
ABSTRACT
Circular RNA (circRNA) is a novel RNA molecule that has become a research focus recently. Although some research indicated that the circRNAs in different subcellular compartments could execute different regulatory functions, a panoramic analysis of the subcellular distribution and the transport mechanism of circRNA is still required. In this study, we comprehensively analyzed the subcellular distribution/characteristics and the transport mechanism, through systemically investigating the circRNA profiles among the subcellular fractions of HepG2 cell (nucleus, cytoplasm, mitochondria, ribosome, cytosol and exosome). CircRNAs were widely distributed among the subcellular fractions except in the mitochondria, with differences in the subcellular distribution/characteristics in terms of classification, length, GC content, alternative circularization and parental gene function. Further analysis indicated this might be due to the selective transportation mediated by the transport-related RNA binding proteins (RBPs). The circRNAs may follow the same transportation mechanism of linear RNAs, in which the RBPs specially recognize/transport the RNAs with the corresponding binding motifs. Interestingly, we found that the exosome could selectively package the circRNAs containing the purine-rich 5ʹ-GMWGVWGRAG-3ʹ motif, with the characteristic of ‘garbage dumping’ and ‘intercellular signaling’ functions. Besides, although we observed numerous circRNAs enriched in the ribosome, we did not reliably identify any unique-peptides from circRNAs using 3D-LC-MS/MS strategy. This suggests that circRNAs rarely function as translation templates in vivo like lincRNA. Our findings not only indicates the differential distributions/characteristics among the subcellular fractions, but also reveals the possible transportation mechanism. This provides an improved understanding of the life history and molecular behavior of circRNA in cells.
Introduction
Circular RNA (circRNA) is a novel type of RNA molecule formed by a covalently closed loop without a 5ʹ-m7G cap and 3ʹ-poly(A) tail, which is generated from the ‘back-splicing’ reaction [Citation1]. Since the 1970s, several circRNAs have been identified in plant viroids as well as yeast and mammalian cells [Citation2–Citation4]; however, these molecules did not attract a great deal of attention because they were usually considered to be the byproducts of the RNA splicing processes based on the low abundance and special structures. However, with the rapid development of high-throughput sequencing technology in recent years, circRNA has become a hotspot in the field of RNA research. Thousands of circRNAs have been discovered in many eukaryotes, and reported to correlate with some diseases including cancer [Citation5–Citation7], cardiovascular disease [Citation8] and nervous system disorders [Citation9].
To date, circRNAs can be mainly classified into three types according to the two ‘back-splicing’ positions in the genome: exonic circRNAs (derived from exonic regions) [Citation1], intronic circRNAs (derived from intronic regions) [Citation10] and exon-intron circRNAs (EIciRNAs: derived from both exonic and intronic regions) [Citation11]. The majority of the detected circRNAs are exonic type, while a minority are intronic and EIciRNAs. Current research indicates that there are two main back-splicing circularization models: lariat- and intron pairing-driven circularization [Citation1,Citation10]. It is worth mentioning that, similar to the alternative splicing of linear RNA, many studies have also indicated the existence of alternative back-splicing of circRNAs, with multiple circRNAs (isogenous circRNAs [ISO circRNAs]) produced from a single gene. For instance, the CSPP1 gene has been reported to produce up to 46 ISO circRNAs [Citation12].
Some studies on circRNAs indicate differential subcellular localization. Electron microscopy, qPCR and RNA-seq analyzes have shown that, similar to linear mRNA, circRNAs are generated in the nucleus, and localized and enriched mainly in the cytoplasm [Citation4,Citation13,Citation14]. The circRNAs residing in the different subcellular compartments can co-localize and interact with different proteins/nucleotides to execute their different regulatory functions.
In the nucleus, two EIciRNAs (circEIF3J and circPAIP2) have been reported to bind to the U1 snRNP, and then to recruit RNA polymerase II to the promoter site of the parental gene, thereby enhancing the expression of both the circRNAs and their homologous mRNAs [Citation11]. Another nuclear circRNA, circANRIL, competitively inhibits the interaction between the pre-rRNA and PES1 protein, thus impairing the processing and maturation of rRNA [Citation8].
Most circRNAs are located and enriched in the cytoplasm, where they exert their functions. Many studies have shown that a variety of circRNAs play important regulatory roles by interacting with miRNAs and proteins in the cytoplasm. First, more than 10 circRNAs, including CDR1as, SRY and circHIPK3, were found to act as ‘miRNA sponges’ [Citation6,Citation7,Citation15,Citation16] by binding competitively to the corresponding miRNAs and further reducing their inhibitory effect on their targets. Among them, CDR1as is the most well-known because of its involvement in the progression of various cancers [Citation6,Citation7]. Second, some circRNAs, such as circFOXO3, have been shown to perform the ‘protein sponge’ function [Citation17].
Interestingly, a large number of circRNAs are also found in the exosome, and differ significantly from those in the nucleus and cytoplasm of the corresponding cells. Furthermore, research has indicated the differential circRNA profiles of the serum exosomes derived from patients with different diseases, thus implicating exosomal circRNA as targets for disease diagnosis and treatment [Citation18,Citation19]. In addition, some recent studies have shown that several circRNAs can be bound to the ribosome, and translated into endogenous peptides [Citation20–Citation22].
In summary, although previous studies have indicated the subcellular localizations and functions of some circRNAs, a comprehensive analysis of the subcellular distribution and the transport mechanism of circRNA is still required. Therefore, we systemically investigated the circRNA profiles among the subcellular fractions (nucleus, cytoplasm, mitochondria, ribosome, cytosol and exosome) of HepG2 cells. Our studies revealed the wide distribution of circRNAs among the subcellular fractions, except the mitochondria. Further comparative analysis indicated the different subcellular distribution and characteristics of the circRNAs in terms of expression, length, GC content, classification, alternative circularization and function of the parental genes. Analysis of the binding motif distribution of the transport-related RBPs among the subcellular circRNAs indicated that the different subcellular distribution characteristics of circRNAs might be due to the RBP-mediated selective transportation. It also implied that the circRNAs follow the same transportation mechanism as that of linear RNAs, in which the RBPs specifically recognize, bind to, and transport the RNAs with the corresponding binding motifs, regardless of circular or linear RNAs. Our research not only provides an improved understanding of the life history and molecular behavior of circRNAs in a cell, but also contributes to the exploration of new biological functions.
Results
CircRNA profiling reveals differential distribution among the subcellular fractions
To explore the subcellular distributions of circRNAs, we fractionated the HepG2 cells into six subcellular fractions, including nucleus, cytoplasm, mitochondria, ribosome, cytosol and exosome. The purity and the integrity of the six subcellular fractions were validated by western blotting and transmission electron microscopy (TEM) ( and Figure S1). The exosome was also identified by NTA and immuogold electron microscopy experiments ( and Figure S2). We then performed whole-transcriptome sequencing of the HepG2 cells and their subcellular fractions. Using two common circRNA algorithms, CIRCexplorer [Citation1] and CIRI [Citation23], we identified a total of 11,232 circRNAs in all of the HepG2 cell fractions. CircRNA expression profile analysis indicated the wide distribution of circRNAs among all the subcellular fractions except the mitochondria, especially in cytoplasm, cytosol and ribosome (). Since we only detected 118 circRNAs with low abundance in the mitochondria, we excluded the mitochondria from the subsequent analyzes. Systematic genome-wide subcellular circRNA profiling was then performed in terms of expression, classification, length, GC content, alternative back-splicing, function of the parental genes and sequence preference.
Figure 1. Western blot and representative TEM images for the subcellular fractions of HepG2 cells and Immunogold electron microscopy images showing the localization of exosomes. (a) Western blot analysis of the purified subcellular fractions. The six purified subcellular fractions of HepG2 cells were verified by western blot analysis using specific detection antibodies (Lamin A: nuclear marker; AKR1B1 and tubulin: cytoplasm marker; UQCRC1 and VDAC1: mitochondria marker; LAMP1 and CD63: exosome marker; RPS5 and RPL24: ribosome marker). (b) Representative TEM images of the purified subcellular fractions (Scale bar: 1 µm for the nucleus, 100 nm for all others). (c) The first TEM image shows the localization of exosomes derived from HepG2 cells as controls. The other three TEM images show the localization of exosomes by TEM coupled to immunogold labeling of CD9, CD63 and Hsp70. The arrows indicate the golden particles.
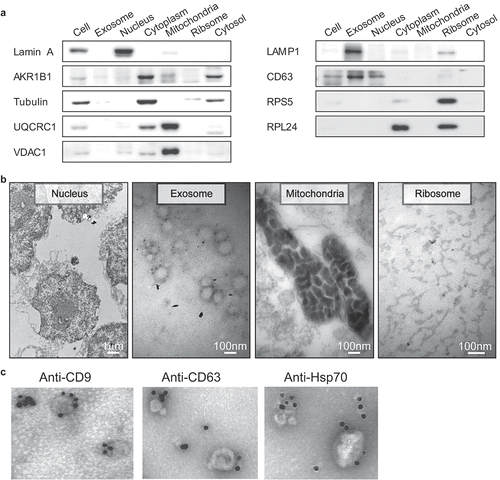
Figure 2. Comparative transcriptome analysis of circRNAs among the subcellular fractions. (a) CircRNA expression profile among the subcellular fractions. The number of circRNAs expressed at various cutoff expression levels is represented by the indicated colors. (b) Correlation matrix of the circRNA expression among the fractions. The size and color intensity of red circles indicate the relative strength of the correlation between the fractions.
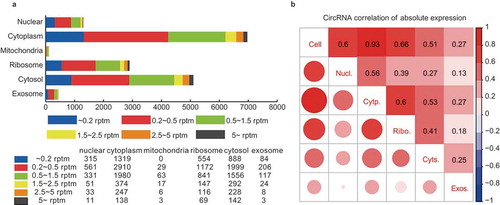
The subcellular expression profile was first analyzed by the correlation matrix among the fractions (). By analyzing the correlations of the circRNA expressions between the HepG2 cell and the subcellular fractions, we found that the cellular circRNAs displayed the highest correlation (0.93) with the cytoplasmic ones. It demonstrated that circRNAs were mainly enriched in the cytoplasm, though they were generated in the nucleus, which is consistent with previous reports [Citation4,Citation13,Citation14]. Further analysis of the circRNA correlations among the five subcellular fractions revealed the differential and uneven distribution of subcellular circRNAs. The weak correlation (0.56) was identified between the nuclear and cytoplasmic circRNAs. Since the circRNAs were generated in the nucleus and enriched in the cytoplasm, this weak correlation indicated the selective nuclear export of circRNAs. The circRNAs also showed differential distribution in the ribosome and cytosol, with a correlation of only 0.41. The nuclear export circRNAs will first enter cytosol, so the weak correlation between ribosomal and cytosolic circRNAs also suggested the selective adsorption or enrichment of certain circRNAs by the ribosome. In addition, the weak correlations (<0.3) between the exosomal circRNAs and those of the other fractions indicated strong selective translocation of circRNAs into the exosome. We also performed the correlation analysis of mRNA expressions among the fractions (Figure S3). The different correlation patterns between circRNAs and mRNAs suggested a selective linear-independent distribution of the circRNAs.
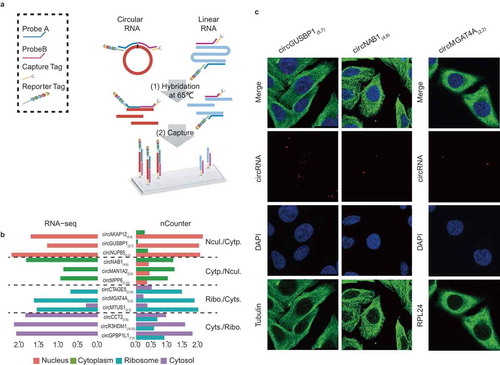
To validate the subcellular distribution of circRNAs, we developed a novel strategy based on the Nanostring nCounter technology [Citation24]. We designed sets of capture and reporter probes to specifically recognize and detect the flanking exonic sequences of targeted circRNA junction points (). One advantage of this strategy is to eliminate the bias introduced by enzymatic reactions [Citation24]. In this study, we used nCounter technology to test 12 highly expressed circRNAs in four subcellular fractions, which displayed similar distribution patterns compared with those detected by RNA-seq (). A circRNA (circHIPK3(2,2)) was selected as positive control, which was detected in nucleus, cytoplasm, ribosomes and cytosol by both RNA-Seq and nCounter analyzes (Figure S4). Their subcellular localizations were further confirmed by the real-time quantitative polymerase chain reactions (qPCR) with divergent primers (Figure S5 and Table S1). Moreover, the subcellular localizations of three specific and highly expressed circRNAs were detected using quantum dot fluorescence in situ hybridization (FISH) (). In addition, five highly expressed exosome circRNAs were verified by nCounter data analysis (Table S2).
Figure 3. Validation of subcellular localization of circRNAs by Nanostring nCounter technology and RNA FISH. (a) Schematic diagram showing circRNA validation based on Nanostring nCounter technology. The ‘nCounter ElementsTM Reagents’ include Probe A, Probe B, Reporter Tag and Capture Tag (shown in the box). Probe A and Probe B are designed to specifically recognize the flanking exonic sequences of targeted circRNA junction points. The Reporter and Capture Tags are designed to bind Probe A and Probe B, respectively. (1) The reaction mixture (containing ‘nCounter ElementsTM Reagents’ and the target RNAs) were first hybridized at 65°C overnight. (2) The Capture Tag anchors the hybrid duplexes to the streptavidin-modified surface of the nCounter cartridge. After repeated washes, the anchored fluorescent barcodes on the cartridge can be used to measure the target-circRNA abundance. (b) Pair-wise comparison of subcellular exonic circRNA expression between RNA-seq (left panel) and nCounter analysis results (right panel). Nucl./Cytp.: the circRNAs preferentially expressed in the nucleus versus the cytoplasm (shown as salmon pink and green bars, respectively); Cytp./Nucl.: the circRNAs preferentially expressed in the cytoplasm versus the nucleus (shown as green and salmon pink bars, respectively); Ribo./Cyts.: the circRNAs preferentially expressed in the ribosome versus the cytosol (shown as blue and purple bars, respectively); Cyts./Ribo.: the circRNAs preferentially expressed in the cytosol versus the ribosome (shown as purple and blue bars, respectively). (c) Subcellular localization images of circRNAs using quantum dot FISH (scale bar: 25 μm). Here we selected three circRNAs (circGUSBP1, circNAB1, and circMGAT4A) that were highly expressed specifically in the nucleus, cytoplasm and ribosome, respectively. These were detected by the Qdot 585 probe FISH (shown as red dots). The nucleus, cytoplasm and ribosome are stained with DAPI (blue), and antibodies against Tubulin (green) and RPL24 (green), respectively.
Analysis of the distribution characteristics of the subcellular circRNAs
To explore which circRNAs are located and enriched in each subcellular faction, we further analyzed the distribution characteristics of the subcellular circRNAs. The comparative analysis revealed the differences in the distribution of characteristics of the subcellular circRNAs, such as classification, length, GC content, alternative back-splicing, and functions of the parental genes. Here we mainly focused on the three pair-wise comparisons (nucleus versus cytoplasm, ribosome versus cytosol, exosome versus other fractions).
Classification
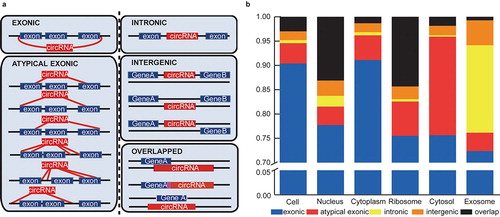
According to the two ‘back-splicing’ positions in the genome, circRNAs were classified into five types: exonic, atypical exonic, intronic, overlap and intergenic types (). 10 highly expressed circRNAs from the five types were validated by identification of the back-splice junctions (Figure S6 and Table S3). Further bioinformatics analysis indicated the differential circRNA-type distribution among the subcellular fractions, although the exonic type circRNAs represented the highest proportion among all the fractions. showed that the proportion of exonic circRNAs in the cytoplasm (91.11%) was higher than in the nucleus (77.74%), ribosomes (75.51%) and cytosol (75.63%). As we known, the cytoplasm consists of cytosolic fraction and the other subcellular fractions such as mitochondria, ribosomes, plasma membrane and membrane encapsulated organelles (endoplasmic reticulum, Golgi, et al), therefore the difference of exonic type circRNAs in different subcellular fractions indicated its main localization in plasma membrane or membrane encapsulated organelles. Moreover, the exosomes had a much higher proportion of intronic circRNAs (17.99%) than that in the other fractions (cell: 0.62%, nucleus: 2.21%, cytoplasm: 0.64%, ribosome: 0.48%, cytosol: 0.28%). In addition, we also found that the ribosome contained a much higher proportion of overlap-type circRNAs (11.99%) than that in the cytoplasm (1.23%). Incidentally, the similar proportion between the cytoplasmic and cellular circRNAs again indicates that circRNAs are mainly enriched and located in the cytoplasm.
Figure 4. Subcelluar circRNA classification based on the two back-splicing positions (start and end) in the genome. (a) CircRNA classification according to the start and end genomic positions (five types: exonic, intronic, atypical exonic, overlap and intergenic types). The red and blue boxes represent the circRNAs and corresponding parental genes, respectively. (b) The proportions of five types of circRNA expressed in the six subcellular fractions. Each type of circRNA is represented by the indicated color box.
Length and GC content
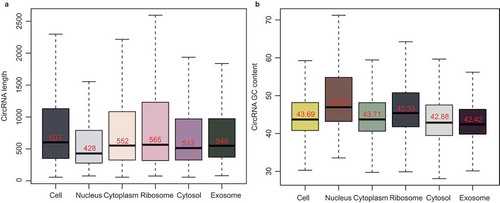
The analysis of the distribution characteristics showed the differences in the lengths of exonic circRNAs in the subcellular fractions. Among them, the length of the nuclear exonic circRNAs was much shorter than that in the other fractions (). Previous research showed that longer transcripts have a higher probability of being bound by RBPs and are more easily transported out of the nucleus [Citation25,Citation26]. The GC bias of the subcellular exonic circRNAs was then analyzed. shows the different GC contents of exonic circRNAs in the subcellular fractions. Here the GC content of the cytoplasmic exonic circRNAs was lower than that of the nuclear ones, which was consistent with some comparative analyzes of mRNA GC content between cytoplasm and nucleus [Citation27]. We also found that the GC content of the ribosomal exonic circRNAs was higher than that of the cytosolic ones, indicating the selective absorption of the exonic circRNAs with high GC content by the ribosome. In addition, the exosome displayed the lowest GC content among the subcellular fractions, which was likely to result from the selective packaging of the circRNAs with low GC content by exosomes.
Figure 5. The length and GC content of the subcellular circRNAs. (a) Bar chart showing the length of exonic circRNAs in the six subcellular fractions. (b) Bar chart showing the GC content of exonic circRNAs in the six subcellular fractions.
Alternative back-splicing and ISO circRNAs
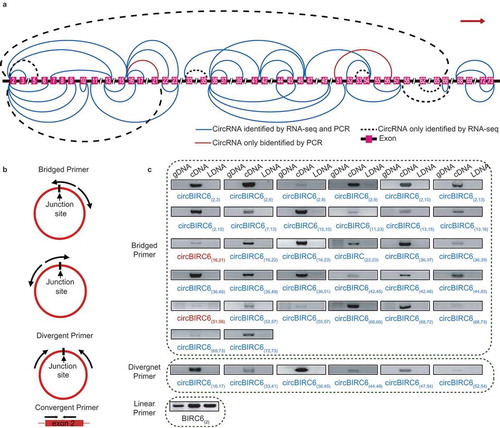
Similar to linear RNAs, it has been reported that alternative back-splicing also occurs in circRNAs, generating ISO circRNAs [Citation1,Citation12]. By analyzing the 9,926 exonic circRNAs detected in HepG2 cells, we found ISO forms of approximately 60% of the exonic circRNAs. Remarkably, 117 genes generated more than 10 ISO circRNAs (Table S4). Among them, we selected three representative genes and verified their ISO circRNAs: BIRC6, which generated the most ISO circRNAs (43 circRNAs), XPO1, which generated ISO circRNAs with the highest average expression (22 circRNA), and ATM, which is a well-known tumor suppressor (16 circRNAs). To efficiently discriminate the ISO circRNAs, we designed a series of bridged primers spanning the junction sites, as well as some conventional divergent primers. The sequencing results of the amplification products validated 68 of 81 ISO circRNAs (83.72%). Moreover, we found seven novel ISO circRNAs that were undetected in the transcriptome data (, Figure S7 and Table S5-S7).
Figure 6. Forty-five isogenous circRNAs derived from the BIRC6 gene. (a) Schematic diagram showing the 45 isogenous circRNAs derived from BIRC6 gene. Forty-three isogenous circRNAs were identified in the RNA-seq analysis. Among them, 36 circRNAs were validated by PCR (described as ‘CircRNAs identified by RNA-seq and PCR’); Seven circRNAs failed to be validated by PCR (described as ‘CircRNAs only identified by RNA-seq’). Two circRNAs were only verified by PCR (described as “CircRNAs only identified by PCR). The colored lines indicate the start and end positions of the circRNAs in the gene. (b) Schematic diagram showing the bridged and divergent primer amplification strategies. The bridged primer is shown to span the exon junctions, so as to only amplify and verify the circRNAs using cDNA templates. The divergent primers are shown to amplify the back-splicing exon junction regions of circRNA; the convergent primers are shown to amplify the exon regions as controls. (c) PCR verification of 38 BIRC6 isogenous circRNAs using bridged and divergent primer amplification strategies. Thirty-two circRNAs were validated by the ‘bridged primer amplification strategy’ using cDNA templates; as controls, the PCR products were not amplified from gDNA and LDNA (the complementary DNA reverse transcribed using oligo dT primers). Among them, 30 circRNAs were also identified by RNA-seq analysis; two circRNAs were novel. Six circRNAs were validated by the ‘divergent primer amplification strategy’. Convergent primers were used to amplify exon 2 as controls.
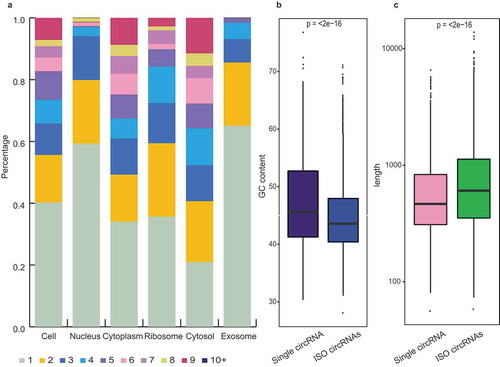
The bioinformatics investigations revealed differential subcellular distributions of the ISO circRNAs, which were mainly enriched in the cytoplasm and its sub-fractions (ribosome and cytosol), with lower abundance in the nucleus and exosome (). The characteristic distributions indicated that the ISO circRNAs are specifically identified and transported to the cytoplasm after their generation in the nucleus, and prefer to retain in the cytoplasm rather than being packaged into the exosome.
Figure 7. Isogenous circRNAs. (a) Percentage bar charts showing the expression distribution of the ISO circRNAs in six fractions. The ISO circRNA numbers are represented by the indicated colors. (b) Comparison of GC content distribution between the single circRNAs (purple) and ISO circRNAs (blue). The P-values are shown at the top of the box. (c) Comparison of length distribution between the single circRNAs (pink) and ISO circRNAs (green). The P-values are shown at the top of the box.
We then compared the expressions of 24 ISO circRNAs from six representative genes in the subcellular fractions. They exhibited differential expression patterns in the subcellular fractions (Figure S8A). To assess the expression patterns of the ISO circRNAs among the subcellular fractions, we further measured the correlation-coefficients for the ISO circRNAs with or without common splice sites (Figure S8B and S8C). The ISO circRNAs with common splice sites had the highest correlation-coefficients, indicating the most similar expression patterns among the subcellular fractions. It also demonstrated that those with common splice sites are more likely to locate in the same subcellular fraction. Incidentally, we found that the cumulative abundance of the ISO circRNAs was higher than that of the single ones generated from the same gene, which increased with increases in the number of ISO forms (Figure S9).
Moreover, the subcellular distribution of the ISO circRNAs was also correlated with other features, such as length and GC content. Compared with the single circRNAs, the ISO forms were longer in length, with lower GC contents (). Our investigation of circRNAs and previous studies on linear RNAs showed that the long transcripts with low GC content were more likely to be transported out of the nucleus [Citation25–Citation27]. These characteristics may be both the cause and consequence of selective transport of ISO circRNAs.
Functions of the parental genes
By investigating the functions of the circRNA parental genes by ‘Ingenuity Pathway Analysis’ (IPA), we found that the circRNAs also exhibited differential subcellular distribution in the parental gene functions. The IPA results showed that the circRNA parental genes in cytoplasm were mainly enriched in four IPA functional clusters: ‘tissue development’, ‘cell fate decisions’, and ‘bio-macromolecule disposal and modification’. In contrast, the nuclear circRNA parental genes displayed enrichment in the ‘material metabolism’, ‘hematopoiesis’ and ‘immune response’ clusters (Figure S10A). The IPA results also showed enrichment of the circRNA parental genes associated with ‘tissue development’ and ‘hematopoiesis and immune response’ in the ribosome fraction, while the cytosolic circRNA parental genes were mainly clustered in the ‘bio-macromolecule disposal and modifications’ functional categories (Figure S10B). Interestingly, the exosome circRNA parental genes exhibited enrichment in the ‘signal transduction’ categories compared with that of the cell, suggesting that exosomes play a role in intercellular signaling (Figure S10C), which is in accordance with the findings of previous studies [Citation28].
RBP-mediated selective transportation may be responsible for the differential subcellular distribution of circRNAs
To explore the potential mechanism responsible for the differential subcellular distributions of circRNAs, we analyzed the binding motif distribution of the transport-related RBPs among the subcellular circRNAs. First, we investigated the potential mechanism of the differential distribution between the nuclear and cytoplasmic circRNAs by analyzing the binding motif distribution of nuclear export RBPs. In this study, we compared the distribution of 29 motifs (recognized by 19 main nuclear export RBPs) [Citation29–Citation31] in nuclear and cytoplasmic exonic circRNAs. The results indicated the enrichment of the circRNAs with these motifs in the cytoplasm (P < 0.05), especially for the 21 motifs from 14 nuclear export RBPs (, Figure S11 and Table S8).
Figure 8. Binding motif analysis of the subcellular circRNAs. (a) Enrichment comparison of the 29 binding motifs of 19 nuclear export RBPs in cytoplasmic (blue) and nuclear circRNAs (green). P-values are shown at the top of the box. (b) Enrichment comparison of the Kozak sequence between ribosomal (carmine) and cytosol (yellow) circRNAs. The p-value are shown at the top of the box. (c) The sequence logo for the preferred motifs of exosomal circRNAs predicted from our data (left panel) and Huang’s RNA-seq data (right panel).
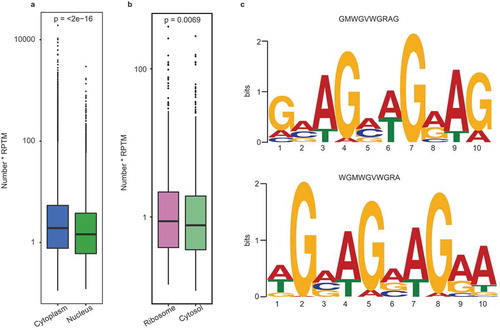
We then investigated the cause of the differential distribution between the ribosomal and cytosolic circRNAs by analyzing the Kozak and internal ribosome entry site (IRES) sequences. These two sequences are recognized by ribosomes as the translation initiation site (TIS) [Citation32,Citation33]. demonstrated the enrichment of the circRNAs with Kozak sequence in the ribosome. Here seven of the top-10 highly expressed circRNAs in the ribosome possessed Kozak sequence (Table S9). In addition, it is known that the IRES element can also bind to the ribosome via distinct secondary structures [Citation32]. In this study, three of the top-10 highly expressed circRNAs in the ribosome contained IRES elements (Figure S12).
To uncover the selective mechanism by which circRNAs are translocated into the exosome, we used MEME software [Citation34] to predict the motif preference of the exosomal exonic circRNAs. The results showed that the circRNAs containing the 5ʹ-GMWGVWGRAG-3ʹ motif were enriched in the exosomes; this was confirmed by predicting the motif preference using the circRNA-seq data from human blood exosomes [Citation18] (). It can be speculated that some RBPs specifically recognize and package circRNAs with the purine-rich motifs into exosomes.
CircRNA-encoded protein was not be detected in the HepG2 cells
Our analysis revealed that numerous circRNAs were enriched in the ribosome fraction, many of which carried the Kozak and IRES sequences for translation initiation. To explore the ability of endogenous circRNAs to encode proteins, we sequenced the endogenous proteins using a 3D-LC-MS/MS strategy (Figure S13). Although we obtained high quality deep MS data, we did not reliably identify any unique peptides for circRNAs.
In total, we identified 448,140 spectra, corresponding to the 2,661 mRNA-encoded proteins with at least two unique peptides. Their average coverage reached 35.85%, indicating that the high quality of our data. To identify the circRNA-derived peptides, we first selected the unique peptides in accordance with the circRNA sequences. The results of MFP-FDR tests of these peptides should not exceed 0.01 in two independent biological replicates. To exclude the shared spectra with mRNA-encoded proteins, we retained only the number one peptide for each spectrum, and then removed those matching the known mRNA-derived proteins using the BLASTP algorithm. After this filtration, we were unable to screen out the peptides encoded by circRNA reliably. This demonstrated that the major role of circRNAs is unlikely to be as translation templates, at least in the HepG2 cells, although thousands of circRNAs can recognized and adsorbed by the ribosome. However, non-detection of such peptides does not rule out translation of circRNAs, since the ORF does not have to include the circjunction.
Discussion
Previous researches have shown that circRNAs are generated in the nucleus and enriched in the cytoplasm, which is consistent with the mechanism associated with linear RNAs [Citation4,Citation13,Citation14]. However, the mechanism by which circRNAs are transported from nucleus to cytoplasm remains to be elucidated. Our studies indicate that the RBP-mediated selective transportation may be responsible for the nuclear export of circRNAs, thus leading to the differential nucleo-cytoplasmic distribution of circRNAs in terms of multiple features, including expression, classification, length, GC content, alternative back-splicing and the functions of parental genes. In this study, we demonstrated that the cytoplasmic circRNAs were longer in length, with lower GC content, the highest proportion of the exonic type, and a higher proportion of ISO circRNAs (). These characteristics are mainly due to positive selection by RBP-mediated nuclear export. Some previous studies also indicated the existence of positive correlations between these characteristics and the nuclear export of linear RNAs [Citation25,Citation26,Citation35–Citation37]. In contrast, the nuclear circRNAs have the shortest length, the highest GC content, a higher proportion of non-exonic type, a lower proportion of ISO circRNAs, and more parental genes associated with ‘material metabolism’, and ‘hematopoiesis and immune response’ functional categories (). This may be due to negative selection by the RBP-mediated nuclear export of circRNAs.
Table 1. Characteristics of the circRNAs among the fractions.
We further explored the subcellular enrichment of circRNAs after nuclear export. Although a large number of circRNAs were discovered in both the ribosome and cytosol, we observed a significant differential distribution between the two fractions (correlation: 0.41). Since the ribosomes are contained within the cytosol, we deduce that the circRNAs first enter the cytosol after nuclear export, with some then selectively adsorbed and enriched by the ribosome. Our results demonstrated that the Kozak or IRES sequences might result in the selective adsorption, leading to the distinct characteristics of the ribosomal circRNAs, which have the longest average length, a higher average GC content, a higher proportion of overlapped types, a higher proportion of ISO circRNAs, and more parental genes associated with ‘tissue development’ functional categories () [Citation38–Citation40].
Interestingly, some circRNAs are packaged into the exosome. These exosomal circRNAs were found to display differential distribution compared with the other subcellular fractions, with even the highest correlation being only 0.27. Analysis of the characteristics of exosomal circRNAs indicated that the packaging of circRNAs into the exosome is a selective process, resulting in a characteristic distribution of circRNAs of moderate length, the lowest GC content, the highest proportion of intronic type, a lower proportion of ISO circRNAs, and more parental genes associated with ‘signal transduction’ functional categories. These characteristics also indicate that the exosome plays roles in ‘intercellular signaling’ and ‘garbage dumping’, which is consistent with previous reports [Citation28]. Further bioinformatics analysis indicated that the selective packaging is mediated by a specific RBP that mediates the selective removal of circRNAs containing the purine-rich 5ʹ-GMWGVWGRAG-3ʹ motif by the exosome. Villarroya-Beltri et al. [Citation41] also described a GA-rich exosome-motif (GAGG) of miRNAs, indicating that the exosome might selectively package the purine-rich RNAs. In addition, previous research also demonstrated that exosome preferentially packaged the shorter circRNAs [Citation42], which is in agreement with our study (Figure S14). It should also be mentioned that we did not reliably identify any circRNA-derived peptides using the deep 3D-LC-MS/MS strategy, although a large number of circRNAs have been discovered in the ribosome. This indicates that ribosomal circRNAs rarely function as translation templates in vivo, though translation of several circRNAs into proteins in vivo has been reported in recent years [Citation20–Citation22]. CircRNAs lack an effective mechanism of degradation and ribosome-dissociation in vivo and therefore, exhibit a constitutive expression pattern once they begin translation.
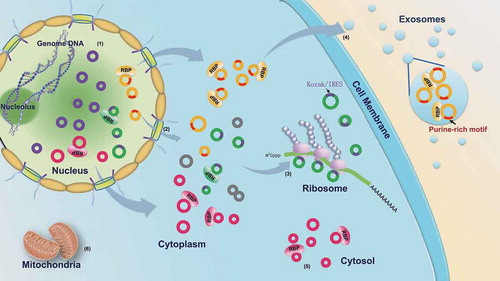
Overall, as a novel type of non-coding RNA, little is known about the life cycle of circRNAs in vivo. Our investigation of the subcellular circRNAs not only indicates differences in the distribution and characteristics among the subcellular fractions, but also reveals the possible transportation mechanism. Similar to the mechanism of linear RNA transportation, a series of RBPs may mediate the selective transportation of circRNAs between the subcellular fractions (). These RBPs selectively recognize and transport the RNAs (circular or linear) with the corresponding binding motifs. Our investigation provides new insights into the distribution, characteristics and life cycle of circRNAs, as well as an understanding of the molecular behavior of circRNAs in the cell.
Figure 9. RBP-mediated transportation of circRNAs among the subcellular fractions. (1) The circRNAs are generated in the nucleus via the back-splicing reaction. (2) The majority of circRNAs are recognized by nuclear export-related RBPs and transported into the cytoplasm; the others without the RBP binding motifs are retained in the nucleus by negative selection. (3) The cytoplasmic circRNAs with Kozak or IRES sequences are specifically adsorbed by the ribosome. (4) The cytoplasmic circRNAs with a purine-rich motif (5ʹ-GMWGVWGRAG-3ʹ) may be recognized by some RBPs and packaged into the exosome. (5) The other cytoplasmic circRNAs are retained in the cytosol by the negative selection. (6) The circRNAs cannot enter the mitochondria. The purple circles represent the circRNAs retained in the nucleus. The green circles represent the circRNAs adsorbed in the ribosome; the purple arcs on the green circles represent the Kozak or IRES sequences. The yellow circles represent the circRNAs packaged into the exosome; the red arcs on the yellow circles represent the purine-rich motif. The pink circles represent the circRNAs accumulated in the cytosol fraction.
Materials and methods
Subcellular fractionation by differential centrifugation
Subcellular fractionation of HepG2 cells was performed according to the standard procedure described by Zeng [Citation43]. The cells were first incubated for 1 h in ice-cold homogenization buffer (210 mM sucrose, 70 mM mannitol, 1 mM EDTA, 1 mM EGTA, 10 mM KCl, 1.5 mM MgCl2, 1% protease inhibitor cocktail (P8340, Sigma) and 10 mM HEPES-KOH at pH 7.4). Subsequently, the cells were homogenized by applying 20 strokes with a pre-chilled 10 ml Dounce homogenizer. The homogenates were then centrifuged at 300 × g for 5 min to remove unbroken cells. The subcellular fractions were obtained by different centrifugation speeds at 4°C: 1) the crude nuclear fraction was obtained from the pellet by centrifugation at 1,000 × g for 10 min; 2) the cytoplasmic fraction was obtained from the supernatant by centrifugation at 1,000 × g for 10 min; 3) the crude mitochondrial fraction was obtained from the pellet by centrifugation of the cytoplasmic fraction at 10,000 × g for 15 min; 4) the cytosolic fraction was obtained from the supernatant by centrifugation at 150,000 × g for 2 h. To acquire high quality preparation of the nucleus and mitochondria, the corresponding crude fractions were re-suspended with homogenization buffer, passed through a 21 G blunt-ended needle 10 times before re-centrifugation.
Ribosome isolation
The Animal Cells and Tissues Ribosome Separation Kit (GenMed Scientific, Inc., USA) was used for preparation of ribosomes according to the manufacturer’s protocol. The cells were homogenized by applying 20 strokes with a pre-chilled 10 ml Dounce homogenizer. After centrifugation at 10,000 × g for 10 min, the supernatant (containing the ribosome fraction) was carefully dropped onto the surface of Reagent C. By ultracentrifugation at 260,000 × g for 2 h (Beckman Optima L-100 XP), the pellet of high-purity ribosomes was harvested and stored at −20°C.
Exosome isolation
HepG2 cells were first cultured for 72 h in the medium containing 10% exosome-depleted FBS. The culture supernatant was subjected to sequential centrifugation at 2,000 × g and 16,500 × g at 4°C to remove suspended cells, cell debris and microvesicles. The supernatant was then filtered using 0.22 µm filter and exosomes were collected by ultracentrifugation at 120,000 × g for 2.5 h. The resulting pellet was washed three times in PBS, and stored at −80°C.
Protein extraction and western blot analysis
Proteins from HepG2 cells and its subcellular fractions were extracted with lysis buffer containing 8 M urea, 30 mM HEPES and 1% protease inhibitor cocktail at 4°C overnight. The extracts were separated by 10%–15% SDS-PAGE. The subcellular fractions were identified by Western blot analysis using antibodies for the detection of the following specific markers: Lamin A (nucleus), UQCRC1 and VDAC1 (mitochondria), RPL24 and RPS5 (ribosome), tubulin and AKR1B1 (cytoplasm) (all Santa Cruz Biotech, Santa Cruz, CA, USA), and CD63 (EXOAB-CD63A-1, SBI) and LAMP1 (exosomes). Unless otherwise stated, all antibodies were purchased from Proteintech (Wuhan, China).
TEM analysis
TEM was employed to visualize organelle preparations as previously reported [Citation44]. For the nucleus, mitochondria and ribosomes, the isolated organelles were fixed sequentially in 0.1 M PBS containing 3% glutaraldehyde (pH 7.4) for 2 h and in 0.1 M PBS containing 1% osmium tetroxide (pH 7.2) for 1.5 h at 4°C. The preparations were then stained with uranyl acetate and following dehydration in graded series of ethanol and acetone, the specimens were embedded into Ultracut (Leica, Germany) and ultrathin sections (thickness, 60 nm) were prepared. Sections were then stained with lead citrate, and examined using a JEM-1230 TEM (JEOL, Japan).
We further identified exosomes using immunogold approach based on the reported protocol [Citation45]. The PBS suspended pellets were placed on formvar carbon-coated nickel grids (200 mesh), blocked, and incubated with rabbit anti-human antibodies (anti-CD9 (EXOAB-CD9A-1, SBI), anti-CD63 (EXOAB-CD63A-1, SBI) and anti-Hsp70 (EXOAB-Hsp70A-1, SBI), followed by incubation with the goat anti-rabbit secondary antibody conjugated with protein A-gold particles (5 nm) (Bioss, China). Finally, the exosomes were negatively stained with 3% phosphotungstic acid for 10 min. The exosome-containing grids were air-dried and observed using a JEM-1400 TEM (JEOL, Japan).
Nanoparticle tracking analysis (NTA)
We determined the size of HepG2 cell-derived exosomes using a NanoSight NS300 (Malvern, UK). The instrument is equipped with particle-tracking and video capture software to characterize the size and concentration of target particles.
RNA-seq library preparation
Total RNAs from HepG2 cells and their subcellular fractions were extracted using RNAiso Plus reagent (9108, TaKaRa, Dalian, China). RNAs (except exosomal RNAs) were treated with the Ribo-Zero Gold rRNA Removal Kit (MRZG12324, Illumina, CA, USA) to remove ribosomal RNA before constructing RNA-seq libraries. Strand-specific RNA-seq libraries were prepared using Next® Ultra™ Directional RNA Library Prep Kit for Illumina (E7420, NEB, MA, USA) according to the manufacturer’s instructions. Library quality was assessed on a Bioanalyzer 2100 (Agilent Technologies, CA, USA).
CircRNA identification, quantification and correlation matrix
The libraries were sequenced using an Illumina HiSeq 4000 Genome Analyzer. Raw reads were filtered, and subsequently mapped to the human reference genome GENCODE v23. The unmapped reads were then extracted to identify junction sites of circRNAs according to two common algorithms (CIRI and CIRCexploer) using the default parameters [Citation1,Citation23]. The circRNAs were named based on the parental gene names and the exonic number for the two back-splicing sites. Only the circRNAs detected in both replicates of each fraction were used in the subsequent bioinformatics analysis.
CircRNA normalization was performed against the number of back-splice junction spanning reads per ten million clean reads (RPTM). The average expression values were obtained from the two replicates using the CIRI and/or CIRCexploer algorithms. The correlation matrix of circRNA expression among the fractions was calculated and visualized using the Corrplot package in the R statistical programming environment (three extremums were eliminated).
CircRNA annotation and classification
Circular RNAs were annotated and classified according to the two back-splicing positions (start and end) in the genome and then divided into five types (): 1) Exonic circRNA: the circRNAs had two classical splicing sites, both of which were located at the ends of exons in a same gene. 2) Atypical exonic circRNA: the circRNAs had at least one non-classical splicing site, and the two splicing sites occurred in the same gene at any location. 3) Intronic circRNA: the two splicing sites were located in the same intron. 4) Intergenic circRNA: the two splicing sites were located in the same intergenic region. 5) Overlap circRNA: the circRNAs spanned the junctions between the protein-coding gene and intergenic region. In this study, the circRNA annotation and classification were based only on the protein coding genes, which may not represent the ideal choice in some situations.
Nanostring ncounter assay of circRNA expression
Some highly-expressed subcellular circRNA candidates were validated using nCounter Nanostring technology. The ‘nCounter ElementsTM Reagents’ include Probe A, Probe B, Reporter Tag and Capture Tag ( and Table S10). Probe A and Probe B are designed to specifically recognize the flanking exonic sequences of targeted circRNA junction points. The Reporter and Capture Tags are designed to bind Probe A and Probe B, respectively. The reaction mixture (containing ‘nCounter ElementsTM Reagents’ and the target RNAs) were first hybridized at 65°C overnight, and then loaded in the nCounter cartridge for the subsequent analysis using the ‘nCounter Analysis System’. In this system, the Capture Tag anchors the hybrid duplexes to the streptavidin-modified surface of the nCounter cartridge. After repeated washes, the anchored fluorescent barcodes on the cartridge were to measure the target-circRNA expression using a digital analyzer. Once the reaction was terminated, the Nanostring nSolver platform was adopted to quantify the expression of circRNAs.
circRNA verification by PCR and quantitative RT-PCR
The cDNA was obtained by reverse transcription of total RNAs using random hexamer primers. Linear DNAs (LDNAs) was obtained by reverse transcription using oligo (dT) primers (RevertAid First-Strand cDNA Synthesis Kit, Thermo Scientific, USA). The concentration of PCR templates (cDNA and LDNA) were measured using a Qubit® 2.0 Flurometer (Life Technologies, CA, USA). Divergent and bridged primers were designed to amplify the circRNAs and convergent primers were used to amplify the exon regions as controls. The qPCR reactions were performed with SYBR Premix Ex Taq II kit (TaKaRa Bio Inc. Dalian, China). PCR reactions were performed using Q5 High-Fidelity polymerase (NEB, MA, USA). Each PCR experiment was performed in three technical replicates.
Quantum dot FISH analysis of the subcellular localization of three circRNAs
To detect the subcellular localization of circRNAs in situ, a set of probes were designed (Table S11) containing a NH2 group at the 5ʹ terminal (Sangon, Shanghai, China). The probes were then covalently conjugated to Qdot 585 (Qdot® ITK™ Carboxyl Quantum Dots, Eugene, OR, USA) based on the carboxyl-linked amino method [Citation46]. Subsequently, the QD-ssDNA probes were diluted in hybridization buffer (25% deionized formamide, 2× standard sodium chloride-sodium citrate, 200 ng/μL sheared salmon sperm DNA, 5× Denhardt’s, 1 mM EDTA and 50 mM PBS at pH 7.0) to a final concentration of 1–5 ng/μL.
Before hybridization, the HepG2 cells were fixed with 4% formaldehyde, followed by permeabilization with 0.25% Triton-X100 in PBS for 15 min. Hybridization with QD-labeled probes was carried out at 37°C in the dark for 12 h. After several washes with 2× SSC and 1× SSC, the nucleus was stained with DAPI for 5 min. The cells were then incubated with antibodies against tubulin and RPL24 (Proteinech) overnight at 4°C, followed by the incubation of Dylight633-conjugated goat anti-rabbit IgG (ThermoFisher) for 2 h at 37°C. Fluorescence signals were observed with Leica TCS SP5 confocal microscope (Leica Microsystems Inc., Exton, PA, USA).
Binding motif distribution of some RBPs among the subcellular circRNAs
We calculated the binding motif numbers of the transport-related RBPs (including the 29 motifs from 19 nuclear export RBP and Kozak sequences) in the subcellular circRNAs using a webserver RBPmap (http://rbpmap.technion.ac.il/) [Citation29]. Two thresholds were used: ‘Stringency level’ was set to ‘Medium stringency’ and ‘Apply conservation filter’ was also used in this assay. In addition, we considered the expression of the circRNAs (RPTM). Overall, the binding motif enrichment in the subcellular circRNA was determined by the products of the binding motif number and the corresponding circRNA expression (enrichment = number × RPTM). The IRES structures in the ribosomal circRNAs were predicted with a webserver IRESPred (http://196.1.114.46:1800/IRESPred/IRESPred.html) [Citation47]. The preferred motif of exosomal circRNAs was predicted by MEME (P < 0.05; parameters: -mod zoops -nmoitfs 5 -minw 4 -maxw 10 -revcomp) [Citation34].
CircRNA length calculation
For the exonic circRNAs, the length was determined by summing up the remaining exon lengths after deleting the introns between the two splicing sites of the circRNAs.
Ingenuity pathway analysis (IPA)
IPA online software (IPA, QIAGEN Redwood City, CA, USA) was employed to detect the ‘Bio Functions’ participating in the parental genes of the selected circRNAs (Fold Change ≥ 2). The disease-related functions were eliminated from this assay.
3D-LC-MS/MS identification of the circRNA-derived peptides
Approximately 108 HepG2 cells were lysed with 1 mL lysis buffer (8 M urea, 30 mM HEPES and 1% protease inhibitor cocktail) at 4°C overnight. Dissolved proteins (3.5 mg) were separated by 12% SDS-containing polyacrylamide gels (50 μg per lane), and stained with Coomassie Blue. Six bands between 10 kD and 34 kD corresponding to the theoretical molecular weights of most of the circRNA ORFs were excised from the gel (Figure S13). The gel bands were subsequently de-stained, dehydrated, reduced, alkylated, and digested with trypsin at 37°C for 16 h.
Using the Agilent 1100 HPLC system, the extracted peptides were loaded onto a RP-C18 column, and eluted with an ACN gradient (2%–80%) over 40 min at a flow rate of 1 mL/min. The fractionated peptides were then concentrated, desalted, and injected into the nanoLC Eksigent 425 system for the secondary RP separation, and analysis using the TripleTOF 6600 mass spectrometer (AB SCIEX).
Raw MS/MS data were converted into an MGF format using ProteinPilot™ software. The exported MGF files were searched with Mascot v 2.3.01 against the human SwissProt database combined with all the ORFs from the identified circRNAs in HepG2 cells. Carbamidomethylation was considered as a fixed modification, whereas oxidation (M) and Gln->pyro-Glu (N-term Q) were considered to be variable modifications. The mass tolerance for MS and MS/MS was 0.05 Da and 0.1 Da, respectively. Peptides beyond the threshold score were further filtered according to the criteria previously mentioned.
Statistical analysis
Statistical analyzes were performed using R software version 3.4.2. Correlations between groups were analyzed by calculating the Pearson’s correlation coefficient. Statistically significant differences were calculated using wilcox.test, p-values < 0.05 were considered statistically significant.
Data availability
RNA sequencing raw data have been deposited at Gene Expression Omnibus (GSE107169).
Supplemental Material
Download MS Word (3.2 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Zhang XO, Wang HB, Zhang Y, et al. Complementary sequence-mediated exon circularization. Cell. 2014;159(1):134–147.
- Sanger HL, Klotz G, Riesner D, et al. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci U S A. 1976;73(11):3852–3856.
- Arnberg AC, Van Ommen GJB, Grivell LA, et al. Some yeast mitochondrial RNAs are circular. Cell. 1980;19(2):313–319.
- Salzman J, Gawad C, Wang PL, et al. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PloS One. 2012;7(2):e30733.
- Bachmayr-Heyda A, Reiner AT, Auer K, et al. Correlation of circular RNA abundance with proliferation - exemplified with human normal, benign and malignant tissues. Sci Rep. 2015;5:8057.
- Yu L, Gong X, Sun L, et al. The circular RNA cdr1as act as an oncogene in Hepatocellular Carcinoma through targeting miR-7 Expression. PloS One. 2016;11(7):e0158347.
- Tang W, Ji M, He G, et al. Silencing CDR1as inhibits colorectal cancer progression through regulating microRNA-7. Onco Targets Ther. 2017;10:2045–2056.
- Holdt LM, Stahringer A, Sass K, et al. Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans. Nat Commun. 2016;7:12429.
- Floris G, Zhang L, Follesa P, et al. Regulatory role of circular RNAs and neurological disorders. Mol Neurobiol. 2017;54(7):5156–5165.
- Zhang Y, Zhang XO, Chen T, et al. Circular intronic long noncoding RNAs. Mol Cell. 2013;51(6):792–806.
- Li Z, Huang C, Bao C, et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol. 2015;22(3):256.
- Dang Y, Yan L, Hu B, et al. Tracing the expression of circular RNAs in human pre-implantation embryos. Genome Biol. 2016;17(1):130.
- Hsu M-T, Coca-Prados M. Electron microscopic evidence for the circular form of RNA in the cytoplasm of eukaryotic cells. Nature. 1979;280(5720):339–340.
- Jeck WR, Sorrentino JA, Wang K, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19(2):141–157.
- Hansen TB, Jensen TI, Clausen BH, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495(7441):384–388.
- Zheng Q, Bao C, Guo W, et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun. 2016;7:11215.
- Du WW, Yang W, Chen Y, et al. Foxo3 circular RNA promotes cardiac senescence by modulating multiple factors associated with stress and senescence responses. Eur Heart J. 2017;38(18):1402–1412.
- Li Y, Zheng Q, Bao C, et al. Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res. 2015;25(8):981–984.
- Dou Y, Cha DJ, Franklin JL, et al. Circular RNAs are down-regulated in KRAS mutant colon cancer cells and can be transferred to exosomes. Sci Rep. 2016;6:37982.
- Yang Y, Fan X, Mao M, et al. Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res. 2017;27(5):626–641.
- Legnini I, Di Timoteo G, Rossi F, et al. Circ-ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol Cell. 2017;66(1):22–37 e9.
- Pamudurti NR, Bartok O, Jens M, et al. Translation of CircRNAs. Mol Cell. 2017;66(1):9–21 e7.
- Gao Y, Wang J, Zhao F. CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015;16:4.
- Geiss GK, Bumgarner RE, Birditt B, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26(3):317–325.
- Erkmann JA, Sanchez R, Treichel N, et al. Nuclear export of metazoan replication-dependent histone mRNAs is dependent on RNA length and is mediated by TAP. RNA. 2005;11(1):45–58.
- Benoit Bouvrette LP, Cody NAL, Bergalet J, et al. CeFra-seq reveals broad asymmetric mRNA and non-coding RNA distribution profiles in Drosophila and human cells. RNA. 2017. DOI:10.1261/rna.063172.117.
- Pastro L, Smircich P, Di Paolo A, et al. Nuclear compartmentalization contributes to stage-specific gene expression control in Trypanosoma cruzi. Front Cell Dev Biol. 2017;5:8.
- Lotvall J, Valadi H. Cell to cell signalling via exosomes through esRNA. Cell Adh Migr. 2007;1(3):156.
- Paz I, Kosti I, Ares M Jr., et al. RBPmap: a web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 2014;42(WebServer issue):W361–7.
- Twyffels L, Gueydan C, Kruys V. Shuttling SR proteins: more than splicing factors. FEBS J. 2011;278(18):3246–3255.
- Williams T, Ngo LH, Wickramasinghe VO. Nuclear export of RNA: different sizes, shapes and functions. Semin Cell Dev Biol. 2017. DOI:10.1016/j.semcdb.2017.08.054
- Pelletier J, Sonenberg N. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature. 1988;334(6180):320–325.
- Kozak M. An analysis of 5ʹ-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 1987;15(20):8125–8148.
- Bailey TL, Boden M, Buske FA, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37(WebServer issue):W202–8.
- Stewart M. Nuclear export of mRNA. Trends Biochem Sci. 2010;35(11):609–617.
- Eckner R, Ellmeier W, Birnstiel ML. Mature mRNA 3ʹ end formation stimulates RNA export from the nucleus. EMBO J. 1991;10(11):3513.
- Lykke-Andersen J, Shu MD, Steitz JA. Communication of the position of exon-exon junctions to the mRNA surveillance machinery by the protein RNPS1. Science. 2001;293(5536):1836–1839.
- Dmitriev SE, Andreev DE, Terenin IM, et al. Efficient translation initiation directed by the 900-nucleotide-long and GC-rich 5ʹ untranslated region of the human retrotransposon LINE-1 mRNA is strictly cap dependent rather than internal ribosome entry site mediated. Mol Cell Biol. 2007;27(13):4685–4697.
- Niazi F, Valadkhan S. Computational analysis of functional long noncoding RNAs reveals lack of peptide-coding capacity and parallels with 3ʹ UTRs. RNA. 2012;18(4):825–843.
- Carlevaro-Fita J, Rahim A, Guigo R, et al. Cytoplasmic long noncoding RNAs are frequently bound to and degraded at ribosomes in human cells. RNA. 2016;22(6):867–882.
- Villarroya-Beltri C, Gutierrez-Vazquez C, Sanchez-Cabo F, et al. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun. 2013;4:2980, PubMed PMID: 24356509. Pubmed Central PMCID: 3905700
- Preusser C, Hung LH, Schneider T, et al. Selective release of circRNAs in platelet-derived extracellular vesicles. J Extracell Vesicles. 2018;7(1):1424473. PubMed PMID: 29359036. Pubmed Central PMCID: 5769804.
- Jiang XS, Zhou H, Zhang L, et al. A high-throughput approach for subcellular proteome: identification of rat liver proteins using subcellular fractionation coupled with two-dimensional liquid chromatography tandem mass spectrometry and bioinformatic analysis. Mol Cell Proteomics. 2004;3(5):441–455.
- Yu C, Wang L, Lv B, et al. TMEM74, a lysosome and autophagosome protein, regulates autophagy. Biochem Biophys Res Commun. 2008;369(2):622–629.
- Thery C, Amigorena S, Raposo G, et al. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006. Chapter 3:Unit3.22. DOI:10.1002/0471143030.cb0322s30
- Medintz IL, Uyeda HT, Goldman ER, et al. Quantum dot bioconjugates for imaging, labelling and sensing. Nat Mater. 2005;4(6):435–446.
- Kolekar P, Pataskar A, Kulkarni-Kale U, et al. IRESPred: web server for prediction of cellular and viral internal ribosome entry site (IRES). Sci Rep. 2016;6:27436.