
ABSTRACT
The recently discovered clustered regularly interspaced short palindromic repeats (CRISPR)-Cpf1 system expands the genome editing toolbox. This system exhibits several distinct features compared to the widely used CRISPR-Cas9 system, but has reduced gene editing efficiency. To optimize the CRISPR-Cpf1 (Cas12a) system, we report the inclusion of self-cleaving ribozymes that facilitate processing of the crRNA transcript to produce the precise guide molecule. Insertion of the 3ʹ-terminal HDV ribozyme boosted the gene editing activity of the CRISPR-Cpf1 system ranging from 1.1 to 5.2 fold. We also demonstrate that this design can enhance CRISPR-based gene activation. We thus generated an improved CRISPR-Cpf1 system for more efficient gene editing and gene regulation.
Introduction
The bacterial CRISPR-Cas system has been repurposed for genome editing jobs in various organisms [Citation1–Citation4]. The widely used CRISPR-Cas9 system requires the Streptococcus pyogenes Cas9 (spCas9) enzyme and a guide RNA (gRNA) to edit genomic regions that have a G-rich protospacer adjacent motif (PAM) sequence. Recently, the CRISPR-Cpf1 system was reported to expand the genome editing possibilities [Citation5,Citation6]. CRISPR-Cpf1 offers several unique features: the Cpf1 nuclease and the matching CRISPR RNA (crRNA) are smaller than the Cas9 counterparts, which is favorable for gene delivery; Cpf1 targets a T-rich PAM sequence, thus expanding the potential target sequences; Cpf1 produces a sticky DNA end that was suggested to favor DNA recombination; Cpf1 has RNase activity for crRNA processing that can be employed for multiplex gene editing [Citation5,Citation6]. Cpf1 was also reported to exhibit high sequence-specificity, thus reducing the chance of off-target effects [Citation7,Citation8]. However, a serious disadvantage of CRISPR-Cpf1 is that it exhibits reduced editing activity compared to CRISPR-Cas9 [Citation7–Citation9], which restricts the potential applications.
In an attempt to optimize CRISPR-Cpf1, we focused on Cpf1 orthologs from Acidaminococcus sp (AsCpf1) and Lachnospiraceae bacterium (LbCpf1) that have been used for genome editing in human cells [Citation5,Citation8]. There remains some uncertainty on the exact 5ʹ and 3ʹ end of the matching crRNA molecules, which may affect their activity. For both crRNA molecules (, the first nucleotide was initially reported to be U [Citation5] but subsequent studies suggested that this nucleotide, due to the RNase activity of Cpf1, is not part of the mature crRNA [Citation10,Citation11]. The effect of the presence/absence of this 5ʹ-terminal U (in between brackets in ) on crRNA activity is not known. As the commonly used RNA polymerase III (Pol III) promoters for small RNA expression prefer to starts with a pyrimidine (G/A) [Citation12,Citation13], expression of the variant with 5ʹ-U may be less efficient. At the 3ʹ end, Pol III will terminate at a heterogeneous position within a T-stretch, thus creating crRNAs with a variable U-tail of 1–6 nucleotides [Citation14]. This U-tail is juxtaposed to the guide sequence that recognizes the DNA target and one study suggested a negative effect of this 3ʹ U-tail on the crRNA activity of AsCpf1 [Citation14]. Therefore, expressing the exact crRNA molecule might be critical for optimal Cpf1 activity. In this study, we attempted to generate more exact crRNA molecules by using the self-cleaving hammerhead (HH) and hepatitis delta virus (HDV) ribozymes that instruct precise RNA processing (). The effect of ribozyme addition on crRNA production and activity was systematically investigated. We demonstrate that the 3ʹ-positioned HDV element can significantly boost the CRISPR-Cpf1 activity. We also demonstrate that this crRNA-HDV design enhanced the performance of CRISPR-based gene activation systems.
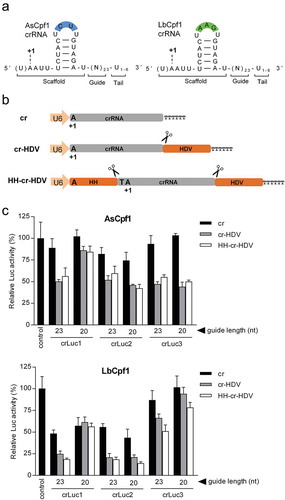
Figure 1. Ribozyme-processed crRNA enhances the Luc knockdown activity of CRISPR-Cpf1. (a) The crRNA structures of the As and LbCpf1 systems. Both crRNA molecules consist of a ~ 20-nt scaffold and a 23-nt guide (N23). The variable U1-6 tail at the 3ʹ-end is generated when a standard Pol III promoter cassette is used. The variable loop nucleotide positions are marked in blue and green boxes. The first crRNA nucleotide is marked as +1A, but the upstream U (in brackets) has also been implicated in the transcription initiation process. (b) Schematic of three crRNA expression constructs. The Pol III human U6 promoter drives crRNA transcription up to the T6 (TTTTTT) termination signal. The HH and HDV ribozymes were introduced to guide crRNA processing exacty at the crRNA border (marked as scissor). The +1A represents the first crRNA nucleotide. (c) Luc knockdown activity of CRISPR-Cpf1. An equimolar amount of crLuc constructs (equivalent to 50 ng cr vector) together with their cognate Cpf1 plasmids (equivalent to 100 ng AsCpf1 vector) were co-transfected into HEK293T cells with 200 ng Luc reporter and 2 ng Renilla luciferase plasmid to control for the transfection efficiency. The empty cr plasmid served as negative control. The relative Luc activity normalized for Renilla expression was determined at two days post-transfection and the control Luc activity was arbitrarily set at 100%. The results are shown as mean values ± standard deviation (SD, n = 3).
Results
The 5ʹ and 3ʹ ribozymes enhance the crRNA activity
To express crRNA molecules with a precise 5ʹ and 3ʹ end, we inserted the HH and HDV ribozymes [Citation15] that can mediate precise intramolecular RNA cleavage (). Three different As/Lb crRNA expression vectors were compared: the standard cr construct and the new designs cr-HDV and HH-cr-HDV. The original cr construct uses the U6 promoter to drive crRNA transcription up to the T6 Pol III termination signal. To generate crRNA transcripts without U-tail, the cr-HDV construct was created by positioning the HDV ribozyme in between the crRNA and the T6 signal. To generate crRNA molecules that start with U at the 5ʹ-end and without U-tail, the HH-cr-HDV construct was made that encodes a 5ʹ-terminal HH ribozyme immediately upstream of the initiator U residue. The HH and HDV ribozymes mediate self-cleavage exactly at the border of the ribozyme-crRNA fusion (), thus releasing the wild-type crRNA.
To evaluate if the inclusion of flanking ribozymes benefits the activity of the CRISRP-Cpf1 system, we performed a Firefly-luciferase (Luc) reporter knockdown assay in transiently transfected HEK293T cells. Three crRNAs (crLuc1, 2 and 3) with different anti-Luc guides were designed, each with 23 or 20-nt guides and all six crRNA designs were tested in the As and Lb context. Equimolar amounts of the crLuc constructs were co-transfected with the matching Cpf1 plasmid and a fixed amount of the Luc target plasmid into HEK293T cells. A Renilla luciferase plasmid was included in all transfections to control for variation in the transfection efficiency. Two days post-transfection, cells were harvested, lysed and the Luc reporter assay was performed to determine the relative Luc activity (Firefly/Renilla).
For AsCpf1 (, upper panel), no significant suppression was induced by the cr constructs compared to the control transfection that was set at 100%. But all cr-HDV and HH-cr-HDV constructs showed significant Luc inhibition. This improvement was obtained regardless of guide RNA length (23 or 20-nt), except for crLuc1 for which the 23-nt guide outperformed the 20-nt guide. Collectively, the cr-HDV design with 23-nt guide seems the best choice for optimal DNA cleavage of the AsCfp1 system.
Efficient Luc inhibition was observed for most cr constructs in LbCpf1 encoding crLuc1, 2 and – to a lesser extent – crLuc3 (, lower panel). LbCpf1 generally mediated more robust Luc knockdown than AsCpf1, consistent with previous reports [Citation7,Citation16]. Most importantly, improved knockdown was achieved by the cr-HDV and in particular the HH-cr-HDV design compared to the cr control, confirming the predominant role of the 3ʹ-terminal HDV motif in this improvement. The 23-nt guide outperformed the 20-nt guide for crLuc1 and 3 but not for crLuc2, cautiously suggesting that the 23-nt guide is the optimal design for the HH-cr-HDV backbone in the LbCpf1 system.
Titration of the CRISPR-Cpf1 components
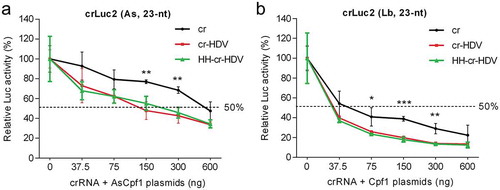
We next tested the dosage effect of CRISPR-Cpf1 components for the crLuc2 set with the 23-nt guide. The three As and Lb constructs (cr, cr-HDV and HH-cr-HDV) and the matching Cpf1 plasmids were titrated – at a fixed ratio – and co-transfected with fixed amounts of the Luc and Renilla vectors into HEK293T cells. The relative Luc activity was determined two days post-transfection. A clear dose-dependent Luc knockdown was apparent for all crLuc2 constructs (). Importantly, the original cr constructs in both the As and Lb contexts were outperformed by the novel cr-HDV and HH-cr-HDV designs. Overall, the LbCpf1 system is more active than AsCpf1, reaching more than 50% knockdown at the lowest plasmid DNA concentration (37.5 ng) and at least 80% knockdown at higher levels.
Figure 2. Titration of CRISPR-Cpf1 components. CRISPR-Cpf1 constructs were titrated in the 37.5–600 ng range and co-transfected into HEK293T cells with 200 ng Luc reporter and 2 ng Renilla luciferase plasmid. The titration was performed with a fixed 1:2 stoichiometry of the cr plasmid and the matching Cpf1 plasmid. The relative Luc activity was determined two days post-transfection and all data are presented as the mean value ± SD (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001; one-way ANOVA.
Expression and processing of the crRNA-ribozyme transcripts
In order to examine crRNA expression from the different vector backbones, we performed Northern blotting analysis. We first compared the As and Lb constructs for the complete crLuc2 set. The same molar amount of the crLuc2 constructs were co-transfected into HEK293T cells with a fixed molar amount of the cognate Cpf1 plasmids. Total cellular RNA was extracted two days post-transfection and a fixed amount (5 µg) was subjected to Northern blotting with the Luc2 probe that targets the crLuc2 guide region (, left panel). A prominent band of ~40-nt that corresponds to the processed crRNAs was apparent for all crLuc2 constructs (, right panel). As expected, a slightly longer and more diffuse crRNA transcript was made from the cr construct compared to cr-HDV and HH-cr-HDV. This result confirms the presence of the variable U-tail in cr and precise 3ʹ end formation by HDV-mediated self-cleavage, which correlates with improved Luc inhibition by the HDV-containing constructs (). A minor signal of >100-nt was detected for several HDV-containing constructs, which reflects the non-processed crRNA-HDV precursor transcript, indicating ~20% incomplete HDV cleavage. However, incomplete cleavage was not observed for the crLuc2-(Lb)-20 constructs, suggesting a sequence-dependent effect. No HH-crRNA-HDV precursor transcript was observed, suggesting complete cleavage by the HH ribozyme.
Figure 3. crRNA expression from the crRNA-ribozyme cassettes. HEK293T cells in a 6-well plate were transfected with the same molar amount of indicated crRNA constructs (equivalent to 1 µg cr vector) and a fixed molar amount of their cognate Cpf1 plasmids (equivalent to 2 µg AsCpf1 vector). Total cellular RNA was harvested two days post-transfection and 5 µg was subjected to Northern blotting using the Luc2 probe targeting the crLuc2 guide region (panel A) and the Lb probe targeting Lb crRNA scaffold (panel B). The crRNA and precursor crRNA-HDV transcripts are marked. An RNA size marker (nt) was included. Ethidium bromide staining of 5S RNA is shown at the bottom as loading control. Quantitation of crRNA normalized by 5S RNA is plotted below the blot and the cr signal of crLuc2 (As) was arbitrarily set at 10. The results were produced in two independent experiments that showed similar trends.
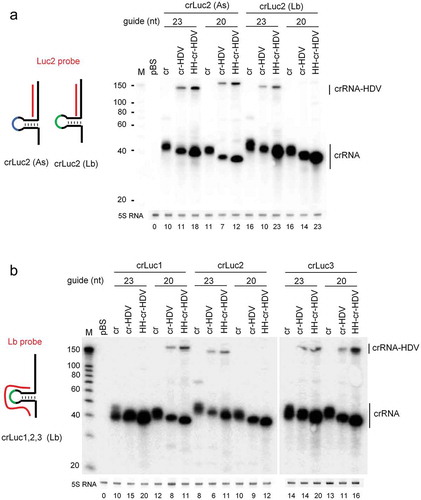
Intriguingly, for both As and LbCpf1, we noticed that crRNA generated by HH-cr-HDV are smaller than the transcripts produced by cr-HDV (, right panel), which is contrary to the expectation that the HH-cr-HDV-generated crRNA starts 1-nt upstream of +1A at the U, thus making 1-nt longer transcripts than the crRNA of cr-HDV (). However, this result is consistent with the recent finding that the RNase activity of AsCpf1 generates crRNAs starting with +1A, with some promiscuous cleavage further downstream [Citation11]. Combining these results, we conclude that the U upstream of +1A is not part of the crRNA.
Quantitation of the signals in indicated generally increased crRNA production for the Lb over the As system, which seems to correlate with the more efficient Luc inhibition by the former system. The two cr-HDV constructs with 23-nt guide exhibited similar crRNA production, yet demonstrated a profound difference in activity (), strongly indicating that the more efficient DNA cleavage activity of LbCpf1 is not due to increased crRNA production. HH-cr-HDV seems to produce most crRNA, which may relate to the difference in nucleotide identity around the transcription initiation site of the U6 promoter, which is a key determinant of the transcription efficiency [Citation12,Citation13]. Indeed, the cr and cr-HDV constructs are identical, but HH-cr-HDV has a unique initiation sequence of the HH ribozyme ().
We next assessed the Lb crLuc constructs with the Lb probe that binds the crRNA scaffold, thus allowing the comparison of crRNA molecules within different guide sequences (, left panel). A dominant ~40-nt mature crRNA band was apparent for all constructs and as expected the cr transcripts are a few nt longer than the cr-HDV and HH-cr-HDV transcripts (, right panel), consistent with previous results ( and ). We observed that the cr-HDV design improved the activity by removal of the U-tail ( lower panel). The absence of the cr-HDV precursor transcripts for some constructs confirms a sequence-dependent effect on HDV cleavage, possibly through misfolding of the ribozyme moiety. Again, the crRNA made by cr-HDV is slightly longer than that made by HH-cr-HDV, indicating RNase-mediated trimming by LbCpf1. The HH-cr-HDV construct seems the most potent crRNA expressor.
Collectively, the data indicate that crRNA molecules can be expressed correctly from cr-HDV and HH-cr-HDV backbones, consistent with the increased Luc knockdown activity when compared to the standard cr construct. LbCpf1 induced more efficient Luc knockdown than AsCpf1, suggesting that it is a better tool for gene editing. Based on the similar crRNA production and Luc knockdown activity of cr-HDV and HH-cr-HDV, we conclude that the HH moiety is less important for CRISPR-Cpf1 optimization, but that the 3ʹ-HDV addition plays a critical role. Collectively, the LbCpf1 system with the cr-HDV design seems most suited for gene editing applications.
Chromosomal gene editing: cr versus cr-hdv
We next wanted to validate the improvement measured for cr-HDV in the context of chromosomal gene editing. We chose the most efficient Lb system with the 23-nt guide crRNA. The crLuc (1, 2 and 3) containing a 23-nt guide in the cr and cr-HDV backbones were used to target the chromosomally integrated Luc gene in the HeLa X1/6 cell line. To do so, the same molar amount of cr or cr-HDV construct plus a fixed amount of LbCpf1 and EGFP plasmids were co-transfected. The empty cr construct was included as negative control. Two days post-transfection, the EGFP-positive cells were isolated by fluorescence-activated cell sorting (FACS) and used for DNA extraction. The editing efficiency was evaluated by the Surveyor nuclease assay that provides information on insertions and deletions (indels) (). A major unedited full-length amplification product of ~850-bp was generated for all samples. All three cr and three cr-HDV molecules were able to induce Luc cleavage and the cleavage products are marked by black arrows. The Luc gene editing efficiency was calculated and the results are generally consistent with the Luc knockdown efficiency: cr-HDV significantly boosted the editing efficiency of cr in Luc1 (18.7% vs. 7.8%) and Luc2 (16.0% vs. 4.2%), but slightly reduced Luc editing for Luc3 (10.3% vs. 13.7%).
Figure 4. The cr-HDV design enhances the gene editing efficiency. (a) Comparison of the cr and cr-HDV constructs for chromosomal Luc gene editing. The same molar amounts of cr and cr-HDV plasmids (equivalent to 100 ng cr vector) and 200 ng LbCpf1 plasmid were co-transfected into HeLa X1/6 cells. Two days post-transfection, the cells were harvested for the Surveyor nuclease assay to estimate the Luc editing efficiency. The empty cr construct served as negative control. The unedited Luc gene amplicon is marked by an open triangle and the black triangles mark the Surveyor nuclease-cleaved bands due to Luc gene editing by Cpf1. The indel percentage (%) was calculated and is plotted below. The graph represents one of two independent experiments that yielded similar results. (b) Comparison of cr and cr-HDV for CCR5 gene editing. An equimolar amount of cr/cr-HDV plasmid (equivalent to 100 ng cr) and 200 ng LbCpf1 plasmid were co-transfected into TZM-bl cells with 100 ng JS-1 plasmid (a GFP expression vector) serving as transfection control. Two days post-transfection, cells were stained with anti-CCR5 antibody and the CCR5 expression was determined by FACS analysis. The empty cr construct servesd as negative control. The y axis represents forward-scattered light (SSC) and the x axis represents CCR5 fluorescence intensity. The CCR5 disruption efficiency was measured for the GFP-positive cell fraction. The fold increase (×) in editing efficiency of cr-HDV versus cr was calculated. The results are presented as mean value ± SD of three independent experiments. The raw data are shown in Figure S1.
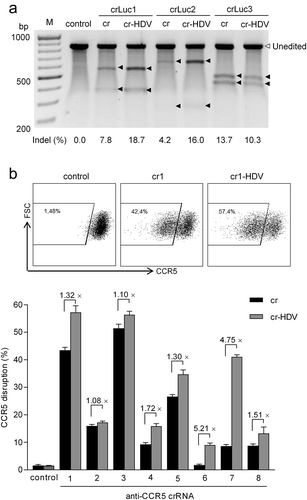
To confirm HDV-mediated improvement of crRNA activity for another target, we designed eight crRNAs against the HIV-1 receptor-encoding CCR5 gene. The cr or cr-HDV constructs were transfected into TZM-bl cells together with a fixed amount of LbCpf1 plasmid. Two days post-transfection, CCR5 gene disruption was determined by FACS analysis of CCR5 protein expression at the cell surface (). The eight crRNAs varied in activity, but we consistently measured superior CCR5 knockout efficiency for the cr-HDV over the cr design. The improvement ranged from a minimal 8% (crRNA2) to ~5-fold (crRNA6 and 7). Taken together, we demonstrated that the cr-HDV design can significantly enhance the genome-editing efficiency of the CRISPR-LbCpf1 system.
The cr-HDV design also improves CRISPR-based activation systems
A DNase-dead Cpf1 (dCpf1) mutant was fused to transcription activation or repression domains to allow the regulation of gene expression in a sequence-specific manner [Citation17–Citation19]. We wanted to test if the cr-HDV design is also superior over cr in CRISPR activation (CRISPRa) systems. To do this, we employed the doxycycline (dox)-inducible Tet-On luciferase reporter system that is present in the HeLa X1/6 cell line [Citation20]. We first made dCpf1-VP64 constructs by fusing As/Lb-dCpf1 to the VP64 transcriptional activation domain (). Six Tet-On targeting crRNAs were designed, four (crRNA1-4) against the tetracycline response element (TRE) upstream of the minimal CMV promoter (CMVmin) and two (crRNA5 and 6) against the leader of the Luciferase gene (). The same molar amount of the crRNA constructs were transfected into cells together with a fixed molar of the cognate Cpf1 plasmids. The canonical Luc-induction method, consisting of dox addition and transfection of a plasmid encoding the rtTA transactivator, served as positive control. A Renilla plasmid was co-transfected to correct for the transfection efficiency. Two days post-transfection, the relative Luc expression representing the CRISPRa activity was determined (). Only 3 crRNAs were active in the As system and 4 of 6 crRNAs in the Lb context. Two As crRNAs (1 and 4) induced Luc expression more robustly than the standard induction method with dox + rtTA. Most importantly, the cr-HDV activity at least paralleled that of cr, but a significant improvement was measured for 3 crRNAs in the As context and 2 crRNAs in the Lb system. Thus, we conclude that the cr-HDV design can also enhance CRISPRa activity.
Figure 5. The cr-HDV design also increases the CRISPRa activity. (a) Schematic of the dCpf1-VP64 construct. (b) Schematic of Tet-On inducible Luc reporter cassette (7xTRE-CMVminimal-Luc) that is chromosomally integrated in HeLa X1/6 cells. The TRE and the location of the crRNA targets are indicated, crRNA1-4 target the 7× TRE repeat and crRNA5 and 6 the Luc gene leader sequence. (c) Luc induction by the CRISPR-based systems. Dox addition and transfection of a rtTA expressing plasmid was used as positive control. For CRISPR-based Tet-On induction, an equimolar amount of cr/cr-HDV construct (equivalent to 100 ng cr vector) and a fixed amount of the cognate dCpf1-VP64 plasmid (equivalent to 200 ng dAsCpf1-VP64 vector) were transfected into HeLa X1/6 cells with a Renilla plasmid to control for transfection efficiency. The empty crRNA plasmid acted as negative control. The relative Luc expression normalized by Renilla was determined at two days post-transfection. The results are represented as mean value ± SD (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001; unpaired t test.
An analysis of the HDV-mediated improvement versus the relative crRNA activity in (gene editing) and (gene activation) indicates that the least active crRNAs benefit most from the cr-HDV design (). For genes with many target sites, it may suffice to simply select the best crRNA for the gene editing/activation job. But for genes with few target sites, the cr-HDV design may be more valuable to achieve efficient gene editing/activation.
Figure 6. The least active crRNAs benefit the most from the HDV design. We plotted the fold increase by the HDV insertion versus the relative crRNA activity (%) of the original cr construct. The data on gene editing and gene activation was taken from and , respectively. The Pearson’s correlation coefficient (r) was calculated.
Discussion
The recently discovered Cpf1 system significantly expands the CRISPR-based genome editing tools. This system exhibits several distinct features, but it also exhibits a reduced editing efficiency compared to the Cas9 system. Here, we attempted to optimize the CRISPR-Cpf1 system by expressing more exact crRNA guide molecules by means of self-cleaving ribozymes. We demonstrate that the 3ʹ-terminal HDV fusion boosts the efficiency of both gene editing and gene activation by CRISPR-Cpf1 systems.
The classical Pol III promoter cassette produces a crRNA with a variable 3ʹ-terminal U-tail, which extends the crRNA guide sequence that is required for target DNA recognition. A negative effect of this U-tail on Cpf1 activity can be expected as guide sequence extension – even when extending the target complementarity – reduced the Cpf1 editing efficiency [Citation8,Citation11]. We demonstrated that this U-tail can be removed by inclusion of the 3ʹ-terminal HDV ribozyme and the cr-HDV design enhanced the gene editing and activation activity of As and LbCpf1 systems. An initial report suggested U to be the first nucleotide of As and Lb crRNAs[Citation5]. Our results indicate that a slightly longer crRNA is produced by cr-HDV than HH-cr-HDV, suggesting that the first U of is not part of crRNA, as was recently shown for AsCpf1 crRNA [Citation11]. Thus, the 5ʹ-terminal HH ribozyme is dispensable for crRNA formation.
The RNase activity of Cpf1 has been employed for processing of multiplexed crRNA transcript into the individual crRNA units [Citation11]. However, this RNase-mediated crRNA processing is imprecise, producing individual crRNAs with additional nucleotides at the 3ʹ-terminal end of crRNA guide region. A negative effect of these 3ʹ-nucleotides on Cpf1 activity is possible as was demonstrated for the 3ʹ-terminal U-tail. Indeed, no gain of gene editing activity was scored for such multiplexed crRNAs [Citation11]. We propose that inclusion of HDV in the crRNA array [crRNA-(HDV-crRNA)n] may improve the gene editing efficiency.
Apart from the 3ʹ-terminal nucleotide, the guide length may be another important factor that affects the editing efficiency of CRISPR-Cpf1 as suggested by comparison of the 20 and 23-nt guides (). It may thus be of interest to perform a detailed survey to identify the optimal guide length for different Cpf1 systems. Our results also showed that the gene editing efficiency of CRISPR-Cpf1 varies significantly among different crRNA molecules (), indicating the importance of crRNA selection for efficient gene editing applications.
The generally more efficient DNA cleavage activity of the Lb over the As system confirms that the former Cpf1 system is the more efficient gene editing tool, as previously suggested across various species [Citation8,Citation16,Citation18]. Despite conflicting results in literature regarding to the CRISPRa potency of these two systems [Citation17,Citation18], our results demonstrate greater CRISPRa activity of the As system, which makes it the candidate of choice for CRISPR-based gene regulation. Inclusion of the HDV ribozyme to facilitate exact 3ʹ-end formation of the crRNA increases both the gene editing and gene activation activity. As the HDV ribozyme is an autonomous element that does not utilize cell type-dependent cofactors, we envisage that the cr-HDV design will be useful for CRISPR-Cpf1 gene editing and regulation applications in a wide range of cell types.
Materials and methods
Vector construction
The plasmids pY010 (addgene# 69,982) and pY016 (addgene# 69,988) that express the human codon-optimized As and LbCpf1, respectively, were kindly donated by Feng Zhang [Citation5]. The plasmids WN10150 (addgene# 80,443) and WN10150 (addgene# 80,441) encoding the human codon-optimized dAsCpf1 and dLbCpf1 were gifts from Ervin Welker. The pSilencer2.0-U6 vector is used as the crRNA expression backbone. The U6-cr, U6-cr-HDV and U6-HH-cr-HDV DNA fragments containing two BsmBI enzyme sites for crRNA cloning were synthesized by IDT and cloned into the pSilencer2.0-U6 vector using the PmII and HindIII restriction enzyme sites by the Gibson cloning method (NEB). The DNA oligonucleotides encoding the crRNA targeting sequences were annealed and inserted into crRNA expression vectors via the BsmBI sites. All crRNAs () were designed via the online design tool Benchling (https://www.benchling.com). To create dCpf1-VP64 constructs, the DNA fragment encoding three tandem HA epitope tags (3xHA) and one VP64 transcriptional activation domain was synthesized by IDT and cloned into WN10150 and WN10150 plasmids using the BamHI and EcoRI sites by Gibson cloning (NEB). All constructs were verified by sequencing using the BigDye Terminator v1.1 Cycle Sequencing Kit (ABI).
Table 1. Target Sequence (PAM Underlined).
Cell culture
Human embryonic kidney (HEK) 293T cells, HeLa X1/6 cells and TZM-bl cells were grown as a monolayer in Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies, Invitrogen) supplemented with 10% fetal calf serum (FCS), minimal essential medium nonessential amino acids, penicillin (100 U/ml) and streptomycin (100 μg/ml) at 37°C and 5% CO2.
Dual-luciferase reporter assay
One day prior to transfection, 3 × 105 HEK293T cells were seeded per well in 12-well plate. Equimolar amount of crRNA constructs (equivalent to 100 ng cr plasmid) and a fixed molar amount of their cognate Cpf1 plasmids (equivalent to 200 ng AsCpf1 plasmid) were co-transfected with 200 ng pGL3 Luc reporter into cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Two ng of Renilla luciferase plasmid was co-transfected in all tests to control for the transfection efficiency. Two days post-transfection, luciferase expression was measured with the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) according to the manufacturer’s protocol. The ratio of Firefly to Renilla was calculated as the relative Luc activity. Three independent transfections were performed.
Northern blotting analysis
One day prior to transfection, 9 × 105 HEK293T cells were seeded per well in a 6-well plate. Equimolar amount of crRNA constructs (equivalent to 1 μg of cr vector) and a fixed molar amount of the matching Cpf1 constructs (equivalent to 2 μg AsCpf1 plasmid) were co-transfected into cells using Lipofectamine 2000 (Invitrogen). Total cellular RNA was extracted 48h post-transfection using mirVana miRNA isolation kit (Ambion). Five μg of total RNA was heated for 5 min at 95°C and then resolved in a 15% denaturing polyacrylamide gel (Precast Novex TBU gel, Life Technologies). The [γ-32P]-labeled decade RNA marker (Life Technologies) was used for size estimation. To check for equal sample loading, the gel was stained in 2 μg/ml ethidium bromide for 20 min and visualized under UV light. The RNA in the gel was transferred to a positively charged nylon membrane (Boehringer Mannheim, GmbH). Locked nucleic acid (LNA) oligonucleotides were 5ʹ end-labeled with the kinaseMax kit (Ambion) in the presence of 1 μl [γ-32P]-ATP (0.37 MBq/μl, Perkin Elmer). Sephadex G-25 spin columns (Amersham Biosciences) were used to remove the unincorporated nucleotides. We used the following oligonucleotides (LNA-positions are underlined): Luc2 probe (5ʹ-GAGATCCTATTTTTGGCAA-3ʹ) and Lb probe (5ʹ-TCTACACTTAGTAGAAATT-3ʹ). The membrane was incubated with labeled LNA oligonucleotides in 10 ml ULTRAhyb hybridization buffer for overnight at 42°C. The membrane was washed with low stringency and high stringency buffers. The signals were captured by Typhoon FLA 9500 (GE Healthcare Life Sciences) and quantitated using Image J software.
Indels detection by the surveyor nuclease assay
One day prior to transfection, 4.5 × 105 HeLa X1/6 cells were seeded per well in a 6-well plate. Equimolar amount of crRNA constructs (equivalent to 1 μg of cr plasmid) and 2 μg LbCpf1 plasmid were co-transfected using Lipofectamine 2000 (Invitrogen). Two days after transfection, the genomic DNA was extracted using the QIAamp DNA Mini Kit (QIAGEN). The genomic region (840 bp) flanking the designed cleavage site was amplified using two primers: forward (5ʹ-AAGACGCCAAAAACATAAAGAAAG-3ʹ) and reverse (5ʹ-AAGAGGTGCGCCCCCAGAAG-3ʹ). The PCR fragments were gel-purified and subjected to surveyor nuclease using the Surveyor Mutation Detection Kit (Integrated DNA Technologies) according to the manufacturer’s instructions. The products were visualized on a 1% agarose gel by ethidium bromide staining. The indel percentage (%) was calculated as described[Citation2].
Determination of the CCR5 knockout efficiency
One day prior to transfection, TZM-bl cells (1.5 × 105 per well) were seeded into 12-well plates. An equimolar amount of crRNA constructs (equivalent to 500 ng cr vector) was co-transfected with 1 μg LbCpf1 plasmid and 500 ng JS-1 plasmid (GFP expressing vector) using Lipofectamine 2000. The culture medium was replaced after incubating for 6h. Two days post-transfection, cells were incubated with pre-warmed trypsin for detachment and then mixed with culture medium. The cells were washed two times with PBS before staining with PE/Cy7 anti-human CD195 (CCR5) antibody (cat#359,108, Biolegend). The stained cells were washed, fixed and followed by analysis with a FACScanto II cell analyzer (BD Bioscience). Only GFP-positive cells (GFP is a marker expressed from the co-transfected JS-1 vector) were analyzed for CCR5 knockout efficiency. The data were analyzed by the FlowJo_V10 software package.
Determination of the CRISPRa activity
One day before transfection, 1.5 × 105 HeLa X1/6 cells were seeded per well into 12-well plate. To achieve Luc-induction, dox (1 µg/ml) was added to the culture medium, while 40 ng of pCMV-rtTA-V10 plasmid expressing rtTA [Citation20] and 2 ng Renilla luciferase plasmid were co-transfected using Lipofectamine 2000. To test Luc induction by the CRISPR-based system, equimolar amount of crRNA constructs (equivalent to 100 ng cr vector) and a fixed molar amount of dCpf1-VP64 constructs (equivalent to 200 ng dAsCpf1 plasmid) were co-transfected with 2 ng Renilla Luciferase plasmid to control for transfection efficiency. The transfection of the empty cr vector and the dCpf1-VP64 vectors served as negative control. Dual luciferase reporter assays were performed 48h post-transfection and the relative Luc activity (Firefly/Renilla) was calculated. Three independent transfection assays were performed.
Statistical analysis
All statistical analyses were performed using Prism 7 (GraphPad Software). The specifics of statistical analysis are noted in the figure legends.
Author Contributions
ZG, EHC and BB designed the experiments. ZG and BB drafted the manuscript. ZG conducted the experiments and all authors analyzed the data and approved the final version.
Supplemental Material
Download MS Word (572.5 KB)Acknowledgments
This research was supported by ZonMw (Translational Gene Therapy program). ZG is supported by a scholarship from the China Scholarship Council (CSC).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826.
- Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819-823.
- Jiang W, Zhou H, Bi H, et al. Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res. 2013;41:e188–e.
- Yin H, Xue W, Chen S, et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol. 2014;32:551.
- Zetsche B, Gootenberg JS, Abudayyeh OO, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–771.
- Fagerlund RD, Staals RH, Fineran PC. The Cpf1 CRISPR-Cas protein expands genome-editing tools. Genome Biol. 2015;16:251.
- Kim D, Kim J, Hur JK, et al. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat Biotechnol. 2016;34:863.
- Kleinstiver BP, Tsai SQ, Prew MS, et al. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat Biotechnol. 2016;34:869.
- Port F, Bullock SL. Augmenting CRISPR applications in Drosophila with tRNA-flanked sgRNAs. Nat Methods. 2016;13:852.
- Fonfara I, Richter H, Bratovič M, et al. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature. 2016;532:517.
- Zetsche B, Heidenreich M, Mohanraju P, et al. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat Biotechnol. 2017;35:31.
- Ma H, Wu Y, Dang Y, et al. Pol III promoters to express small RNAs: delineation of transcription initiation. Mol Ther Nucleic Acids. 2013;3:e161.
- Gao Z, Harwig A, Berkhout B, et al. Mutation of nucleotides around the+ 1 position of type 3 polymerase III promoters: the effect on transcriptional activity and start site usage. Transcription. 2017;8:275–287.
- Gao Z, Herrera-Carrillo E, Berkhout B. Delineation of the exact transcription termination signal for type 3 polymerase III. Mol Ther Nucleic Acids. 2018;10:36–44.
- Ferré-D’Amaré AR, Scott WG. Small self-cleaving ribozymes. Cold Spring Harb Perspect Biol. 2010;2:a003574.
- Moreno-Mateos MA, Fernandez JP, Rouet R, et al. CRISPR-Cpf1 mediates efficient homology-directed repair and temperature-controlled genome editing. Nat Commun. 2017;8:2024.
- Tak YE, Kleinstiver BP, Nuñez JK, et al. Inducible and multiplex gene regulation using CRISPR-Cpf1-based transcription factors. Nat Methods. 2017;14:1163.
- Tang X, Lowder LG, Zhang T, et al. A CRISPR-Cpf1 system for efficient genome editing and transcriptional repression in plants. Nat Plants. 2017;3:17018.
- Zhang X, Wang J, Cheng Q, et al. Multiplex gene regulation by CRISPR-ddCpf1. Cell Discov. 2017;3:17018.
- Kleibeuker W, Zhou X, Centlivre M, et al. A sensitive cell-based assay to measure the doxycycline concentration in biological samples. Hum Gene Ther. 2009;20:524–530.