
ABSTRACT
Prp16 is a DEAH box pre-mRNA splicing factor that triggers a key spliceosome conformational switch to facilitate second step splicing in Saccharomyces cerevisiae. However, Prp16 functions are largely unexplored in Schizosaccharomyces pombe, an attractive model with exon-intron architecture more relevant to several other eukaryotes. Here, we generated mis-sense alleles in SpPrp16 whose consequences on genome-wide splicing uncover its nearly global splicing role with only a small subset of unaffected introns. Prp16 dependent and independent intron categories displayed a striking difference in the strength of intronic 5ʹ splice site (5’SS)-U6 snRNA and branch site (BS)-U2 snRNA interactions. Selective weakening of these interactions could convert a Prp16 dependent intron into an independent one. These results point to the role of SpPrp16 in destabilizing 5’SS-U6snRNA and BS-U2snRNA interactions which plausibly trigger structural alterations in the spliceosome to facilitate first step catalysis. Our data suggest that SpPrp16 interactions with early acting factors, its enzymatic activities and association with intronic elements collectively account for efficient and accurate first step catalysis. In addition to splicing derangements in the spprp16F528S mutant, we show that SpPrp16 influences cell cycle progression and centromeric heterochromatinization. We propose that strong 5’SS-U6 snRNA and BS-U2 snRNA complementarity of intron-like elements in non-coding RNAs which lead to complete splicing arrest and impaired Seb1 functions at the pericentromeric loci may cumulatively account for the heterochromatin defects in spprp16F528S cells. These findings suggest that the diverse Prp16 functions within a genome are likely governed by its intronic features that influence splice site–snRNA interaction strength.
Introduction
Pre-mRNA splicing is a central eukaryotic RNA processing event which contributes to a cell’s functional transcriptome. Splicing occurs by the recognition and removal of introns through the concerted action of over 150 conserved proteins and five snRNAs (U1, U2, U4, U5 and U6) that associate to form the spliceosome [Citation1]. The recognition of key intronic elements like the 5′ splice site (5′SS), branch site (BS) and 3′ splice site (3′SS) by multiple spliceosomal factors is critical for the alignment of splice-sites for the two transesterification reactions that generate mature spliced mRNA. During splicing, the spliceosome undergoes several compositional and structural changes resulting in formation and disruption of various RNA-protein, RNA-RNA and protein–protein interactions mediated by non-snRNP ATP dependent DExD/H box helicases [Citation2]. Biochemical and genetic studies in budding yeast Saccharomyces cerevisiae demonstrated functions for eight such helicases at distinct steps in spliceosome assembly, catalysis and disassembly [Citation3]. The DEAD box proteins Prp5 and Prp28 function in spliceosome assembly [Citation4–Citation6]. Prp2 coalesces with the activated assembled spliceosome (Bact complex), destabilizes early assembly factors and promotes first step catalysis by generating binding sites for first step factors Cwc25 and Yju2 [Citation7,Citation8]. After the first step cleavage reaction, Prp16 triggers a conformational change in the spliceosome to dissociate Cwc25 and Yju2 thereby allowing for the binding of second step factors [Citation9,Citation10]. Prp16 also destabilizes the U2-U6 helix I to reconfigure the catalytic centre for second step catalysis after 5′SS cleavage by displacing NTC components Cwc2 and Isy1 [Citation11–Citation13]. Prp22 helicase promotes exon-exon ligation and release of spliced mRNA from the spliceosome [Citation14]. Brr2 activity is necessary for both spliceosome activation (Bact) and disassembly [Citation15,Citation16].
The ATPase activity of these helicases in budding yeast ensures splicing fidelity by preventing substrates with suboptimal cis elements from undergoing productive splicing [Citation17–Citation19]. Prp28 and Prp5 scrutinize the strength of 5′SS-U6 snRNA and BS-U2 snRNA interactions, respectively, during early spliceosome assembly [Citation5,Citation20–Citation22]. Prp2 proofreads the catalytic core before first step catalysis, Prp16 ensures branch site fidelity and Prp22 scrutinizes 3′SS fidelity during second step exon ligation [Citation23–Citation25]. In case of substrates with suboptimal splice sites, the ATPase activities of these DExD/DExH proteins trigger a conformational change in spliceosomes prior to the occurrence of an upstream event. This untimely switch due to kinetically slow progression of sub-optimal substrates through an earlier event leads to splicing failure [Citation9,Citation23,Citation25]. In the normal splicing context, S. cerevisiae Prp16 binds to spliceosomes only after the first step catalysis [Citation26]. However, two recent studies report an earlier spliceosomal association of Prp16 can occur before first step splicing in substrates with invariant branch residue A to C mutation and also when substrates have suboptimal 5′SS which undergo slow first step cleavage [Citation9,Citation27]. ATP hydrolysis by Prp16 rejects these substrates to a Prp43 dependent discard pathway [Citation25,Citation27]. In the normal splicing context, Prp43 promotes spliceosome disassembly on completion of an error-free splicing cycle [Citation28]. Additionally, in substrates with sub-optimal splice-sites S. cerevisiae Prp16 and Prp22 enable the utilization of alternate BS and 3’SS, respectively [Citation29].
Schizosaccharomyces pombe (fission yeast) splicing factors share higher sequence similarity to homologues in mammals and other eukaryotes than budding yeast S. cerevisiae factors [Citation30,Citation31]. Further, its genome encodes transcripts with multiple short introns (average length 89 nucleotides) and degenerate splice signals as in genomes of other fungi, Drosophila melanogaster, Caenorhabditis elegans, protozoan parasites and plants [Citation32]. Therefore, although our understanding of pre-mRNA splicing was pioneered in S. cerevisiae, features of S. pombe make it a promising model to understand splicing mechanisms relevant to many other unicellular and multicellular organisms. Prior studies from our laboratory on some fission yeast splicing factors homologous to budding yeast second step factors, indicate that in S. pombe their functions and interactions are important for first step catalysis [Citation33,Citation34]. These underscore the co-evolution of spliceosome assembly and splice-site recognition mechanisms with changes in exon-intron architectures. Among splicing factors that are ATP dependent RNA helicases, a recent study indicates that fission yeast SpPrp22 and SpPrp43 act to release spliced products, in an unconventional intron of the non-coding telomeric RNA [Citation35]. Thus, these helicases serve a conserved function in late post-catalytic spliceosomes as observed for their budding yeast counterparts. However, the functions of other S. pombe ATP dependent RNA helicases during spliceosome assembly, activation and catalysis are largely unexplored.
Here, we generated mis-sense mutants in the helicase domain of fission yeast SpPrp16 and uncovered its nearly global splicing functions through transcriptome analysis. We deduce that the strength of 5’SS-U6 snRNA and BS-U2 snRNA interactions are intronic risk factors that together govern dependence on SpPrp16 for splicing. Further, our genetic studies provide insights on its interactions in the activated spliceosome. Interestingly, we find a role for SpPrp16 in cell cycle progression and in heterochromatinization of centromeres and telomeres. Based on our results presented here, we infer that the strength of these splice sites – snRNA interactions in genome-wide transcripts determines whether the major Prp16 function in that organism is in splicing and/or gene silencing.
Results
The carboxyl-terminal of Prp16 is highly conserved and a mis-sense allele at SpPrp16 C-terminal abrogates splicing of several cellular transcripts
Schizosaccharomyces pombe Prp16 (SpPrp16) is an essential DExD/H box splicing factor [Citation36] that shares significant conservation at the C-terminal domain with its human (69.2% similarity) and budding yeast homologues (69.4% similarity) (Supplementary Figure S1(a)). Comparative modelling of SpPrp16 with ScPrp43 confirm its structural similarity to DEAH splicing factors having signatory walker motifs that perform ATP binding, hydrolysis and RNA duplex unwinding (Supplementary Figure S1(b)). The pre-mRNA splicing role of SpPrp16 is however unexplored. To gain functional insights on SpPrp16, we tested the consequence of mis-sense mutants in the residues G515 and F528 homologous to S. cerevisiae G373 and Y386 residues, as mutants in these residues were instrumental in understanding of S. cerevisiae Prp16 functions [Citation23,Citation37] (Supplementary Figure S1(a)). Mutants in SpPrp16 were generated by random substitutions at each of these residues followed by the integration of the mutant alleles at the leu1 locus of a strain where the endogenous, essential prp16+ locus was disrupted (Supplementary Figure S1(c)) and was supported by expressing the wild-type protein from a plasmid (pREP4Xspprp16+). Replacements, in prp16G515 and in prp16F528 that are expressed from the leu1 locus and sustain viability of cells after eviction of the pREP4Xspprp16+ plasmid were selected. Sequence analysis of prp16 allele that was integrated at the leu1 locus was determined and we identified spprp16G515A, spprp16F528S and spprp16F528H mis-sense alleles (). The spprp16F528S and spprp16F528H cells were slow growing at all temperatures as compared to a control strain where the wild-type spprp16+allele was expressed from the leu1 locus. The spprp16F528S mutant was notably cold sensitive at 23°C. The spprp16G515A mutant growth was comparable to the wild type at all three temperatures (). To gain further insights on SpPrp16 function in fission yeast, we exploited the two mis-sense mutants spprp16F528S and spprp16G515A, with differing growth phenotypes.
Figure 1. spprp16F528S mutant affects splicing of introns with diverse features. (a) Growth kinetics of wild-type (WT) and mutants (F528S, F528H and G515A) on EMM complete media at 23°C, 30°C and 37°C analysed by spotting 10-fold serial dilutions of cultures grown to OD595 of 0.7. Schematic representation of the wild-type (leu1:spprp16+) and mutant strains (leu1:spprp16) generated is shown. (b) Semi-quantitative RT-PCRs showing the splicing of tfIId+I1 in WT, spprp16G515A and spprp16F528S cells grown at 30°C. Comparison of tfIId+I1 splicing in the WT and spprp16F528S mutant at 30°C and 23°C is also shown. (C and D) Semi-quantitative RT-PCRs showing the splicing of mdm35+I1 (c) and nda3+I4 (d) in WT, spprp16G515A and spprp16F528S cells grown at 30°C. Reverse transcription for each transcript was done with the reverse primer (RP) corresponding to the downstream exon. PCR on the cDNA was performed with the same RP in combination with upstream exonic forward primer (FP). RT-PCR on the intronless actin transcript (act1+) served as normalization control. P- pre-mRNA, M – mRNA and g. DNA – genomic DNA control as indicated. Bar graphs represent data from three biological replicates. In this and other quantitative assays, the statistical significance was determined by unpaired student’s t-test . *** = p < 0.001, ** p < 0.005, * = p < 0.05 and ns = non-significant.
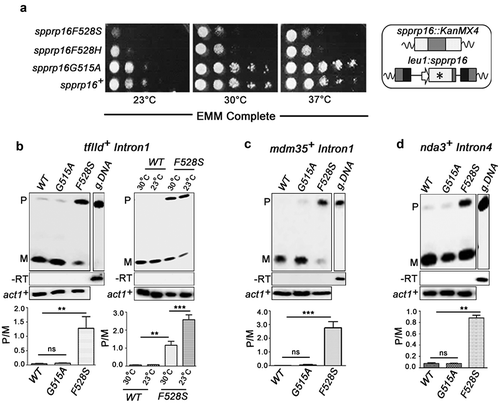
We first tested a few cellular transcripts for the splicing of introns with diverse lengths and splice site consensus elements – tfIId+Intron1 (tfIID+I1), mdm35+Intron1 (mdm35+I1) and nda3+Intron4 (nda3+I4) (), Supplementary Table S2; for intron characteristics). All these introns were poorly spliced in spprp16F528S even in cells grown at the optimal temperature 30°C. The tfIId+Intron1 splicing phenotype was aggravated at 23°C agreeing with the cold sensitivity of spprp16F528S (, right panel). In wild-type cells, splicing was efficient at both temperatures. In the spprp16G515A mutant all three introns were spliced normally ().
Genome-wide analysis of the dependency of introns on SpPrp16 for splicing
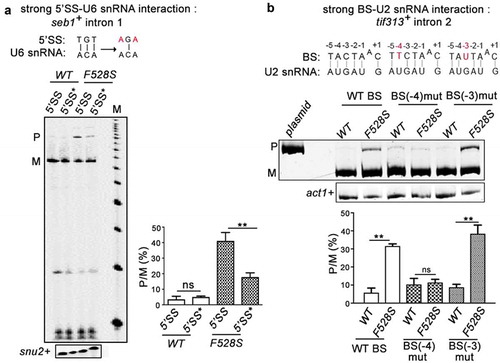
To assess the genome-wide dependency of introns on SpPrp16, a global splicing profile was generated by deep sequencing of RNA (RNA-seq) from spprp16+ and spprp16F528S strains grown at 30°C; 96% of the obtained reads which aligned to the S. pombe genome were mapped to a virtual dataset of 70 bp sequence stretches representing all S. pombe 5′exon-intron (EI) and exon-exon (EE) junctions (see Materials and Methods for filters applied). The splice index (SI) of each intron was computed as log2 (EE/EI) where EE represents reads from exon-exon junctions and EI represents reads from 5ʹexon-intron junctions. Of the total 5,122 introns analysed, 4,040 were statistically significant between the two biological replicates. The differential SI was calculated as the ratio of wild-type SI by mutant SI. Splicing of 3,853 introns were affected at differential SI cut-off of 1.5 fold while splicing of 187 introns were unaffected in the spprp16F528S mutant (; Dataset S1). The transcriptome sequencing data was validated by semi-quantitative RT-PCR for representative introns from the SpPrp16 dependent (seb1+I1 and dga1+ I2) and independent (dga1+ I3) categories (Supplementary Table S2). Consistent with the RNA-seq data set, splicing of seb1+I1 was severely impaired while dga1+I3 was efficiently spliced in spprp16F528S cells (). Multi-intron containing transcripts like dga1+ (dga1+I3 and dga1+I2; Supplementary Table S2) had introns with contrasting splicing phenotypes in the spprp16F528S mutant (, compare the middle and right panels). Such intron-specific differential splicing factor dependence is also reported in other fission yeast splicing factor mutants [Citation33,Citation34,Citation38,Citation39] suggesting that individual intronic features determine dependency on a splicing factor. Integrative genomics viewer representation of RNA seq reads spanning the exon-intron-exon regions of these candidate introns are shown (Supplementary Figure S3). Reads that correspond to the intronic region of seb1+I1 and dga1+ I1 and I2 were enriched in the sprp16F528S mutant when compared to the reads in wild type cells. On the other hand, for reads mapping to intron 3 of the dga1+ transcript, the variation between the wild type and mutant cells was not statistically significant. We analysed for global intronic signatures that distinguish SpPrp16 dependent and independent intron categories. No remarkable difference was observed with respect to sequence consensus of 5′SS, polypyrimidine tract, BS and 3′SS. Other general features like intron length, AU content, 5′ SS to BS and BS to 3′SS distances were also similar between the two categories (Supplementary Figure S2(a-f)). However, analysis of the log odds ratio revealed the occurrence of a rare nucleotide at the +4 position of 5’SS in the SpPrp16 dependent intron category (Supplementary Table S3). We also analysed the U6 snRNA-5′SS and U2 snRNA-BS base pairing interactions as they are known to be critical in the formation of the catalytic centre for first step splicing [Citation2,Citation13,Citation40] (). The +4, +5 and +6 nucleotides of the 5′SS in SpPrp16 dependent and independent introns differed in the strength of interaction with the – ACA invariant residues of U6 snRNA ACAGAGA box. A larger percentage of SpPrp16 dependent introns had complete 5’SS-U6 snRNA complementarity for all three base-pairs (+ + +). Conversely, loss of complementarity at these positions (– – –) was relatively enriched in the independent intron category (). To validate this correlation, we tested whether weakening the 5’SS-U6 interaction can bypass the requirement of SpPrp16 for splicing. Primer extension was done on RNA from spprp16+ wild type and spprp16F528S mutant cells transformed with pDBlet seb1 E1-I1-E2 minigene construct with the wild type 5’SS sequence (TGT at +4 to +6 positions) and another construct where TGT was mutated to AGA to weaken the intronic interaction with U6 snRNA. In agreement with the prediction that the strength of 5’SS-U6 snRNA interaction could be a feature linking splicing efficiency of an intron with SpPrp16 activity, we observe that the poor splicing of seb1+ intron1 seen in cells of the spprp16F528S mutant is partially rescued on mutating the 5’SS in this intron (). The partial rescue of splicing defects instigated us to check whether the complementarity between BS and U2 snRNA could be an additional determinant contributing to SpPrp16 requirement for splicing. Interestingly, Principal Component Analysis (PCA) of intron subsets indicate a significant correlation between the strength of 5’SS-U6 snRNA interaction and BS-U2 snRNA complementarity for SpPrp16 independent introns ( and S2(g-i)). The lack of 5’SS-U6 interactions at all three 5’SS positions +4, +5 and +6 (– – –) was associated with weakened BS-U2 snRNA complementarity at −3 and −1 residues of the BS. Other SpPrp16 independent introns whose 5ʹ SS +4 and +5 residues (– – +) cannot base-pair with U6 had potentially weakened BS-U2 pairing based on nucleotide variations at BS −4 and +1 positions. Also, the absence of complementarity at +4 position (– + +) of 5’SS in certain introns co-occurred with loss of complementarity at −3 residue of BS. These results suggest that weakened 5’SS-U6 and BS-U2 interactions can cumulatively pre-dispose these introns to be spliced independent of SpPrp16 activity. To validate these observations, we chose another candidate intron tif313+ intron 2 as an example of an intron with complete BS-U2 snRNA complementarity, very weak 5’SS-U6 snRNA base-pairing strength and where transcriptome data indicate dependence on SpPrp16 for splicing. These features make it an appropriate candidate to test the individual contribution of BS-U2 snRNA interaction strength and requirement for SpPrp16 activity. The splicing of the mini-gene transcript comprising tif313 exon2-intron2-exon3 with wild type BS, or two other mini-genes mutated either at the BS −4 (A to T) or at the −3 position (C to U) were assessed. These mutations reduce its BS complementarity with U2 snRNA. All three mini-transcripts were tested in wild-type and in spprp16F528S mutant cells. These mutations reduce this intron’s BS complementarity with U2 snRNA. The poor splicing of the wild type tif313+ intron 2 mini-transcript in spprp16F528S cells as compared to wild-type cells confirmed that splicing of this intron is dependent on SpPrp16. The BS −4 mutant mini-transcript was rendered as an efficiently spliced substrate even in spprp16F528S cells. However, the splicing inefficiency of tif313 intron 2 was exacerbated when the mini-transcript had BS mutation at the −3 residue as indicated by increased levels of unspliced pre-mRNA even in spprp16+ wild-type cells wherein it was efficiently spliced otherwise (). These results suggest that the −4 residue of tif313+ intron 2 BS plays a critical role in its interaction with snRNA and its destabilization requires Prp16 function. These data are supported by the co-relations we observed between the splicing efficiencies of introns within other cellular transcripts with natural sequence variations influence their 5’SS-U6 snRNA or BS-U2 snRNA interactions (Supplementary tables S4A and B). We validated this observation by semi-quantitative RT-PCR to examine splicing efficiency of introns. These data on sec6102+ intron 3 and intron 5 splicing demonstrate that their respective SpPrp16 dependence and independence correlates with variations in their 5’SS-U6 snRNA strength (Supplementary Figure S4(a)). Similarly, the SpPrp16 dependent splicing of intron 4 in apl5+ is correlated with strong BS-U2 snRNA interaction, whereas intron 2 splicing in the same transcript is SpPrp16 independent and is correlated with weaker BS-U2 snRNA interactions (Supplementary Figure S4(b)).
Figure 2. SpPrp16 has nearly ubiquitous splicing functions. (a) Frequency histogram representing the number of introns plotted against their respective log2 differential splice index (SI). Introns with log2 differential SI ≥ 1 (i.e., ≥2 fold difference between the wild-type and mutant) are considered strongly dependent on SpPrp16 for splicing and those with log2 differential SI < 0.6 (i.e. <1.5 fold difference between the wild-type and mutant) are considered unaffected in the spprp16F528S mutant. The introns with marginally affected differential SI values (log2 differential splice index between 0.6 and 1) were excluded from this representation. (b) Semi-quantitative RT-PCR validation of the transcriptome-based splicing phenotype at 30°C for SpPrp16 dependent seb1+ intron1 (left panel), dga1+ intron2 (middle panel) and independent intron dga1+ intron3 (right panel). (c) Schematic of a S. pombe transcript with global sequence consensus for the intronic 5’SS, BS and 3’SS elements. Base-pairing of the 5’SS and the BS with U6 snRNA and U2 snRNA, respectively, are indicated by dotted lines. (d) Graph representing the number of introns from SpPrp16 dependent and independent classes categorized based on the complementarity of 5’SS +4, +5 and +6 nucleotides with the U6 snRNA sequence – ACA. Base pairing at each position is denoted by a ‘+’sign and no base pairing by ‘–’ sign. The number of introns with complete complementarity indicated as ‘+++’ is significantly different between the dependent and independent intron categories at p = 0.03. (e) Principal Component Analysis (PCA) showing variance between the number of dependent and independent introns (Y axis) with loss of BS-U2 snRNA base-pairing for each indicated BS residue (X axis). The lines (Z axis) represent data for intron subsets with varying 5’SS-U6 snRNA strength. Green (– – –), red (– – +) and blue (– + +) lines indicates three intron subsets categorized based on 5’SS-U6 complementarity at the 5’SS +4, +5 and +6 positions. The invariant branch residue A was positioned as ‘0’ and other BS nucleotides are numbered with respect to this residue.
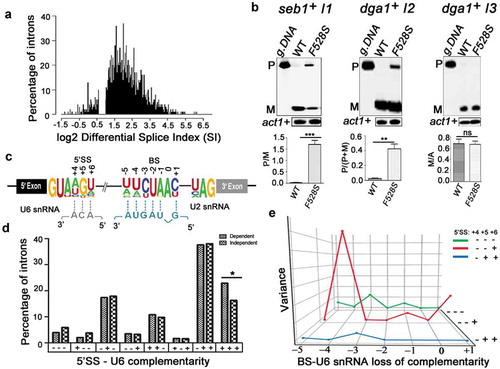
Figure 3. SpPrp16 destabilizes 5’SS-U6 snRNA and BS-U2 snRNA interactions. (a) Primer extension to assess the splicing of seb1+ E1-I1-E2 mini-transcripts having wild-type or mutant 5’SS (depicted as 5’SS and 5’SS* respectively) in the WT (leu1:spprp16+) and F528S (leu1:spprp16F528S) mutant cells. The mutations introduced in the 5’SS (highlighted in red) and positions which can base-pair with U6 snRNA are shown (black lines). Primer extension on snu2+ transcripts served as the loading control. Lane M- 100 nts to 1000 nts DNA size marker (b). Semi-quantitative RT-PCRs to analyse the splicing of tif313+ intron2 in mini-transcripts comprising exon2-intron2-exon3 with wild-type or mutant branch site (BS) in the WT (leu1:spprp16+) and F528S (leu1:spprp16F528S) mutant strains. The mutations introduced (highlighted in red) and the positions that can base-pair with U2 snRNA are shown (black lines).
SpPrp16 facilitates first step splicing catalysis
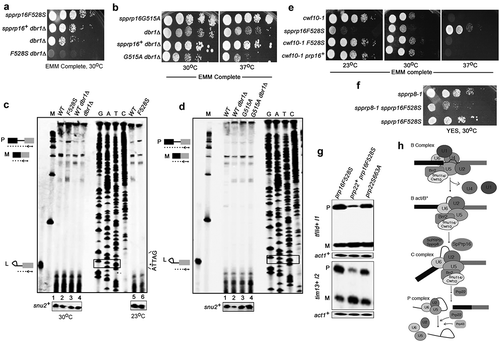
The budding yeast scprp16 mutants (prp16-1, prp16-2, prp16-201 to prp16-204 and prp16-302 alleles) largely accumulate lariat intron-3′exon splicing intermediates indicating a second step splicing arrest or inefficient transition of these mutant spliceosomes from first to second step catalytic confirmation [Citation23,Citation41–Citation43]. To assess if equivalent mutants in the fission yeast SpPrp16 protein also allowed first step catalysis, we took up primer extension assays. Further, as the inactivation of lariat debranching enzyme dbr1+ could stabilize the lariat intron-3′exon RNA species by impeding its predicted rapid turnover [Citation44], we generated the fission yeast double mutants spprp16F528S dbr1Δ and spprp16G5151A dbr1Δ to carry out growth kinetics and splicing analyses in these strains. The spprp16F528S dbr1Δ double mutant spores were extremely poor growing indicating a synthetic lethal interaction (, bottom row) while the spprp16G515A dbr1Δ double mutant was viable and mildly temperature sensitive at 37°C (, bottom row). Primer extension assays were done on RNA from spprp16+, spprp16F528S, spprp16+dbr1Δ and dbr1Δ strains to assess levels of pre-mRNA, mRNA and lariat intron-3′exon species from tfIId+I1 splicing. spprp16F528S cells had high pre-mRNA and reduced spliced mRNA levels, i.e. ~15% as compared to mRNA in the wild-type prp16+ strain (, compare lane 2 with 1). As expected, splicing was unaffected in the control strains spprp16+dbr1Δ and dbr1Δ (, lanes 3 and 4). Interestingly, lariat intron-3′exon species was not detected in the spprp16F528S mutant grown at 30°C or even in cells grown at 23°C (, lanes 5 and 6). These data indicate a predominant arrest prior to first step catalysis in the spprp16F528S mutant. However, the corresponding mutant in S. cerevisiae, prp16-1, showed high levels of lariat intron-3′exon and precursor mRNA marking a strong arrest after first step catalysis and failure of only the second reaction [Citation41]. Primer extension assays in the spprp16G515A dbr1Δ double mutant strain showed neither first nor second step splicing defects and splicing efficiency was comparable to spprp16G515A single mutant and control strains ().
Figure 4. SpPrp16 facilitates first step splicing catalysis. (a and b) Genetic interaction of spprp16F528S and spprp16G515A with dbr1Δ. Comparative growth profile of double mutants with respective single mutants at the indicated temperatures. (c) Primer extensions to estimate the levels of pre-mRNA, mRNA and lariat-exon2 species from tfIId+I1 splicing in spprp16F528S and appropriate control strains grown at 30°C and 23°C. The schematic to the left marks the expected position of extension products from pre-mRNA (P), mRNA (M) and lariat exon-2 (L) species. RNA from WT (leu1:spprp16+), F528S (leu1:spprp16F528S), WTdbr1 (leu1:spprp16+ dbr1Δ) and dbr1Δ strains grown at 30°C were used in lanes 1 to 4. For lanes 5 and 6, RNA from WT and F528S cultures grown at 23°C was the input. The DNA sequencing ladder (lanes GATC) was prepared using the tfIId exon2 RP on pDBlet tfIId E1-I1-E2 plasmid as the template. ATTAG is the reverse complement of BS sequence and asterisk marks the position for invariant branch residue A. Lane M indicates DNA size markers in the range of 100–1000 nts. (d) Primer extension reactions to assess tfIId+intron1 splicing in the spprp16G515A dbr1Δ double mutant grown at 37°C as compared to the single mutant and wild-type control strains. RNA from WT (leu1:spprp16+), WT dbr1Δ (leu1:prp16+ dbr1Δ), G515A (leu1:spprp16G515A) and G515A dbr1Δ (leu1:spprp16G515A dbr1Δ) were assessed in lanes 1 to 4. (E and F) Genetic interaction of spprp16F528S with splicing factor mutants cwf10-1 (e) and spprp8-1 (f). Growth of respective double mutants was compared with single mutant strains at the indicated temperatures. Ten-fold serial dilutions of each culture from an initial inoculum grown to equal OD595 were spotted. (g) Semi-quantitative RT-PCR assessment of tfIId+I1 and tim13+I2 splicing in spprp16F528S cells that overexpress spprp22+. (h) The proposed juncture of SpPrp16 function in the splicing pathway based on the genetic interaction of spprp16F528S with other splicing factor mutants.
SpPrp16 shows strong genetic interactions with other pre-catalytic splicing factors
We used genetic approaches to understand fission yeast SpPrp16 functions in spliceosome assembly and its catalytic transitions that lead to first and subsequently second step splicing reactions. Genetic interactions of spprp16F528S with conditional mutants in other essential splicing factors were tested. Cwf10 is an U5 snRNP GTPase and studies with the fission yeast N-terminal deletion mutant cwf10-ΔNTE, suggested its role in spliceosome activation as was already known for Snu114, its S. cerevisiae orthologue [Citation45,Citation46]. We generated the fission yeast double mutant of cwf10-1 (temperature sensitive allele in the guanosine triphosphate-binding domain of Cwf10 – C323Y [Citation47],) and spprp16F528S and found that the cold sensitivity of the spprp16F528S allele at 23°C was robustly suppressed in the double mutant. However, temperature sensitivity of cwf10-1 cells remained unsuppressed in the double mutant (). The retention of cwf10-1 temperature sensitivity while rescue of spprp16F528S cold sensitivity in the double mutant hints at SpPrp16 functions in the activated spliceosome, downstream to Cwf10 action. SpPrp8 is the orthologue of S. cerevisiae Prp2, an ATP dependent RNA helicase that remodels the catalytic centre of the activated spliceosome to promote the first splicing reaction [Citation48]. The synthetic sickness of the spprp16F528S spprp8-1 double mutant suggests a role for SpPrp16 probably at the juncture of Bact to C transition (). As the activity of the splicing helicase Prp22 is required for spliceosome disassembly [Citation49,Citation50] we tested whether overexpression of fission yeast Prp22 could improve splicing in spprp16F528S cells. The pBG1 plasmid expressing Prp22 under its endogenous promoter [Citation35] was transformed into spprp16F528S and control spprp16+ cells. A partial rescue of tfIId+ I1 and tim13+ I2 splicing defects was indeed observed in spprp16F528S cells that overexpress wild-type spprp22+ (). The suppression of the splicing defect was not noted if catalytically inactive prp22S663A was expressed in spprp16F528S cells. Thus, active Prp22 could enhance recycling of stalled spliceosome complexes in spprp16F528S. Alternatively, a possibility that cannot be ruled out is partial complementation of the loss of SpPrp16 function by SpPrp22. Together, our results imply that the spprp16F528S allele causes strong splicing defects with arrest of spliceosome complexes prior to the first catalytic reaction ().
SpPrp16 interactions with intronic branch nucleotide mutations
Budding yeast Prp16 helicase contributes to splicing fidelity and several ATPase defective scprp16 mutants can suppress the splicing arrest of ACT1 substrates with branch-site mutations [Citation9,Citation23,Citation41]. This instigated us to examine interactions of S. pombe Prp16 wild type and mutant proteins with substrates bearing mutations in the intronic branch nucleotide. In budding yeast, splicing of substrates with the ACT1 BS mutation A259C was arrested at or prior to the first transesterification reaction, while A259G mutant substrate was specifically compromised for second step splicing [Citation23,Citation51]. To test the effects of similar BS substitutions in a fission yeast substrate we created plasmids expressing tfIID E1-I1-E2eGFP mini-transcripts with the wild-type branch residue A (A441) or plasmids with its variants A441C (Br-C) or A441G (Br-G). These were transformed into wild-type, spprp16F528S and spprp16G515A strains () and primer extension assays were done to assess splicing of wild-type and mutant mini-transcripts. The wild-type mini-transcript was spliced normally in both spprp16G515A mutant and wild-type cells (, lanes 1 and 5) while splicing of the Br-C substrate was moderately affected in these cells (, lanes 2 and 6). In spprp16F528S mutant the splicing of wild-type mini-transcript itself was inefficient (, compare lane 3 with lanes 1 and 5) and the defects were further exacerbated for the Br-C mutant substrate (, compare lanes 3 and 4) indicating an additive effect. This observation contrasts the findings that budding yeast scprp16-1 allele can suppress the first step arrest of a substrate with Br-C mutation. Intriguingly, the splicing of Br-G substrate was completely arrested prior to first catalysis even in spprp16+ wild-type cells (), which again differed from S. cerevisiae where an identical intronic mutation allowed for first step catalysis and caused arrest before second step. Early arrest in fission yeast of Br-G substrate led us to speculate that the interaction of the first step splicing factor Cwf25 with BS in S. pombe may be weaker than the homologous Cwc25-BS interaction in S. cerevisiae. To examine this, we used the recently solved cryo-electron microscopy structure of the catalytically active budding yeast spliceosome where the N-terminal Cwc25 helix (encompassing residues 3 to 48, ) is in proximity to the intronic branch site [Citation52]. The homology-based modelling of fission yeast Cwf25 with Cwc25 (PDB ID-5LJ5) show their structural identity at this region (). In this structure, the N-terminal amino acid close to the branch residue A is serine in budding yeast Cwc25 whereas glycine occupies this position in the modelled fission yeast Cwf25 ()). The – OH group of serine forms electrostatic interactions with the – NH2 or the = O group at carbon 6 of the purine ring when the branch residue is an A or G. This interaction we predict may be weak in the case of fission yeast Cwf25 as glycine at the homologous position lacks the hydroxyl group (), right panels). We speculate that these weakened Cwf25–BS interactions may cause the early splicing arrest that we note in fission yeast substrate with branch residue G. As the suppression of splicing phenotypes due to branch site mutants in S. cerevisiae is a consequence of compromised ATPase activity of ScPrp16 mutant proteins, we investigated the in vitro enzymatic activities of our fission yeast Prp16 mutants.
Figure 5. Spprp16F528S mutant exacerbates the splicing defect of substrates with branch-site mutations. (a) Schematic of tfIId E1-I1-E2eGFP mini-transcript. The 5′SS, BS with invariant branch nucleotide A or mutants C or G residues and the 3′SS of the mini-transcript are shown. The GFP-RP primer used for reverse transcription is marked by an arrow. (b) Primer extensions to detect the splicing of tfIId mini-transcripts with branch residue A or C in spprp16+, spprp16F528S and spprp16G515A strains. The expected positions of cDNA from pre-mRNA (P), lariat-exon2 (L) and mRNA (M) species are depicted. Primer extension on snu2+ transcripts served as the loading control. Lane M – 100 nts to 1000 nts DNA size marker (c) Primer extension on tfIID mini-transcripts with the invariant branch residue A or the mutant residue G in spprp16 wild-type and mutant strains. (d) Alignment of N-terminal 50 amino acids in S. cerevisiae Cwc25 and S. pombe Cwf25. The red outline marks residues detected in the cryo-electron microscopy structure of budding yeast spliceosome [Citation52]. (e) The structure shows N-terminal region from Cwc25 (magenta) [Citation52] and Cwf25 (modelled based on homology with Cwc25, cyan – magenta overlap) in proximity to the branch site consensus (blue labelled A70 is the invariant branch residue) of the UBC4 pre-mRNA substrate (orange). The close-up images to the right show interactions of the N-terminal most residue in budding yeast Cwc25 (serine, Ser3-orange) and Cwf25 (glycine, Gly3-orange) with the intronic branch residue A. The side chain interactions of serine and glycine (orange) with the -NH2 group (blue) of branch site adenosine are indicated by dotted red lines.
![Figure 5. Spprp16F528S mutant exacerbates the splicing defect of substrates with branch-site mutations. (a) Schematic of tfIId E1-I1-E2eGFP mini-transcript. The 5′SS, BS with invariant branch nucleotide A or mutants C or G residues and the 3′SS of the mini-transcript are shown. The GFP-RP primer used for reverse transcription is marked by an arrow. (b) Primer extensions to detect the splicing of tfIId mini-transcripts with branch residue A or C in spprp16+, spprp16F528S and spprp16G515A strains. The expected positions of cDNA from pre-mRNA (P), lariat-exon2 (L) and mRNA (M) species are depicted. Primer extension on snu2+ transcripts served as the loading control. Lane M – 100 nts to 1000 nts DNA size marker (c) Primer extension on tfIID mini-transcripts with the invariant branch residue A or the mutant residue G in spprp16 wild-type and mutant strains. (d) Alignment of N-terminal 50 amino acids in S. cerevisiae Cwc25 and S. pombe Cwf25. The red outline marks residues detected in the cryo-electron microscopy structure of budding yeast spliceosome [Citation52]. (e) The structure shows N-terminal region from Cwc25 (magenta) [Citation52] and Cwf25 (modelled based on homology with Cwc25, cyan – magenta overlap) in proximity to the branch site consensus (blue labelled A70 is the invariant branch residue) of the UBC4 pre-mRNA substrate (orange). The close-up images to the right show interactions of the N-terminal most residue in budding yeast Cwc25 (serine, Ser3-orange) and Cwf25 (glycine, Gly3-orange) with the intronic branch residue A. The side chain interactions of serine and glycine (orange) with the -NH2 group (blue) of branch site adenosine are indicated by dotted red lines.](/cms/asset/2a4c1c59-056f-4e3c-8df0-48e5c12f842d/krnb_a_1585737_f0005_oc.jpg)
In vitro biochemical activities of the wild-type and mutant helicase proteins
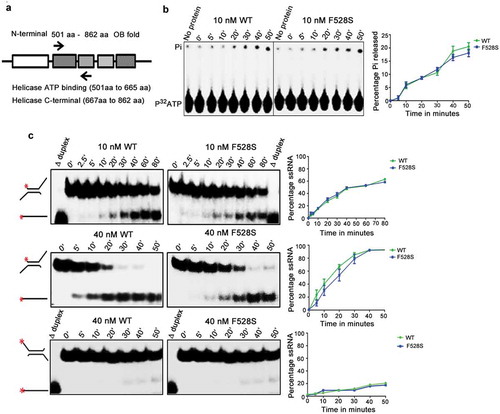
The in vitro helicase and ATPase activity of the spprp16F528S helicase domain was tested to assess if the splicing defects observed in the spprp16F528S mutant could be attributed to the catalytic functions of its helicase domain. The wild-type SpPrp16 and Spprp16F528S helicase domains (amino acids 501 to 862) were purified from bacteria as translational fusions with the Maltose Binding Protein (MBP) tag (). These proteins were first tested for their ability to hydrolyse ATP. Both the wild-type and Spprp16F528S helicase proteins triggered Pi release from γP32 ATP in a time-dependent manner with comparable efficiency (). The interesting observation that the Spprp16F528S helicase protein hydrolysed ATP led us to check if it could efficiently couple this chemical energy into mechanical energy by unwinding in vitro RNA substrates. Two structurally different dsRNA substrates – one with 3′ overhang and the other with 5′ single-stranded overhang were used for determining its potential as a RNA helicase (). The wild-type SpPrp16 helicase domain showed a time and protein concentration dependent conversion of the 3′ overhang containing dsRNA to ssRNA with ~50% unwinding being achieved in ~42 and ~15 min by 10 and 40 nM wild-type protein, respectively (, WT upper and middle panels). The unwinding activity of Spprp16F528S helicase protein was nearly comparable to the wild-type protein for both the concentrations tested (, F528S upper and middle panels). These observations were unexpected, as the spprp16F528S mutant is strongly impaired for in vivo splicing. This in vitro enzymatic competency of Spprp16F528S contradicts the inactivity of the corresponding S. cerevisiae protein Scprp16Y386D. These data can explain the differing response that these Prp16 homologues have on substrates with branch nucleotide mutations. The proficient ATP hydrolysing ability of the Spprp16F528S mutant may enable rejection of tfIID BrC sub-optimal substrate from the splicing cycle and thereby aggravate the splicing defect (). dsRNA with just the 5′ overhang was a poor substrate for both the proteins, confirming SpPrp16 to be a 3′ to 5′ helicase like ScPrp16 (, bottom panel).
Figure 6. Biochemical characterization of Spprp16 helicase domain mutants. (a) Schematic of SpPrp16 domain architecture. Arrow marks indicate primer positions used to amplify the helicase domain (501–862 amino acids) of SpPrp16 wild type and mutant proteins. (b) ATP hydrolysis by wild-type and Spprp16F528S helicase protein upon incubating 10 nM of each protein with 1mM ATP and tracer amounts of γP32ATP at 30°C for the different time points indicated. (c) dsRNA unwinding activity of the wild-type and Spprp16F528S helicase proteins on a 3′ overhang containing RNA duplex labelled at the 5′ end (schematic to the left). The reactions were arrested at the indicated time points (in minutes). The upper and middle panels represent the activity of 10 nM and 40 nM proteins, respectively. Δ duplex indicates heat denatured RNA duplex, a marker for ssRNA. Bottom panel shows helicase activity of the wild-type and Spprp16F528S helicase protein on a 5ʹ overhang containing RNA duplex; 40 nM of wild-type and mutant protein was used in this experiment.
SpPrp16 is required for cell cycle progression and transcriptional gene silencing at the centromeric locus
In addition to the splicing phenotypes, the spprp16F528S cells were also elongated and resembled cell division cycle (cdc) mutants (Supplementary Figure S5(a)). We observed high incidence of fragmented nuclei, a signature of chromosome segregation defects and prevalence of cut (cells untimely torn) cells due to the occurrence of cytokinesis before nuclear division (). These observations suggested mitotic defects in spprp16F528S cells. FACS analysis was done to assess the DNA content in spprp16F528S and wild-type cells (Supplementary Figure S5(b)). Wild type fission yeast cells grown in nitrogen-depleted media for 12 h served as the G1 synchronized cell population [Citation53] and wild-type spprp16+ cells grown in defined complete media served as the G2 control. Majority of spprp16F528S cells had 2C DNA content which together with their elongated morphology suggest an arrest at the G2 or M phase (Supplementary Figure S5(b)). The role of SpPrp16 in cell cycle progression could well be a consequence of impaired splicing of introns such as – cdc2+I2 and cdk8+ I1 in transcripts that encode Cdc2 and Cdk8 kinases required for G2 to mitotic transition (Supplementary Figure S5(c); Supplementary Table S2). Splicing of nda3+I4, whose transcripts encode tubulin, the principal component of mitotic spindle is also affected in spprp16F528S cells (). However, such splicing-dependent cell cycle arrest phenotypes do not exclude possibilities of a more direct role for Prp16 in cell cycle.
Figure 7. SpPrp16 is required for successful mitosis and for efficient transcriptional gene silencing at centromeres and telomeres. (a) Bright field and DAPI stained images of spprp16+, spprp16F528S and spprp16G515A cells. The ‘cut’ cells (white arrowheads at the site of cell separation) and lagging chromosome (fragmented nuclei) in spprp16F528S are shown. Scale bar is 10 µm in all panels. (b) Thiabendazole (TBZ) sensitivity of spprp16 mutants as compared to wild-type and spago1Δ strains at 30°C. Cultures were grown to equivalent OD595 before spotting 10-fold serial dilutions on PMG complete media containing TBZ (at 15 µg/ml concentration). The growth proficiency on PMG complete media (left) served as the control. (c) Normalized transcript levels for centromeric ncRNAs obtained from RNA-seq of spprp16+ (WT) and spprp16F528S mutant cells (left graph). P value significance for upregulated ncRNAs in the spprp16F528S mutant is shown. The semi-quantitative RT-PCR for two centromeric transcripts ncRNA.232 and ncRNA.362 are shown in the right upper and lower panels respectively. For ncRNA.232, ‘*’ to the right side of the gel indicates the unspliced precursor. Control PCR reactions without reverse transcription are shown in the -RT panel. (d) Semi-quantitative RT-PCR to assess the splicing of ncRNA.232 mini-transcript in the WT (leu1:spprp16+) and F528S (leu1:spprp16F528S) mutant strain. (e) Transcriptional silencing of ade6+ centromeric reporter in the wild-type and spprp16F528S mutant cells assessed in media with limiting adenine. cwf10-1 and spago1Δ served as positive controls for defective centromeric heterochromatin.
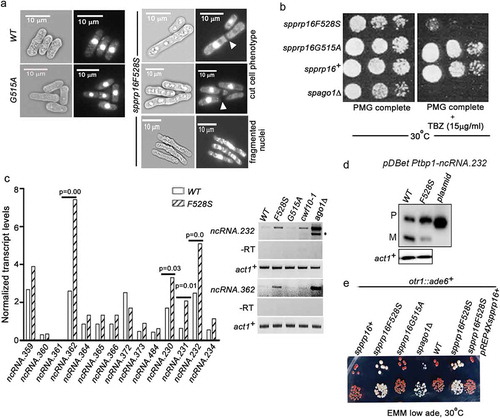
Lagging chromosomes and the thiabendazole (TBZ) sensitivity of spprp16F528S () suggest abnormalities in centromeric heterochromatinization [Citation54]. Interestingly the growth defect of spprp16F528S in the presence of TBZ is similar to that of the heterochromatin defective ago1Δ strain (). To investigate the heterochromatin status in the spprp16F528S mutant, we revisited our transcriptome sequencing data and analysed reads from the centromeric locus. Intriguingly, the spprp16F528S mutant had higher number of reads that mapped to these loci, as compared to that in the wild-type, indicating abnormalities in centromeric transcriptional silencing (Supplementary Figure S6). A closer investigation revealed significantly elevated levels of spncRNA.362, spncRNA.230, spncRNA.231 and spncRNA.232 transcripts in the spprp16F528S mutant (). These abnormal ncRNA levels were confirmed for ncRNA.232 and ncRNA.362 by semi-quantitative RT-PCR (, right panel). The RNAi mutant ago1Δ and the splicing factor mutant cwf10-1 with reported heterochromatin defects [Citation47] served as positive controls for estimating abnormal ncRNA levels. The major and minor amplicons observed for ncRNA.232 in ago1Δ cells correspond to the unspliced and spliced forms resulting from the inclusion or removal of its intron-like element as previously reported [Citation55]. While our study was ongoing, Mutazono et al., 2017 showed that the splicing efficiency of intron-like elements in centromeric dg transcripts was inversely correlated with heterochromatin status. Interestingly, we note that these introns in centromeric transcripts also have features in their 5’SS (ncRNA.232 5’SS-GTAAGT) and BS consensus (ncRNA.232 BS-TACTGAT) that indicate strong base pairing with U5 snRNA and U2 snRNA, respectively. These features likely make them Prp16 dependent for splicing. To further explore the link between centromeric transcription and its heterochromatinization in the context of SpPrp16, we assessed the splicing of intron-like elements in centromeric ncRNAs such as ncRNA.232. To be able to assess the splicing of ncRNA.232 it is required to test these RNAs out of the centromeric context as in wild-type the centromeric locus is transcriptionally silenced. To this end, a mini-gene construct comprising the exon1, intron-like elements and exon2 for ncRNA.232 was generated and transformed into spprp16+ and spprp16F528S cells. The splicing of mini-ncRNA.232 transcript was inefficient even in the wild-type () confirming that these centromeric transcripts are poor splicing substrates. This defect was much exacerbated in the spprp16F528S mutant. This suggests that the partial or inefficient splicing in wild-type cells cause slower progression through splicing which increases the residence time of sub-spliceosome complexes on centromeric transcripts. Such specific sub-spliceosomal complexes may be competent to recruit the silencing machinery. The involvement of specific sub-spliceosomal complexes in this recruitment role reconciles with the fact that not all splicing factor mutants show heterochromatin defects, as these mutants may arrest at different stages. Severe centromeric splicing defect in spprp16F528S cells probably lead to arrest with complexes having entirely unspliced RNA and fail to promote/recruit factors for efficient silencing at the centromeric locus. Together these results suggest that in wild-type cells the intrinsic inefficient splicing of centromeric transcripts and not a complete splicing arrest can promote heterochromatinization. The role of SpPrp16 in centromeric silencing was further validated by generating strains with either spprp16+ or spprp16F528S allele integrated at the leu1 locus and otr1::ade6+ reporter gene at the pericentric repeats of chromosome I. Growth of these strains in media with limiting adenine would reflect the extent of centromeric heterochromatinization. The red colony colour of wild-type cells indicates complete silencing of centromeric ade6+ while the nearly white colour of spprp16F528S cells implies ade6+ expression due to impaired centromeric silencing (). The defective silencing of ade6+ reporter in the spprp16F528S mutant could be completely rescued on complementation with the prp16+ expressing plasmid. The spprp16G515A mutant on the other hand could efficiently silence the centromeric ade6+ reporter gene expression similar to the wild-type. Together, our study uncovers the contribution of splice site–snRNA interaction strength as a feature governing SpPrp16 requirement for splicing and heterochromatinization.
Discussion
Studies on budding yeast and human DExH/DExD box splicing factors are central to our understanding of spliceosomal transitions, organization of catalytic centre conformations and mechanisms underlying splicing fidelity. A homozygous autosomal recessive G332D mutation in DHX38, the human homologue of Prp16 helicase, and dominant mutations in homologues of other splicing factors like Prp4, Prp8, Prp31, etc., are associated with retinitis pigmentosa in humans (RP) [Citation56–Citation58]. Studies testing the effects of homologous Prp8-RP mutations in S. cerevisiae uncover defects in spliceosome activation and in spliceosomal transition from first to second step catalysis [Citation59,Citation60]. While developmental roles for spliceosomal proteins are emerging from several models [Citation61–Citation63], relatively unexplored are the splice–site interactions for accurate recognition and removal of short introns that are abundant in the genomes of several eukaryotes [Citation32]. Remarkably, Arabidopsis Prp16 and Brr2 are required for the splicing of only subsets of introns [Citation62,Citation63] and Prp16 orthologues in Chlamydomonas reinhardtii and Caenorhabditis elegans majorly function in other gene regulatory pathways [Citation64,Citation65].
Here from genome-wide transcriptome analysis, we infer nearly global splicing functions for fission yeast SpPrp16 and find that a small subset of introns with weak 5’SS-U6 snRNA interactions are efficiently spliced in spprp16F528S cells. This identified strong U6 snRNA-5′SS interactions as an intronic risk factor, which individually or in combination with the strength of BS-U2 interaction can dictate dependence on SpPrp16. A significant proportion of fission yeast SpPrp16 dependent introns have uridine (U) at the 5’SS +4 position which is complementary to the invariant A of U6 snRNA ACAGAGA box and thus can participate in U6-5’SS interaction. Interestingly, the global 5′SS consensus in Chlamydomonas introns lacks U at this position [Citation66]. The resulting weak 5′SS-U6 snRNA complementarity in Chlamydomonas introns may facilitate its destabilization in the Chlamydomonas Prp16 mutant mut6 and explain the splicing competence of this mutant. Therefore, we infer that the global dependence on Prp16 for splicing in a genome is dictated by the strength of splice site – snRNA interactions and decipher Prp16 functions at the juncture of 5′SS cleavage in fission yeast. Interestingly, study on human Prp8 also reveals weak 5’SS element in introns as a feature that governs its splice-site utilization across the human transcriptome [Citation67].
We surmise early associations and functions for S. pombe Prp16 based on genetic interactions and primer extension assays. These data are in agreement with reports which show budding yeast Prp16 association with the spliceosome prior to catalysis at instances when substrates undergo slow 5′SS cleavage kinetics or have a Br-C mutation [Citation9,Citation23]. The prevalence of degenerate 5′SS which can establish strong U6 snRNA interactions in genomes like fission yeast plausibly make SpPrp16 early splicing functions more pervasive. Nevertheless, we do not exclude the possibility of obtaining SpPrp16 alleles with defects in second step catalysis in future. As yet, second step splicing arrest in fission yeast either due to mutations in splicing factors or cis intronic mutations in protein-coding transcripts are not known. Intriguingly, the functional telomerase is formed due to uncoupling of first and second step splicing reactions in the TER1 precursor transcript. The cleaved 5ʹ exon generated after the first splicing reaction forms the functional telomerase without the intermediates undergoing the second step [Citation35,Citation68]. Hyper stabilized 5′SS-U6 snRNA or BS-U2 snRNA interactions form the mechanistic basis of this uncoupling among the different fungi tested [Citation35]. TER1 like splice sites although rare in protein-coding S. pombe transcripts are frequent in non-coding RNAs. Analysis of these ncRNA introns in spprp16 mutants could give further leads to the role of SpPrp16 in the second step of splicing.
The efficient in vitro dsRNA unwinding (), and contrasting in vivo splicing functions of Spprp16F528S mutant may be due to its impaired interactions with other splicing factors, or, interactions within the protein that are critical for coupling helicase activity to its in vivo splicing functions. Such opposing effects of compromised in vivo splicing, but near normal in vitro enzymatic activities are also reported for budding yeast Prp22 (S635A and T637A), Prp2 (S378L) and Prp43 (T384A and T384V) mutants [Citation69–Citation71] which together suggest that the ATPase or helicase activities of these proteins do not entirely explain their biological functions. Or in the in vivo contexts, its unwinding activities may be closely coupled to spliceosomal associations and may be influenced by protein co-factors. Supporting the possibility of such protein co-factors, the G patch proteins Spp2 and Ntr1 are known to positively regulate the in vivo activity of budding yeast helicases Prp2 [Citation72,Citation73] and Prp43, respectively, [Citation71,Citation74]. Also, Prp8 C-terminal fragment positively influences the poor in vitro helicase activity of Brr2, and Snu114 regulates Brr2 activity during spliceosome activation and disassembly [Citation15,Citation75]. Physical interactions between Prp16 and Brr2 is supported by the recent cryo-electron microscopy structure of budding yeast which demonstrate Prp16 interaction with the Sec 63–1 helicase module of Brr2 and Jab1 domain of Prp8 [Citation52,Citation76]. We modelled this SpPrp16 domain (amino acids 481 to 1101) into the budding yeast spliceosome structure and found these interactions to be retained even with SpPrp16 (Supplementary Figure S7). Also, S. cerevisiae Prp8 mutations defective in transition from first to second step catalysis and homologous to the human retinitis pigmentosa alleles, map to the Prp8 Jab1 domain and may affect its association with Prp16 [Citation60]. Future genetic or biochemical experiments can substantiate the role of these proteins in modulating helicase functions of SpPrp16.
Splicing phenotypes of BS mutant substrates differ between budding and fission yeasts (). The partial first step arrest of a mutant Br-C substrate is exacerbated in the equivalent spprp16F528S mutant unlike in S. cerevisiae where this first step arrest is suppressed in the scprp16-1 mutant. In budding yeast, first step catalysis commences as Cwc25 binds to the branch site [Citation9,Citation77] and this binding is unstable for substrates with the intronic branch residue mutation Br-C. Therefore, Prp16 ATPase mediated release of the first step splicing factors Cwc25 and Yju2 occurs before 5’SS cleavage leading to catalytic arrest [Citation7,Citation9,Citation52,Citation77]. However, Prp16 mutants defective in ATPase activity allows longer time for Cwc25 to remain associated with the branch site due to the delayed conformational switch thus favouring progression to first step catalysis [Citation9,Citation23]. We show that the Spprp16F528S helicase domain is enzymatically efficient in vitro (). The inability of the spprp16F528S to suppress the first step arrest of the Br-C mutant is therefore explained by its ATPase proficiency that could displace Cwf25 from the branch site thereby prevent first step splicing. This, in turn, contrasts with the ability of the ATPase defective budding yeast mutant scprp16-1(Y386D) protein to suppress splicing of substrates with Br-C mutation. Possibly, the Spprp16F528S protein may also fail to stabilize BrC-Cwf25 interaction in the activated spliceosome, as it is reported for the budding yeast wild-type Prp16 [Citation9]. This may account for the enhanced early splicing arrest in spprp16F528S cells as compared to the moderate level of splicing in wild-type cells presented with a Br-C substrate ().
The high levels of centromeric ncRNA transcripts and aberrant silencing of centromeric reporter genes in spprp16F528S cells highlights SpPrp16 functions in transcriptional gene silencing (). Mutants in Prp5, Prp8, Prp13 and Prp12 also show G2 cell cycle arrest with independent reports on its varying degrees of impaired centromeric heterochromatin [Citation47,Citation78,Citation79]. Here, we suggest that the unrepressed centromeric chromatin in spprp16F528S cells can result in inadequate H3K9me2 methylation. Other studies have shown diminished levels of H3K9me2 at centromeric loci to affect cohesin deposition and subsequent sister chromatid adhesion vital for properly timed mitotic metaphase to anaphase transition [Citation54,Citation80]. This supports the link between centromeric heterochromatinization and cell cycle progression observed in the context of spprp16F528S mutant. It has been proposed that a subset of splicing factors bind to the cryptic intronic elements in the centromeric transcripts (as in ncRNA.232). Reducing the length of the transcript region between TSS and dg intron, increased splicing and reduced repressive H3K9 di-methylation marks. However, mutation of splice sites which reduced H3K9me2 repressive mark did not improve splicing [Citation55,Citation81–Citation83]. This suggest that splicing efficiency does not always inversely co-relate with heterochromatin status. Our results show that the complete splicing arrest of ncRNA.232 in the spprp16F528S mutant fails to promote heterochromatinization (). We reason that for these ncRNA with cryptic intronic elements, slow splicing progression, albeit inefficient, is the normal intrinsic property contributed by strong 5’ss-U6snRNA and BS-U2snRNA interactions along with other features such as the distance between the TSS and the intron-like element as described by Mutazono et al., 2017. The presence of both pre-mRNA and mRNA forms of ncRNA.232 transcript observed in wild-type cells could be an outcome of slow splicing catalysis rather than a stage-specific splicing arrest. Such slow progression through the splicing pathway helps in spliceosome stalling, proposed to be the pre-requisite for proper heterochromatinization [Citation55,Citation81–Citation83]. However, complete splicing arrest of the centromeric transcript ncRNA.232 observed in the spprp16F528S reconciles with Prp16 requirement for destabilizing 5’SS-U6snRNA and BS-U2snRNA interactions and likely causes a complete block in progression through the splicing pathway. The latter can thereby negatively impact recruitment of the silencing machinery required for heterochromatinization. The ncRNA binding protein Seb1 is required for the recruitment of the SHREC complex comprising deacetylases to peri-centromeric regions. This complex works in parallel to RNAi by promoting a positive feed-back loop with alternate rounds of deacetylation and methylation for heterochromatinization [Citation84]. Seb1 has also been recently shown to nucleate heterochromatin assembly by inducing RNAPII stalls [Citation85]. Splicing of the seb1+cellular transcript is severely compromised in spprp16F528S and thus could contribute to heterochromatin defects. Interestingly, while we demonstrate the effects of fission yeast Prp16 in splicing and transcriptional gene silencing, its orthologues Mut6 and Mog1 in Chlamydomonas reinhardtii and C. elegans identified as mutants defective in transgene silencing and in translational repression during sex determination, respectively, have solely splicing-independent roles [Citation64,Citation65]. Our results suggest a mechanistic link between splicing and gene-silencing and future work on fission yeast Prp16 can shed light on the complex relationship between these regulatory pathways in higher eukaryotes.
Materials and methods
Additional details can be found in Supplementary Materials and Methods.
Yeast strains and growth conditions
The standard genetic and molecular techniques used are as described previously [Citation86,Citation87]. All strains generated and used in this study are listed in Supplementary Table S1.
All primers used in the study are listed in Supplementary Table S5.
RNA-seq data analysis
RNA was isolated from biological replicates of WT and spprp16F528S mutant cultures grown at 30°C. The library was prepared using Illumina Total Prepamp Kit (Cat No: AMIL1791) and sequenced on the Illumina Hi-seq 2500 platform. Paired-end reads of 100 base length were obtained. The total reads were 43,967,073 and 52,543,226 for the wild-type and mutant, respectively, in batch1; and 61,531,918 and 40,993,549 in batch 2 for the two respective strains. The reads were mapped using bowtie 2–2.1.0 [Citation88] to the S. pombe genome database version Schizosaccharomyces_pombe ASM294v2.26 and reads that matched to multiple loci were removed from further analysis. Reads which corresponded to S. pombe genome (96% of total reads) from all the four samples were mapped using bowtie-1.0.0 onto a compiled 70 bp junction sequence (built-in-house) which represented all consecutive exon-intron (EI) and exon-exon (EE) sequences for the S. pombe genome. Only reads which traversed the junction and extended to either sides of the junction by at least 4 bases were considered. If all the read counts (EE and EI) in a biological batch was less than three for any intron, it was excluded from analysis. These two filters applied exempted 72 introns from a total of 5,122 S. pombe introns (Dataset S1). Splice index (SI) was calculated as SI = log2(EE/EI), as described in [Citation46]. For EI reads only those corresponding to upstream 5′exon relative to the intron (5′exon-Intron) were considered. The difference in the SI values between the wild-type and mutant for each intron gave the differential SI. This was computed for all introns in both biological replicates. Statistically significant introns between the biological replicates were identified by the Cochran-Mantel-Haenszel (CMH) chi-square (mantelhaen.test command in R). The false-discovery rate (q-value) was computed using the Bioconductor q-value package, and a cut-off of q < 0.05 was applied. Using this statistical test a dataset of SpPrp16 dependent and independent introns significant between the two biological replicates was derived.
To analyse the differential expression levels of centromeric non-coding RNA in the wild-type and spprp16528S mutant cells the reads were mapped using TopHat [Citation89] were provided as input to Cufflink 2.2.1 [Citation90].
Helicase and ATPase assays
The helicase and ATPase assays of SpPrp16 wild-type and mutant helicase proteins were performed as previously described [Citation91]. RNA duplexes for helicase assays were prepared by annealing chemically synthesized RNA oligos listed in Supplementary Table S5. Detailed methods are in SI Materials and Methods.
Homology modelling
The homology-based model of SpPrp16 was generated using the crystal structure of SpPrp43 (PDB ID-2XAU) [Citation92] in SWISS MODEL [Citation93]. For studies using the cryo-electron microscopy structure of budding yeast spliceosome [Citation52] to predict SpPrp16 spliceosomal interactions, the structure of SpPrp16 C-terminal (residue 481–1101) was modelled based on homology with ScPrp16 (in PDB ID-5LJ5). To understand Cwc25 and Cwf25 interactions with the branch site nucleotide, homology-based model of Cwf25 was generated using 5LJ5 as the template.
The homology models were generated in SWISS MODEL [Citation93] and energy minimized using Swiss-PDB viewer which implements an empirical force field by partial implementation of GROMOS96 in vacuum with default 20 cycles of steepest descent. The models were validated by means of PROCHECK [Citation94] and Verify_3D available at http://nihserver.mbi.ucla.edu/SAVES/ [Citation95]. The quality of all models was also evaluated by ProSA. The obtained models were saved in PDB format and visualized using PYMOL or chimera.
Author contributions
UV and DV conceived and designed the experiments. DV, AKS and RK performed the experiments. DV and AKS performed RT-PCR and primer extension reactions. AKS performed the protein purifications and ATPase assays. RK performed homology modelling studies and assisted in in vitro assays. PB performed the bioinformatic analysis of the RNA-seq datasets and was guided by SS. DV and UV wrote the manuscript.
Supplemental Material
Download Zip (4.2 MB)Acknowledgments
We thank Drs. Robin Allshire, Peter Baumann, Danesh Moazed, Rob Marteinssen, and Kathleen Gould for generously gifting their mutants for use in genetic interaction studies. We thank Dr. Danny Bitton from Prof. Bahler’s lab for his helpful inputs in RNA seq data analysis. Prof. D.N. Rao and lab members, Department of Biochemistry, IISc and Rakesh Ramachandran, Dr. Srinivasan’s lab at MBU, IISc are gratefully acknowledged for their critical inputs in this study. We thank Ganit Labs, IBAB Bangalore for RNA-sequencing services. DST-FIST (SR/FST/LS11-044/2016), UGC-Centre for Advanced Studies F-S-12/2012 (SAP-11) and infrastructure and research support from DBT-IISc Partnership Programme (DBT/BF/PRIns/2011-2012/28.9.2012), Government of India are acknowledged for supporting this work. Scholarship from Council of Scientific and Industrial Research for DV, Scholarship from the Indian Institute of Science to AKS and Research Associate fellowship from Department of Biotechnology to RK are acknowledged.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009 Feb 20;136(4):701–718. PubMed PMID: 19239890; Eng.
- Staley JP, Guthrie C. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell. 1998 Feb 6;92(3):315–326. PubMed PMID: 9476892; eng.
- Schwer B. A new twist on RNA helicases: DExH/D box proteins as RNPases. Nat Struct Biol. 2001 Feb;8(2):113–116. PubMed PMID: 11175897; Eng.
- Chen JY, Stands L, Staley JP, et al. Specific alterations of U1-C protein or U1 small nuclear RNA can eliminate the requirement of Prp28p, an essential DEAD box splicing factor. Mol Cell. 2001 Jan;7(1):227–232. PubMed PMID: 11172727; eng.
- Yang F, Wang XY, Zhang ZM, et al. Splicing proofreading at 5ʹ splice sites by ATPase Prp28p. Nucleic Acids Res. 2013 Apr;41(8):4660–4670. PubMed PMID: 23462954; PubMed Central PMCID: PMCPmc3632134. eng.
- Ruby SW, Chang TH, Abelson J. Four yeast spliceosomal proteins (PRP5, PRP9, PRP11, and PRP21) interact to promote U2 snRNP binding to pre-mRNA. Genes Dev. 1993 Oct;7(10):1909–1925. PubMed PMID: 8405998; eng.
- Warkocki Z, Odenwalder P, Schmitzova J, et al. Reconstitution of both steps of Saccharomyces cerevisiae splicing with purified spliceosomal components. Nat Struct Mol Biol. 2009 Dec;16(12):1237–1243. PubMed PMID: 19935684; Eng.
- Lardelli RM, Thompson JX, Yates JR 3rd, et al. Release of SF3 from the intron branchpoint activates the first step of pre-mRNA splicing. RNA (New York, NY). 2010 Mar;16(3):516–528. PubMed PMID: 20089683; PubMed Central PMCID: PMCPmc2822917. Eng.
- Tseng CK, Liu HL, Cheng SC. DEAH-box ATPase Prp16 has dual roles in remodeling of the spliceosome in catalytic steps. RNA (New York, NY). 2011 Jan;17(1):145–154. PubMed PMID: 21098140; PubMed Central PMCID: PMCPmc3004056. Eng.
- Schwer B, Guthrie C. A conformational rearrangement in the spliceosome is dependent on PRP16 and ATP hydrolysis. EMBO J. 1992 Dec;11(13):5033–5039. PubMed PMID: 1464325; PubMed Central PMCID: PMCPmc556981. eng.
- Hogg R, de Almeida RA, Ruckshanthi JP, et al. Remodeling of U2-U6 snRNA helix I during pre-mRNA splicing by Prp16 and the NineTeen Complex protein Cwc2. Nucleic Acids Res. 2014 Jul;42(12):8008–8023. PubMed PMID: 24848011; PubMed Central PMCID: PMCPmc4081067. Eng.
- Villa T, Guthrie C. The Isy1p component of the NineTeen complex interacts with the ATPase Prp16p to regulate the fidelity of pre-mRNA splicing. Genes Dev. 2005 Aug 15;19(16):1894–1904. PubMed PMID: 16103217; PubMed Central PMCID: PMCPmc1186189. Eng.
- Mefford MA, Staley JP. Evidence that U2/U6 helix I promotes both catalytic steps of pre-mRNA splicing and rearranges in between these steps. RNA (New York, NY). 2009 Jul;15(7):1386–1397. PubMed PMID: 19458033; PubMed Central PMCID: PMCPmc2704075. eng.
- Schwer B, Gross CH. Prp22, a DExH-box RNA helicase, plays two distinct roles in yeast pre-mRNA splicing. EMBO J. 1998 Apr 01;17(7):2086–2094. PubMed PMID: 9524130; PubMed Central PMCID: PMCPmc1170553. eng.
- Maeder C, Kutach AK, Guthrie C. ATP-dependent unwinding of U4/U6 snRNAs by the Brr2 helicase requires the C terminus of Prp8. Nat Struct Mol Biol. 2009 Jan;16(1):42–48. PubMed PMID: 19098916; PubMed Central PMCID: PMCPmc2707180. Eng.
- Hahn D, Beggs JD. Brr2p RNA helicase with a split personality: insights into structure and function. Biochem Soc Trans. 2010 Aug;38(4):1105–1109. PubMed PMID: 20659012; Eng.
- Horowitz DS. The splice is right: guarantors of fidelity in pre-mRNA splicing. RNA (New York, NY). 2011 Apr;17(4):551–554. PubMed PMID: 21357751; PubMed Central PMCID: PMCPmc3062167. eng.
- Query CC, Konarska MM. Splicing fidelity revisited. Nat Struct Mol Biol. 2006 Jun;13(6):472–474. PubMed PMID: 16757943; eng.
- Liu YC, Cheng SC. Functional roles of DExD/H-box RNA helicases in Pre-mRNA splicing. J Biomed Sci. 2015 Jul 16;22:54. PubMed PMID: 26173448; PubMed Central PMCID: PMCPmc4503299. eng.
- Xu YZ, Query CC. Competition between the ATPase Prp5 and branch region-U2 snRNA pairing modulates the fidelity of spliceosome assembly. Mol Cell. 2007 Dec 14;28(5):838–849. PubMed PMID: 18082608; PubMed Central PMCID: PMCPmc2246091. eng.
- Perriman R, Barta I, Voeltz GK, et al. ATP requirement for Prp5p function is determined by Cus2p and the structure of U2 small nuclear RNA. Proc Natl Acad Sci U S A. 2003 Nov 25;100(24):13857–13862. PubMed PMID: 14610285; PubMed Central PMCID: PMCPmc283511. eng.
- Liang WW, Cheng SC. A novel mechanism for Prp5 function in prespliceosome formation and proofreading the branch site sequence. Genes Dev. 2015 Jan 1;29(1):81–93. PubMed PMID: 25561497; PubMed Central PMCID: PMCPmc4281567. eng.
- Burgess SM, Guthrie C. A mechanism to enhance mRNA splicing fidelity: the RNA-dependent ATPase Prp16 governs usage of a discard pathway for aberrant lariat intermediates. Cell. 1993 Jul 2;73(7):1377–1391. PubMed PMID: 8324826; eng.
- Wlodaver AM, Staley JP. The DExD/H-box ATPase Prp2p destabilizes and proofreads the catalytic RNA core of the spliceosome. RNA (New York, NY). 2014 Mar;20(3):282–294. PubMed PMID: 24442613; PubMed Central PMCID: PMCPmc3923124. Eng.
- Mayas RM, Maita H, Staley JP. Exon ligation is proofread by the DExD/H-box ATPase Prp22p. Nat Struct Mol Biol. 2006 Jun;13(6):482–490. PubMed PMID: 16680161; PubMed Central PMCID: PMCPmc3729281. Eng.
- Schwer B, Guthrie C. PRP16 is an RNA-dependent ATPase that interacts transiently with the spliceosome. Nature. 1991 Feb 7;349(6309):494–499. PubMed PMID: 1825134; eng.
- Koodathingal P, Novak T, Piccirilli JA, et al. The DEAH box ATPases Prp16 and Prp43 cooperate to proofread 5ʹ splice site cleavage during pre-mRNA splicing. Mol Cell. 2010 Aug 13;39(3):385–395. PubMed PMID: 20705241; PubMed Central PMCID: PMCPmc3722364. eng.
- Arenas JE, Abelson JN. Prp43: an RNA helicase-like factor involved in spliceosome disassembly. Proc Natl Acad Sci U S A. 1997 Oct 28;94(22):11798–11802. PubMed PMID: 9342317; PubMed Central PMCID: PMCPmc23592. Eng.
- Semlow DR, Blanco MR, Walter NG, et al. Spliceosomal DEAH-box ATPases remodel pre-mRNA to activate alternative splice sites. Cell. 2016 Feb 25;164(5):985–998. PubMed PMID: 26919433; PubMed Central PMCID: PMCPmc4979991. eng.
- Kaufer NF, Potashkin J. Analysis of the splicing machinery in fission yeast: a comparison with budding yeast and mammals. Nucleic Acids Res. 2000 Aug 15;28(16):3003–3010. PubMed PMID: 10931913; PubMed Central PMCID: PMCPmc108416. eng.
- Kuhn AN, Kaufer NF. Pre-mRNA splicing in Schizosaccharomyces pombe: regulatory role of a kinase conserved from fission yeast to mammals. Curr Genet. 2003 Feb;42(5):241–251. PubMed PMID: 12589463; eng.
- Lim LP, Burge CB. A computational analysis of sequence features involved in recognition of short introns. Proc Natl Acad Sci U S A. 2001 Sep 25;98(20):11193–11198. PubMed PMID: 11572975; PubMed Central PMCID: PMCPmc58706. eng.
- Banerjee S, Khandelia P, Melangath G, et al. Splicing functions and global dependency on fission yeast slu7 reveal diversity in spliceosome assembly. Mol Cell Biol. 2013 Aug;33(16):3125–3136. PubMed PMID: 23754748; PubMed Central PMCID: PMCPmc3753915. eng.
- Vijaykrishna N, Melangath G, Kumar R, et al. The fission yeast pre-mRNA processing factor 18 (prp18+) has intron-specific splicing functions with links to G1-S cell cycle progression. J Biol Chem. 2016 Nov 15;291:27387–27402. PubMed PMID: 27875300; eng.
- Kannan R, Hartnett S, Voelker RB, et al. Intronic sequence elements impede exon ligation and trigger a discard pathway that yields functional telomerase RNA in fission yeast. Genes Dev. 2013 Mar 15;27(6):627–638. PubMed PMID: 23468430; PubMed Central PMCID: PMCPMC3613610. eng.
- Kim DU, Hayles J, Kim D, et al. Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol. 2010 Jun;28(6):617–623. PubMed PMID: 20473289; PubMed Central PMCID: PMCPmc3962850. Eng.
- Burgess S, Couto JR, Guthrie C. A putative ATP binding protein influences the fidelity of branchpoint recognition in yeast splicing. Cell. 1990 Mar 9;60(5):705–717. PubMed PMID: 2138057; eng.
- Eckert D, Andree N, Razanau A, et al. Prp4 kinase grants the license to splice: control of weak splice sites during spliceosome activation. PLoS Genet. 2016 Jan;12(1):e1005768. PubMed PMID: 26730850; PubMed Central PMCID: PMCPmc4701394. eng.
- Sridharan V, Heimiller J, Singh R. Genomic mRNA profiling reveals compensatory mechanisms for the requirement of the essential splicing factor U2AF. Mol Cell Biol. 2011 Feb;31(4):652–661. PubMed PMID: 21149581; PubMed Central PMCID: PMCPmc3028654. eng.
- Madhani HD, Guthrie C. A novel base-pairing interaction between U2 and U6 snRNAs suggests a mechanism for the catalytic activation of the spliceosome. Cell. 1992 Nov 27;71(5):803–817. PubMed PMID: 1423631; eng.
- Couto JR, Tamm J, Parker R, et al. A trans-acting suppressor restores splicing of a yeast intron with a branch point mutation. Genes Dev. 1987 Jul;1(5):445–455. PubMed PMID: 2890553; eng.
- Madhani HD, Guthrie C. Genetic interactions between the yeast RNA helicase homolog Prp16 and spliceosomal snRNAs identify candidate ligands for the Prp16 RNA-dependent ATPase. Genetics. 1994 Jul;137(3):677–687. PubMed PMID: 8088513; PubMed Central PMCID: PMCPmc1206027. eng.
- Vijayraghavan U, Company M, Abelson J. Isolation and characterization of pre-mRNA splicing mutants of Saccharomyces cerevisiae. Genes Dev. 1989 Aug;3(8):1206–1216. PubMed PMID: 2676722; eng.
- Nam K, Hudson RH, Chapman KB, et al. Yeast lariat debranching enzyme. Substrate and sequence specificity. J Biol Chem. 1994 Aug 12;269(32):20613–20621. PubMed PMID: 7519612; eng.
- Bartels C, Urlaub H, Luhrmann R, et al. Mutagenesis suggests several roles of Snu114p in pre-mRNA splicing. J Biol Chem. 2003 Jul 25;278(30):28324–28334. PubMed PMID: 12736260; eng.
- Livesay SB, Collier SE, Bitton DA, et al. Structural and functional characterization of the N terminus of Schizosaccharomyces pombe Cwf10. Eukaryot Cell. 2013 Nov;12(11):1472–1489. PubMed PMID: 24014766; PubMed Central PMCID: PMCPmc3837936. eng.
- Bayne EH, Portoso M, Kagansky A, et al. Splicing factors facilitate RNAi-directed silencing in fission yeast. Science. 2008 Oct 24;322(5901):602–606. PubMed PMID: 18948543; PubMed Central PMCID: PMCPMC2585287. eng.
- Kim SH, Lin RJ. Spliceosome activation by PRP2 ATPase prior to the first transesterification reaction of pre-mRNA splicing. Mol Cell Biol. 1996 Dec;16(12):6810–6819. PubMed PMID: 8943336; PubMed Central PMCID: PMCPmc231684. Eng.
- Company M, Arenas J, Abelson J. Requirement of the RNA helicase-like protein PRP22 for release of messenger RNA from spliceosomes. Nature. 1991 Feb 07;349(6309):487–493. PubMed PMID: 1992352; eng.
- Fourmann JB, Schmitzova J, Christian H, et al. Dissection of the factor requirements for spliceosome disassembly and the elucidation of its dissociation products using a purified splicing system. Genes Dev. 2013 Feb 15;27(4):413–428. PubMed PMID: 23431055; PubMed Central PMCID: PMCPmc3589558. Eng.
- Query CC, Konarska MM. Suppression of multiple substrate mutations by spliceosomal prp8 alleles suggests functional correlations with ribosomal ambiguity mutants. Mol Cell. 2004 May 7;14(3):343–354. PubMed PMID: 15125837; eng.
- Galej WP, Wilkinson ME, Fica SM, et al. Cryo-EM structure of the spliceosome immediately after branching. Nature. 2016 Jul 26;537(7619):197–201. PubMed PMID: 27459055; Eng.
- Costello G, Rodgers L, Beach D. Fission yeast enters the stationary phase G0 state from either mitotic G1 or G2 [journal article]. Curr Genet. 1986;11(2):119–125.
- Volpe T, Schramke V, Hamilton GL, et al. RNA interference is required for normal centromere function in fission yeast. Chromosome Res. 2003;11(2):137–146. PubMed PMID: 12733640; eng.
- Chinen M, Morita M, Fukumura K, et al. Involvement of the spliceosomal U4 small nuclear RNA in heterochromatic gene silencing at fission yeast centromeres. J Biol Chem. 2010 Feb 19;285(8):5630–5638. PubMed PMID: 20018856; PubMed Central PMCID: PMCPMC2820790. eng.
- Ajmal M, Khan MI, Neveling K, et al. A missense mutation in the splicing factor gene <em>DHX38</em> is associated with early-onset retinitis pigmentosa with macular coloboma. J Med Genet. 2014;51(7):444–448.
- Růžičková Š, Staněk D. Mutations in spliceosomal proteins and retina degeneration. RNA Biol. 2016;1–9. DOI:10.1080/15476286.2016.1191735
- Mordes D, Luo X, Kar A, et al. Pre-mRNA splicing and retinitis pigmentosa. Mol Vis. 2006;10/26(12):1259–1271. PubMed PMID: PMC2683577.
- Boon KL, Grainger RJ, Ehsani P, et al. prp8 mutations that cause human retinitis pigmentosa lead to a U5 snRNP maturation defect in yeast. Nat Struct Mol Biol. 2007 Nov;14(11):1077–1083. PubMed PMID: 17934474; PubMed Central PMCID: PMCPmc2584834. eng.
- Mayerle M, Guthrie C. Prp8 retinitis pigmentosa mutants cause defects in the transition between the catalytic steps of splicing. RNA (New York, NY). 2016 May;22(5):793–809. PubMed PMID: 26968627; PubMed Central PMCID: PMCPmc4836653. eng.
- McKie AB, McHale JC, Keen TJ, et al. Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13). Hum Mol Genet. 2001 Jul 15;10(15):1555–1562. PubMed PMID: 11468273; eng.
- Tsugeki R, Tanaka-Sato N, Maruyama N, et al. CLUMSY VEIN, the Arabidopsis DEAH-box Prp16 ortholog, is required for auxin-mediated development. Plant J. 2015 Jan;81(2):183–197. PubMed PMID: 25384462; eng.
- Mahrez W, Shin J, Munoz-Viana R. BRR2a Affects Flowering Time via FLC Splicing. PLoS Genet. 2016 Apr;12(4):e1005924. PubMed PMID: 27100965.
- Wu-Scharf D, Jeong B, Zhang C, et al. Transgene and transposon silencing in Chlamydomonas reinhardtii by a DEAH-box RNA helicase. Science. 2000 Nov 10;290(5494):1159–1162. PubMed PMID: 11073454; eng.
- Graham PL, Kimble J. The mog-1 gene is required for the switch from spermatogenesis to oogenesis in Caenorhabditis elegans. Genetics. 1993 Apr;133(4):919–931. PubMed PMID: 8462850; PubMed Central PMCID: PMCPmc1205409. eng.
- Labadorf A, Link A, Rogers MF, et al. Genome-wide analysis of alternative splicing in Chlamydomonas reinhardtii. BMC Genomics. 2010 Feb 17;11:114. PubMed PMID: 20163725; PubMed Central PMCID: PMCPmc2830987. eng.
- Wickramasinghe VO, Gonzalez-Porta M, Perera D, et al. Regulation of constitutive and alternative mRNA splicing across the human transcriptome by PRPF8 is determined by 5ʹ splice site strength. Genome Biol. 2015 Sep 21;16:201. PubMed PMID: 26392272; PubMed Central PMCID: PMCPmc4578845. eng.
- Kannan R, Helston RM, Dannebaum RO, et al. Diverse mechanisms for spliceosome-mediated 3ʹ end processing of telomerase RNA. Nat Commun. 2015 Jan 19;6:6104. PubMed PMID: 25598145; PubMed Central PMCID: PMCPMC4299874. eng.
- Plumpton M, McGarvey M, Beggs JD. A dominant negative mutation in the conserved RNA helicase motif ‘SAT’ causes splicing factor PRP2 to stall in spliceosomes. Embo J. 1994 Feb 15;13(4):879–887. PubMed PMID: 8112301; PubMed Central PMCID: PMCPmc394887. Eng.
- Schneider S, Campodonico E, Schwer B. Motifs IV and V in the DEAH box splicing factor Prp22 are important for RNA unwinding, and helicase-defective Prp22 mutants are suppressed by Prp8. J Biol Chem. 2004 Mar 5;279(10):8617–8626. PubMed PMID: 14688266; Eng.
- Tanaka N, Schwer B. Mutations in PRP43 that uncouple RNA-dependent NTPase activity and pre-mRNA splicing function. Biochemistry. 2006 May 23;45(20):6510–6521. PubMed PMID: 16700561; PubMed Central PMCID: PMCPmc2517181. eng.
- Roy J, Kim K, Maddock JR, et al. The final stages of spliceosome maturation require Spp2p that can interact with the DEAH box protein Prp2p and promote step 1 of splicing. RNA (New York, NY). 1995 Jun;1(4):375–390. PubMed PMID: 7493316; PubMed Central PMCID: PMCPmc1482403. Eng.
- Silverman EJ, Maeda A, Wei J, et al. Interaction between a G-patch protein and a spliceosomal DEXD/H-box ATPase that is critical for splicing. Mol Cell Biol. 2004 Dec;24(23):10101–10110. PubMed PMID: 15542821; PubMed Central PMCID: PMCPmc529041. Eng.
- Tanaka N, Aronova A, Schwer B. Ntr1 activates the Prp43 helicase to trigger release of lariat-intron from the spliceosome. Genes Dev. 2007 Sep 15;21(18):2312–2325. PubMed PMID: 17875666; PubMed Central PMCID: PMCPmc1973145. eng.
- Small EC, Leggett SR, Winans AA, et al. The EF-G-like GTPase Snu114p regulates spliceosome dynamics mediated by Brr2p, a DExD/H box ATPase. Mol Cell. 2006 Aug 04;23(3):389–399. PubMed PMID: 16885028; PubMed Central PMCID: PMCPmc3777414. eng.
- van Nues RW, Beggs JD. Functional contacts with a range of splicing proteins suggest a central role for Brr2p in the dynamic control of the order of events in spliceosomes of Saccharomyces cerevisiae. Genetics. 2001 Apr;157(4):1451–1467. PubMed PMID: 11290703; PubMed Central PMCID: PMCPmc1461596. eng.
- Chiu YF, Liu YC, Chiang TW, et al. Cwc25 is a novel splicing factor required after Prp2 and Yju2 to facilitate the first catalytic reaction. Mol Cell Biol. 2009 Nov;29(21):5671–5678. PubMed PMID: 19704000; PubMed Central PMCID: PMCPmc2772750. eng.
- Habara Y, Urushiyama S, Shibuya T, et al. Mutation in the prp12+ gene encoding a homolog of SAP130/SF3b130 causes differential inhibition of pre-mRNA splicing and arrest of cell-cycle progression in Schizosaccharomyces pombe. RNA (New York, NY). 2001 May;7(5):671–681. PubMed PMID: 11350031; PubMed Central PMCID: PMCPmc1370119. eng.
- Lundgren K, Allan S, Urushiyama S, et al. A connection between pre-mRNA splicing and the cell cycle in fission yeast: cdc28+ is allelic with prp8+ and encodes an RNA-dependent ATPase/helicase. Mol Biol Cell. 1996 Jul;7(7):1083–1094. PubMed PMID: 8862522; PubMed Central PMCID: PMCPMC275960. eng.
- Bernard P, Maure JF, Partridge JF, et al. Requirement of heterochromatin for cohesion at centromeres. Science. 2001 Dec 21;294(5551):2539–2542. PubMed PMID: 11598266; eng.
- Dumesic PA, Natarajan P, Chen C, et al. Stalled spliceosomes are a signal for RNAi-mediated genome defense. Cell. 2013 Feb 28;152(5):957–968. PubMed PMID: 23415457; PubMed Central PMCID: PMCPMC3645481. eng.
- Bayne EH, Bijos DA, White SA, et al. A systematic genetic screen identifies new factors influencing centromeric heterochromatin integrity in fission yeast. Genome Biol. 2014;15(10):481. PubMed PMID: 25274039; PubMed Central PMCID: PMCPmc4210515. eng.
- Lee NN, Chalamcharla VR, Reyes-Turcu F, et al. Mtr4-like protein coordinates nuclear RNA processing for heterochromatin assembly and for telomere maintenance. Cell. 2013 Nov 21;155(5):1061–1074. PubMed PMID: 24210919; PubMed Central PMCID: PMCPMC3974623. eng.
- Marina DB, Shankar S, Natarajan P, et al. A conserved ncRNA-binding protein recruits silencing factors to heterochromatin through an RNAi-independent mechanism. Genes Dev. 2013 Sep 1;27(17):1851–1856. PubMed PMID: 24013500; PubMed Central PMCID: PMCPMC3778239. eng.
- Parsa JY, Boudoukha S, Burke J, et al. Polymerase pausing induced by sequence-specific RNA-binding protein drives heterochromatin assembly. Genes Dev. 2018 Jul 1;32(13–14):953–964. PubMed PMID: 29967291; PubMed Central PMCID: PMCPMC6075038. eng.
- Moreno S, Klar A, Nurse P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1991;194:795–823. PubMed PMID: 2005825; eng.
- Okazaki K, Okazaki N, Kume K, et al. High-frequency transformation method and library transducing vectors for cloning mammalian cDNAs by trans-complementation of Schizosaccharomyces pombe. Nucleic Acids Res. 1990 Nov 25;18(22):6485–6489. PubMed PMID: 2251111; PubMed Central PMCID: PMCPmc332599. eng.
- Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. PubMed PMID: 19261174; PubMed Central PMCID: PMCPmc2690996. eng.
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics (Oxford, England). 2009 May 01;25(9):1105–1111. PubMed PMID: 19289445; PubMed Central PMCID: PMCPmc2672628. eng.
- Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012 Mar 01;7(3):562–578. PubMed PMID: 22383036; PubMed Central PMCID: PMCPmc3334321. eng.
- Wang Y, Wagner JD, Guthrie C. The DEAH-box splicing factor Prp16 unwinds RNA duplexes in vitro. Curr Biol. 1998 Apr 09;8(8):441–451. PubMed PMID: 9550699; eng.
- He Y, Andersen GR, Nielsen KH. Structural basis for the function of DEAH helicases. EMBO Rep. 2010 Mar;11(3):180–186. PubMed PMID: 20168331; PubMed Central PMCID: PMCPmc2838688. eng.
- Arnold K, Bordoli L, Kopp J, et al. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics (Oxford, England). 2006 Jan 15;22(2):195–201. PubMed PMID: 16301204; eng.
- Laskowski RA, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J Mol Biol. 1993 Jun 20;231(4):1049–1067. PubMed PMID: 8515464; eng.
- Luthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992 Mar 05;356(6364):83–85. PubMed PMID: 1538787; eng.