
ABSTRACT
RNA binding proteins regulate gene expression through several post-transcriptional mechanisms. The broadly expressed HuR/ELAVL1 is important for proper function of multiple immune cell types, and has been proposed to regulate cytokine and other mRNA 3′ UTRs upon activation. However, this mechanism has not been previously dissected in stable cellular settings. In this study, HuR demonstrated strong anti-apoptotic and activation roles in Jurkat T cells. Detailed transcriptomic analysis of HuR knockout cells revealed a substantial negative impact on the activation program, coordinately preventing the expression of immune response gene categories, including all cytokines. Knockout cells showed a significant defect in IL-2 production, which was rescued upon reintroduction of HuR. Interestingly, the mechanism of HuR regulation did not involve control of the cytokine 3′ UTRs: HuR knockout did not affect the activity of 3′ UTR reporters in 293 cells, and had no effect on IL-2 and TNF 3′ UTRs in resting or activated Jurkats. Instead, impaired cytokine production corresponded with defective induction of the IL-2 promoter upon activation. Accordingly, upregulation of NFATC1 was also impaired, without 3′ UTR effects. Together, these results indicate that HuR controls cytokine production through coordinated upstream pathways, and that additional mechanisms must be considered in investigating its function.
Introduction
HuR/ELAVL1 is an abundant and ubiquitously expressed RNA binding protein (RBP) that is essential for many developmental and homeostatic processes. HuR knockout (KO) mice die in embryogenesis, where HuR is necessary for placental, skeletal and splenic development [Citation1]. Postnatal deletion of HuR causes depletion of bone marrow, thymic, and intestinal progenitor cells through apoptosis [Citation2]. Similarly, HuR is necessary for neuronal survival [Citation3,Citation4] and in spermatogenesis [Citation5], and is upregulated in many cancers [Citation6,Citation7]. In the immune system, HuR is required for thymocyte maturation and exit to the periphery [Citation8], for T cell proliferation and migration [Citation9–Citation11], and for B cell antibody response [Citation12,Citation13]; HuR deletion and overexpression impacts inflammatory responses in vivo [Citation14,Citation15].
The dynamic control of cytokine and other mRNAs upon activation of immune cells has been closely examined both at the transcriptional and post-transcriptional level. Their transcription is robustly induced by the NFATC homologs and other factors [Citation16,Citation17], best characterized at the interleukin (IL)-2 promoter. In addition, IL-2, tumor necrosis factor (TNF), interferon (IFN)-gamma, colony stimulating factor 2 (CSF2) and other mRNAs are extremely labile in resting cells, and are strongly post-transcriptionally stabilized after stimulation [Citation18]. The destabilization is caused by a group of RBPs (AUF1/HNRNPD, TTP/ZFP36 and others) that bind to ‘class II’ AU-rich elements (AREs) – tandem copies of AUUUA – found in many cytokine mRNA 3′ UTRs [Citation19,Citation20].
Several lines of evidence have led to a model where HuR stabilizes cytokine mRNAs through interactions with the 3′ UTR AREs [reviewed in Citation20–Citation22]. HuR is induced in T cells upon activation, translocates from the nucleus to the cytoplasm, and binds to the ARE regions of many cytokine mRNAs. Overexpression of HuR has been shown to upregulate 3′ UTR reporters and to increase the half-life of ARE-containing mRNAs. Thus, HuR has been proposed to positively affect mRNA stability by competing with the destabilizing RBPs mentioned above for binding at the ARE sites. An alternative model for these observations is that HuR promotes decay, but its overexpression titrates away necessary complex components, resulting in protection [Citation22]. However, these and other molecular mechanisms of cytokine mRNA control, as well as HuR’s overall cellular roles, have not been examined in KO T cell line models.
In the context of mouse models, the effects of HuR on cytokine production vary greatly, likely due to the complex in vivo physiological feedback, developmental transitions, and timing of the HuR deletion. Upon conditional knockout of HuR in late-stage thymocytes, activated peripheral CD4+ T cells showed transcriptionally elevated IL-2 levels, but substantially reduced IL-4, IL-5, and IL-13 [Citation10]. In another model, conditional HuR KOs in activated T cells, after Th2 polarization, exhibited transcriptional upregulation of IL-4 and increased IL-2 and IL-13 mRNA stability [Citation23]. Th17 polarization of the same knockouts resulted in decreased IL-17 mRNA stability and protein levels [Citation9]. HuR deletion in early thymocytes did not change IL-3 mRNA levels, and increased those of TNF [Citation8]. Finally, in an inducible mouse overexpression model, upregulation of HuR in macrophages increased TNF mRNA stability, but strongly downregulated its translation [Citation14]. Thus, while animal models are undoubtedly necessary to examine the relevant biological roles of HuR, they provide fewer avenues in deciphering its direct molecular functions.
Like many other RBPs, HuR has been implicated in post-transcriptional control of gene expression through several mechanisms, and their relative importance in regulating each target is mostly unknown. Aside from roles in 3′ UTR control, HuR is strongly associated with splicing: a major portion of HuR binding sites determined by CLIP-seq are intronic, found near splice sites, show sequence conservation, and affect expression of the target gene [Citation24,Citation25]. Alternative splicing of hundreds of mRNAs is suggested to facilitate HuR’s role in ensuring a proper B cell response [Citation12]. HuR also downregulates inclusion of Fas mRNA exon 6, and controls alternative splicing of Eif4enif1 [Citation26,Citation27]. Furthermore, positive and negative effects of HuR on translation have been identified [Citation14,Citation28–Citation31], and HuR has been shown to impact alternative polyadenylation [Citation32,Citation33].
In this study, the cellular role and molecular mechanism of HuR function was addressed using stable KO cell line models. HuR prevents apoptosis and establishes an activated expression program in Jurkat cells; interestingly, it does so without regulating cytokine expression through the 3′ UTR, coordinately controlling upstream transcriptional events instead.
Results
HuR KO Jurkat cells exhibit a strong apoptotic phenotype
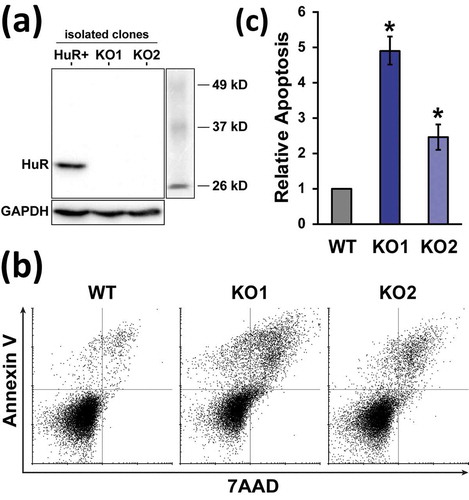
The RNA-binding protein HuR is important for multiple aspects of T cell development and activation, and several specific functions, molecular modes of action, as well as many targeted mRNAs have been identified. To examine its cellular roles and molecular mechanism in a T cell setting in detail, a CRISPR/Cas9-mediated HuR KO model was created in Jurkat cells. Following transient sgRNA/Cas9 plasmid transfection and clonal screening by dot immunoblot, two clones (generated with independent sgRNAs targeting HuR exons 5 and 3) were identified, and validated by western blot to completely lack HuR protein ()). The use of two targeting sites ensures lack of common off-target mutations between the clones that can complicate phenotypic analysis. The resulting cell lines propagated slower than the parental WT, and had a substantial fraction of blebbing, apoptotic cells (Figure S1(a)). These cells resembled the apoptotic process induced in WT Jurkats by staurosporine (Figure S1A). The apoptotic phenotype was reproducibly confirmed by Annexin V/7AAD staining and flow cytometry ()), with the knockout lines demonstrating 2.5–4.9 fold higher percentages of cell death ()). Thus, deletion of HuR in two independent Jurkat clones resulted in substantially higher apoptosis. Extended passage of the KO lines diminished the apoptotic phenotype, indicating ongoing adaptation/selection in the clonal cell lines (Figure S2). In addition to cell death, a smaller fraction of large, multinucleated cells was observed in the populations (Figure S1(a) and Figure S3). For the non-apoptotic KO cells, phase-contrast microscopy and staining of the actin cytoskeleton with phalloidin, along with a nuclear 7AAD stain, revealed an otherwise normal size and morphology (Figure S1 and Figure S3).
Figure 1. Generation and apoptosis phenotype of HuR KO Jurkat cells. (a) Western blot of a HuR-positive clone isolated during KO generation, along with two independent KO clones. (b) Representative measurement of WT and HuR KO apoptosis rates by Annexin V and 7AAD staining, followed by flow cytometry. (c) Relative apoptosis of early-passage HuR KO1 and KO2 clones, n = 4. WT Jurkat cell cultures contained 7.4 ± 1.7% apoptotic cells. Error bars represent standard error of the mean. *, p-value < 0.05, paired two-tailed Student’s t test of apoptotic percentages between WT and KO
HuR controls the expression of a large set of mRNAs
In T cells, HuR is known to bind to a large set of mRNAs [Citation34,Citation35], and to control mRNA fates during the activation program [Citation36]. To elucidate the global and specific effects of HuR on mRNA expression as a function of activation, RNA-Seq measurements were carried out in WT and HUR KO1 cells in resting and PMA/ionomycin-activated states, allowing for multiple pairwise comparisons between the datasets. Consistent with the expected substantial changes in gene expression upon activation, the largest differences between the resulting four groups of samples were attributable to the activation state by principal component analysis (PC1, Figure S4(a)) and hierarchical clustering (Figure S4(b)), while the three biological replicates in each group showed high reproducibility. Interestingly, the second principal component, accounting for 26% of the variance, corresponded to presence of HuR, with particularly significant differences in the activated state (Figure S4(a)). This separation indicated that HuR controls the levels of a large set of mRNAs in Jurkat cells. Accordingly, differential expression (DE) analysis with stringent cutoffs (4-fold change and 5% FDR) identified 553 DE genes in resting KO vs WT cells, and 1398 DE genes in activated KO vs WT cells ()). Notably, these gene sets contained considerably more mRNAs that are upregulated, rather than decreased, in the KO. Together, these observations demonstrate that HuR is a broad post-transcriptional regulatory factor that substantially impacts the gene expression program.
Figure 2. Profiling of mRNA levels in resting vs. activated, WT vs. HuR KO1 cells (n = 3) reveals a substantial defect in the activation program of HuR KOs. (a) Number of differentially expressed (upregulated and downregulated) genes passing a 4-fold change and 5% false discovery rate (FDR) cutoff, comparing the indicated sample groups. (b) Heatmap of mRNA level changes for differentially expressed genes in WT cells between activated and resting states (left), and in the activated state between KO and WT cells (right). (c) Overlap between genes upregulated upon activation in WT cells, and those downregulated in KO relative to WT in activated cells. (d) clusterProfiler analysis of Gene Ontology category enrichment in upregulated and downregulated mRNA sets defined by the comparisons indicated at the bottom
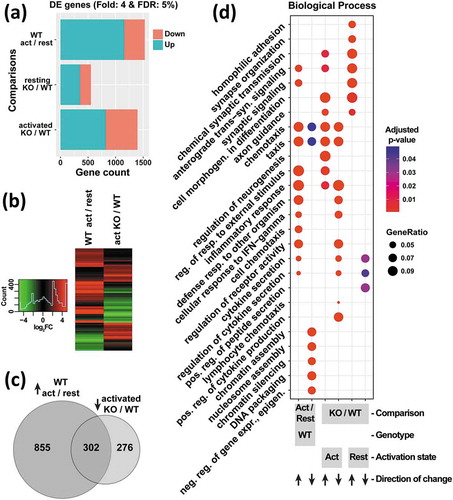
Hur controls a major portion of the activation response
Activation of T cells upon antigen encounter elicits a massive transformation in gene expression leading to changes in proliferation state, morphology, and other characteristics. To assess the involvement of HuR in the activation program, the DE gene sets were examined in detail. As reported above, stimulation of WT Jurkat cells caused extensive expression changes, mostly in the direction of upregulation ()). Strikingly, this activation response was largely negated or prevented in the absence of HuR: genes that were upregulated upon activation in WT cells were significantly downregulated in the activated KO vs WT comparison; conversely, genes that were repressed upon WT cell activation were significantly higher in KO vs WT activated cells ()). The substantial overlap between genes impacted by cell stimulation and HuR KO, and their opposing directions of change ()), indicates involvement of HuR in establishing the activated expression program. Examination of the gene ontology (GO) category enrichment of the DE gene sets provided further insight into the role of HuR in the reprogramming. Stimulation of WT cells upregulated genes involved in immune responses, including chemotaxis and production/secretion of cytokines, as well as their regulatory factors (), first column). Remarkably, nearly all of these categories were also enriched in genes that were downregulated in activated KO vs WT cells (), fourth column). Additionally, genes that positively regulate cytokine production were downregulated in HuR KO cells. In summary, HuR is responsible for enacting a major part of the gene expression changes upon activation in Jurkat cells.
HuR is necessary for cytokine production upon activation
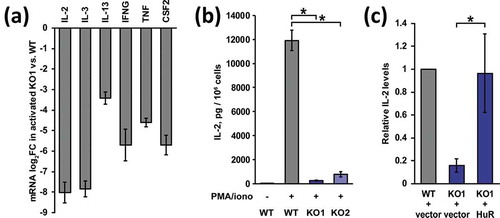
Cytokine production is a key consequence of T cell activation. Prompted by the GO enrichment results, expression of cytokine mRNAs was investigated in HuR KO Jurkat cells. Comparing activated KO to WT cells, RNA-seq data showed a strong reduction of levels for practically all cytokines that are induced by stimulation in this cell line ()), ranging from 11-fold (IL-13) to 259-fold (IL-2) downregulation. To validate the mRNA expression data, secreted IL-2 levels after 24 hours of PMA/ionomycin activation were measured by ELISA ()). Again, the robust activation seen in WT cells was diminished 45-fold in HuR KO1, confirming the RNA-seq results. Furthermore, IL-2 production in the independently derived HuR KO2 was also 15-fold attenuated relative to WT, demonstrating that the phenotype was not a consequence of off-target mutations or clonal differences. It should be noted that the KO cells still underwent morphological changes upon stimulation (rounding up), suggesting that PMA/PKC-mediated aspects of the activation program are intact (Figure S1(b)). Next, HuR’s role in cytokine production was definitively addressed with a rescue experiment. HuR KO1 cells were transduced with a retroviral vector encoding HuR (along with an empty vector control), stable integrant populations were selected, and expression of HuR was confirmed (Figure S5). Measurement of secreted IL-2 levels showed robust rescue of this cytokine’s production upon reintroduction of HuR ()). Overall, these results establish that HuR is essential for cytokine expression upon activation in Jurkat cells.
Figure 3. HuR regulates cytokine production in Jurkat cells. (a) RNAseq mRNA level log2 fold changes (FC) of expressed cytokines in activated KO1 vs. WT Jurkat cells. (b) Secreted IL-2 levels in WT and HuR KO cells after stimulation with PMA/ionomycin, n = 3. (c) Rescue of IL-2 production in HuR KO1 cells transduced with MSCV-MPIG-HuR retrovirus, compared to empty vector retrovirus, n = 3. Error bars represent standard error of the mean. *, p-value < 0.05, paired two-tailed Student’s t test of normalized ELISA spectrometry measurements
HuR does not significantly regulate cytokine mRNA 3′ UTRs in Jurkat and 293 cells
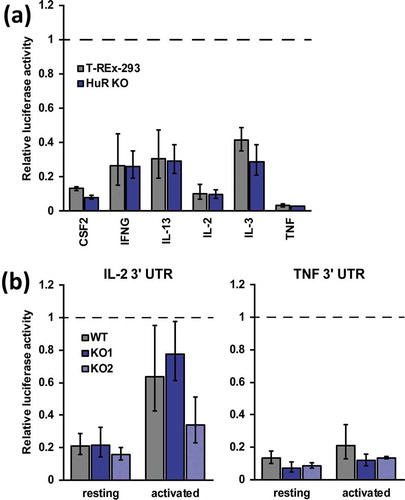
Early studies implicated HuR, along with other ARE-binding proteins, in interactions with the 3′ UTRs of several cytokine mRNAs or their fragments, including IL-2, IL-3, IL-13, CSF2/GMCSF, TNF [Citation37–Citation45]. Overexpression of HuR caused stabilization of CSF2, TNF and IL-13 3′ UTR or IL-3 mRNA-containing reporters [Citation43,Citation44,Citation46,Citation47], and recombinant HuR prevented degradation of TNF ARE RNA in an in vitro assay [Citation42], leading to a model of post-transcriptional mRNA stabilization by binding their 3′ UTRs and competing with other, destabilizing factors. The established knockout cell lines provide a stable system to examine this and other regulatory mechanisms mediating HuR’s role in mRNA expression. To evaluate HuR’s effect on cytokine expression through their 3′ UTRs, full-length UTR sequences were cloned into a dual-luciferase reporter vector. Initially, a panel of six cytokine UTRs was screened in the easily transfectable human embryonic kidney T-REx-293 cells and a derived HuR KO cell line [Citation48]. Previously, we have demonstrated a significant, albeit mild, regulation of an AUUUA-containing reporter by HuR using these lines [Citation49]. Relative to empty vector controls, all of the constructs showed substantial downregulation in T-REx-293 cells, ranging from 2.4-fold (IL-3) to 30-fold (TNF), consistent with the known destabilizing role of the UTRs ()). Strikingly, reporter activity in the absence of HuR was not statistically significantly decreased for any of the UTRs. These results indicate that HuR does not directly or indirectly regulate the tested cytokine 3′ UTRs in 293 cells. Next, activity of the IL-2 and TNF 3′ UTR reporters was tested in resting and activated Jurkat cells, as well as HuR KO derivatives. Both reporters showed substantial repression relative to empty vector controls in resting WT Jurkats, and were de-repressed to varying extents upon activation ()), confirming the previously known activities of these UTRs. However, deletion of HuR in two independent clones did not show consistent differences from WT for the IL-2 UTR, and demonstrated small, statistically insignificant further repression of the TNF UTR. Notably, de-repression of the IL-2 UTR upon activation was still observed in HuR KO cells ()), indicating that HuR is dispensable for stabilization of the reporter. Altogether, HuR does not substantially participate in post-transcriptional regulation of the tested cytokine UTRs in Jurkat or 293 cells.
Figure 4. HuR does not regulate IL-2 and TNF levels through their 3′ UTR. (a) Activity of luciferase reporters containing cytokine mRNA 3′ UTRs measured in WT and HuR KO T-REx-293 cells, n = 2. (b) Activity of IL-2 and TNF 3′ UTR reporters in WT, HuR KO1 and HuR KO2 Jurkat cells in resting and activated states, n = 3. Renilla luciferase activity was normalized to co-expressed firefly luciferase activity and to empty (no 3′ UTR) control reporters
HuR controls NFATC1 levels and IL-2 promoter activity upon activation
Upon activation, T cell cytokine production is upregulated and maintained by transcriptional and post-transcriptional mechanisms. Since HuR affected cytokine levels, but post-transcriptional control through the 3′ UTR was minimal, transcriptional pathways were examined. Stimulation-induced activation of the IL-2 promoter is one of the most characterized features of T cell activation [Citation50]. Accordingly, activity of a murine IL-2 promoter luciferase reporter [Citation51] was tested in WT and HuR KO Jurkat cells. As expected, activation of WT cells elicited a robust IL-2 transcriptional response ()). Interestingly, this response was strongly diminished in the absence of HuR, with 26-fold reduction in KO1 cells, and 2.7-fold decrease in KO2 cells. The reduced activity indicates that HuR is necessary for IL-2 mRNA transcription in Jurkat cells, and explains the impaired endogenous IL-2 secretion in HuR KO cells.
Figure 5. HuR controls IL-2 production transcriptionally and regulates NFATC1 levels upon activation. (a) Activity of a firefly luciferase reporter under the IL-2 gene promoter in WT, HuR KO1 and KO2 Jurkat cells under resting and activated conditions, n = 4. Activity was normalized to co-transfected renilla luciferase and expressed relative to WT activated cells. (b) Representative western blot of NFATC1 levels in resting and activated WT and KO cells. (c) Quantification of NFATC1 protein levels under the above conditions, n = 4. (d) Relative luciferase activity of the NFATC1 3′ UTRs, measured as in , n = 3
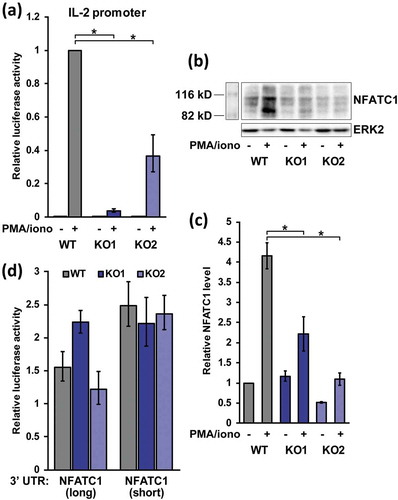
The coordinated attenuation of all expressed cytokines in KO cells suggested that HuR may affect their expression through a common upstream factor, since HuR itself does not have appreciable DNA binding activity [Citation52]. The NFATC1/C2 transcription factors are master regulators of cytokine production upon activation [Citation53,Citation54], and their levels were investigated next. Upon stimulation, NFATC2 protein is activated by dephosphorylation and nuclear import, while NFATC1 mRNA transcription is also turned on in an autoregulatory loop. NFATC2 showed a mild (1.6-fold) but significant upregulation in the RNA-seq mRNA levels in activated KO vs WT cells, but NFATC1 demonstrated a strong, 5.1-fold downregulation, suggesting that HuR may regulate NFATC1 expression. When tested by Western blot, NFATC1 levels were substantially (4.2-fold) increased upon stimulation in WT cells, and this induction was significantly reduced in HuR KO cells ()). Thus, HuR affects the accumulation of the induced NFATC1 transcription factor upon activation in Jurkat cells. Finally, regulation of the NFATC1 3′ UTR by HuR was examined. NFATC1 is transcribed in two UTR isoforms, where the shorter UTR is associated with NFATC1 induction. Interestingly, when both isoforms were tested in luciferase assays in activated Jurkat cells, the presence or absence of HuR had no statistically significant effect on activity ()), indicating that HuR does not impact NFATC1 expression through its 3′ UTR.
Discussion
HuR is necessary for many biological processes, and has been found to act through several molecular mechanisms. In this study, knockout cell lines were used as defined, tractable models to characterize its cellular roles, and perhaps more importantly, its mechanisms of action. Two independent KO lines in Jurkat cells demonstrated increased cell death and defective activation phenotypes ( and ). The anti-apoptotic role of HuR is consistent with its regulation of apoptosis/survival-relevant transcripts [Citation55], as well as results from mouse KO models: deletion of HuR shows increased apoptosis in placental development [Citation1] and mature neurons [Citation4]. Additionally, recent studies in pancreatic cancer cell lines indicated that HuR knockdown reduces proliferation and migration [Citation56], and HuR KO leads to apoptosis and abrogates xenograft tumor growth [Citation57]. The significance of the observed phenotypes is also in line with the large number of mRNAs bound by HuR in T cells [Citation34]. Accordingly, stable elimination of HuR in Jurkat cells lead to profound differences in the mRNA expression profile, comparable in extent to the changes due to stimulation ()). The higher number of upregulated (compared to downregulated) genes upon HuR KO, both in resting and activated states, suggests that HuR may carry an underappreciated repressive function for many mRNAs, or it indirectly affects mRNA levels through other factors. In this regard, most of the indirect changes are not likely to be mediated by several known post-transcriptional factors, since their levels are largely unperturbed in the RNA-seq data (Supplementary Table 1).
Strikingly, many of the genes affected by activation in WT cells were also differentially expressed, but with the opposite direction of change, in the HuR KO vs WT comparison in the activated state ()). In other words, the activation expression program is in large part controlled by HuR, and does not properly ensue in its absence. Stimulation-induced genes involved in cytokine secretion and its regulation, as well as inflammatory/defensive responses and chemotaxis, are among the transcripts that fail to be upregulated in HuR KO cells ()). The lack of upregulation was particularly noticed for the cytokines themselves ()). Confirming the mRNA level observations, IL-2 secretion was severely disrupted in HuR KO Jurkat cells, and rescued by reintroduction of HuR ()).
Early studies pointed to a positive role for HuR in cytokine control, in general agreement with the observed defect in cytokine expression signatures and the IL-2 production phenotype in HuR KO Jurkat cells ( and ). However, the regulatory mechanism in Jurkat and 293 cells is a surprise that is inconsistent with some of the previous data: HuR does not appreciably regulate the 3′ UTRs of CSF2, IFNG, IL-2, IL-3, IL-13, and TNF mRNAs in 293 cells, and the major IL-2 and TNF cytokine mRNAs in Jurkats. Previously, HuR was shown to bind AU-rich sequences in TNF, CSF2 and IL-3 UTRs [Citation38–Citation43,Citation45,Citation58] and HuR immunoprecipitation was shown to enrich IL-2, IL-13, INFg, and TNF mRNA from lysates relative to IgG controls [Citation8,Citation35,Citation44,Citation59,Citation60]. Together with reporter data demonstrating that overexpression of HuR causes stabilization of TNF, IL-13, and CSF2 UTR fragment or IL-3 whole mRNA reporters [Citation43,Citation44,Citation46,Citation47], these observations generated a prevailing model that HuR regulates mRNA stability through the 3′ UTR. HuR has also been shown to bind and/or stabilize several other mRNAs [Citation28,Citation61–Citation64]. It should be noted that the majority of the earlier studies necessarily used overexpression as a means of modulating HuR levels, and its effect on 3′ UTR reporter assays may be more difficult to interpret than a knockout state. Nevertheless, HuR knockdown in a fibrosarcoma line caused reduction of IL-3 UTR reporter levels, and decreased half-lives of the reporter mRNA [Citation65]. HuR KD in Jurkat cells prevented activation-induced stabilization of INFg mRNA and decreased IL-13 mRNA half-life, although it is not established whether it occurred directly through the 3′ UTR [Citation45,Citation66].
However, other experimental evidence suggests that HuR does not substantially regulate certain cytokine 3′ UTRs. For example, overexpression of HuR did not stabilize CSF2 and IL-3 ARE reporters, while a fos ARE reporter was stabilized [Citation67]. Knockdown of HuR in Jurkat cells produced a very mild (1.2 fold) destabilizing effect on a IL-3 UTR reporter [Citation58], likely insufficient to explain the drastic decrease in its mRNA levels seen in the HuR KO. Studies with IL-2 showed that HuR overexpression failed to stabilize GFP-IL-2 reporter half-life, and HuR did not bind to IL-2 mRNA affinity resin [Citation68]. HuR also didn’t bind IL-2 UTR fragments efficiently [Citation39]. An additional report found that HuR binds the IL-2 3′ UTR, but this binding event does not impact IL-2 mRNA stability [Citation37]. Finally, a transcriptome-wide method to detect HuR-associated mRNAs in Jurkat cells identified no enrichment for cytokine gene categories, and showed strong depletion for IL-2 and IL-3 mRNAs (although some binding of CSF2 and TNF), and none of the above cytokine mRNAs were enriched in a RIP-seq study in activated T cells [Citation34,Citation35]. It should be noted that HuR-bound transcripts as a group do show slower decay rates upon activation [Citation36]. However, when the role of HuR in controlling the cytokine UTRs was directly assessed by comparison of UTR reporter activities in two stable and tractable WT vs HuR KO cell systems in this study (), no regulation was observed. Thus, HuR does not always lead to direct stabilization of transcripts containing ARE elements, and other mechanisms should be investigated. Some of the above discrepancies likely stem from the differences in the interrogated cell types.
Instead of regulating the cytokine response through the 3′ UTR in Jurkat cells, the data indicates that HuR exerts broad control through upstream factors. The coordinated and profound lack of cytokine induction pointed to a transcriptional response, and the induction of the IL-2 promoter was indeed strongly impaired in HuR KO cells ()). Transcriptional control of the CCL5 chemokine has been shown to depend on HuR in MCF-7 cells [Citation69]. Induction of the major cytokine transcription factor NFATC1 was impaired upon stimulation in HuR KO cells ()), strongly suggesting that the IL-2 transcriptional response was abrogated at least in part due to this deficiency. In similarity to the cytokine mRNA results, 3′ UTR isoforms of NFATC1 were not coherently regulated by HuR ()), indicating regulation through other modes. Here, control of splicing is a likely mechanism, since HuR is found to associate with intronic sites as much as or more than 3′ UTRs, comprising 30–35% of CLIP sites in HeLa cells, and 40–45% in 293T cells [Citation24,Citation49]. Additionally, HuR regulates alternative splicing of Fas pre-mRNA [Citation26]. Interestingly, differential exon usage analysis of the RNA-seq data indicates that the NFATC1 mRNA undergoes substantial alternative splicing upon activation in WT cells (Figure S6, left), and this splicing program is largely negated (not enacted) in the absence of HuR (Figure S6, right). Thus, it can be hypothesized that HuR directly affects alternative splicing of NFATC1 upon activation, with subsequent effects on its mRNA and/or protein stability and function. However, other post-transcriptional modes that do not involve the UTR, including regulation of target mRNA translation, cannot be ruled out and require further examination. Overall, further insight into HuR’s modes of action and targets of regulation will be gained as additional knockout models amenable to mechanistic dissection become available, such as the pancreatic and colon cancer cell lines [Citation57].
Materials and methods
CRISPR/Cas9-mediated generation of HuR KO clones
Knockouts in Jurkat cells were obtained by selection-free immunoblot screening as previously described [Citation48]. Jurkat cells were cultured in RPMI1640 media with 10% FBS and 1x penicillin/streptomycin at 37°C with 5% CO2. Three pSpCas9(BB) plasmids co-expressing Cas9 and sgRNAs targeting the third, fourth, and fifth exons of HuR were independently transiently transfected into cells by electroporation in a 0.4 mL cuvette (300V, infinite resistance, 960 µF capacitance). Clonal populations were grown and screened by dot immunoblot with anti-HuR antibodies (Santa Cruz Biotechnology, clone 3A2, sc-5261). HuR KO1 (clone 1A8, targeting the 5th exon) and HuR KO2 (clone 1F3, targeting the 3rd exon) were validated by western blot. KO cells were cultured on WT Jurkat-conditioned media to reduce adaptation to the absence of HuR.
Cellular and molecular characterization of KO clones
For confocal microscopy, cells were resuspended in 50 µL PBS, fixed at room temperature for 20 minutes by addition of 250 µL 4% PFA in PBS, washed in PBS + 1% BSA, and permeabilized in 0.3% Triton-X100 in PBS + 1% BSA at r.t. for 10 minutes. Cells were spun and resuspended in PBS + 1% BSA, and 50 µL of suspension were labeled with 1 µL of 50 µg/mL 7AAD (Invitrogen) and 2 µL of 200 U/mL Phalloidin-CF488A (Biotium) at r.t. for 20 minutes. For RNAseq material collection and IL-2 production measurements, WT and HuR KO early-passage cells at a density of 0.5 × 106 cells/mL were activated with 20 ng/mL PMA + 500 ng/mL ionomycin or left untreated for 24 hours. Total RNA from cell pellets was collected with RiboZol. Three biological replicates were performed. RNAseq libraries were produced using the NEBnext Ultra Directional RNA library prep kit and sequenced on an Illumina NextSeq instrument. Note that the RNA-seq data confirms biallelic deletion at the sgRNA-targeted site (Figure S7), and a 5–7-fold reduction in HuR transcript levels in the activated and resting states, respectively. Read processing and R analysis was carried out using the systemPiper [Citation70] workflow. Transcriptome and genome alignment was performed by tophat2/bowtie2 [Citation71,Citation72] to the Gencode v27/GRCh38 annotations. Differential expression analysis was performed using DESeq2 [Citation73] and DEXSeq [Citation74]. Gene ontology enrichment analyses were carried out using the GOstats [Citation75] and clusterProfiler [Citation76] packages. The raw and processed RNA-seq data are deposited in GEO under accession GSE121966. For IL-2 production measurement, supernatant media after cell activation were spun down and frozen. IL-2 amounts were measured by ELISA (R&D Systems Quantikine) using the kit standard controls. The following antibodies were used in western blot: HuR (Santa Cruz, clone 3A2), GAPDH (ThermoFisher, clone 6C5), NFATC1 (Biolegend, clone 7A6), ERK2 (Santa Cruz).
Re-expression of HuR in KO cells
HuR ORF was cloned into a modified retroviral MSCV-PIG vector (containing an MluI site between XhoI and EcoRI) using BglII-XhoI. MSCV-PIG (Puro IRES GFP) was a gift from Scott Lowe (Addgene plasmid # 18751). HuR-encoding retrovirus (along with empty vector control retrovirus) was produced using Phoenix-AMPHO packaging cells co-transfected with VSVG pseudotyping plasmid. WT and HuR KO1 Jurkat cells were transduced with the viral media by spinfection at 800 rpm for 1 hour at r.t. with 8 µg/mL polybrene. Two to three days later, cells were placed on selection with 4 µg/mL puromycin to obtain stable integration populations.
Luciferase reporter assays
For 3′ UTR testing, all full-length UTR regions except NFATC1(long) were cloned into a modified psiCHECK-2 (Promega) vector containing BsmBI sites [Citation49] using Golden Gate cloning (see Supplementary File 1 for oligonucleotide sequences). The NFATC1(long) 3′ UTR, which contained BsmBI sites, was analogously cloned using SapI. First, an existing SapI site in psiCHECK-2 was altered by site-directed mutagenesis (NEB, Q5), and SapI sites were introduced into the MCS by a short insert with XhoI/NotI ends. Luciferase assays in T-REx-293 and derivative HuR KO cells were performed at 50–70% confluency, transfected with 10 ng plasmid using Mirus Trans-IT-LT1 as previously described [Citation49]. Luciferase reporter activity of the 3′ UTR constructs in Jurkat and derivative HuR KO cells was also measured in a 96 well format. Cell aliquots (100 µL of cells at 0.5 × 106 cells/mL) were transfected with Trans-IT-LT1 (1 µL of 30 ng/µL of reporter plasmid, 0.3 µL Trans-IT-LT1, 9 µL Opti-MEM media). Eighteen hours after transfection, cells were activated with 20 ng/mL PMA + 500 ng/mL ionomycin or left untreated, and 24 hours after transfection cells were harvested and lysed in 10 µL passive lysis buffer. Each biological replicate consisted of three technical replicates (wells). For promoter activity testing, the IL-2 promoter luciferase plasmid was a gift from Anjana Rao (Addgene plasmid # 12194). Due to the lower firefly luciferase activity of the plasmid, transfections were carried out by electroporation of 2.5 × 106 cells in 250 µL of Opti-MEM + 2 µg IL-2 promoter plasmid + 2 µg renilla luciferase transfection control plasmid (pRL-TK) in a 0.4 mL cuvette (280V, infinite resistance, 960 µF capacitance). Twenty four hours after transfection, cells were split into two 100 µL wells, and activated with PMA/ionomycin as above or left untreated for 6 hours. Cells were lysed in 20 µL of passive lysis buffer, and 7–15 µL of lysate were assayed with 50 µL of Promega Dual-Luciferase reagents on a Turner 20/20 luminometer with 10 second integration. Averaging and statistical analysis (two-tailed paired Student’s t-test) were done after logarithmic transformation. Data in graphs are displayed in the original scale.
Supplemental Material
Download Zip (9 MB)Acknowledgments
I am thankful to the Rasmussen lab for confocal microscopy assistance. This work was supported by the University of California Cancer Research Coordinating Committee, Grant ID# CRN-18-524844.
Disclosure statement
No potential conflict of interest was reported by the author.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Katsanou V, Milatos S, Yiakouvaki A, et al. The RNA-binding protein Elavl1/HuR is essential for placental branching morphogenesis and embryonic development. Mol Cell Biol. 2009 May;29(10):2762–2776. PubMed PMID: 19307312; PubMed Central PMCID: PMCPMC2682039.
- Ghosh M, Aguila HL, Michaud J, et al. Essential role of the RNA-binding protein HuR in progenitor cell survival in mice. J Clin Invest. 2009 Dec;119(12):3530–3543. PubMed PMID: 19884656; PubMed Central PMCID: PMCPMC2786787.
- Skliris A, Papadaki O, Kafasla P, et al. Neuroprotection requires the functions of the RNA-binding protein HuR. Cell Death Differ. 2015 May;22(5):703–718. PubMed PMID: 25301069; PubMed Central PMCID: PMCPMC4392069.
- Sun K, Li X, Chen X, et al. Neuron-specific HuR-deficient mice spontaneously develop motor neuron disease. J Immunol. 2018 Jul 1;201(1):157–166. PubMed PMID: 29760195; PubMed Central PMCID: PMCPMC6008238.
- Chi MN, Auriol J, Jegou B, et al. The RNA-binding protein ELAVL1/HuR is essential for mouse spermatogenesis, acting both at meiotic and postmeiotic stages. Mol Biol Cell. 2011 Aug 15;22(16):2875–2885. PubMed PMID: 21737689; PubMed Central PMCID: PMCPMC3154883.
- Abdelmohsen K, Gorospe M. Posttranscriptional regulation of cancer traits by HuR. Wiley Interdiscip Rev RNA. 2010 Sep-Oct;1(2):214–229. . PubMed PMID: 21935886; PubMed Central PMCID: PMCPMC3808850.
- Brody JR, Dixon DA. Complex HuR function in pancreatic cancer cells. Wiley Interdiscip Rev RNA. 2018 May;9(3):e1469. . PubMed PMID: 29452455; PubMed Central PMCID: PMCPMC6040811.
- Papadaki O, Milatos S, Grammenoudi S, et al. Control of thymic T cell maturation, deletion and egress by the RNA-binding protein HuR. J Immunol. 2009 Jun 1;182(11):6779–6788. PubMed PMID: 19454673.
- Chen J, Cascio J, Magee JD, et al. Posttranscriptional gene regulation of IL-17 by the RNA-binding protein HuR is required for initiation of experimental autoimmune encephalomyelitis. J Immunol. 2013 Dec 1;191(11):5441–5450. PubMed PMID: 24166976; PubMed Central PMCID: PMCPMC3831112.
- Techasintana P, Ellis JS, Glascock J, et al. The RNA-binding protein HuR posttranscriptionally regulates IL-2 homeostasis and CD4(+) Th2 differentiation. Immunohorizons. 2017 Aug 1;1(6):109–123. PubMed PMID: 30035254; PubMed Central PMCID: PMCPMC6052877.
- Chen J, Martindale JL, Cramer C, et al. The RNA-binding protein HuR contributes to neuroinflammation by promoting C-C chemokine receptor 6 (CCR6) expression on Th17 cells. J Biol Chem. 2017 Sep 1;292(35):14532–14543. PubMed PMID: 28684423; PubMed Central PMCID: PMCPMC5582845.
- Diaz-Munoz MD, Bell SE, Fairfax K, et al. The RNA-binding protein HuR is essential for the B cell antibody response. Nat Immunol. 2015 Apr;16(4):415–425. PubMed PMID: 25706746; PubMed Central PMCID: PMCPMC4479220.
- DeMicco A, Naradikian MS, Sindhava VJ, et al. B Cell-Intrinsic Expression of the HuR RNA-Binding Protein Is Required for the T Cell-Dependent Immune Response In Vivo. J Immunol. 2015 Oct 1;195(7):3449–3462. PubMed PMID: 26320247; PubMed Central PMCID: PMCPMC4575876.
- Katsanou V, Papadaki O, Milatos S, et al. HuR as a negative posttranscriptional modulator in inflammation. Mol Cell. 2005 Sep 16;19(6):777–789. PubMed PMID: 16168373.
- Yiakouvaki A, Dimitriou M, Karakasiliotis I, et al. Myeloid cell expression of the RNA-binding protein HuR protects mice from pathologic inflammation and colorectal carcinogenesis. J Clin Invest. 2012 Jan;122(1):48–61. PubMed PMID: 22201685; PubMed Central PMCID: PMCPMC3248801.
- Macian F, Lopez-Rodriguez C, Rao A. Partners in transcription: NFAT and AP-1. Oncogene. 2001 Apr 30;20(19):2476–2489. . PubMed PMID: 11402342.
- Graef IA, Chen F, Crabtree GR. NFAT signaling in vertebrate development. Curr Opin Genet Dev. 2001 Oct;11(5):505–512. PubMed PMID: 11532391.
- Lindstein T, June CH, Ledbetter JA, et al. Regulation of lymphokine messenger RNA stability by a surface-mediated T cell activation pathway. Science. 1989 Apr 21;244(4902):339–343. PubMed PMID: 2540528.
- Chen CY, Shyu AB. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci. 1995 Nov;20(11):465–470. PubMed PMID: 8578590.
- Vlasova-St Louis I, Bohjanen PR. Post-transcriptional regulation of cytokine signaling by AU-rich and GU-rich elements. J Interferon Cytokine Res. 2014 Apr;34(4):233–241. . PubMed PMID: 24697201; PubMed Central PMCID: PMCPMC3976587.
- Meisner NC, Filipowicz W. Properties of the regulatory RNA-binding protein hur and its role in controlling miRNA repression. Adv Exp Med Biol. 2011;700:106–123. . PubMed PMID: 21755477.
- Brennan CM, Steitz JA. HuR and mRNA stability. Cell Mol Life Sci. 2001 Feb;58(2):266–277. 10.1007/PL00000854. PubMed PMID: 11289308.
- Gubin MM, Techasintana P, Magee JD, et al. Conditional knockout of the RNA-binding protein HuR in CD4(+) T cells reveals a gene dosage effect on cytokine production. Mol Med. 2014 Mar;20(20):93–108. . PubMed PMID: 24477678; PubMed Central PMCID: PMCPMC3960399.
- Lebedeva S, Jens M, Theil K, et al. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol Cell. 2011 Aug 5;43(3):340–352. PubMed PMID: 21723171.
- Mukherjee N, Corcoran DL, Nusbaum JD, et al. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell. 2011 Aug 5;43(3):327–339. PubMed PMID: 21723170; PubMed Central PMCID: PMCPMC3220597.
- Izquierdo JM. Hu antigen R (HuR) functions as an alternative pre-mRNA splicing regulator of Fas apoptosis-promoting receptor on exon definition. J Biol Chem. 2008 Jul 4;283(27):19077–19084. . PubMed PMID: 18463097.
- Chang SH, Elemento O, Zhang J, et al. ELAVL1 regulates alternative splicing of eIF4E transporter to promote postnatal angiogenesis. Proc Natl Acad Sci U S A. 2014 Dec 23;111(51):18309–18314. PubMed PMID: 25422430; PubMed Central PMCID: PMCPMC4280608.
- Mazan-Mamczarz K, Galban S, Lopez de Silanes I, et al. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc Natl Acad Sci U S A. 2003 Jul 8;100(14):8354–8359. PubMed PMID: 12821781; PubMed Central PMCID: PMCPMC166233.
- Kullmann M, Gopfert U, Siewe B, et al. ELAV/Hu proteins inhibit p27 translation via an IRES element in the p27 5ʹUTR. Genes Dev. 2002 Dec 1;16(23):3087–3099. PubMed PMID: 12464637; PubMed Central PMCID: PMCPMC187493.
- Kawai T, Lal A, Yang X, et al. Translational control of cytochrome c by RNA-binding proteins TIA-1 and HuR. Mol Cell Biol. 2006 Apr;26(8):3295–3307. PubMed PMID: 16581801; PubMed Central PMCID: PMCPMC1446930.
- Lal A, Kawai T, Yang X, et al. Antiapoptotic function of RNA-binding protein HuR effected through prothymosin alpha. Embo J. 2005 May 18;24(10):1852–1862. PubMed PMID: 15861128; PubMed Central PMCID: PMCPMC1142594.
- Zhu H, Zhou HL, Hasman RA, et al. Hu proteins regulate polyadenylation by blocking sites containing U-rich sequences. J Biol Chem. 2007 Jan 26;282(4):2203–2210. PubMed PMID: 17127772.
- Dai W, Zhang G, Makeyev EV. RNA-binding protein HuR autoregulates its expression by promoting alternative polyadenylation site usage. Nucleic Acids Res. 2012 Jan;40(2):787–800. . PubMed PMID: 21948791; PubMed Central PMCID: PMCPMC3258158.
- Mukherjee N, Lager PJ, Friedersdorf MB, et al. Coordinated posttranscriptional mRNA population dynamics during T-cell activation. Mol Syst Biol. 2009;5:288. . PubMed PMID: 19638969; PubMed Central PMCID: PMCPMC2724974.
- Techasintana P, Davis JW, Gubin MM, et al. Transcriptomic-wide discovery of direct and indirect HuR RNA targets in activated CD4+ T cells. PLoS One. 2015;10(7):e0129321. . PubMed PMID: 26162078; PubMed Central PMCID: PMCPMC4498740.
- Blackinton JG, Keene JD. Functional coordination and HuR-mediated regulation of mRNA stability during T cell activation. Nucleic Acids Res. 2016 Jan 8;44(1):426–436. . PubMed PMID: 26490963; PubMed Central PMCID: PMCPMC4705648.
- Seko Y, Azmi H, Fariss R, et al. Selective cytoplasmic translocation of HuR and site-specific binding to the interleukin-2 mRNA are not sufficient for CD28-mediated stabilization of the mRNA. J Biol Chem. 2004 Aug 6;279(32):33359–33367. PubMed PMID: 15020598.
- Ma WJ, Cheng S, Campbell C, et al. Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J Biol Chem. 1996 Apr 5;271(14):8144–8151. PubMed PMID: 8626503.
- Raghavan A, Robison RL, McNabb J, et al. HuA and tristetraprolin are induced following T cell activation and display distinct but overlapping RNA binding specificities. J Biol Chem. 2001 Dec 21;276(51):47958–47965. PubMed PMID: 11602610.
- Vakalopoulou E, Schaack J, Shenk T. A 32-kilodalton protein binds to AU-rich domains in the 3ʹ untranslated regions of rapidly degraded mRNAs. Mol Cell Biol. 1991 Jun;11(6):3355–3364. PubMed PMID: 1903842; PubMed Central PMCID: PMCPMC360189.
- Myer VE, Fan XC, Steitz JA. Identification of HuR as a protein implicated in AUUUA-mediated mRNA decay. Embo J. 1997 Apr 15;16(8):2130–2139. . PubMed PMID: 9155038; PubMed Central PMCID: PMCPMC1169815.
- Ford LP, Watson J, Keene JD, et al. ELAV proteins stabilize deadenylated intermediates in a novel in vitro mRNA deadenylation/degradation system. Genes Dev. 1999 Jan 15;13(2):188–201. PubMed PMID: 9925643; PubMed Central PMCID: PMCPMC316394.
- Dean JL, Wait R, Mahtani KR, et al. The 3ʹ untranslated region of tumor necrosis factor alpha mRNA is a target of the mRNA-stabilizing factor HuR. Mol Cell Biol. 2001 Feb;21(3):721–730. PubMed PMID: 11154260; PubMed Central PMCID: PMCPMC86664.
- Casolaro V, Fang X, Tancowny B, et al. Posttranscriptional regulation of IL-13 in T cells: role of the RNA-binding protein HuR. J Allergy Clin Immunol. 2008 Apr;121(4):853–9 e4. PubMed PMID: 18279945; PubMed Central PMCID: PMCPMC2666917.
- Wang JG, Collinge M, Ramgolam V, et al. LFA-1-dependent HuR nuclear export and cytokine mRNA stabilization in T cell activation. J Immunol. 2006 Feb 15;176(4):2105–2113. PubMed PMID: 16455966.
- Fan XC, Steitz JA. Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. Embo J. 1998 Jun 15;17(12):3448–3460. . PubMed PMID: 9628880; PubMed Central PMCID: PMCPMC1170681.
- Ming XF, Stoecklin G, Lu M, et al. Parallel and independent regulation of interleukin-3 mRNA turnover by phosphatidylinositol 3-kinase and p38 mitogen-activated protein kinase. Mol Cell Biol. 2001 Sep;21(17):5778–5789. PubMed PMID: 11486017; PubMed Central PMCID: PMCPMC87297.
- Estep JA, Sternburg EL, Sanchez GA, et al. Immunoblot screening of CRISPR/Cas9-mediated gene knockouts without selection. BMC Mol Biol. 2016 Apr 2;17:9. 10.1186/s12867-016-0061-0. PubMed PMID: 27038923; PubMed Central PMCID: PMCPMC4818936.
- Li Y, Estep JA, Karginov FV. Transcriptome-wide identification and validation of interactions between the miRNA machinery and HuR on mRNA targets. J Mol Biol. 2018 Feb 2;430(3):285–296. PubMed PMID: 29273203.
- Liao W, Lin JX, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity. 2013 Jan 24;38(1):13–25. PubMed PMID: 23352221; PubMed Central PMCID: PMCPMC3610532.
- Macian F, Garcia-Rodriguez C, Rao A. Gene expression elicited by NFAT in the presence or absence of cooperative recruitment of Fos and Jun. Embo J. 2000 Sep 1;19(17):4783–4795. PubMed PMID: 10970869; PubMed Central PMCID: PMCPMC302068.
- Kim HS, Wilce MC, Yoga YM, et al. Different modes of interaction by TIAR and HuR with target RNA and DNA. Nucleic Acids Res. 2011 Feb;39(3):1117–1130. PubMed PMID: 21233170; PubMed Central PMCID: PMCPMC3035456.
- Muller MR, Rao A. NFAT, immunity and cancer: a transcription factor comes of age. Nat Rev Immunol. 2010 Sep;10(9):645–656. PubMed PMID: 20725108.
- Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005 Jun;5(6):472–484. PubMed PMID: 15928679.
- Abdelmohsen K, Lal A, Kim HH, et al. Posttranscriptional orchestration of an anti-apoptotic program by HuR. Cell Cycle. 2007 Jun 1;6(11):1288–1292. PubMed PMID: 17534146.
- Jimbo M, Blanco FF, Huang YH, et al. Targeting the mRNA-binding protein HuR impairs malignant characteristics of pancreatic ductal adenocarcinoma cells. Oncotarget. 2015 Sep 29;6(29):27312–27331. PubMed PMID: 26314962; PubMed Central PMCID: PMCPMC4694992.
- Lal S, Cheung EC, Zarei M, et al. CRISPR knockout of the HuR gene causes a xenograft lethal phenotype. Mol Cancer Res. 2017 Jun;15(6):696–707. 10.1158/1541-7786.MCR-16-0361. PubMed PMID: 28242812; PubMed Central PMCID: PMCPMC5466444.
- Gonzalez-Feliciano JA, Hernandez-Perez M, Estrella LA, et al. The role of HuR in the post-transcriptional regulation of interleukin-3 in T cells. PLoS One. 2014;9(3):e92457. . PubMed PMID: 24658545; PubMed Central PMCID: PMCPMC3962401.
- Meisner NC, Hackermuller J, Uhl V, et al. mRNA openers and closers: modulating AU-rich element-controlled mRNA stability by a molecular switch in mRNA secondary structure. Chembiochem. 2004 Oct 4;5(10):1432–1447. PubMed PMID: 15457527.
- Ramgolam VS, DeGregorio SD, Rao GK, et al. T cell LFA-1 engagement induces HuR-dependent cytokine mRNA stabilization through a Vav-1, Rac1/2, p38MAPK and MKK3 signaling cascade. PLoS One. 2010 Dec 29;5(12):e14450. PubMed PMID: 21206905; PubMed Central PMCID: PMCPMC3012057.
- Peng SS, Chen CY, Xu N, et al. RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. Embo J. 1998 Jun 15;17(12):3461–3470. PubMed PMID: 9628881; PubMed Central PMCID: PMCPMC1170682.
- Levy NS, Chung S, Furneaux H, et al. Hypoxic stabilization of vascular endothelial growth factor mRNA by the RNA-binding protein HuR. J Biol Chem. 1998 Mar 13;273(11):6417–6423. PubMed PMID: 9497373.
- Wang W, Furneaux H, Cheng H, et al. HuR regulates p21 mRNA stabilization by UV light. Mol Cell Biol. 2000 Feb;20(3):760–769. PubMed PMID: 10629032; PubMed Central PMCID: PMCPMC85192.
- Wang W, Caldwell MC, Lin S, et al. HuR regulates cyclin A and cyclin B1 mRNA stability during cell proliferation. Embo J. 2000 May 15;19(10):2340–2350. PubMed PMID: 10811625; PubMed Central PMCID: PMCPMC384372.
- Raineri I, Wegmueller D, Gross B, et al. Roles of AUF1 isoforms, HuR and BRF1 in ARE-dependent mRNA turnover studied by RNA interference. Nucleic Acids Res. 2004;32(4):1279–1288. . PubMed PMID: 14976220; PubMed Central PMCID: PMCPMC390274.
- Stellato C, Gubin MM, Magee JD, et al. Coordinate regulation of GATA-3 and Th2 cytokine gene expression by the RNA-binding protein HuR. J Immunol. 2011 Jul 1;187(1):441–449. PubMed PMID: 21613615; PubMed Central PMCID: PMCPMC5801757.
- Chen CY, Xu N, Shyu AB. Highly selective actions of HuR in antagonizing AU-rich element-mediated mRNA destabilization. Mol Cell Biol. 2002 Oct;22(20):7268–7278. PubMed PMID: 12242302; PubMed Central PMCID: PMCPMC139819.
- Shim J, Lim H, Yates JR, et al. Nuclear export of NF90 is required for interleukin-2 mRNA stabilization. Mol Cell. 2002 Dec;10(6):1331–1344. PubMed PMID: 12504009.
- Brauss TF, Winslow S, Lampe S, et al. The RNA-binding protein HuR inhibits expression of CCL5 and limits recruitment of macrophages into tumors. Mol Carcinog. 2017 Dec;56(12):2620–2629. PubMed PMID: 28731284.
- Backman TWH, Girke T. systemPipeR: NGS workflow and report generation environment. BMC Bioinformatics. 2016 Sep 20;17:388. 10.1186/s12859-016-1241-0. PubMed PMID: 27650223; PubMed Central PMCID: PMCPMC5029110.
- Kim D, Pertea G, Trapnell C, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013 Apr 25;14(4):R36. PubMed PMID: 23618408; PubMed Central PMCID: PMCPMC4053844.
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012 Mar 4;9(4):357–359. . PubMed PMID: 22388286; PubMed Central PMCID: PMCPMC3322381.
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. . PubMed PMID: 25516281; PubMed Central PMCID: PMCPMC4302049.
- Anders S, Reyes A, Huber W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012 Oct;22(10):2008–2017. . PubMed PMID: 22722343; PubMed Central PMCID: PMCPMC3460195.
- Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007 Jan 15;23(2):257–258. . PubMed PMID: 17098774.
- Yu G, Wang LG, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012 May;16(5):284–287. PubMed PMID: 22455463; PubMed Central PMCID: PMCPMC3339379.