage or translational inhibition[Citation1–Citation3]. Since the discovery of the first two mammalian microRNAs, mounting evidences have shown that miRNAs are involved in a variety of physiological and pathological processes[Citation4]. Many major cellular functions such as development, differentiation, growth and metabolism are known to be regulated by miRNAs[Citation5]. In addition, it has been suggested that miRNAs play vital roles in the pathogenesis of human diseases. For instance, by digging into the miRNA expression profiles of 93 primary human breast tumors, Blenkiron et al. identified a number of miRNAs that were differentially expressed between different molecular tumor subtypes[Citation6]. Recently, Zhang et al. identified miRNA-26a as a key regulon that inhibits progression and metastasis of c-Myc/EZH2 double high advanced hepatocellular carcinoma[7]. Consequently, many studies aim at identifying key miRNAs as diagnostic and therapeutic biomarkers for human diseases. It is thus of great significance to uncover the potential associations between miRNAs and various diseases.
Many efforts made to predict potential disease-related miRNAs using experimental approaches have been proven successful, such as qRT-PCR and microarray profiling. Although reliable, experimental based methods are generally time-consuming and labor-intensive[Citation8]. With the increasing amount of available biological data, a great number of computational models have been developed by taking advantage of multiple data sources to effectively and efficiently predict associations between miRNAs and diseases[Citation9–Citation11]. Under the assumption that miRNAs with similar functions tend to be associated to phenotypically similar diseases[Citation12,Citation13]. Jiang et al. proposed the first computational model based on hypergeometric distribution to predict new miRNA-disease associations[Citation14], in which they integrated the phenotypic similarity network of diseases, the miRNA functional similarity network as well as the known human disease-miRNA association networks. Xu et al. introduced a network-centric approach to prioritize candidate disease miRNAs by constructing four topological features that are distinguishable between prostate cancer (PC) and non-PC miRNAs[Citation15]. Xuan et al. proposed a model named HMDP which calculated miRNA-disease associations based on the functional similarities of k most similar neighbors of disease-associated miRNAs[Citation16]. Specifically, miRNAs within the same clusters or families were assigned higher weights since they were more likely to be related to similar diseases when calculating the miRNA functional similarity matrix. Nevertheless, HDMP cannot be applied to diseases without any known related miRNAs since it is based on local similarity measures. To solve this issue, Chen et al. developed a novel computational approach called HGIMDA which integrates miRNA functional similarity, disease semantic similarity, kernel similarity of Gaussian interaction profile, and experimentally validated miRNA-disease associations to predict potential miRNA-disease associations[Citation17]. They further constructed a heterogeneous graph to iteratively update the association scores between unconfirmed miRNAs and diseases. Based on the assumption that miRNAs with targets related to a given disease were also likely to be associated with that disease, Shi et al. developed a computational framework to identify the miRNA-disease associations by conducting random walk with restart (RWR) algorithm on protein-protein interaction (PPI) networks[Citation18]. Chen et al. proposed a method named WBSMDA to uncover the potential miRNAs related with multiple complex diseases by calculating a within score and a between score to obtain the final relevance scores for the unconfirmed miRNA-disease associations. Besides, WBSMDA could also be applied to diseases without any known related miRNAs[Citation19].
Recently, several studies taking advantage of network topological structures have been proposed to prioritize disease-related miRNAs. Sun et al. developed NTSMDA to predict potential disease-miRNA associations by calculating the network topological similarity for both miRNAs and diseases. Nevertheless, since NTSMDA only utilized the known miRNA-disease association network to compute the network topological similarities, it is quite sensitive to the quality of the input data and cannot be applied to diseases without any known associated miRNAs[Citation20]. You et al. developed a path-based model named PBMDA for miRNA-disease association prediction by integrating various biological data. Concretely, PBMDA adopted a depth-first search algorithm to search paths of certain lengths for given miRNA-disease pairs on a heterogeneous graph and obtained comparable performance. However, the computational complexity of PBMDA could be extremely high in large networks[Citation21]. Chen et al. proposed NDAMDA to predict miRNA-disease associations based on network distance analysis. The highlight of their method lies in that two types of distances were considered, i.e. the direct distance and average distance. The direct distance represented a distance between two miRNAs (diseases) and the average distance represented the mean network distances of all miRNAs (diseases)[Citation22].
In addition, several machine learning-based models were proposed to predict the potential miRNA-disease associations. Jiang et al. adopted the support vector machine (SVM) to predict the associations between miRNAs and diseases. They first extracted a set of features for each positive and negative miRNA-disease association, and then trained the SVM classifier with the constructed features to classify candidate disease-related miRNAs[Citation23]. Chen et al. developed RBMMMDA which can not only predict the new associations between miRNAs and diseases, but also obtain the type of corresponding association[Citation24]. Zou et al. introduced a biased SVM which was trained by a bagging algorithm to classify miRNA-disease pairs[Citation25]. Liu et al. first constructed a heterogeneous network by connecting disease similarity network, miRNA similarity network as well as known miRNA-disease associations. They then extended random walk with restart to predict miRNA-disease associations in the heterogeneous network[Citation26]. Li et al. utilized the matrix completion algorithm to update the adjacency matrix of known miRNA-disease associations and then predicted the potential miRNA-disease associations[Citation27]. Chen et al. proposed another computational model called RKNNMDA which utilized the SVM ranking model to obtain reliable k-nearest-neighbors for each miRNA and disease. Specifically, it can be used to predict potential miRNAs for diseases without any known miRNAs[Citation28]. They further proposed another model named MKRMDA to discover the potential miRNA-disease associations[Citation29]. The innovation of MKRMDA was that it could automatically optimize the multiple kernel combinations of disease and miRNA. Chen et al. presented a computational model named LRSSLMDA, which projected miRNAs/diseases’ statistical feature profile and graph theoretical feature profile to a common subspace. It used Laplacian regularization to preserve the local structures of the training data and a L1-norm constraint to select important miRNA/disease features for prediction[Citation30]. Xiao et al. proposed a graph regularized non-negative matrix factorization method for identifying miRNA-disease associations and their method was robust to the noises existing in the current datasets[Citation31]. Zeng et al. derived a structural perturbation method to predict potential associations between miRNAs and diseases by using structural consistency as an indicator to estimate the link predictability of related networks[Citation32]. Chen et al. developed the first decision tree learning-based model named EGBMMDA by employing Extreme Gradient Boosting Machine[Citation33]. They constructed an informative feature vector by incorporating statistical measures, graph theoretical measures as well as matrix factorization results. Generally, a limitation of the machine learning-based algorithms is that there are no validated negative samples for miRNA-disease associations. They further used ensemble learning to combine rank results obtained by three classic similarity-based algorithms to predict miRNA-disease associations[Citation34]. Recently, Chen et al. proposed a novel computational model to predict miRNA-disease associations based on bipartite network projection, which achieved comparable results in different cross-validation frameworks[Citation35].
Although existing computational methods have been greatly improved in many details, they still have limitations. Therefore, developing novel methods to efficiently and reliably excavate the potential miRNA-disease associations is significant for human health and medical advance. In this study, we propose a novel method for MiRNA-Disease Association prediction based on Global Linear Neighborhoods (GLNMDA). Specifically, GLNMDA linearly reconstructs each miRNA (disease) by weighted combinations of its direct neighbors and indirect neighbors that can be reached by any steps of random walks. To demonstrate the effectiveness of our method, we implement leave-one-out cross-validation (LOOCV) and five-fold cross-validation for GLNMDA. As a result, GLNMDA obtained global AUC value of 0.929, local AUC value of 0.867 and 5-fold cross validation value of 0.926, respectively. Moreover, we compared our method with four state-of-the-art methods and the results indicated that our method consistently outperformed the other methods. In addition, three types of case studies were performed on five common cancers to verify the reliability and robustness of GLNMDA. Together, GLNMDA is an effective method for predicting potential miRNA-disease associations.
Results
Performance evaluation
In this section, we applied LOOCV and 5-fold cross-validation to test the prediction performance of our method based on known miRNA-disease associations from HMDD v2.0 databases[Citation36]. LOOCV could be carried out in two manners: global and local LOOCV. In both frameworks, each known miRNA-disease association was left in turn as a test sample and other known miRNA-disease associations were regarded as training samples[Citation37]. The only difference between global LOOCV and local LOOCV was that whether all the diseases were investigated simultaneously. In the global LOOCV, the test sample was compared and ranked with all candidate miRNAs, whereas in the local LOOCV, the test sample is compared and ranked with the miRNAs only associated with the specific disease. We also implemented 5-fold cross validation to evaluate the performance of GLNMDA. In the framework of 5-fold cross validation, all the known miRNA-disease associations were randomly divided into five disjoint parts, where each part was picked out as test samples in turn and the other four parts were treated as training samples. In addition, Receiver Operating Characteristics (ROC) curves were plotted by calculating the true positive rate (TPR) and the false positive rate (FPR) at varying thresholds[Citation38]. The prediction performance of GLNMDA can be quantitatively evaluated by calculating the Area Under the ROC Curve (AUC). Specifically, the value of AUC is from 0 to 1 and the larger the AUC values, the better the predicted results. As shown in , GLNMDA achieved AUC values of 0.929, 0.867 and 0.926 in global LOOCV, local LOOCV and 5-fold cross-validation, respectively, which clearly demonstrated the superior performance of our method.
Figure 1. Flowchart of potential disease-miRNA association prediction based on the computational model of GLNMDA.
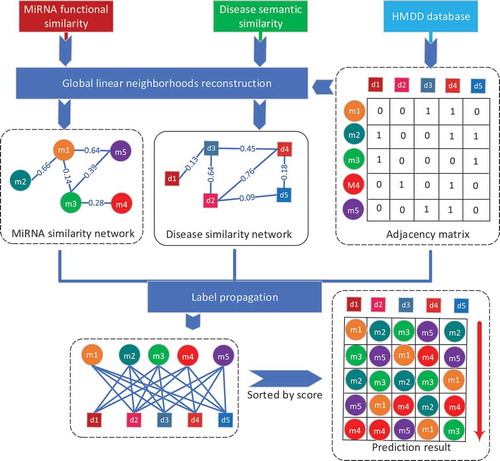
Figure 2. The comparison results between GLNMDA and the othe1`r four computational models in the framework of global LOOCV.
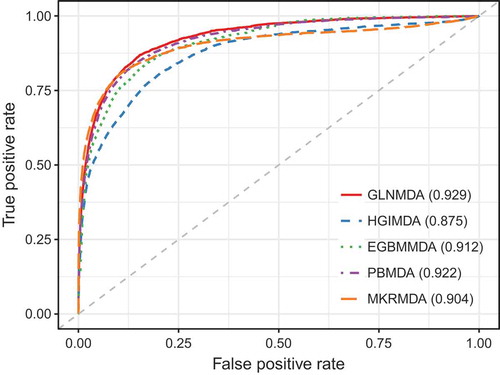
We further compared GLNMDA with four state-of-the-art methods (i.e. HGIMDA[Citation17], EGBMMDA[Citation33], PBMDA[Citation21], MKRMD[Citation29]), all of which have also achieved excellent performances in predicting potential miRNA-disease associations. As mentioned above, HGIMDA was an efficient prediction framework based on heterogeneous graph inference. Both EGBMMDA and MKRMDA were machine learning-based approaches with different feature extraction schemas. PBMDA was a depth-first model which took network topology into account. As shown in , HGIMDA, EGBMMDA, PBMDA and MKRMDA obtained AUCs of 0.875, 0.912, 0.922 and 0.904 in global LOOCV, respectively. Similarly, they obtained AUCs of 0.823, 0.807, 0.853 and 0.827 in the local LOOCV framework, respectively (). For 5-fold cross-validation, they achieved AUCs of 0.867, 0.904, 0.916 and 0.884, respectively (). Obviously, GLNMDA consistently outperformed the four methods in all three cross-validation frameworks. In conclusion, GLNMDA could serve as a reliable tool to predict the potential associations between miRNAs and diseases.
Figure 3. The comparison results between GLNMDA and the other four computational models in terms of local LOOCV.
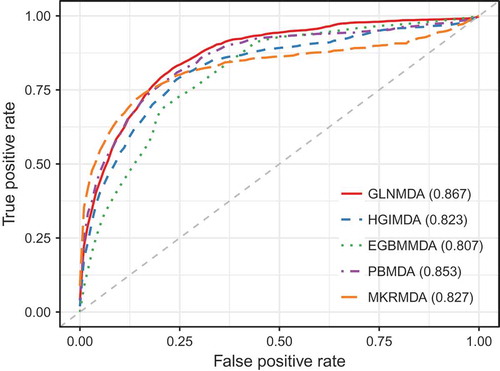
Figure 4. The comparison results between GLNMDA and the other four computational models in the framework of 5-fold cross-validation.
Parameter analysis
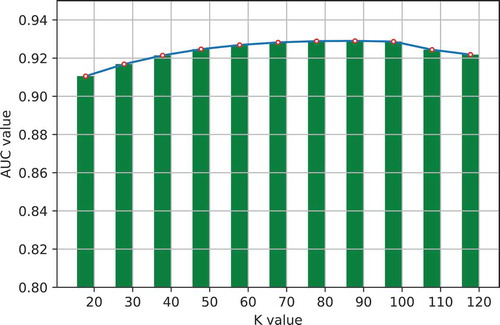
One important step in GLNMDA is to learn a rank-k non-negative symmetric matrix to reconstruct the miRNA similarity network and disease similarity network from miRNA space and disease space, respectively. To test whether different values of k would affect the final prediction results, we selected eleven values of k ranging from 20 to 120 with an interval of 10 and then compared the prediction accuracy in all three cross-validation frameworks. As illustrated in –, GLNMDA obtained the worst performance in all the cross validations when k = 20 while the performance remains relatively stable when k > 20. Therefore, we can conclude that different values of k only have minor effects on the final results.
Figure 5. The effects of different values of k in global cross validation.
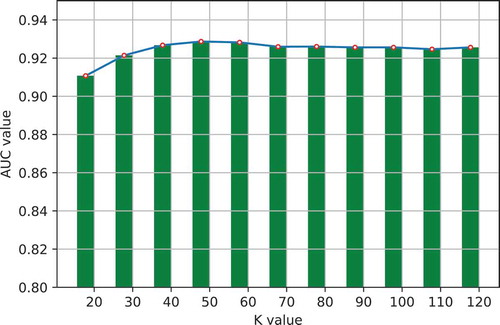
Figure 6. The effects of different values of k in local cross validation.
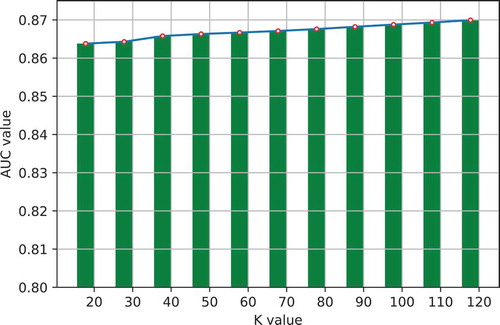
Figure 7. The effects of different values of k in 5-fold cross validation.
Case studies
To further demonstrate the predictive power of GLNMDA, we conducted three types of case studies on five common human diseases. Specifically, we selected 16 common diseases among the four databases (i.e. dbDEMC[Citation39], miR2Disease[Citation40], miRwayDB[Citation41] and PhenomiR[Citation42]) for the subsequent case studies and validated the prediction results across all the databases. The 16 common diseases are Breast Neoplasms, Cervical Intraepithelial Neoplasia, Colorectal Neoplasms, Hepatocellular Carcinoma, Lymphoma, Lung Neoplasms, Leukemia, Nasopharyngeal Neoplasms, Liver Neoplasms, Ovarian Neoplasms, Pancreatic Neoplasms, Prostatic Neoplasms, Stomach Neoplasms, Thyroid Neoplasms, Urinary Bladder Neoplasms and Uterine Cervical Neoplasms. Due to space limitations, we provided the validation results of 5 diseases in the main text and put the results of the other diseases on Github(https://github.com/ShengPengYu/GLNMDA/tree/master/CaseStudy). The first type of case study was implemented for Lung Neoplasm (LN), Hepatocellular Carcinoma (HC) and Breast Neoplasms (BN), in which we prioritized the top 50 predicted miRNAs for the given diseases based on the known disease-miRNA associations from HMDD v2.0. The prediction results were then verified by another four databases recording experimentally validated disease-related miRNAs.
Lung Neoplasms (LN) characterized by high mortality and high concurrency is one of the most common cancers and have caused a serious threat to human health especially in male[Citation43]. It has been reported that untreated patients with small cell lung cancer will quickly deteriorate and eventually die in 12 weeks[Citation44,Citation45]. Increasing evidence has suggested that miRNAs can not only be utilized to classify LNs, but also have the potential to be biomarkers for early diagnosis and clinical treatment of LN[Citation46–Citation49]. As shown in , 45 out of the top 50 candidate miRNAs were confirmed to be associated with LN. For instance, the hsa-let-7 family which regulates the cell cycle and the hsa-mir-200 family that induces cell death and cell proliferation were all differentially expressed in LN tumor samples[Citation50]. Among the five unconfirmed miRNAs, hsa-miR-499 has been found that the rs3746444T> C polymorphism in its mature sequence could contribute to poor prognosis by modulating cancer-related gene expression and thus involve in the tumorigenesis of LN[Citation51]. Besides, studies have shown that miR-103 was able to promote proliferation of small cell lung cancer cells through targeting MED26 mRNA 3ʹ-UTR[Citation52].
Table 1. Top 50 predicted miRNAs associated with Lung Neoplasms based on known associations in HMDD. I, II, III and IV represent dbDEMC, miR2Disease, miRwayDB and PhenomiR, respectively. The first and third columns record the 1–25 and 26–50 related miRNAs, respectively.
Hepatocellular Carcinoma (HC) is a primary malignancy of the liver and occurs predominantly in patients with underlying chronic liver disease and cirrhosis. Accumulating evidences have shown that the expression patterns of certain miRNAs were significantly different between HC and normal tissues, which might serve as a diagnostic tool for HC[Citation53]. For instance, the ectopic expression of hsa-mir-101 could dramatically suppress the ability of hepatoma cells to form colonies in vitro and to develop tumors in nude mice[Citation54]. The top 50 HC-related miRNAs predicted by our method was listed in . As a result, 46 of the top 50 predicted miRNAs were confirmed to be associated with the given disease by at least one database from dbDEMC, miR2Disease, miRwayDB and PhenomiR. As a matter of fact, one of the unconfirmed miRNAs, hsa-mir-34a, has been to shown to inhibit migration and invasion by down-regulation of c-Met expression in human hepatocellular carcinoma cells[Citation55].
Table 2. Top 50 predicted miRNAs associated with Hepatocellular Carcinoma based on known associations in HMDD. I, II, III and IV represent dbDEMC, miR2Disease, miRwayDB and PhenomiR, respectively. The first and third columns record the 1–25 and 26–50 related miRNAs, respectively.
Breast Neoplasms (BN) is one of the most common female cancers that threatens women’s physical and mental health, accounting for 22% of female cancers[Citation56]. Recent research on miRNAs has implicated that the loss of tumor suppressor miRNAs or overexpression of oncogenic miRNAs can lead to breast cancer tumorigenesis or metastasis. Our prediction results showed that 47 of top 50 candidate miRNAs were confirmed by experimental findings recorded in at least one of the four databases dbDEMC, miR2Disease, miRwayDB and PhenomiR (). For example, the overexpression of hsa-mir-21 (ranked 1st in the prediction list) in human breast cancer is associated with advanced clinical stage, lymhp node metastasis and patient poor prognosis. Moreover, solid evidence has been provided that the C allele of hsa-mir-146a (ranked 2nd in the prediction list) is associated with early familial breast tumor development[Citation57].
Table 3. Top 50 predicted miRNAs associated with Breast Neoplasms based on known associations in HMDD. I, II, III and IV represent dbDEMC, miR2Disease, miRwayDB and PhenomiR, respectively. The first and third columns record the 1–25 and 26–50 related miRNAs, respectively.
In addition, to test the ability of GLNMDA in predicting for diseases without any known associated miRNAs, we conducted the second type of case study on Colorectal Neoplasms (CN). It is reported that more than 1 million individuals will develop colorectal cancer every year worldwide and the disease-specific mortality rate is nearly 33% in the developed world[Citation58]. Firstly, we removed all known associations related with CN and we then used GLNMDA to predict the potential associations between miRNAs and diseases. As a result, 49 of top 50 predicted candidate miRNAs have been confirmed by at least one database from dbDEMC, miR2Disease, miRwayDB and PhenomiR or HMDD (). The only unconfirmed miRNA was hsa-mir-199a. As a matter of fact, evidences have demonstrated that hsa-mir-199a plays a critical role in the cell biological behaviors of colorectal cancer through its target genes[Citation59]. Our prediction results were consistent with existing findings and provided computational evidence for its association with CN.
Table 4. Top 50 predicted miRNAs associated with Colorectal Neoplasms based on known associations in HMDD. I, II, III and IV represent dbDEMC, miR2Disease, miRwayDB and PhenomiR, respectively. The first and third columns record the 1–25 and 26–50 related miRNAs, respectively.
Lastly, we conducted the third type of case studies for Lymphoma where the older version of HMDD was used to prioritize miRNAs with the given disease and the latest version of HMDD v2.0 was adopted to evaluate the prediction results. Due to the distribution characteristics of the lymphatic system, lymphoma is a systemic disease which can invade almost any tissue and organ in the body[Citation60]. miRNAs have also been shown to act as potential biomarkers for the diagnosis of Lymphoma. For example, the under-expression of hsa-mir-150 will increase the incidence of apoptosis and reduced cell proliferation in normal cells[Citation61]. Here, we implemented GLNMDA based on the older version of HMDD which included 1395 associations between 271 miRNAs and 137 diseases. As a result, 49 out of the top 50 predicted miRNAs were confirmed by the HMDD v2.0 and/or the other four databases (). Only hsa-mir-199a was not confirmed. The results showed that GLNMDA is a reliable method to predict the potential miRNA-disease associations.
Table 5. Top 50 predicted miRNAs associated with Lymphoma based on known associations in the older version of HMDD. I, II, III and IV represent dbDEMC, miR2Disease, miRwayDB and PhenomiR, respectively. The first and third columns record the 1–25 and 26–50 related miRNAs, respectively.
Discussion
The identification of novel associations between miRNAs and diseases plays a crucial role in understanding the disease pathogenesis at the miRNA level. In this study, considering the sparsity and incompleteness of disease semantic similarity matrix and miRNA functional similarity matrix, we presented a novel method for miRNA-disease association prediction based on global linear neighborhoods. To demonstrate the effectiveness of the proposed method, we applied global LOOCV, local LOOCV and 5-fold cross-validation to evaluate the prediction performance. GLNMDA achieved AUCs of 0.929, 0.867 and 0.926 in the three frameworks, respectively. We further compared GLNMDA with four state-of-the-art methods and the results confirmed the superior performance of GLNMDA over the other methods. Besides, three types of case studies were implemented on five common human diseases to further validate the utility of GLNMDA. As a result, GLNMDA could uncover novel miRNA-disease associations as expected.
The success of GLNMDA could be largely attributed to the following factors. Firstly, we used the global neighborhoods information to reconstruct the miRNA similarity matrix and disease similarity matrix, which alleviated the sparsity and incompleteness problem existing in the current datasets. Secondly, known experimentally verified miRNA-disease information were used as the benchmark dataset in the cross-validation schema and the initial dataset for predicting latent human miRNA-disease association. Lastly, the known information was propagated by label propagation algorithm iteratively to the whole network according to the similarities reconstructed by GLNMDA.
Nevertheless, there are still limitations in the current version of GLNMDA. Our approach can be improved in the following directions. Firstly, the performance of GLNMDA can be further improved by integrating more available experimentally-verified human miRNA-disease associations. Secondly, multiple information sources can be integrated properly to measure the functional similarity between miRNAs, such as the information of their target genes. In essence, construction for reliable miRNA similarity matrix as well as the disease similarity matrix would help improve the accuracy of GLNMDA.
Materials and methods
Human mirna-disease associations
The human microRNA disease database (HMDD), which contains 5340 experimentally verified links between 495 miRNAs and 383 diseases, is a reliable database[Citation36]. We downloaded miRNA-disease associations information from HMDD database directly. Furthermore, we constructed an adjacent matrix R, of which the element was defined as follows: Rij = 1 if disease d(i) have an interaction with miRNA m(j), and 0 otherwise. Our goal is to confirm the uncertain associations between miRNAs and diseases.
miRNA functional similarity
The miRNA functional similarity used in this paper was calculated by Wang et al. and can be downloaded directly at (http://www.cuilab.cn/files/images/cuilab/misim.zip) [Citation62]. We used M to denote the miRNA functional similarity network, where each element Mij represents the functional similarity score between miRNA m(i) and m(j).
Disease semantic similarity model
Mesh database (http://www.ncbi.nlm.nih.gov/) is a strict system for disease classification and is a credible dataset for effectively researching the relationship between different diseases[Citation62]. The relationship between different diseases can be described through a structure of Directed Acyclic Graph (DAG). A disease A can be described as DAG(A) = (A, T(A), E(A)), where T(A) represents all its ancestors and itself, and E(A) contains edge information including the direct edges linking parent nodes to child nodes. The contribution of disease di in DAG(A) to the semantic value of disease A was defined as follows:
Here, ∆ is the semantic contribution factor and we set ∆ = 0.5 in this paper. For disease di, the contribution of itself is 1, while the contribution of another disease dj decreases as the distance between di and dj increases. Hence, the semantic value of disease A can be calculated according to the contribution of ancestor diseases and disease A itself [Citation63]:
Taken together, the semantic similarity of disease di and disease dj can be calculated as follows:
According to Equation (3), we can construct an overall disease semantic similarity matrix D where Dij represents the semantic similarity between disease di and disease dj.
GLNMDA
In this work, we present a novel framework named GLNMDA to predict potential disease-related miRNAs based on global linear neighborhoods reconstruction. The key assumption of GLNMDA is that each miRNA (disease) can be linearly reconstructed by weighted combinations of its direct neighbors and indirect neighbors which can be reached by any steps of random walk. GLNMDA mainly consists of three steps: Firstly, we reconstruct the miRNA similarity network and disease similarity network based on the known miRNA-disease associations. Secondly, we utilize label propagation algorithm to prioritize novel interactions based on the reconstructed networks, respectively. Lastly, we obtain the final prediction results by combining the output from both miRNA space and disease space. An overall workflow is illustrated in .
Feature representation for miRNAs and diseases
Generally, the reconstruction algorithm is conducted on feature vectors. Therefore, the first step of our algorithm is to construct the feature vectors for both diseases and miRNAs. As presented in the previous work[Citation64], we adopted ‘interaction profile’ to build the features for miRNAs and diseases. Specifically, suppose the miRNA-disease interaction network consists of m RNAs and n diseases, where (M1, M2, M3, …, Mm) and (D1, D2, D3, …, Dn) represent the miRNA set and disease set, respectively. As stated above, if miRNA Mi is related with disease Dj, the entry in the corresponding adjacency matrix is 1 and 0 otherwise. As a result, we could take each column as the feather vector for a given disease and each row as the feature vector for a given miRNA. Obviously, the adjacency matrix R is the disease feature matrix and the transpose of R represents the miRNA feature matrix.
Reconstruction of similarity matrix for diseases and miRNAs
With the rapid development of bio-technology, an increasing amount of biological data is now available for miRNA-disease association studies, including various similarity datasets for diseases and miRNAs. However, due to the limitation of current experimental conditions as well as the inherent noises in these datasets, the miRNA functional similarity matrix M and disease semantic similarity matrix D obtained were in general sparse and incomplete, which might greatly affect the accuracy of prediction results. To address this problem, we here use global linear neighborhoods reconstruction (GLNR) to rebuild the miRNA similarity network and disease similarity network. We assume that each miRNA (disease) can be linearly reconstructed by weighted combinations of its direct neighbors and indirect neighbors which can be reached by any steps of random walk[Citation65]. Let X be the n × m data matrix where xi(i = 1,2,…,n) is the i-th data point in X. According to GLNR, xi can be reconstructed as follows:
where g(xi) is the global neighborhood of xi. Let W be the symmetric n × n similarity matrix between the data points to be learned. Instead of explicitly selecting k neighbors to make W sparse[Citation66], we propose to learn a rank-k non-negative symmetric matrix W = UUT by the following objective function:
where U is a n × k feature matrix. In this paper, for a more general description, X could be either miRNA feature matrix RT or disease feature matrix R. To solve the optimization problem, we first calculated the derivative of Equation (5) with respect to U and we have:
Since X contains only non-negative data, we could obtain the multiplicative update rule as follows:
It is worth noting that to guarantee the convergence of the iterative update rule, we need to normalize our training data in advance[Citation67,Citation68]. Besides, to get an informative value of k for matrix factorization, we employed the clusterONE algorithm accordingly[Citation69], a method for detecting potentially overlapping protein complexes from protein-protein interaction networks. Specifically, clusterONE builds on the concept of the cohesiveness score and uses a greedy growth process to find groups in protein-protein interaction networks that are likely to correspond to protein complexes. It has also been widely adopted to identify cohesive clusters in other types of biological networks due to its simplicity and efficiency[Citation70]. By substituting M into Equation (7), a miRNA clustering matrix was learned as follows:
We then reconstructed the miRNA similarity matrix based on the learned clustering matrix
:
Where is a diagonal matrix with its (i,i)-th element equal to the sum of the ith row of
. Similarly, we could get the disease clustering matrix
by substituting D into Equation (7) as follows:
The reconstructed disease similarity matrix was then obtained by:
Where is a diagonal matrix with its (i,i)-th element equal to the sum of the ith row of
.
After and
were learned, we combined them with existing similarity matrices as follows:
Eventually, we obtained the final disease similarity matrix SD and miRNA similarity matrix SM according to Equation (12) and Equation (13).
Label propagation
After the reconstructed miRNA similarity matrix and disease similarity matrix were obtained, we applied label propagation to predict miRNA-disease associations in miRNA space and disease space, respectively. Generally, a traditional label propagation problem can be presented as follows:
where t is the time step and Zt+1 represents the iteration results after t + 1 steps of label propagation. is a hyper-parameter, Y is a binary matrix encoding the initial label information of data points against each class[Citation65]. The label information of the vertices propagates iteratively between adjacent vertices and the propagation process will eventually converge to a unique global optimization quadratic criterion. Equation (14) has a closed-form solution: Z = (1-α)(I-αL)−1Y, where I is an identity matrix,
is the Laplacian matrix of W and D is a diagonal matrix with its (i, i)-th element equal to the sum of the i-th row of W, i.e.
.
We will use Equation (14) to update the label of each data object until convergence since the closed-form solution to Equation (14) has high computational complexity due to the matrix inversion operation. Here, ‘convergence’ means that the predicted labels of unlabeled data does not change in successive iterations. Therefore, we can predict miRNA-disease association from both disease space and miRNA space:
where FD and FM represent the prediction results from disease space and miRNA space, respectively. Parameter was used to allocate the weight rate of its neighbors while (1-α) represents the probability of receiving its initial label information. The final prediction result F was obtained by combining the results from both miRNA space and disease space:
Parameter β was used to balance the prediction results from disease space and miRNA space, and we simply set β = 0.5. The procedure of GLNMDA is summarized in Algorithm 1. In addition, the source code of GLNMDA could be freely downloaded at https://github.com/ShengPengYu/GLNMDA .
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Ardekani AM, Naeini MM. The role of micrornas in human diseases. Avicenna J Med Biotechnol. 2010 Oct;2(4):161–179. PubMed PMID: 23407304; PubMed Central PMCID: PMC3558168.
- Miska EA. How microRNAs control cell division, differentiation and death. Curr Opin Genet Dev. 2005 Oct;15(5):563–568. . PubMed PMID: 16099643.
- Iorio MV, Ferracin M, Liu CG, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005 Aug 15;65(16):7065–7070. PubMed PMID: 16103053.
- Ambros V. The functions of animal microRNAs. Nature. 2004 Sep 16;431(7006):350–355. . PubMed PMID: 15372042.
- Tang W, Wan SX, Yang Z, et al. Tumor origin detection with tissue-specific miRNA and DNA methylation markers. Bioinformatics. 2018 Feb 1;34(3):398–406. PubMed PMID: WOS:000423978700006; English.
- Blenkiron C, Goldstein LD, Thorne NP, et al. MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biol. 2007;8(10):R214. . PubMed PMID: 17922911; PubMed Central PMCID: PMC2246288
- Zhang X, Zhang X, Wang T, et al. MicroRNA-26a is a key regulon that inhibits progression and metastasis of c-Myc/EZH2 double high advanced hepatocellular carcinoma. Cancer Lett. 2018;426:98–108. . PubMed PMID: WOS:000432877900011; English
- Zeng X, Zhang X, Zou Q. Integrative approaches for predicting microRNA function and prioritizing disease-related microRNA using biological interaction networks. Brief Bioinform. 2016 Mar;17(2):193–203. . PubMed PMID: 26059461.
- Tang W, Liao ZJ, Zou Q. Which statistical significance test best detects oncomiRNAs in cancer tissues? An exploratory analysis. Oncotarget. 2016 Dec 20;7(51):85613–85623. . PubMed PMID: WOS:000391353200147; English.
- Chen X, Yan CC, Zhang X, et al. Long non-coding RNAs and complex diseases: from experimental results to computational models. Brief Bioinform. 2017 Jul;18(4):558–576. PubMed PMID: WOS:000405717400002; English.
- Liao ZJ, Li DP, Wang XR, et al. Cancer diagnosis through isomir expression with machine learning method. Curr Bioinf. 2018;13(1):57–63. PubMed PMID: WOS:000425531200008; English.
- Lu M, Zhang Q, Deng M, et al. An analysis of human microRNA and disease associations. Plos One. 2008;3(10):e3420. . PubMed PMID: 18923704; PubMed Central PMCID: PMCPMC2559869.
- Zou Q, Li JJ, Song L, et al. Similarity computation strategies in the microRNA-disease network: a survey. Brief Funct Genomics. 2016 Jan;15(1):55–64. PubMed PMID: WOS:000370155900008; English.
- Jiang Q, Hao Y, Wang G, et al. Prioritization of disease microRNAs through a human phenome-microRNAome network. Bmc Syst Biol. 2010 May 28;4(Suppl 1):S2. PubMed PMID: 20522252; PubMed Central PMCID: PMCPMC2880408. English.
- Xu J, Li CX, Lv JY, et al. Prioritizing candidate disease miRNAs by topological features in the miRNA target-dysregulated network: case study of prostate cancer. Mol Cancer Ther. 2011 Oct;10(10):1857–1866. PubMed PMID: 21768329.
- Xuan P, Han K, Guo M, et al. Prediction of microRNAs associated with human diseases based on weighted k most similar neighbors. Plos One. 2013 Aug 8;8(8):e70204. PubMed PMID: 23950912; PubMed Central PMCID: PMCPMC3738541. English.
- Chen X, Yan CC, Zhang X, et al. HGIMDA: heterogeneous graph inference for miRNA-disease association prediction. Oncotarget. 2016 Oct 4;7(40):65257–65269. PubMed PMID: WOS:000387281000057; English.
- Shi H, Xu J, Zhang G, et al. Walking the interactome to identify human miRNA-disease associations through the functional link between miRNA targets and disease genes. Bmc Syst Biol. 2013 Oct;8(7):101. . PubMed PMID: 24103777; PubMed Central PMCID: PMCPMC4124764. English.
- Chen X, Yan CC, Zhang X, et al. WBSMDA: within and between score for miRNA-disease association prediction. Sci Rep. 2016 Feb;16(6):21106. . PubMed PMID: 26880032; PubMed Central PMCID: PMCPMC4754743. English.
- Sun DD, Li A, Feng HQ, et al. NTSMDA: prediction of miRNA-disease associations by integrating network topological similarity. Mol Biosyst. 2016;12(7):2224–2232. . PubMed PMID: WOS:000378395000020; English.
- You ZH, Huang ZA, Zhu Z, et al. PBMDA: A novel and effective path-based computational model for miRNA-disease association prediction. Plos Comput Biol. 2017 Mar;13(3):e1005455. PubMed PMID: 28339468; PubMed Central PMCID: PMCPMC5384769. English.
- Chen X, Wang LY, Huang L. NDAMDA: network distance analysis for MiRNA-disease association prediction. J Cell Mol Med. 2018 May;22(5):2884–2895. . PubMed PMID: WOS:000430392700032; English.
- Jiang QH, Wang GH, Jin SL, et al. Predicting human microRNA-disease associations based on support vector machine. Int J Data Min Bioin. 2013;8(3):282–293. . PubMed PMID: WOS:000324166600002; English.
- Chen X, Yan CC, Zhang X, et al. RBMMMDA: predicting multiple types of disease-microRNA associations. Sci Rep. 2015 Sep;8(5):13877. . PubMed PMID: 26347258; PubMed Central PMCID: PMCPMC4561957. English.
- Zou Q, Li J, Hong Q, et al. Prediction of MicroRNA-disease associations based on social network analysis methods. Biomed Res Int. 2015;2015:810514. . PubMed PMID: 26273645; PubMed Central PMCID: PMCPMC4529919. English
- Liu YS, Zeng XX, He ZY, et al. Inferring MicroRNA-disease associations by random walk on a heterogeneous network with multiple data sources. Ieee Acm T Comput Bi. 2017 Jul-Aug;14(4):905–915. PubMed PMID: WOS:000407464700014; English.
- Li JQ, Rong ZH, Chen X, et al. MCMDA: matrix completion for MiRNA-disease association prediction. Oncotarget. 2017 Mar 28;8(13):21187–21199. PubMed PMID: WOS:000397642400057; English.
- Chen X, Wu QF, Yan GY. RKNNMDA: ranking-based KNN for MiRNA-disease association prediction. Rna Biology. 2017;14(7):952–962. . PubMed PMID: WOS:000407258600015; English.
- Chen X, Niu YW, Wang GH, et al. MKRMDA: multiple kernel learning-based Kronecker regularized least squares for MiRNA-disease association prediction. J Transl Med. 2017 Dec 12;15(1):251. PubMed PMID: 29233191; PubMed Central PMCID: PMCPMC5727873. English.
- Chen X, Huang L. LRSSLMDA: laplacian regularized sparse subspace learning for mirna-disease association prediction. Plos Comput Biol. 2017 Dec;13(12):e1005912. . PubMed PMID: 29253885; PubMed Central PMCID: PMCPMC5749861. English.
- Xiao Q, Luo JW, Liang C, et al. A graph regularized non-negative matrix factorization method for identifying microRNA-disease associations. Bioinformatics. 2018 Jan 15;34(2):239–248. PubMed PMID: WOS:000419593000008; English.
- Zeng XX, Liu L, Lu LY, et al. Prediction of potential disease-associated microRNAs using structural perturbation method. Bioinformatics. 2018 Jul 15;34(14):2425–2432. PubMed PMID: WOS:000438248700012; English.
- Chen X, Huang L, Xie D, et al. EGBMMDA: extreme gradient boosting machine for mirna-disease association prediction. Cell Death Dis. 2018 Jan 5;9(1):3. PubMed PMID: 29305594; PubMed Central PMCID: PMCPMC5849212. English.
- Chen X, Zhou Z, Zhao Y, ELLPMDA: ensemble learning and link prediction for miRNA-disease association prediction. RNA Biol. 2018 May 25:1–12. DOI:10.1080/15476286.2018.1460016. PubMed PMID: 29619882.
- Chen X, Xie D, Wang L, et al. BNPMDA: bipartite network projection for miRNA-disease association prediction. Bioinformatics. 2018 Apr 25. DOI:10.1093/bioinformatics/bty333. PubMed PMID: 29701758.
- Li Y, Qiu CX, Tu J, et al. HMDD v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014 Jan;42(D1):D1070–D1074. PubMed PMID: WOS:000331139800157; English.
- Wong TT. Performance evaluation of classification algorithms by k-fold and leave-one-out cross validation. Pattern Recogn. 2015 Sep;48(9):2839–2846. . PubMed PMID: WOS:000356112400007; English.
- Linden A. Measuring diagnostic and predictive accuracy in disease management: an introduction to receiver operating characteristic (ROC) analysis. J Eval Clin Pract. 2006 Apr;12(2):132–139. . PubMed PMID: 16579821.
- Yang Z, Wu LC, Wang AQ, et al. dbDEMC 2.0: updated database of differentially expressed miRNAs in human cancers. Nucleic Acids Res. 2017 Jan 4;45(D1):D812–D818. PubMed PMID: WOS:000396575500113; English.
- Jiang Q, Wang Y, Hao Y, et al. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009 Jan;37(Database issue):D98–104. PubMed PMID: 18927107; PubMed Central PMCID: PMCPMC2686559.
- Das SS, Saha P, Chakravorty N. miRwayDB: a database for experimentally validated microRNA-pathway associations in pathophysiological conditions. Database (Oxford). 2018 Jan 1;2018. doi:10.1093/database/bay023. PubMed PMID: 29688364; PubMed Central PMCID: PMCPMC5829561. English.
- Ruepp A, Kowarsch A, Schmidl D, et al. PhenomiR: a knowledgebase for microRNA expression in diseases and biological processes. Genome Biol. 2010 Jan 20;11(1):R6. PubMed PMID: 20089154; PubMed Central PMCID: PMCPMC2847718. English.
- Yanaihara N, Caplen N, Bowman E, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006 Mar;9(3):189–198. PubMed PMID: 16530703.
- Yu SL, Chen HY, Chang GC, et al. MicroRNA signature predicts survival and relapse in lung cancer. Cancer Cell. 2008 Jan;13(1):48–57. PubMed PMID: 18167339.
- Walker S. Updates in small cell lung cancer treatment. Clin J Oncol Nurs. 2003 Sep-Oct;7(5):563–568. . PubMed PMID: 14603554.
- Raponi M, Dossey L, Jatkoe T, et al. MicroRNA classifiers for predicting prognosis of squamous cell lung cancer. Cancer Res. 2009 Jul 15;69(14):5776–5783. PubMed PMID: 19584273.
- Seike M, Goto A, Okano T, et al. MiR-21 is an EGFR-regulated anti-apoptotic factor in lung cancer in never-smokers. Proc Natl Acad Sci U S A. 2009 Jul 21;106(29):12085–12090. PubMed PMID: 19597153; PubMed Central PMCID: PMCPMC2715493.
- Patnaik SK, Kannisto E, Knudsen S, et al. Evaluation of microRNA expression profiles that may predict recurrence of localized stage i non-small cell lung cancer after surgical resection. Cancer Res. 2010 Jan 1;70(1):36–45. PubMed PMID: WOS:000278404300007; English.
- Croce CM. Causes and consequences of microRNA dysregulation in cancer. Eur J Cancer. 2012 Jul;48:S8–S9. PubMed PMID: WOS:000313036500033; English.
- Inamura K, Ishikawa Y. MicroRNA In Lung Cancer: novel biomarkers and potential tools for treatment. J Clin Med. 2016 Mar 9;5(3). DOI:10.3390/jcm5030036 PubMed PMID: 27005669; PubMed Central PMCID: PMCPMC4810107. English.
- Fm Q, Yang L, Xx L, et al. Sequence variation in mature microRNA-499 confers unfavorable prognosis of lung cancer patients treated with platinum-based chemotherapy. Clin Cancer Res. 2015 Apr 1;21(7):1602–1613. PubMed PMID: WOS:000352076700015; English.
- Zhu XX, Zhang X, Wang HF, et al. MTA1 gene silencing inhibits invasion and alters the microRNA expression profile of human lung cancer cells. Oncol Rep. 2012 Jul;28(1):218–224. PubMed PMID: WOS:000304638900031; English.
- Murakami Y, Yasuda T, Saigo K, et al. Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene. 2006 Apr 20;25(17):2537–2545. PubMed PMID: 16331254.
- Su H, Yang JR, Xu T, et al. MicroRNA-101, down-regulated in hepatocellular carcinoma, promotes apoptosis and suppresses tumorigenicity. Cancer Res. 2009 Feb 1;69(3):1135–1142. PubMed PMID: 19155302.
- Li N, Fu H, Tie Y, et al. miR-34a inhibits migration and invasion by down-regulation of c-Met expression in human hepatocellular carcinoma cells. Cancer Lett. 2009 Mar 8;275(1):44–53. PubMed PMID: 19006648.
- Al-Hajj M, Wicha MS, Benito-Hernandez A, et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003 Apr 1;100(7):3983–3988. PubMed PMID: 12629218; PubMed Central PMCID: PMCPMC153034.
- Pastrello C, Polesel J, Della Puppa L, et al. Association between hsa-mir-146a genotype and tumor age-of-onset in BRCA1/BRCA2-negative familial breast and ovarian cancer patients. Carcinogenesis. 2010 Dec;31(12):2124–2126. PubMed PMID: WOS:000284953900013; English.
- Ogino S, Giannakis M. Immunoscore for (colorectal) cancer precision medicine. Lancet. 2018 May 26;391(10135):2084–2086. . PubMed PMID: WOS:000433257200007; English.
- Han Y, Kuang YT, Xue XF, et al. NLK, a novel target of miR-199a-3p, functions as a tumor suppressor in colorectal cancer. Biomed Pharmacother. 2014 Jun;68(5):497–505. PubMed PMID: WOS:000342667800001; English.
- Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. Ca-Cancer J Clin. 2017 Jan-Feb;67(1):7–30. . PubMed PMID: WOS:000393807800003; English.
- Watanabe A, Tagawa H, Yamashita J, et al. The role of microRNA-150 as a tumor suppressor in malignant lymphoma. Leukemia. 2011 Aug;25(8):1324–1334. PubMed PMID: WOS:000293778900012; English.
- Wang D, Wang JA, Lu M, et al. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics. 2010 Jul 1;26(13):1644–1650. PubMed PMID: WOS:000278967500010; English.
- Qu Y, Zhang HX, Liang C, et al. KATZMDA: prediction of miRNA-disease associations based on katz model. Ieee Access. 2018;6:3943–3950. . PubMed PMID: WOS:000426286900001; English
- Zhang W, Qu QL, Zhang YQ, et al. The linear neighborhood propagation method for predicting long non-coding RNA - protein interactions. Neurocomputing. 2018 Jan;17(273):526–534. . PubMed PMID: WOS:000414762100049; English.
- Zhang W, Chen Y, Li D. Drug-target interaction prediction through label propagation with linear neighborhood information. Molecules. 2017 Nov 25;22(12). DOI:10.3390/molecules22122056 PubMed PMID: 29186828; English.
- Zhu L, Shen JL, Xie L, et al. Unsupervised topic hypergraph hashing for efficient mobile image retrieval. Ieee T Cybernetics. 2017 Nov;47(11):3941–3954. PubMed PMID: WOS:000413003100037; English.
- Zhu L, Shen JL, Jin H, et al. Landmark classification with hierarchical multi-modal exemplar feature. Ieee T Multimedia. 2015 Jul;17(7):981–993. PubMed PMID: WOS:000356522300006; English.
- Wong KC. MotifHyades: expectation maximization for de novo DNA motif pair discovery on paired sequences. Bioinformatics. 2017 Oct 1;33(19):3028–3035. PubMedPMID: WOS:000411514100008; English.
- Nepusz T, Yu HY, Paccanaro A. Detecting overlapping protein complexes in protein-protein interaction networks. Nat Methods. 2012 May 9;5:471–U81. PubMed PMID: WOS:000303544800024; English.
- Li Y, Liang C, Wong KC, et al. Mirsynergy: detecting synergistic miRNA regulatory modules by overlapping neighbourhood expansion. Bioinformatics. 2014 Sep 15;30(18):2627–2635. PubMed PMID: WOS:000342913000012; English.
Table