
ABSTRACT
Most single-molecule techniques observing RNA in vitro or in vivo require fluorescent labels that have to be connected to the RNA of interest. In recent years, a plethora of methods has been developed to achieve site-specific labelling, in many cases under near-native conditions. Here, we review chemical as well as enzymatic labelling methods that are compatible with single-molecule fluorescence spectroscopy or microscopy and show how these can be combined to offer a large variety of options to site-specifically place one or more labels in an RNA of interest. By either chemically forming a covalent bond or non-covalent hybridization, these techniques are prerequisites to perform state-of-the-art single-molecule experiments.
Introduction
In the last decades, the view on the role of RNA inside cells has fundamentally changed. From its anticipated function as a mainly passive linker between genetic information (DNA) and enzymatic activity (proteins), in form of messenger-, ribosomal- and transfer-RNA, it is now known that RNA is actively involved in many other essential functions in all domains of life, such as gene regulation (e.g. riboswitches, microRNAs, small nucleolar RNAs) and enzymatic function (ribozymes). Even to date, new non-coding RNAs involved in many cellular processes are found. The fact that RNAs can be highly versatile molecules regarding structure and function led to the ‘RNA World’ hypothesis, which states that RNA may have evolved prior to DNA and proteins.
To obtain a complete understanding of life on the cellular level it is imperative to elucidate the function of RNA on a structural and dynamical basis. Experimental methods like crystallization, cryo-electron microscopy and nuclear magnetic resonance spectroscopy provided tremendous insights into the field of RNA. However, these techniques are to some extent based on ensemble type experiments or integrate over a large number of molecules. Thus, during or after the actual measurement minor populations might or will inevitably get lost by averaging over the ensemble of all populations. To completely understand the role of RNAs in cells it is not only necessary to understand how they function in an ensemble, but also on a single-molecule level, to see how even minor states and populations of RNA-complexes might influence the overall function of the cell.
In recent years several new approaches to investigate biomolecules have been established. Since the first observation of nucleic acids on a single molecular level [Citation1–Citation3] fluorescence-based methods became an effective tool for single-molecule studies. The fact that highly sensitive cameras and detectors became affordable in the last years, allowed researchers to gain new insights in biophysical and biochemical processes on a molecular sphere. Additionally, dyes were developed that showed significantly increased brightness and stability, further strengthening these approaches.
In single-molecule fluorescence spectroscopy, two general approaches have been established over the last 15 years. For localization of RNAs and other biomolecules inside the cell super-resolution microscopy (SRM) has become the state-of-the-art application [Citation4]. Structural changes within the complex of interest or between several molecules can be observed with the Förster Resonance Energy Transfer (FRET), which is also routinely performed on a single molecular level [Citation5].
In 2014, William Moerner, Eric Betzig and Stefan Hell were awarded with chemistry Nobel Prize for key contributions in the field of super-resolution microscopy [Citation6–Citation8]. In SRM the refraction limit of conventional light microscopy (~200 nm) can be surpassed by the introduction of fluorophores into macromolecules and utilization of various photophysical effects.
Popular SRM methods are Stimulated Emission Depletion Microscopy (STED), Photo-Activated Localization Microscopy (PALM) and Stochastic Optical Reconstruction Microscopy (STORM). All these methods allow highly resolved images (down to 10 nm) far beyond the refraction limit of light.
While SRM is used to localize certain molecules without any knowledge about the structure, FRET can be used to study dynamics, kinetics and structures of small or large biomolecules. FRET is a distance-dependent, radiationless energy transfer usually between two fluorophores (donor and acceptor) in close proximity (ca. 1–10 nm). The donor is excited by a laser and transfers its energy by dipole–dipole interaction to the acceptor. Since the efficiency of this energy transfer (E-FRET) is highly dependent on the distance of the dyes, structural data can be obtained by determination of E-FRET.
In the last decade, major advances have been made in the field of single-molecule fluorescence spectroscopy, making SRM, single-molecule FRET (smFRET) and other single-molecule methods much more controllable and manageable. An absolute prerequisite for these studies is efficient, specific, and robust fluorescent labelling of the molecules of interest. And while some of the methods require only one fluorescent label without restrictions on the placement of the label, multicolour and in particular FRET-based techniques require very precise, site-specific modification of the RNA construct. Therefore, prior to performing fluorescence-based methods on biomolecules, several considerations have to be made: Which label do I use, where can I label my sample, how can I label my sample, and does the label interfere with my sample? All these are major considerations which have to be taken care of before setting up fluorescence experiments.
On both the chemical and biochemical side, several key advances have been made in recent years, which provide a significant number of different approaches for the site-specific fluorescence labelling of biomolecules in general, and RNA in particular.
In this review we will try to provide a guideline for scientists on how to choose a suitable labelling strategy, and what approaches are available to perform robust and well-interpretable single-molecule experiments. The focus will of course be labelling of the RNA, but we will also touch on DNA and protein labelling, as many questions dictate the analysis of RNA-DNA or RNA-protein complexes.
Single-molecule spectroscopy
Super-resolution fluorescence microscopy
In SRM fluorophores are used for high resolution (below the Abbe diffraction limit) images, giving insides in cellular structures and processes that could not be obtained by regular light microscopy. To receive high resolved pictures two different approaches are commonly used. In STED the sample is scanned with a confocal laser and the effective excitation volume is reduced with a depletion laser [Citation9]. In PALM and STORM, the sample is illuminated on a wide field and the fluorophores stochastically switch between a dark and a bright state over a certain period of time [Citation3,Citation10]. Afterwards, the centre of each fluorophore is determined by computational analysis. These three methods allow live imaging of living cells at a time resolution of seconds for PALM and STORM or down to milliseconds for STED [Citation11,Citation12]. Overall, SRM is an ideal tool for localization and tracking biomolecules on a single molecular level and can readily be used in vivo [Citation13,Citation14].
Common methods for fluorophore labelling in SRM are the use of fusion proteins like GFP or antibody-based immunostaining. These approaches are targeted for proteins, but for a complete understanding of the molecular mechanisms in cells the selective labelling of other biomolecules, especially DNA and RNA, is essential. However, selective RNA labelling proves much more challenging than protein staining, mostly due to labelling selectivity.
Single-molecule FRET
A common tool for detection of dynamics and interactions in or between molecules is based on FRET, a radiationless energy transfer between two fluorophores (donor and acceptor) with overlapping emission and absorption spectra. The efficiency of this energy transfer is highly dependent on the distance of the two dyes. Hence, structural data – normally between 1 and 10 nm, varying with used dyes and their electrochemical surrounding – can be obtained by determination of E-FRET. FRET has been used in ensemble type experiments for decades and also single-molecule approaches have become very popular among scientists in the last years. In general, there are three ways to perform smFRET experiments; solution-type confocal, surface-based confocal laser scanning (CLSM) or total internal reflection fluorescence (TIRF) microscopy. In confocal smFRET a freely diffusing sample is observed. By measuring the donor and acceptor fluorescence in a short time frame E-FRET can be determined, and conclusions about structure, dynamical states or conformational changes in the range of the labels can be made. By confocal smFRET a time resolution below milliseconds is possible [Citation15]. However, usually, the molecules can only be observed for the short time window while they diffuse through the excitation beam. Hence, transition between states or conformational changes that take milliseconds to seconds – which are quite frequent in biomolecular processes – cannot be detected with this confocal method.
Long-term observation of single molecules and their dynamics can be achieved by wide-field excitation. To minimize background radiation and increase fluorophore lifetime TIRF microscopy is used. Hereby, the sample is immobilized on a glass slide and the excitation beam meets the glass in a critical angle that leads to total reflection. At this spot, an evanescent wave is created which excites the acceptor dyes, and FRET can be detected. For smFRET on a TIRF setup, the sample has to be immobilized, requiring an extra modification. In case of proteins, affinity tags like the His-tag can be used for immobilization [Citation16,Citation17], while for nucleic acids it is common to insert a covalently bound biotin molecule into the DNA or RNA, to form a highly stable biotin-streptavidin/neutravidin complex on the glass slide. Due to this immobilization, a larger number (up to several hundreds) of molecules can be observed simultaneously over a longer time period. Usually, videos from seconds to a few minutes are recorded when using this method, but under optimized conditions measurements for over an hour are possible [Citation18]. Even though recent progress in CMOS technology has made faster cameras available, compared to confocal smFRET microscopy the time resolution in TIRF microscopy is limited to the millisecond range, and additionally; modification of the labelled complex is required for immobilization [Citation19]. While most of the experiments are performed in vitro, recent progress has been made to also measure FRET inside living cells [Citation20], which comes at the expense of more elaborate delivery techniques, i.e. microinjection.
Two-colour coincidence detection
With two-colour coincidence detection, corresponding events of a complex comprised of two differently labelled molecules against a background of chance coincidence events of fluorescent unassociated molecules are detected. Rather than using one laser and detect FRET, both dyes are excited simultaneously with two different lasers. Due to this approach, the fluorophores do not need to be placed in FRET range of each other, but can rather be placed at any convenient labelling site within each of the molecules. This method is particularly useful when no structural data of the complex of interest is available. Since cross-talk between channels is minute, sub-femtomole quantities of the complex can be used due to high sensitivity. However, chance coincidence events need to be considered and subtracted from the measured data [Citation21,Citation22].
Chemoenzymatic labelling methods
For either of the above-mentioned approaches, the RNA has to be decorated with suitable fluorophores. For most localization approaches, the precise position of the fluorescent dye within the RNA is not crucial for the information derived (even though this ideally has to be verified experimentally). Additionally, commercially available options that are optimized for certain applications have become available, relieving the researcher of the task to prepare labelled RNA. However, especially for smFRET methods, the labelling position has to be chosen according to either an a priori structural and functional model or ideally based on available structural data. This immediately narrows down possible labelling sites or combinations thereof, as attachment of the fluorophores has to fulfil three main requirements: 1) Minimal structural and functional changes to the RNA under investigation (a point that is ideally proven by direct experimental comparison against an unlabelled RNA), 2) Suitability with respect to distance and changes in FRET efficiency upon conformational changes, and 3) minimal perturbations of photophysical properties of the fluorophores. As points 1) and 3) at least in part are also applicable to SRM, the following labelling approaches are in principle also suitable for these methodologies, even though most of them have not been employed until now.
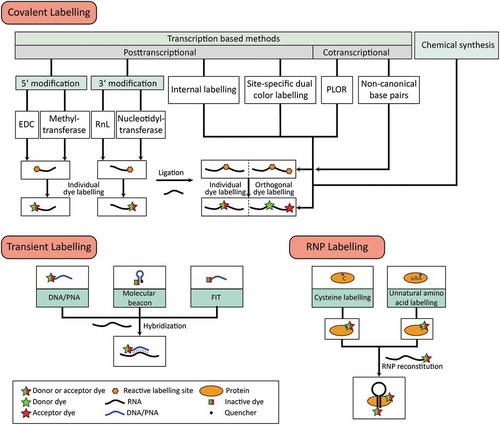
In order to have the most freedom to place the fluorescent dyes, we have collected a number of methods that allow to place fluorescent dyes (two or more) within one RNA molecule, ideally using chemical labelling strategies that are orthogonal and thus support position-specific labelling with the dye of choice. We also describe recently developed and available chemical coupling reactions, and list a number of commercially available dyes that have been frequently used in single-molecule approaches and are compatible with the methods described.
As many of the methods can also be used in a convergent synthesis-like strategy, provides a detailed overview about these methods and possible combinations thereof.
Figure 1. Overview of covalent and transient labelling techniques for RNA and RNP labelling. For each approach, the methods chosen can be combined to obtain RNAs or RNPs with multiple site-specific modifications.
Solid phase synthesis
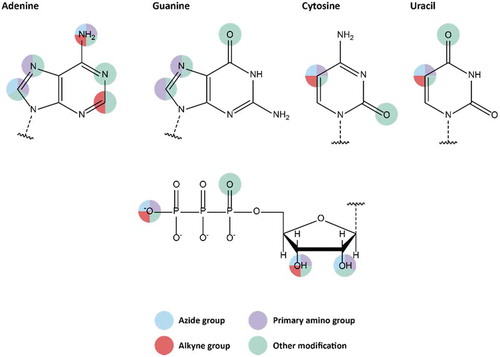
The most common chemical approach to introduce modifications into oligonucleotides is solid phase synthesis of the desired DNA or RNA strand. Step by step incorporation of modified and protected nucleosides via, e.g. phosphoramidite chemistry, allows its user to site-specifically insert modifications and labels at sugar, base or phosphate backbone of the nucleic acid (). However, due to side reactions taking place during solid phase synthesis, the length of the DNA or RNA strand is limited to ca. 100 bases [Citation23,Citation24].
Figure 2. Available modifiable building blocks for solid-phase synthesis. Attachment sites for all four natural nucleotides are shown, as well as attachment sites on the ribose moiety.
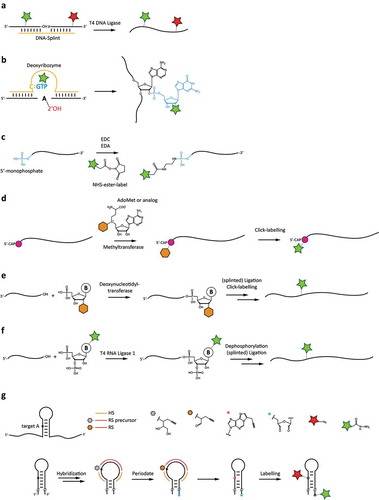
Splinted ligation
A well-established method for RNA modification is based on the RNA ligation ability of T4-DNA-ligase [Citation25–Citation27]. Therefore, the RNAs have to be hybridized to one complementary DNA strand and the hydroxyl at the 3ʹ-end of one RNA is ligated to the monophosphate at the 5ʹ-end of a second RNA (). Usage of splint supported T4-DNA-ligase for RNA ligation is well understood, and since ligation can only occur when the DNA:RNA-hybrid is formed, fewer side products are possible compared to standard RNA ligases.
Figure 3. Chemoenzymatic labelling techniques for site-specifically attaching fluorophore labels to RNAs. a: Splinted ligation of commercially synthesized, fluorophore-labelled RNAs. b: DNAzyme-mediated addition of modified GTP. c: 5ʹ-end-labeling using carbodiimide-based chemistry. d: 5ʹ-guanosine-labeling using the mRNA modifying methyltransferase with modified S-adenosylmethionine analogues. e: Labelling using a terminal deoxynucleotidyl transferase, and subsequent ligation. f: Internal labelling by 3ʹ-modification using a modified pNp substrate, and subsequent ligation. g: Dual-colour labelling using targeting strand for internal labelling, together with periodate-based functionalization of the 3ʹ-end.
Posttranscriptional labelling
The most immediate and most widespread method to obtain homogenous RNA is in vitro transcription. This method is fast and comparably cheap but does not immediately offer options for site-specific introduction of labels or modifiable residues. There are however several methods available that use transcribed RNA and offer means for site-specific posttranscriptional labelling.
After transcription via T7 RNA polymerase has been achieved and the required purification steps have been finished, the labelling methods described below can be used to modify an RNA strand at a desired position. The RNA can either be labelled internally or at the 5ʹ or 3ʹ end, offering a quite versatile set of tools for the strategic placement of labels. These methods focus on the use of different chemical and enzymatic reactions to achieve attachment of a fluorophore probe to an RNA.
Internal labelling with modified GTP
This method utilizes a deoxyribozyme as a functional single-stranded DNA catalyst (). The deoxyribozyme 10DM24 is able to attach mononucleotides to the ribose 2ʹ-hydroxyl group of internal adenosines of transcribed RNA. Its catalytic core contains a cytidine that recognizes guanosine triphosphate (GTP) via Watson-Crick base-pairing, and forms a three-helix junction with the substrate RNA via hybridization, placing the GTP in the middle of this complex. The GTP is covalently connected to the target adenosine via a 2ʹ,5ʹ-phosphodiester bond, releasing pyrophosphate. Using chemically modified GTPs, this approach can be used to attach internal fluorescent dyes or bioorthogonal groups that can then be ‘clicked’ with functional dye molecules. Alternatively, spin labels, cross-linkers and other structural probing agents can be added to the RNA [Citation28].
Carbodiimide-based functionalization
Functionalization of the 5ʹ-end of nucleic acids can be achieved by chemical modification with carbodiimides (). The most common representative is 1-Ethyl-3(3-dimethylaminopropyl)carbodiimide (EDC), due to its water solubility as hydrochloride salt. EDC can be used to activate carboxylic acids as well as organic phosphates [Citation29]. Thus, monophosphates are required at the 5ʹ-end for RNA modification. This can be accomplished either by doing a T7 in vitro transcription in excess of GMP to GTP or after transcription by dephosphorylation and subsequent phosphorylation. The 5ʹ-monophosphate end of the RNA can then be functionalized in a buffered one pot reaction with EDC, imidazole and ethylenediamine. This results in a primary amine modified phosphate, which can be used for further selective coupling with N-hydroxysulfosuccinimide (NHS ester). Alternatively, the monophosphate can be coupled directly to a label carrying a primary amine in the presence of EDC. While this approach is suitable for larger quantities of RNA, partial degradation of the RNA usually occurs during functionalization, making further purification necessary. Furthermore, this method is limited to modification of the free phosphate on the 5ʹ end, which has to be considered during construct design.
5ʹ-Cap labelling employing a methyltransferase
An efficient enzymatic labelling strategy for 5ʹ RNA labelling is the utilization of methyltransferases (). Under normal conditions, these enzymes transfer a methyl group from their co-substrate S-Adenosyl-L-methionine (AdoMet) to a target molecule. Cap (guanine-N7-)-methyltransferases specifically methylate the N7 position of the 5ʹ-cap of RNAs. Using artificial AdoMet analogues, a bioorthogonal functional group gets transferred to the 5ʹ-cap and can be used to couple fluorescent labels to the RNA, i.e. via click chemistry. Recent studies show [Citation30,Citation31] that the methyltransferase Ecm1 even transfers sterically demanding groups like vinylbenzyl- or azidobutenyl groups with good yield, while other methyltransferases struggle to incorporate such bulky groups. One problem that may arise is the abolishment of RNA-function after incorporation of bulky residues, which can impair the protein-binding abilities of the 5ʹ-cap. Since the 5ʹ-cap of the RNA is crucial for translation, this process is abrogated in most cases after the substitution of the methyl group.
3ʹ-Labelling employing a nucleotidyl transferase
Nucleotidyltransferases like terminal deoxynucleotidyl transferase (TdT) and poly(A) polymerase (PAP) can be used to enzymatically label the 3ʹ-end of a post-transcriptional RNA strand [Citation32]. This approach can be used to introduce 2ʹ- or 3ʹ-modified nucleotides which bear a reactive group that can be coupled with a fluorescent dye (). Directly introducing fluorophores with this method proved unsuccessful until now, presumably due to the sterical hindrance of the bulky dyes, which also may impede subsequent ligation steps. Introducing a nucleotide with an azide or alkyne group opens up the possibility to use click-chemistry to attach a dye to the 3ʹ RNA end directly or after splinted ligation with another RNA strand, 3ʹ-adapter ligation or addition of poly(A)-tails. Recent work shows that this method can be used to incorporate modifications of all four NTPs, but incorporation efficiency is different for each one [Citation33,Citation34]. However, a significant downside of this approach is the difficult control for incorporation of a single NTP; multiple nucleotides can get incorporated, even at low NTP concentration. To counter this problem, an adjustment of reaction time is possible. Additionally, using 3ʹ-modified NTPs completely blocks the incorporation of additional NTPs after the first one.
3ʹ-labelling with T4 RNA ligase 1
The enzyme T4 RNA ligase 1 can be used for single nucleotide extensions at the 3ʹ-end of a transcribed RNA [Citation32]. A nucleoside 3ʹ, 5ʹ-biphosphate, bearing a wide range of modifications at the sugar-, phosphate- or base-site, can be attached to the RNA (). The 3ʹ-phosphate group then blocks further incorporations, ensuring single nucleotide extension. In a recently developed approach, this 3ʹ-phosphate can be removed using shrimp alkaline phosphatase (rSAP) and the modified RNA can be purified with, e.g. RP-HPLC. Afterwards, a second RNA strand can be attached after dephosphorylation by means of splinted ligation, using either T4 DNA Ligase or RNA ligase 2. This way, a 3ʹ-modification can be turned into an internal modification. The method is relatively cost- and time-saving; it is however quite sensitive to reaction conditions, so in order to gain acceptable yields optimization steps are necessary. However, as either of the unmodified RNAs can be obtained from transcription or solid phase synthesis, this convergent approach offers a very broad range of modifications in RNAs and additionally renders long RNAs available for site-specific modifications [Citation35].
Site-specific dual colour labelling
A recently developed approach by the groups of Sigel and Freisinger allows in vitro dual colour labelling at the 3ʹ-end and at a specific cytosine or adenine (only reported for adenine so far) in a loop region of the RNA under oxidative conditions, yielding an RNA suitable for FRET experiments () [Citation36].
To label an adenosine of the in vitro transcribed btuB riboswitch (275mer) from Escherichia coli, they used a designed DNA reactive strand (RS) carrying a diol and a propargyl moiety as functional groups. RS is hybridized to the RNA with the help of two specific DNA helping strands (HS), leaving two nucleotides space between the specific and the functional group of RS. After hybridization of both RS and HS, the target adenine and the 3ʹ-end are activated by oxidation with sodium periodate. The diol group of RS is oxidized to an aldehyde, which reacts with the free amine of the target adenine attaching the functional propargyl group to the RNA, while the two hydroxyls (at the 2ʹC-3ʹC-bond) of the 3ʹ-end are oxidized to a dialdehyde. Both activated nucleotides can then be labelled orthogonally in a one-pot reaction. Fluorophores with an azide group can be coupled to the adenine via standard click reaction, while a hydrazide-modified label can be coupled to the 3ʹ-end.
Generally, this new dual labelling approach can be used for RNAs up to a few hundred base pairs (bp) in length (>300). In principle, any A or C within the RNA can be labelled with high positional specificity. After a first successful labelling, an additional second label could even be introduced at another A or C in the same manner, giving access to diverse labelling schemes in the desired RNA. The dialdehyde at the 3ʹ-end presents an additional option for further functionalization (e.g. immobilization).
One limitation of this technique is that labeling of the nucleobase employs both nitrogens at the Watson-Crick interface, preventing base pairing of the labelled nucleotide, and thus potentially interfering with structure formation. This has to be taken into account during construct design, and may render placement of the labels outside of helical regions less disruptive. It may have to be considered that periodate chemistry is used, which due to its oxidative nature might interfere with other modifications.
Cotranscriptional functionalization/modification
To date, most methods used to covalently label RNAs are applied posttranscriptionally or after solid phase synthesis. This of course requires additional experimental steps, i.e. coupling and purification, before the desired modified RNA is obtained. These additional steps can be time-consuming and may partially degrade the RNA, which inevitably leads to undesired impurities and reduced yields. Nevertheless, for shorter RNAs, posttranscriptional modification is well established and comparatively convenient. However, particularly for longer RNAs (several hundred nucleotides and more), it would be ideal to site-specifically modify the RNA during enzymatic production. For quite some time, it was attempted to implement ways to achieve site-specific labelling of RNAs during standard in vitro transcription. Here we will discuss two promising methods for modification of the RNA during transcription.
Genetic alphabet expansion
One approach to functionalize RNAs cotranscriptionally relies on the expansion of the genetic code during standard in vitro transcription. To achieve this, an unnatural base pair has to be incorporated into the DNA amplification and RNA transcription system. Since the first enzymatic incorporation of a new base pair into nucleic acids in 1989 [Citation37], many different non-canonical base pairs have been developed and introduced into nucleic acids [Citation38–Citation40]. Among many other potential unnatural base pairs, Romesberg et al. [Citation41,Citation42] discovered the hydrophobic dTPT3:dNaM () pair to be a highly promising candidate for genetic alphabet expansion, with low unspecific incorporation rates of the canonical bases to the unnatural base.
Figure 4. Cotranscriptional functionalization or modification approaches. a: Modification based on genetic code expansion using non-natural base pairs. b: Reaction scheme of the PLOR technique employing reinitiated transcription reactions. c: Integration of RNA sequences binding profluorophores for labelling.
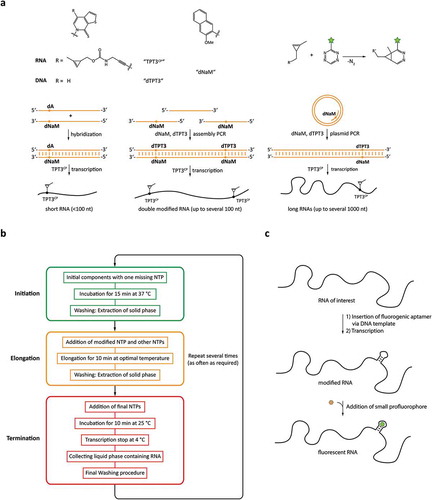
Based on the work of Romesberg et al. Kath-Schorr and coworkers synthesized a methyl cyclopropene-modified unnatural triphosphate (TPT3CP) for site-specific incorporation during in vitro transcription and subsequent fluorophore labelling [Citation43]. Prior to transcription with the modified unnatural RNA base, dNaM has to be introduced site specifically into the desired DNA template. For short RNAs (<100 nt) dNaM can be introduced into the DNA template by solid phase synthesis at any position. For standard T7 transcription hybridization of the modified template to a complementary DNA strand with dA opposing to dNaM is sufficient. To generate the transcription template of a long non-coding RNA (401 nt) with one modification site a dNaM modified primer was used in a plasmid PCR in the presence of dTPT3 triphosphate. For a 185mer with two modification sites, assembly PCR with two different oligomers containing dNaM and a third unmodified oligomer was performed. In vitro transcriptions were done with T7 polymerase in the presence of TPT3CP. RNA labelling was achieved with a copper-free click reaction between the methyl cyclopropane group of TPT3CP and tetrazine-modified fluorophore. In theory, other orthogonal labelling systems can be used for this RNA modification approach. To our knowledge, so far only one or several fluorophores of the same type have been introduced into RNA by genetic alphabet expansion. In theory, it should however also be possible to introduce two different non-canonical base pairs into the DNA template by assembly PCR with subsequent transcription of a dually modified RNA. By carefully choosing the two reactive groups an orthogonal labelling system for two different fluorophores can be achieved, rendering this technique attractive for FRET users, working with longer RNAs that cannot be synthesized by splinted ligation. Furthermore, the required enzymes needed for this labelling approach are standard equipment in every RNA laboratory. However, the access to the modified unnatural bases is a significant hindrance since a commercialized system is not yet available.
Position-selective labelling of RNA (PLOR)
Another approach to modify RNAs cotranscriptionally is based on the ‘pause-restart’ ability of T7 RNA polymerase. Pausing of transcription elongation has been investigated for some years [Citation44–Citation46]. Based on the ability to stall RNA elongation by exclusion of an NTP the Stagno and Wang groups developed a hybrid solid-liquid phase method for site-selective RNA synthesis during in vitro transcription () [Citation47,Citation48]. The whole process is carried out on an automated platform able to exchange the liquid phase during synthesis. DNA templates are immobilized on beads (solid phase) via biotin–streptavidin interaction. The transcription is initiated in absence of one specific NTP, leading to a stable DNA-RNA-polymerase complex stalled at the position of the missing NTP. The solid phase is extracted several times for complete removal of the initial building blocks. This step is critical since incomplete removal of NTPs leads to unlabelled full-length transcription of the DNA template. Elongation is done by addition of the missing NTP (and up to two other NTPs). This can be any modified base adequate for incorporation by T7 polymerase. At the next pause site, the washing procedure is repeated. The elongation process can be repeated several times until all desired modifications are inserted, and the RNA has been transcribed to its desired length. For termination, the reaction vessel is cooled down and the liquid phase is collected. Reinitiation can be prevented by the addition of an NTP mix without GTP or addition of heparin – a well-known T7 polymerase initiation inhibitor – at the last elongation cycle. To increase RNA yield, this process can be repeated several times with the same bead-bound DNA.
PLOR allows its user to highly modify RNAs during in vitro transcription. A distinct advantage of this new method is that it can be used for short and long RNAs in the same manner. Similar to solid phase synthesis, in PLOR single or multiple modifications are introduced right during RNA construction. Purification of the modified RNA is performed like in any other in vitro transcription, without greater effort.
When doing PLOR several things have to be considered. For selective positioning, the chosen labelling sites must not have consecutive bases (e.g. AA or CCC). Otherwise, the modified nucleotide would be incorporated at undesired positions. The used NTPs have to be highly purified, since even minor impurities of another NTP will cause the polymerase to keep transcribing instead of pausing. Overall PLOR is a promising method for synthesis of long modified RNAs that cannot be generated by solid-phase synthesis and other techniques. However, this method either requires an automated platform or a lot of handiwork, rendering it difficult for many labs. If becoming commercially available, PLOR might be the method of choice for the synthesis of modified long RNAs in the near future.
Fluorogenic aptamers
Visualization of fluorescently labelled RNAs in vivo has been challenging for researchers for years. For observation of proteins inside cells, different methods have been well established. One popular method is the use of fusion proteins, green fluorescent protein (GFP) and its derivatives have been widely used to label and track proteins in vivo [Citation49–Citation51]. In contrast, imaging RNA in cellular environments is rather difficult and only few tools are available. Fluorescence in situ hybridization (FISH) – explained in a chapter below – is one of the most popular among them, but requires invasive methods like microinjection, electroporation or membrane permeabilization via cell penetrating substances for in vivo imaging, which may lead to cell damages. Further drawbacks of FISH may be inhomogeneous probe distribution and inefficient annealing to the target RNA [Citation52].
For RNAs, a similar minimally invasive approach is based on the use of fluorogenic RNA aptamers. They bind and drastically enhance the fluorescence of small profluorophores [Citation53], and can be inserted to the RNA of interest by cloning, allowing transcription of the RNA with the additional aptamer domain right inside the cell (). Furthermore, the small organic dyes required for staining are much more cell permeable compared to the relatively large fluorescent probes used in FISH.
Since their first discovery in 2003 [Citation54] fluorescent RNA aptamers had to surpass several challenges, like RNA folding, low binding affinity of the fluorophore to the RNA, cell toxicity – in case of the malachite green aptamer [Citation54] – or the development of bright GFP-like fluorescent systems with high contrast between bound and unbound state of the fluorophore to the aptamer [Citation55]. In recent years several fluorescent RNA aptamers have been reported that overcome most of these challenges; namely the Spinach [Citation55], Broccoli [Citation56] and Mango [Citation57] systems, all of which have been used for in vitro and in vivo studies and the Mango aptamer was already detected on single molecular level [Citation57]. For example, a tandem array of the Spinach aptamer was used to study mRNAs in living bacterial cells [Citation58], and an RNA-FRET system based on the aptamers Mango and Spinach was expressed in E. coli and showed that such RNA aptamer-FRET devices can be used to study conformational changes of RNAs in vivo [Citation59]. Especially the recently developed aptamers Mango II – IV, which bind the small dyes TO1-biotin (excitation 510 nm) and TO3-biotin (excitation 637 nm), show dissociation constants in the low nanomolar range and partially even higher fluorescence than enhanced GFP, rendering this aptamer group a highly interesting tool for RNA research.
When using fluorogenic aptamers placement of the dye binding domain should be well reasoned in regard to the complex of interest. While for large RNAs like mRNAs introduction of aptamers should be less invasive, especially for small RNAs the insertion of a large aptamer domain with several dozen nucleotides might interfere with the function of the RNA. Nevertheless, the relatively small Mango aptamer has been used to image small non-coding RNAs in mammalian cells [Citation60], suggesting fluorogenic aptamers as a promising tool for in vivo studies of single RNA molecules.
Transient labelling methods
In transient labelling methods, not the RNA of interest, but rather a molecule that transiently binds to the target RNA is coupled with a spectroscopic probe (). Fluorescence in situ hybridization (FISH) techniques are common methods to visualize RNA of a known sequence. The basic idea of these techniques is the utilization of a nucleic acid strand complementary to the RNA of interest. This nucleic acid is labelled with a fluorophore and hybridizes to the RNA via Watson-Crick base pairing. Here, some transient labelling methods utilizing FISH techniques are presented.
Hybridization with deoxyribonucleic acids (DNAs)
A transient labelling technique is the hybridization of a short DNA oligomer carrying a fluorophore to the complementary RNA of interest. Preferred sites for hybridization are internal loops in the RNA, which may artificially be inserted into the RNA. To achieve sequence-specific hybridization, the RNA/DNA duplex should at least contain 12 base pairs, although a higher number of base pairs may be required to tune both specificity and stability. It is important that the RNA loop chosen for DNA hybridization is not involved in the formation of the tertiary RNA structure, so as to not interrupt folding and functional activity of the RNA upon DNA binding. Two wild type loops can be replaced by different artificial loops, to achieve binding of differently labelled DNA oligomers to either loop [Citation61].
Hybridization with peptide nucleic acids (PNAs)
A similar approach to hybridizing a short fluorescently labelled DNA strand to the RNA strand of interest is the use of PNAs. These non-natural, nuclease-resistant analogues of DNA contain an uncharged polyamide backbone instead of a sugar-phosphate backbone and bind much more strongly to RNA, even at low salt concentrations. Due to this stronger hybridization, shorter oligomers can be used, which in turn relieves the need of RNA-modification. This positively affects the labelling strategy, since more flexibility is possible in choosing an appropriate labelling site and the RNA is less disturbed in its structure as well as folding pathway. Another advantage of the high binding affinity is the use of moderate annealing temperatures, which can prevent decomposition of the fluorophore label. As only an equimolar amount of PNA is required for efficient hybridization, the amount of unbound labelled nucleic acid is reduced, resulting in decreased background fluorescence. As PNAs have a higher mismatch sensitivity than DNAs, labelling is more selective. Hybridization of PNAs gives similar spectroscopic results as with hybridization of DNAs, with the only apparent disadvantage being availability of such PNAs [Citation62].
Hybridization with molecular beacons
Molecular beacons can consist of either DNA or PNA. DNA molecular beacons consist of an internal probe sequence, flanked by two complementary sequences forming a helix. At the end of this helix, a fluorophore as well as a suitable quencher are attached to either strand. The DNA forms a stem-loop structure while in solution, effectively putting the 5ʹ- and 3ʹ-ends into close proximity, which quenches the fluorescence of the dye. In some cases, DNA hybridization can be enforced by introducing bridged LNA nucleotides. PNA molecular beacons on the other hand fold on themselves due to their strong hydrophobic properties and do not require a complementary sequence to form stem-loop structures. Additionally, opposite charges of amino acids on the 5ʹ- and 3ʹ-end can further ensure that fluorophore and quencher are put into close proximity. Since a part of the molecular beacon sequence is complementary to the RNA of interest, upon the presence of the target RNA, the molecular beacon stem-loop is opened and the molecular beacon hybridizes with the RNA. Since dye and quencher are now spatially maximally separated, the dye emits fluorescence. Due to this only-upon-binding fluorescence, background fluorescence is lowered and posthybridization washing steps to remove unbound probes are avoided. PNA molecular beacons have some advantages over DNA molecular beacons due to reasons already explained: faster hybridization, reduced non-specific binding, and higher signal-to-noise ratio [Citation63,Citation64]
Hybridization with forced intercalation (FIT)-probes
The forced intercalation method relies on DNA or PNA single strands, in which a certain nucleobase is replaced by an intercalator dye. Upon hybridization with a target RNA molecule, the dye intercalates between the Watson-Crick base pairs of the hybrid, and fluorescence is emitted. Some requirements for these probes are a high responsiveness of fluorescence, especially if unbound probes cannot be washed out from the sample. Also, high brightness aids detection against an autofluorescent background. Commonly used fluorophores for this method are dyes of the thiazole orange (TO) family, but also dyes that emit in the blue and green spectra have been used for FIT. There are also TO dyes, which can discriminate between full and single-mismatched hybridization adjacent to the intercalation site. Incorporating two spectrally overlapping fluorophores can increase the signal brightness [Citation65–Citation67].
Chemical coupling approaches
As nucleic acid constructs for several approaches have become commercially available, the need for chemical coupling steps to be performed by the researcher has significantly decreased. For the applications where coupling is still to be performed, approaches based on several different coupling chemistries are available, as most of these methods are based on the incorporation of a bioorthogonal reactive group rather than the direct incorporation of a fluorophore. This approach has some advantages, since the dye may interfere with further reactions, i.e. ligation [Citation33]. With the incorporation of a relatively small reactive group into the RNA strand, the RNA structure does not get perturbed and the RNA can be processed normally. Afterwards, a dye molecule bearing a complementary reactive group can be coupled with the RNA strand. The chemical reactions employed are compatible with both RNA folding and stability (). The [3 + 2]-cycloaddition between an azide and an alkyne moiety is a popular ‘click’ reaction often used in chemical biology as well as organic chemistry [Citation68]. The coupling of the 1,3-dipolar with the dipolarophile has a strong thermodynamic driving force and forms a 1,2,3-triazole [Citation69,Citation70]. The alkyne and azide reactive groups can either be linked to the RNA or the dye molecule; building blocks for either SPS or transcription are available, as well as suitable dye derivatives (see ).
Table 1. Commercially available fluorescent dye for different modification strategies and chemical coupling approaches.
Figure 5. Chemical coupling reactions for fluorophore attachment to RNAs. a and b: Cu-catalyzed 3 + 2-cycloaddition, with azide moiety either in the RNA or on the fluorophore. c: 3 + 2-cycloaddition with strained alkyne. d: Cycloaddition using a strained diene (i.e. norbornene) and tetrazine. e: N-hydroxy-succinimide coupling to primary amines.
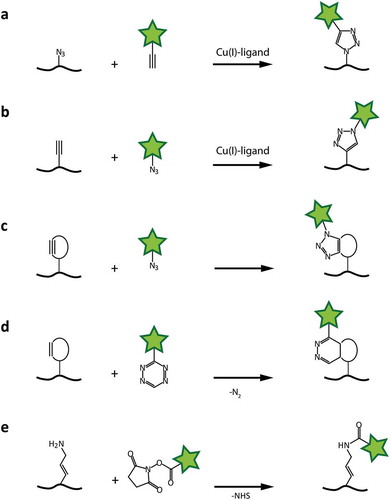
One variation of this reaction is the copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) [Citation71,Citation72]. The copper(I), in biological systems usually derived from reduced copper(II)-sulphate, forms a complex with a ligand like THPTA or TBTA to prevent reoxidation. The azide and alkyne supposedly form an intermediate 6-ring-complex with the copper(I), before a 5-ring is formed and the catalytic copper is released [Citation71].
Another form of this reaction is the strain-promoted azide-alkyne cycloaddition (SPAAC) [Citation73,Citation74]. It utilizes the geometric deformation of an alkyne in a closed ring system [Citation75]. The normally linear geometry of the triple bond cannot be maintained in a closed system, which puts a heavy strain onto the ring [Citation73,Citation76]. This strain is sufficient to act as activation energy for the cycloaddition, in which the more favourable ring geometry is restored. The reaction acts without any form of metal catalyst, making it convenient for biochemical applications [Citation77–Citation79].
The reaction of a strained dienophile with a 1,2,4,5-tetrazine provides a very fast bioorthogonal coupling method. This strain-promoted inverse electron-demand Diels-Alder cycloaddition (SPIEDAC) also utilizes the strain of cyclic alkenes and alkynes. Due to the fast reaction and low toxicity, this method is especially useful for in vivo studies [Citation80].
Primary amine groups can be introduced internally, as well as at the 5ʹ-end of RNAs via EDC functionalization. These groups can be coupled with NHS ester-activated dye molecules under physiologic conditions. A stable amide bond is formed with the release of NHS. With the addition of a sulphonate group to the NHS ester, water solubility can be increased. However, NHS esters are prone to hydrolysis in aqueous solution over time, while higher buffer pH values and temperature increase the rate of hydrolysis. Specificity for aliphatic amines over aromatic ones (which is present in every nucleobase) is achieved by elevating the pH of the reaction mixture.
Labelling RNA-binding proteins
Ribonucleoproteins (RNPs) consist of an RNA and one or several RNA-binding proteins (RBPs). They span all domains of life and are involved in diverse roles, including regulation, replication and post-transcriptional modifications [Citation81]. Due to the strong complex formation with their corresponding RNA, RBPs make up for a useful labelling target for fluorophore probes. Labelling of RBPs allows studying of their interaction with RNA. Combining labelled proteins with labelled RNA allows monitoring of the complex subunits, giving insight on binding, structure and dynamics of complex formation and enzymatic activity of the reconstituted complex. Here, some standard methods of protein labelling will be explained. Analogous to RNA labelling, CuAAC, SPAAC and SPIEDAC reactions can be employed for proteins.
Coupling of cysteine residues
A well-established method is the labelling of cysteine residues within a protein. The thiol residue of this amino acid makes for a coupling site, using maleimide dye derivatives, disulphide or haloacetyl compounds. The cysteine needs to be situated on the protein surface, thus being solvent-accessible. Either a native or via site-directed mutagenesis inserted cysteine can be used for labelling. Although cysteine is relatively rare in the proteome, additional native cysteines in the protein sometimes need to be mutated to achieve site-specific labelling, preferably without perturbation of the proteins folding and function [Citation82–Citation85].
Since cysteines tend to form intramolecular disulphide bonds, rendering them inactive to coupling reactions, the protein needs to be available in a reduced state, using additives like 2-ME, TCEP or DTT. However, these agents must be removed before conjugation with a coupling dye, to prevent competition between protein and additive thiol groups. An interesting approach to circumvent the reduction problematic is the use of solid-phase-based labelling, where the proteins are precipitated and the coupling reaction is performed with reduced proteins in the solid state [Citation83].
Incorporation and coupling of unnatural amino acids
Genetic code expansion is a powerful tool to incorporate unnatural amino acids (uAAs) into proteins at a desired position. For this method, a biorthogonal aminoacyl-tRNA synthetase (aaRS)/tRNA pair needs to be introduced into the host cell [Citation86–Citation88]. For bioorthogonality, the aaRS should not be able to charge endogenous tRNAs with uAAS, nor should the introduced tRNA be charged with canonical amino acids. To achieve these conditions, the amber codon suppression system for pyrrolysine from Methanococcus jannaschii, Methanosarcina mazei or Methanosarcina barkeriis is often used [Citation79,Citation88–Citation91]. The PylRS/tRNAPyl recognizes the amber stop codon UAG (the least abundant termination codon in E. coli) as a sense codon and can incorporate a wide spectrum of uAAs into the peptide chain at a desired, with site-directed mutagenesis introduced, labelling site. These uAAs can bear a reactive side chain, like an alkyne, strained alkyne or strained alkene. With an azide dye, the protein can then be labelled in a copper-catalyzed or strain promoted alkyne-azide cycloaddition, with a tetrazine dye a strain-promoted inverse electron-demand Diels-Alder cycloaddition can be performed [Citation92].
Combination with transient DNA labelling via click-paint
Attaching a docking strand DNA to a protein gives the opportunity to then label the docking strand with a dye-labelled imaging strand DNA. For this purpose, antibody-based immunocytochemistry was used to bind a biotinylated antibody to a protein, which in turn was able to bind a biotinylated docking strand via a streptavidin linker. Some problems can arise due to this labelling method because of the relatively large size of antibodies. A more recent method utilizes an incorporated unnatural amino acid in a protein to ‘click’ a functionalized docking strand to the protein. This docking strand can then be hybridized with a large number of imaging strands [Citation93,Citation94].
Summary
Novel strategies to introduce site-specific modifiers into RNA that have been developed over the last years can be used to covalently or transiently introduce fluorescent dyes into virtually any RNA of interest. While not all of these strategies have already been applied to single-molecule spectroscopy yet, any technique that overcomes the size limitation imposed by classic solid phase synthesis is a valuable tool when site-specific modifications are required. As most of the enzymatic tools can easily be combined with other chemical or chemoenzymatic approaches (see ), the labelling schemes that have become possible are much more diverse than using any technique alone. This also holds true for experimental approaches that work under near-native conditions. Furthermore, the last years have seen experimental scenarios that require both RNA and protein labelling, which has been aided by developments that again allow mild labelling conditions. Looking at fluorescent dyes, the diversity regarding coupling chemistry of commercially available fluorophores has kept pace with newly developed approaches. With these new methods, and combinations with established protocols, the experimental possibilities in single-molecule techniques investigating RNA as well as RNPs are now greater than ever.
Acknowledgments
This work has been supported by the DFG, CRC 902 “Molecular Principles of RNA-based regulation”. M.H. is a member of the Cluster of Excellence EXC115 “Macromolecular complexes in action”. The authors thank Prof. Harald Schwalbe for constant support.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Ha T, Enderle T, Ogletree DF, et al. Probing the interaction between two single molecules: fluorescence resonance energy transfer between a single donor and a single acceptor. Proc Natl Acad Sci U S A. 1996;93:6264–6268.
- Russell R, Zhuang X, Babcock HP, et al. Exploring the folding landscape of a structured RNA. Proc Natl Acad Sci U S A. 2002;99:155–160.
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods. 2006;3:793–795.
- Huang B, Bates M, Zhuang X. Super-resolution fluorescence microscopy. Annu Rev Biochem. 2009;78:993–1016.
- Zhuang X. Single-molecule RNA science. Annu Rev Biophys Biomol Struct. 2005;34:399–414.
- Betzig E. Single molecules, cells, and super-resolution optics (nobel lecture). Angew Chem. 2015;54:8034–8053.
- Hell SW. Nanoscopy with focused light (nobel lecture). Angew Chem. 2015;54:8054–8066.
- Moerner WE. Single-molecule spectroscopy, imaging, and photocontrol: foundations for super-resolution microscopy (nobel lecture). Angew Chem. 2015;54:8067–8093.
- Hell SW, Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Lett. 1994;19:780–782.
- Betzig E, Patterson GH, Sougrat R, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645.
- Hanne J, Zila V, Heilemann M, et al. Super-resolved insights into human immunodeficiency virus biology. FEBS Lett. 2016;590:1858–1876.
- Sydor AM, Czymmek KJ, Puchner EM, et al. Super-resolution microscopy: from single molecules to supramolecular assemblies. Trends Cell Biol. 2015;25:730–748.
- Ishigaki M, Iketani M, Sugaya M, et al. STED super-resolution imaging of mitochondria labeled with TMRM in living cells. Mitochondrion. 2016;28:79–87.
- Spahn C, Glaesmann M, Gao Y, et al. Sequential super-resolution imaging of bacterial regulatory proteins: the nucleoid and the cell membrane in single, fixed E. coli cells. Methods Mol Biol. 2017;1624:269–289.
- Kim JY, Kim C, Lee NK. Real-time submillisecond single-molecule FRET dynamics of freely diffusing molecules with liposome tethering. Nat Commun. 2015;6:6992.
- Pal P, Lesoine JF, Lieb MA, et al. A novel immobilization method for single protein spFRET studies. Biophys J. 2005;89:L11–L13.
- Lata S, Reichel A, Brock R, et al. High-affinity adaptors for switchable recognition of histidine-tagged proteins. J Am Chem Soc. 2005;127:10205–10215.
- Hengesbach M, Kim NK, Feigon J, et al. Single-molecule FRET reveals the folding dynamics of the human telomerase RNA pseudoknot domain. Angew Chem. 2012;51:5876–5879.
- Roy R, Hohng S, Ha T. A practical guide to single-molecule FRET. Nat Methods. 2008;5:507–516.
- Sustarsic M, Kapanidis AN. Taking the ruler to the jungle: single-molecule FRET for understanding biomolecular structure and dynamics in live cells. Curr Opin Struct Biol. 2015;34:52–59.
- Ashbridge B, Orte A, Yeoman JA, et al. Single-molecule analysis of the human telomerase RNA.dyskerin interaction and the effect of dyskeratosis congenita mutations. Biochemistry. 2009;48:10858–10865.
- Orte A, Clarke R, Balasubramanian S, et al. Determination of the fraction and stoichiometry of femtomolar levels of biomolecular complexes in an excess of monomer using single-molecule, two-color coincidence detection. Anal Chem. 2006;78:7707–7715.
- Helm M, Porcher S. Chemical synthesis of DNA and RNA containing modified nucleotides. In: Grosjean H, editor. DNA and RNA modification enzymes: comparative structure, mechanism, functions, cellular interactions and evolution. Austin: Landes Bioscience; 2009. p. 550–559.
- Scaringe SA, Wincott FE, Caruthers MH. Novel RNA synthesis method using 5‘-O-Silyl-2‘-O-orthoester protecting groups. J Am Chem Soc. 1998;120:11820–11821.
- Akiyama BM, Stone MD. Assembly of complex RNAs by splinted ligation. Methods Enzymol. 2009;469:27–46.
- Hengesbach M, Kobitski A, Voigts-Hoffmann F, et al. RNA intramolecular dynamics by single-molecule FRET. Curr Protoc Nucleic Acid Chem. 2008;34: Chapter 11:Unit 11 2.
- Kershaw CJ, O’Keefe RT. Splint ligation of RNA with T4 DNA ligase. Methods Mol Biol. 2012;941:257–269.
- Buttner L, Javadi-Zarnaghi F, Hobartner C. Site-specific labeling of RNA at internal ribose hydroxyl groups: terbium-assisted deoxyribozymes at work. J Am Chem Soc. 2014;136:8131–8137.
- Nakajima N, Ikada Y. Mechanism of amide formation by carbodiimide for bioconjugation in aqueous media. Bioconjug Chem. 1995;6:123–130.
- Holstein JM, Anhauser L, Rentmeister A. Modifying the 5ʹ-Cap for click reactions of eukaryotic mRNA and to tune translation efficiency in living cells. Angew Chem. 2016;55:10899–10903.
- Muttach F, Masing F, Studer A, et al. New AdoMet analogues as tools for enzymatic transfer of photo-cross-linkers and capturing RNA-Protein interactions. Chemistry. 2017;23:5988–5993.
- Hartmann RK, Bindereif A, Schön A, et al. Handbook of RNA biochemistry. Weinheim: Wiley-VCH; 2015.
- Winz ML, Samanta A, Benzinger D, et al. Site-specific terminal and internal labeling of RNA by poly(A) polymerase tailing and copper-catalyzed or copper-free strain-promoted click chemistry. Nucleic Acids Res. 2012;40:e78.
- Martin G, Keller W. Tailing and 3ʹ-end labeling of RNA with yeast poly(A) polymerase and various nucleotides. Rna. 1998;4:226–230.
- Keyhani S, Goldau T, Blumler A, et al. Chemo-enzymatic synthesis of position-specifically modified RNA for biophysical studies including light control and NMR spectroscopy. Angew Chem. 2018;57:12017–12021.
- Zhao M, Steffen FD, Borner R, et al. Site-specific dual-color labeling of long RNAs for single-molecule spectroscopy. Nucleic Acids Res. 2018;46:e13.
- Piccirilli JA, Krauch T, Moroney SE, et al. Enzymatic incorporation of a new base pair into DNA and RNA extends the genetic alphabet. Nature. 1990;343:33–37.
- Malyshev DA, Romesberg FE. The expanded genetic alphabet. Angew Chem. 2015;54:11930–11944.
- Seo YJ, Matsuda S, Romesberg FE. Transcription of an expanded genetic alphabet. J Am Chem Soc. 2009;131:5046–5047.
- Someya T, Ando A, Kimoto M, et al. Site-specific labeling of RNA by combining genetic alphabet expansion transcription and copper-free click chemistry. Nucleic Acids Res. 2015;43:6665–6676.
- Dhami K, Malyshev DA, Ordoukhanian P, et al. Systematic exploration of a class of hydrophobic unnatural base pairs yields multiple new candidates for the expansion of the genetic alphabet. Nucleic Acids Res. 2014;42:10235–10244.
- Li L, Degardin M, Lavergne T, et al. Natural-like replication of an unnatural base pair for the expansion of the genetic alphabet and biotechnology applications. J Am Chem Soc. 2014;136:826–829.
- Eggert F, Kulikov K, Domnick C, et al. Iluminated by foreign letters - Strategies for site-specific cyclopropene modification of large functional RNAs via in vitro transcription. Methods. 2017;120:17–27.
- Edenberg ER, Downey M, Toczyski D. Polymerase stalling during replication, transcription and translation. Curr Biol. 2014;24:R445–R452.
- Guo Q, Nayak D, Brieba LG, et al. Major conformational changes during T7RNAP transcription initiation coincide with, and are required for, promoter release. J Mol Biol. 2005;353:256–270.
- Lyakhov DL, He B, Zhang X, et al. Pausing and termination by bacteriophage T7 RNA polymerase. J Mol Biol. 1998;280:201–213.
- Liu Y, Holmstrom E, Zhang J, et al. Synthesis and applications of RNAs with position-selective labelling and mosaic composition. Nature. 2015;522:368–372.
- Liu Y, Holmstrom E, Yu P, et al. Incorporation of isotopic, fluorescent, and heavy-atom-modified nucleotides into RNAs by position-selective labeling of RNA. Nat Protoc. 2018;13:987–1005.
- Dedecker P, Mo GC, Dertinger T, et al. Widely accessible method for superresolution fluorescence imaging of living systems. Proc Natl Acad Sci U S A. 2012;109:10909–10914.
- Moen I, Jevne C, Wang J, et al. Gene expression in tumor cells and stroma in dsRed 4T1 tumors in eGFP-expressing mice with and without enhanced oxygenation. BMC Cancer. 2012;12:21.
- Tiwari DK, Nagai T. Smart fluorescent proteins: innovation for barrier-free superresolution imaging in living cells. Dev Growth Differ. 2013;55:491–507.
- Bao G, Rhee WJ, Tsourkas A. Fluorescent probes for live-cell RNA detection. Annu Rev Biomed Eng. 2009;11:25–47.
- Warner KD, Chen MC, Song W, et al. Structural basis for activity of highly efficient RNA mimics of green fluorescent protein. Nat Struct Mol Biol. 2014;21:658–663.
- Babendure JR, Adams SR, Tsien RY. Aptamers switch on fluorescence of triphenylmethane dyes. J Am Chem Soc. 2003;125:14716–14717.
- Paige JS, Wu KY, Jaffrey SR. RNA mimics of green fluorescent protein. Science. 2011;333:642–646.
- Filonov GS, Moon JD, Svensen N, et al. Broccoli: rapid selection of an RNA mimic of green fluorescent protein by fluorescence-based selection and directed evolution. J Am Chem Soc. 2014;136:16299–16308.
- Dolgosheina EV, Jeng SC, Panchapakesan SS, et al. RNA mango aptamer-fluorophore: a bright, high-affinity complex for RNA labeling and tracking. ACS Chem Biol. 2014;9:2412–2420.
- Zhang J, Fei J, Leslie BJ, et al. Tandem spinach array for mRNA imaging in living bacterial cells. Sci Rep. 2015;5:17295.
- Jepsen MDE, Sparvath SM, Nielsen TB, et al. Development of a genetically encodable FRET system using fluorescent RNA aptamers. Nat Commun. 2018;9:18.
- Autour A, Jeng S, Cawte A, et al. Fluorogenic RNA Mango aptamers for imaging small non-coding RNAs in mammalian cells. Nat Commun. 2018;9:656.
- Smith GJ, Sosnick TR, Scherer NF, et al. Efficient fluorescence labeling of a large RNA through oligonucleotide hybridization. Rna. 2005;11:234–239.
- Schmitz AG, Zelger-Paulus S, Gasser G, et al. Strategy for internal labeling of large RNAs with minimal perturbation by using fluorescent PNA. Chembiochem Eur J Chem Biol. 2015;16:1302–1306.
- Xi C, Balberg M, Boppart SA, et al. Use of DNA and peptide nucleic acid molecular beacons for detection and quantification of rRNA in solution and in whole cells. Appl Environ Microbiol. 2003;69:5673–5678.
- Zheng J, Yang R, Shi M, et al. Rationally designed molecular beacons for bioanalytical and biomedical applications. Chem Soc Rev. 2015;44:3036–3055.
- Hovelmann F, Gaspar I, Ephrussi A, et al. Brightness enhanced DNA FIT-probes for wash-free RNA imaging in tissue. J Am Chem Soc. 2013;135:19025–19032.
- Kohler O, Jarikote DV, Seitz O. Forced intercalation probes (FIT Probes): thiazole orange as a fluorescent base in peptide nucleic acids for homogeneous single-nucleotide-polymorphism detection. Chembiochem Eur J Chem Biol. 2005;6:69–77.
- Mannack LV, Eising S, Rentmeister A. Current techniques for visualizing RNA in cells. F1000Res. 2016;5:775.
- Kolb HC, Finn MG, Sharpless KB. Click chemistry: diverse chemical function from a few good reactions. Angew Chem. 2001;40:2004–2021.
- Huisgen R. 1,3‐dipolar cycloadditions. Past and future. Angew Chem. 1963;2:565–632.
- Huisgen R. Kinetics and reaction mechanisms: selected examples from the experience of forty years. Pure Appl Chem. 1989;61:613–628.
- Rostovtsev VV, Green LG, Fokin VV, et al. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem. 2002;41:2596–2599.
- Tornoe CW, Christensen C, Meldal M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J Org Chem. 2002;67:3057–3064.
- Agard NJ, Prescher JA, Bertozzi CR. A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J Am Chem Soc. 2004;126:15046–15047.
- Laughlin ST, Baskin JM, Amacher SL, et al. In vivo imaging of membrane-associated glycans in developing zebrafish. Science. 2008;320:664–667.
- Meier H, Petersen H, Kolshorn H. Chem Ber. 1980;113:2398.
- Turner RB, Jarrett AD, Goebel P, et al. Heats of hydrogenation. IX. Cyclic acetylenes and some miscellaneous olefins. J Am Chem Soc. 1973;95:790.
- Evans HL, Slade RL, Carroll L, et al. Copper-free click–a promising tool for pre-targeted PET imaging. Chem Comm. 2012;48:991–993.
- Neef AB, Schultz C. Selective fluorescence labeling of lipids in living cells. Angew Chem. 2009;48:1498–1500.
- Plass T, Milles S, Koehler C, et al. Genetically encoded copper-free click chemistry. Angew Chem. 2011;50:3878–3881.
- Horner KA, Valette NM, Webb ME. Strain-promoted reaction of 1,2,4-triazines with bicyclononynes. Chemistry. 2015;21:14376–14381.
- Hogan DJ, Riordan DP, Gerber AP, et al. Diverse RNA-binding proteins interact with functionally related sets of RNAs, suggesting an extensive regulatory system. PLoS Biol. 2008;6:e255.
- Joo C, Ha T. Labeling proteins for single-molecule FRET. Cold Spring Harb Protoc. 2012;2012:1009–1012.
- Kim Y, Ho SO, Gassman NR, et al. Efficient site-specific labeling of proteins via cysteines. Bioconjug Chem. 2008;19:786–791.
- Adio S, Sharma H, Senyushkina T, et al. Dynamics of ribosomes and release factors during translation termination in E. coli. eLife. 2018;7:e34252.
- Lai WC, Ermolenko DN. Ensemble and single-molecule FRET studies of protein synthesis. Methods. 2018;137:37–48.
- Anderson JC, Wu N, Santoro SW, et al. An expanded genetic code with a functional quadruplet codon. Proc Natl Acad Sci U S A. 2004;101:7566–7571.
- Liu DR, Schultz PG. Progress toward the evolution of an organism with an expanded genetic code. Proc Natl Acad Sci U S A. 1999;96:4780–4785.
- Young TS, Ahmad I, Yin JA, et al. An enhanced system for unnatural amino acid mutagenesis in E. coli. J Mol Biol. 2010;395:361–374.
- Chen PR, Groff D, Guo J, et al. A facile system for encoding unnatural amino acids in mammalian cells. Angew Chem. 2009;48:4052–4055.
- Chin JW, Santoro SW, Martin AB, King DS, Wang L, Schultz PG. Addition of p-azido-L-phenylalanine to the genetic code of Escherichia coli. J Am Chem Soc. 2002;124:9026–9027.
- Nguyen DP, Lusic H, Neumann H, et al. Genetic encoding and labeling of aliphatic azides and alkynes in recombinant proteins via a pyrrolysyl-tRNA Synthetase/tRNA(CUA) pair and click chemistry. J Am Chem Soc. 2009;131:8720–8721.
- Nikic-Spiegel I. Genetic code expansion- and click chemistry-based site-specific protein labeling for intracellular DNA-PAINT imaging. Methods Mol Biol. 2018;1728:279–295.
- Jungmann R, Avendano MS, Woehrstein JB, et al. Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nat Methods. 2014;11:313–318.
- Nikic I, Estrada Girona G, Kang JH, et al. Debugging eukaryotic genetic code expansion for site-specific click-PAINT super-resolution microscopy. Angew Chem. 2016;55:16172–16176.