
ABSTRACT
CsrA is a widely conserved, abundant small RNA binding protein that has been found in E. coli and other Gram-negative bacteria where it is involved in the regulation of carbon metabolism, biofilm formation and virulence. CsrA binds to single-stranded GGA motifs around the SD sequence of target mRNAs where it inhibits or activates translation or influences RNA processing. Small RNAs like CsrB or CsrC containing 13–22 GGA motifs can sequester CsrA, thereby abrogating the effect of CsrA on its target mRNAs. In B. subtilis, CsrA has so far only been found to regulate one target, hag mRNA and to be sequestered by a protein (FliW) and not by an sRNA. Here, we employ a combination of in vitro and in vivo methods to investigate the effect of CsrA on the small regulatory RNA SR1 from B. subtilis, its primary target ahrC mRNA and its downstream targets, the rocABC and rocDEF operons. We demonstrate that CsrA can promote the base-pairing interactions between SR1 and ahrC mRNA, a function that has so far only been found for Hfq or ProQ.
Abbreviations: aa, amino acid; bp, basepair; nt, nucleotide; PAA, polyacrylamide; SD, Shine Dalgarno.
Introduction
Posttranscriptional regulation of gene expression is exerted by small regulatory RNAs (sRNAs) (rev. in [Citation1–Citation4]). SR1 is a dual-function sRNA from Bacillus subtilis [Citation5]. It base-pairs with ahrC mRNA encoding the transcriptional activator of the arginine catabolic operons rocABC and rocDEF and inhibits its translation [Citation6–Citation8]. In addition, SR1 codes for the small peptide SR1P that interacts with GapA [Citation9–Citation11]. Both functions of SR1 are remarkably conserved over one billion years of evolution [Citation12]. Previously, we found that the abundant RNA chaperone Hfq bound both SR1 and ahrC mRNA, but did not stabilize either RNA nor did it promote the interaction between both complementary molecules. Instead, Hfq was required for ahrC translation by opening a double-stranded region near the SD sequence [Citation8]. So far, no other RNA-binding protein was found in Gram-positive bacteria that could replace Hfq in promoting the interaction between partially complementary sRNAs and their target RNAs.
CsrA (carbon storage regulator A) is a widely conserved, abundant small (≈60 aa) RNA binding protein that is involved in the regulation of carbon metabolism, biofilm formation and virulence (rev. in [Citation13,Citation14]). CsrA binds to 2–6 sites containing single-stranded GGA motifs around the SD sequence of target mRNAs [Citation15] where it inhibits [Citation16,Citation17] or activates translation [Citation18], influences RNA processing [Citation19] or alters transcript elongation [Citation20]. A recent integrated transcriptomics approach revealed a global role of CsrA in E. coli [Citation21]. Interestingly, not only the expression of 87 transcriptional regulators and 11 sensor kinases was found to be affected by CsrA, but also the abundance of 11 base-pairing sRNAs, among them GadY, Spot42, GcvB and MicL. In Gram-negative bacteria, small RNAs like CsrC or CsrB containing 13–22 CsrA binding motifs can sequester CsrA, thereby relieving repression or reversing activation of corresponding target mRNAs. In B. subtilis, CsrA has so far only been found to regulate one target, hag mRNA, and is not sequestered by an sRNA, but by a protein (FliW) [Citation22]. Thereby, FliW interacts with a C-terminal extension of CsrA and allosterically antagonizes CsrA in a non-competitive manner thus preventing CsrA binding to hag mRNA [Citation23].
Here, we investigate the effect of CsrA on SR1, its primary target ahrC mRNA and its downstream targets, the rocABC and rocDEF operons.
Results
CsrA binds SR1 and ahrC mRNA in nanomolar range
Using the SELEX procedure, a consensus binding site for E. coli CsrA was identified. This sequence contains a critical GGA motif with less conserved nucleotides upstream and downstream. The GGA motif in natural CsrA binding sites is typically found in the loop of a short hairpin or in a single-stranded region [Citation15]. The secondary structure of SR1 [Citation8] displays three GGA motifs, motif 1 in the large bulge interrupting stem-loop I, motif 2 in the single-stranded central region and motif 3 at the base of the small central stem-loop II (). To investigate, whether CsrA can bind SR1, plasmid pGPPM1 was constructed allowing the purification of C-terminally Strep-tagged B. subtilis CsrA from E. coli. To exclude co-purification of Hfq, an E. coli ∆hfq strain was used. CsrAStrep was employed in EMSA (electrophoretic mobility shift assay) with internally labelled wild-type SR1 (SR1WT). As shown in , an RNA-protein complex was observed upon increasing concentrations of CsrAStrep. A Kd value of 183 nM was calculated for the SR1WT/CsrA complex (Fig. S2).
Figure 1. Secondary structures of SR1 and ahrC483 mRNA.
Secondary structures of SR1 (a) and ahrC mRNA (b) as determined before [Citation8] are shown. GGA motifs are highlighted in black and numbered. Complementary regions A to G in SR1 and A’ to G’ in ahrC mRNA are highlighted in grey. The SD sequence of ahrC is boxed. Mutations in GGAs of SR1 and ahrC are indicated.
![Figure 1. Secondary structures of SR1 and ahrC483 mRNA.Secondary structures of SR1 (a) and ahrC mRNA (b) as determined before [Citation8] are shown. GGA motifs are highlighted in black and numbered. Complementary regions A to G in SR1 and A’ to G’ in ahrC mRNA are highlighted in grey. The SD sequence of ahrC is boxed. Mutations in GGAs of SR1 and ahrC are indicated.](/cms/asset/57edb3e6-1474-4b84-97c3-3477d51b7204/krnb_a_1605811_f0001_b.gif)
Figure 2. CsrA binds to SR1 and ahrC mRNA.
EMSA with purified internally α 32-P-[UTP] labelled RNA and increasing concentration of purified CsrA. 0.15 fmol of labelled wild-type or mutated SR1 or ahrC mRNA species of different size were incubated with CsrA at the indicated concentrations in a total volume of 10 µl (final RNA concentration 0.015 nM) for 10 min, followed by separation on 4% (ahrC RNA) or 8% (SR1) native PAA gels. The investigated mutations are indicated. Autoradiograms of the gels are shown. (a) EMSAs with wild-type and mutated SR1 species. (b) Competition EMSA with heterologous RNAIII (left) or unlabelled SR1 species (right). Above, the fold excess of the competitor RNA is indicated. (c) EMSAs with wild-type full-length ahrC483 mRNA containing 11 GGA* motifs (left) and shortened (nt 1–136) ahrC136 mRNA containing the three 5ʹ GGA*s (centre) as well as ahrC136 RNA mutated in the GGA*s (right) are shown. (d) EMSAs with ahrC483 mRNA mutated in addition to GGA1*-3* in one other GGA*.
![Figure 2. CsrA binds to SR1 and ahrC mRNA.EMSA with purified internally α 32-P-[UTP] labelled RNA and increasing concentration of purified CsrA. 0.15 fmol of labelled wild-type or mutated SR1 or ahrC mRNA species of different size were incubated with CsrA at the indicated concentrations in a total volume of 10 µl (final RNA concentration 0.015 nM) for 10 min, followed by separation on 4% (ahrC RNA) or 8% (SR1) native PAA gels. The investigated mutations are indicated. Autoradiograms of the gels are shown. (a) EMSAs with wild-type and mutated SR1 species. (b) Competition EMSA with heterologous RNAIII (left) or unlabelled SR1 species (right). Above, the fold excess of the competitor RNA is indicated. (c) EMSAs with wild-type full-length ahrC483 mRNA containing 11 GGA* motifs (left) and shortened (nt 1–136) ahrC136 mRNA containing the three 5ʹ GGA*s (centre) as well as ahrC136 RNA mutated in the GGA*s (right) are shown. (d) EMSAs with ahrC483 mRNA mutated in addition to GGA1*-3* in one other GGA*.](/cms/asset/f28646b2-a064-45b4-89f7-663874274eda/krnb_a_1605811_f0002_b.gif)
To analyse the role of the individual CsrA binding motifs, SR1 species with point mutations in motif 1 (GGA to GCA, SR1mut1), motif 2 (GGA to GAA; SR1mut2) and motif 3 (GGAto GCA, SR1mut3) were tested (). SR1mut1 was bound by CsrAStrep with nearly the same affinity as wild-type SR1. No complex was observed with SR1mut2, and intermediate levels of complex were observed with SR1mut3 suggesting that the GGA2 motif in SR1 is important. To confirm the specificity of the SR1-CsrA interaction, unlabelled heterologous RNAIII of plasmid pIP501 [Citation24,Citation25] was used as competitor (, left). Whereas a 10-fold excess of unlabelled SR1 could outcompete labelled SR1, a 104-fold excess of RNAIII was required (, left). When the mutated SR1 species were used in a competition assay, a 10-fold excess of SR1mut1 and SR1mut3 competed with labelled SR1WT, whereas a 100-fold excess of SR1mut2 was not able to compete with SR1WT (, right). This further corroborated the importance of GGA motif 2 in SR1.
The primary target of SR1, ahrC mRNA, carries 11 GGA motifs (), with six of them (1, 3, 7, 8, 10 and 11) in fully or partially (2 free nt) single-stranded regions. In the following, we designate the ahrC GGA motifs GGA* to distinguish them from the SR1 GGA motifs. Using EMSAs, CsrA binding to full-length ahrC483 RNA and a 3ʹ truncated species, ahrC136 RNA, was tested. shows that both species bound CsrA with about the same affinity with a Kd of 405 nM (Fig. S2). ahrC136 RNA contains three 5ʹ GGA* motifs, with two (GGA2* and GGA3*) adjacent to G’, which is one of the seven regions (A’ to G’) in ahrC mRNA that are complementary to SR1 regions A to G (see ). The initial contact between SR1 and ahrC mRNA occurs between G and G’ [Citation7]. Three mutated ahrC136 species were tested in EMSAs. Whereas ahrC136mut1 and ahrC136mut2/3 bound with an efficiency comparable to the wild-type, only ahrC136 carrying mutations in all three GGA*s (GGA1*-3* to GCA) was unable to bind CsrA (). However, full-length ahrC483 carrying mutations in GGA1*-3* still bound CsrA (). Therefore, we mutated GGA*s 6, 7, 8 and 10 individually to GCA in ahrC483 species lacking GGA1* to 3* and assayed these mutant RNAs for CsrA binding (). No decrease in binding was detected. This suggests that several GGA* motifs may be required for CsrA binding.
CsrA induces slight structural changes in ahrC mRNA
To analyse whether binding of CsrA induces structural alterations in SR1, a detailed footprinting experiment with RNases T1, T2 and A was performed (). Except the expected protection at GGA2, no structural changes were observed. The importance of single GGA motifs for CsrA binding was further investigated by comparative footprinting with RNase T1 on 5ʹ-labelled SR1WT, SR1mut1 and SR1mut2 in the presence or absence of CsrA. Only SR1 GGA2, but not GGA1 or GGA3, was protected by CsrA () corroborating the EMSA results. As expected, no protection in SR1mut2 by CsrA was observed (). An RNase T1, T2, A and nuclease S1 footprinting experiment was also carried out with ahrC136 RNA containing the SD sequence, start codon and GGA1* to 3* (). All GGA*s showed a clear protection by CsrA which agreed with the EMSA results. No alterations were visible at the SD sequence (not shown). By contrast, distinct changes were observed upstream of GGA2*: C113 and U118 that are base-paired in the absence of CsrA, became single-stranded in its presence as shown by RNase A, and, to a lower extent, nuclease S1 and RNase T2 cuts. This makes the 6 nt long region G’, which is base-paired in the absence of CsrA more accessible for the initial interaction with SR1 region G. The data are summarized in . To study possible further structural changes, full-length ahrC483 RNA was subjected to partial digestion with RNase T1 in the presence and absence of CsrA (Fig. S3). Although the resolution of this gel does not allow visualization of each individual GGA*, no alterations in the general RNase T1 cleavage pattern could be observed at the GGA motifs downstream of GGA3*.
Figure 3. RNase footprinting of SR1 and ahrC136 mRNA at increasing concentrations of CsrA.
Secondary structure probing of 5ʹ labelled SR1 with RNases T1, T2 and A (a) and wild-type (b) and two SR1 mutants with RNases T1 (c) after binding of increasing concentrations of CsrA was performed as described in Materials and Methods. (d) Secondary structure probing of 5ʹ labelled ahrC136 mRNA with RNases T1, T2, A and nuclease S1 after binding of increasing amounts of CsrA. Red dots indicate enhanced protection, green dots better accessibility upon CsrA binding. Autoradiograms of the gels are shown. TL and T1L, T1 cleavage under denaturing conditions. OH, alkaline ladder. Bars indicate the positions of the GGA motifs. (e) Secondary structure of ahrC136 RNA as probed previously [Citation8]. The SD sequence is boxed; CsrA binding motifs are highlighted in black and region G’ for the initial interaction with SR1 in grey. Red and green residues indicate enhanced protection or accessibility, respectively, according to the autoradiogram shown in (d).
![Figure 3. RNase footprinting of SR1 and ahrC136 mRNA at increasing concentrations of CsrA.Secondary structure probing of 5ʹ labelled SR1 with RNases T1, T2 and A (a) and wild-type (b) and two SR1 mutants with RNases T1 (c) after binding of increasing concentrations of CsrA was performed as described in Materials and Methods. (d) Secondary structure probing of 5ʹ labelled ahrC136 mRNA with RNases T1, T2, A and nuclease S1 after binding of increasing amounts of CsrA. Red dots indicate enhanced protection, green dots better accessibility upon CsrA binding. Autoradiograms of the gels are shown. TL and T1L, T1 cleavage under denaturing conditions. OH, alkaline ladder. Bars indicate the positions of the GGA motifs. (e) Secondary structure of ahrC136 RNA as probed previously [Citation8]. The SD sequence is boxed; CsrA binding motifs are highlighted in black and region G’ for the initial interaction with SR1 in grey. Red and green residues indicate enhanced protection or accessibility, respectively, according to the autoradiogram shown in (d).](/cms/asset/b59669cf-d204-4df9-a189-d9f0c2816130/krnb_a_1605811_f0003_c.jpg)
CsrA does not affect the stability of SR1 or ahrC mRNA
A recent transcriptomics study revealed that CsrA affects the stability of more than 200 RNAs in E. coli [Citation21]. We analysed the influence of CsrA binding on the stability of SR1 or ahrC mRNA. To determine the SR1 half-life in the presence and absence of CsrA we performed Northern blotting with B. subtilis DB104 and its isogenic ∆csrA strain. In both strains, the half-life was about 3.5 to 4 min indicating that CsrA does not affect the stability of SR1 (). As ahrC mRNA was not detectable in Northern blots, we applied qRT-PCR to determine its half-life in DB104, DB104(∆csrA) and DB104(∆sr1). The half-life in DB104 and DB104(∆csrA) was 1.5 to 2 min (). In addition, qRT-PCR corroborated our previous result [Citation7] that binding of SR1 does not promote the degradation of ahrC mRNA. Only a slight (1.5-fold) effect on the half-life of ahrC mRNA was detectable in DB104(∆sr1).
Figure 4. Effects of CsrA on the stability of both SR1 and ahrC mRNA and on ahrC translation.
(a) The half-life of SR1 was determined in wild-type strain DB104 and in the isogenic ∆csrA strain after rifampicin addition and Northern blotting as described in Materials and Methods. (b) The half-life of ahrC mRNA was determined by qRT-PCR as described in Materials and Methods. In both cases, cultures were grown in complex TY medium to OD600 = 4.5. The average values obtained from three biological replicates with standard deviations are shown. (c) In vitro translation of ahrC in the presence and absence of CsrA and/or SR1. In vitro translation was performed as described in Materials and Methods. Used amounts of CsrA, SR1start to stop and ahrC RNA are indicated. Below, the amounts of synthesized protein relative to the reaction only containing the ahrC template are shown. (d) Translational ahrC-lacZ fusions under the constitutive promoter pI were integrated into the amyE locus of B. subtilis DB104, DB104 (∆csrA), DB104 (∆sr1) as well as DB104 (∆sr1 pWSR1) and DB104 (∆sr1 ∆csrA pWSR1). β-galactosidase activities were measured after growth in TY medium at OD600 = 4.5. In all cases, the indicated values are the results of three biological replicates. Error bars represent standard deviations. Control, vector pGAB1 with promoterless lacZ integrated into the amyE locus. Induction, sr1 transcription from pWSR1 was induced with 0.2 µg/ml anhydrotetracycline.
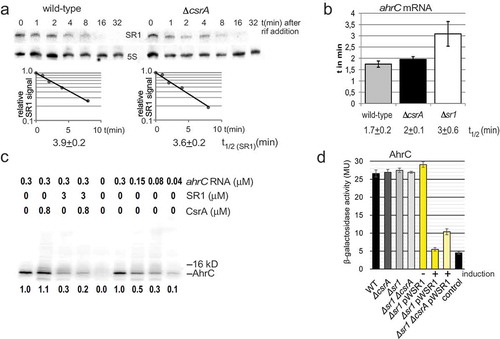
CsrA does not influence the translation of ahrC mRNA directly
Since CsrA bound to ahrC mRNA and SR1 but did not alter their stability, we asked whether it directly affects ahrC translation. To investigate this issue, we employed in vitro translation (). In vitro transcribed and purified ahrC mRNA was used as template and either in vitro transcribed and purified SR1start to stop or purified CsrAStrep or both were added. We used an amount of CsrA that almost completely bound ahrC483 RNA in the previous EMSA (). No significant differences in the amounts of AhrC protein were observed with or without CsrAStrep. As expected from our previous data [Citation7], addition of SR1 resulted in a three-fold decrease of AhrC confirming that SR1 directly inhibits ahrC translation. Lanes 6 to 9 show a direct correlation between the amounts of added ahrC mRNA and the obtained translation product. In summary, CsrA does not appear to influence the translation of ahrC mRNA directly.
To substantiate this result in vivo, B. subtilis wild-type strain DB104 with a translational ahrC483-lacZ fusion in the amyE locus was constructed as described in Materials and Methods. The ahrC483-lacZ fusion contains all 11 GGA*s and all regions complementary to SR1. Since the native ahrC promoter is extremely weak (ahrC mRNA is not detectable in Northern blots, see above) and overexpression of ahrC is toxic, the moderate constitutive copR promoter pI [Citation24,Citation26] was employed for ahrC transcription. DB104 (amyE::pI-ahrC-lacZ) was used as recipient strain for transformation with chromosomal DNA from ∆csrA or the ∆sr1 strains and with sr1 overexpression plasmid pWSR1 (≈50 copies/cell). Five transformants of each strain were grown in parallel in TY medium to OD600 = 4.5 and β-galactosidase activities measured (). No alterations in the ∆csrA strain were observed compared to the wild-type strain, suggesting that CsrA does not affect the translation of ahrC. No difference was seen in the case of the ∆sr1 strain because of the higher strength of pI compared to the native ahrC promoter. The amount of SR1 transcribed from its native promoter appears to be too low to significantly affect translation of the ahrC mRNA from amyE::pI-ahrC-lacZ. However, using pWSR1 for transcription of SR1 in excess over ahrC mRNA, a five- to six-fold reduction in β-galactosidase activity was observed (). In the ∆csrA background, with pWSR1, only a three-fold decreased β-galactosidase activity was measured (). Induction vs. no induction of sr1 transcription from pWSR1 confirms that the effect on ahrC translation is indeed due to SR1. Taken together, both in vitro and in vivo data show that CsrA does not affect the translation of ahrC mRNA directly whereas SR1 does.
CsrA promotes the base-pairing interaction between SR1 and ahrC mRNA
SR1 and its primary target ahrC mRNA share seven short stretches of complementarity designated A to G and A’ to G’, respectively ([Citation6,Citation7], ). As CsrA was able to bind both RNAs with a similar affinity (), we asked whether it promotes their base-pairing. To this end, SR1/ahrC mRNA complex formation was studied by EMSA in the presence and absence of 1 µM CsrA. Labelled SR1 and increasing concentrations (between 25 and 400 nM) of unlabelled ahrC RNA were used (). In the second experiment, labelled ahrC mRNA and the same increasing concentrations of unlabelled SR1 were employed (). In the absence of CsrA, only about 33% of the labelled RNA were present in the complex at the highest concentration of unlabelled complementary RNA. By contrast, in the presence of CsrA, the amount of the SR1/ahrC mRNA complex increased in both experiments significantly, two-fold for labelled SR1, and two- to three-fold for labelled ahrC mRNA. Thus, CsrA appears to bind both RNAs and facilitates their base-pairing.
Figure 5. SR1/ahrC mRNA complex formation in the presence and absence of CsrA.
In both cases, 1 µM CsrA was employed if indicated and complex formation monitored in 6% native PAA gels as described in Materials and Methods. The labelled RNAs (0.15 fmol per reaction) were used in at least 100-fold lower equimolar amounts than the unlabelled RNAs. (a) Complex formation between internally α-32P-[UTP]-labelled SR1 and unlabelled full-length ahrC483 mRNA. Used ahrC mRNA concentrations are indicated. (b) Complex formation between internally α-32P-[UTP]-labelled full-length ahrC483 mRNA and unlabelled SR1. Used SR1 concentrations are indicated.
![Figure 5. SR1/ahrC mRNA complex formation in the presence and absence of CsrA.In both cases, 1 µM CsrA was employed if indicated and complex formation monitored in 6% native PAA gels as described in Materials and Methods. The labelled RNAs (0.15 fmol per reaction) were used in at least 100-fold lower equimolar amounts than the unlabelled RNAs. (a) Complex formation between internally α-32P-[UTP]-labelled SR1 and unlabelled full-length ahrC483 mRNA. Used ahrC mRNA concentrations are indicated. (b) Complex formation between internally α-32P-[UTP]-labelled full-length ahrC483 mRNA and unlabelled SR1. Used SR1 concentrations are indicated.](/cms/asset/69902a1a-8643-4d8b-8e0c-be2fb8fcc688/krnb_a_1605811_f0005_b.gif)
The analysis ofAhrC target promoters procABC and procDEF demonstrates the concerted action of CsrA, ahrC mRNA and SR1 in vivo
AhrC is the transcriptional activator of the arginine catabolic operons rocABC and rocDEF [Citation27]. Previously, we have shown that in the absence of SR1, the amount of the downstream targets rocABC and rocDEF mRNA was four-fold and seven-fold increased, respectively [Citation7]. This was due to SR1 inhibiting the translation of ahrC mRNA [Citation8]. To investigate whether CsrA has an effect on the transcription of both roc operon mRNAs by promoting SR1/ahrC base-pairing, transcriptional procABC- and procDEF-lacZ fusions were constructed and integrated into the amyE locus of DB104, DB104(∆csrA), DB104(∆ahrC), DB104 (∆ahrC ∆csrA), DB104(∆sr1), DB104(∆sr1 ∆csrA), DB104(∆sr1 pGKSR1) as well as DB104(∆sr1 ∆csrA pGKSR1). Cells were grown in complex TY medium to OD600 = 4.5 where both sr1 and csrA levels are high ([Citation8]; Fig. S4) and β-galactosidase activities measured (). The activities of procABC and procDEF were approximately 4.5-fold higher in the csrA knockout strain. In the ∆ahrC ∆csrA strain, promoter activities decreased to wild-type level suggesting that CsrA exerts its effect via ahrC RNA. In the sr1 knockout strain, promoter activities were five- to six-fold higher than in the wild-type. This agrees with a previous report that levels of both roc-operon mRNAs are enhanced in the absence of SR1 [Citation7]. In the ∆sr1 ∆csrA strain, no further increase of procABC and procDEF activities was observed compared to either single-knockout indicating that both SR1 and CsrA act on the same regulatory level. Overexpression of sr1 from pGKSR1 partially compensated for the lack of CsrA. Promoter activities were two-fold lower than in the ∆sr1 ∆csrA strain, which is in good agreement with the data shown in . High SR1 concentrations resulted in high amounts of SR1/ahrC RNA complex in the absence of CsrA whereas at low SR1 concentrations, CsrA was required to obtain comparable amounts of SR1/ahrC RNA complex. The results of these β-galactosidase measurements demonstrate that the effect of CsrA on SR1/ahrC complex formation measured in vitro is also important in vivo. In addition, we compared the procABC and procDEF activities in the presence and absence of CsrA in CSE minimal medium in the presence of L-arginine, since AhrC is only active when L-arginine is present. As shown in Fig. S5, the activity of both promoters is about 2.5-fold higher in the absence of CsrA, confirming the effects observed in TY medium as well as the increase in the amount of SR1/ahrC mRNA complex in the presence of CsrA ().
Figure 6. Effects of CsrA on the transcription from rocABC and rocDEF promoters.
Transcriptional procABC-lacZ and procDEF-lacZ fusions were integrated into the amyE locus of B. subtilis and β-galactosidase activities measured after growth in TY medium until stationary phase at OD600 = 4.5. The indicated values are the results of three biological replicates. Error bars represent standard deviations. (a) DB104 wild-type and the indicated isogenic knockout and overexpression strains were used. (b) Effects of mutations in SR1 GGA2 and ahrC GGA1* and GGA3*. The indicated strains were used. VC, vector pMG16 with promoterless lacZ integrated into the amyE locus.
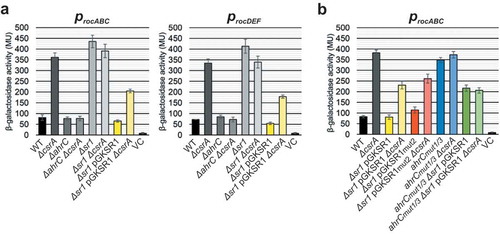
In we have shown that CsrA binds to GGA1* to GGA3* in ahrC mRNA and to GGA2 in SR1. All 5ʹ GGA*s in ahrC mRNA are located in the coding sequence. Therefore, only motifs GGA1* and 3*, but not GGA2*, can be mutated without altering the ahrC ORF. To confirm the effect of CsrA on ahrC in vivo and to analyse the effect of GGA1* and 3*, GGA1* was replaced by AGA and GGA3* by GGG. These mutations ensure that the ahrC ORF is maintained, the SR1 binding sites are not affected and the structure of the stem-loop in ahrC mRNA is not altered. Mutations were introduced into the chromosomal ahrC gene expressed under its native promoter yielding strain DB104 (ahrCmut1/3). procABC activities of this strain were investigated in the presence and absence of CsrA with or without sr1 overexpression from pGKSR1 (). For comparison, the relevant strains analysed in were grown in parallel. Although identical AhrC proteins were synthesized, procABC activities were 4.5-fold higher in the presence of mutated ahrC compared to wild-type ahrC indicating that either SR1 or CsrA bound less efficiently to ahrCmut1/3 mRNA, inhibiting its translation. When CsrA was absent, no significant differences between wild-type and ahrCmut1/3 were visible suggesting that CsrA was responsible. This was confirmed for procABC in the presence and absence of CsrA when GGA1* and GGA3* were mutated in ahrC. Overexpression of sr1 in the presence of CsrA resulted in procABC activities that were three-fold lower in the ahrC wild-type than in the ahrCmut1/3 strain. No significant differences were found in the ahrCmut1/3 strain in the presence or absence of CsrA whereas a three-fold difference was observed when ahrC RNA carried the wild-type CsrA binding motifs suggesting that the absence of csrA has the same effect as the loss of two 5ʹ CsrA binding sites in ahrC. These data provide compelling evidence that CsrA binds to ahrC mRNA in vivo and that CsrA binding sites GGA1* and GGA3* play an important role.
To investigate the in vivo role of GGA2 in SR1, pGKSR1mut2 was used to analyse procABC transcriptional fusions. In the absence of CsrA, pGKSR1mut2 showed an intermediate effect comparable to pGKSR1 (). A comparison between the sr1 and the sr1mut2 overexpression strains in the presence of CsrA revealed a ≈ 1.5-fold higher procABC activity, indicating that binding of SR1mut2 to ahrC mRNA or to CsrA was slightly affected. When csrA was deleted in both strains, a similar (2.7-fold vs. 2.2-fold) increase in procABC activity was observed for pGKSR1 compared to pGKSR1mut2. This suggests that binding of CsrA to motif 2 in SR1 is not crucial for the regulation of ahrC downstream targets.
Taken together, these data show that SR1, ahrC mRNA and CsrA act in concert to regulate the arginine catabolic operons. Thereby, CsrA binding to ahrC mRNA is required for SR1-dependent regulation of the roc operons whereas CsrA binding to SR1 plays only a minor role.
CsrA has no effect on the translation of rocA, rocD or ccpN
To exclude additional direct effects of CsrA on the translation of rocA or rocD, the first ORFs of the rocABC and rocDEF operons, respectively, translational rocA-lacZ and rocD-lacZ fusions were constructed under control of constitutive heterologous promoter pIII [Citation24] (). They were integrated into the amyE locus of DB104 and DB104 (∆csrA) and β-galactosidase activities measured in TY medium. Upon using heterologous promoter pIII it is apparent that CsrA does not affect rocA or rocD translation ().
Figure 7. Effects of CsrA on the translation of rocA, rocD and ccpN.
(a) Schema of the pIII-rocA-lacZ fusion. Heterologous promoter pIII used to replace procABC is followed by the 5ʹ UTR of rocA (rocD, ccpN), the SD and the first 17 codons of the corresponding ORF fused in frame to the 2nd lacZ codon. (b) Translational rocA-lacZ, rocD-lacZ, and ccpN-lacZ fusions were integrated into the amyE locus of B. subtilis DB104 or DB104 (∆csrA) and β-galactosidase activities measured after growth in TY medium until OD600 = 4.5. In all cases, the indicated values are the results of three biological replicates. Error bars represent standard deviations.
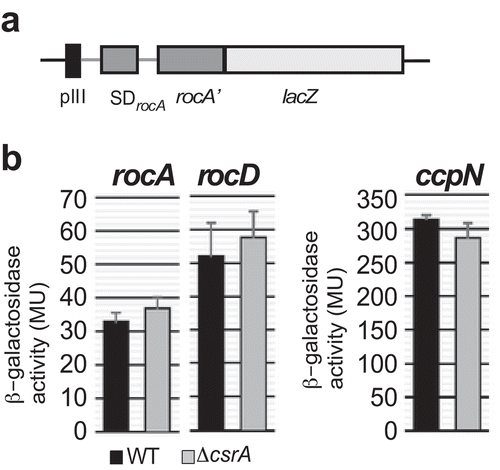
SR1 is regulated by CcpN, a transcription factor that represses sr1 transcription under glycolytic conditions [Citation6] by binding to two sites upstream of the −35 and overlapping the −10 box of the sr1 promoter [Citation28–Citation30]. To rule out that the SR1 levels were additionally altered by direct binding of CsrA to ccpN mRNA, a translational pIII-ccpN-lacZ fusion was constructed and measured as above. Again, no difference in ccpN translation was detected between wild-type and csrA knockout strain ().
Therefore, we can conclude that CsrA has only an effect on the promoter activity of the downstream target operons of SR1, but does not influence the translation of their genes directly.
sr1 overexpression impairs the use of arginine as carbon source, but additional deletion of csrA restores wild-type growth
The target of the sr1/ahrC/csrA regulatory system is arginine catabolism. To answer the questions, whether sr1 overexpression impairs the use of L-arginine as carbon source and whether deletion of csrA can compensate this effect, we analysed growth in liquid minimal media with different carbon sources. B. subtilis wild-type, ∆ahrC, ∆sr1, ∆csrA strains, a strain for constitutive overexpression of sr1 from plasmid pGKSR1 in the ∆sr1 background and a ∆csrA ∆sr1 pGKSR1 strain were grown in glucose-free CSE minimal medium and in CSE with L-arginine (CSEA) or glucose (CSEG) as additional carbon sources. In all cases, succinate and glutamate were provided to allow basal growth. The growth curves in clearly show that all strains grew slowly in CSE and quickly in CSEG. In CSEA, the ∆ahrC strain and the ∆sr1 pGKSR1 strain (downregulation of ahrC by sr1 overexpression) showed impaired growth after exhaustion of succinate and glutamate suggesting that L-arginine cannot be used as sole carbon source. Deletion of csrA in the sr1 overexpression strain (∆csrA ∆sr1 pGKSR1) compensated for the sr1 overexpression effect and restored wild-type growth. These results are consistent with the in vitro effects of CsrA on the interaction between SR1 and ahrC mRNA.
Figure 8. Effects of CsrA and sr1 overexpression on the growth of B. subtilis in CSE minimal medium with different carbon sources.
(a) B. subtilis strain DB104 and its isogenic ∆sr1, ∆ahrC, ∆csrA mutants as well as ∆ahrC/∆csrA, ∆sr1(pGKSR1) and ∆sr1/∆csrA(pGKSR1) strains were inoculated at OD600 = 0.1 into CSE minimal medium or CSE supplemented with either glucose (CSEG) or arginine (CSEA) as additional carbon source and growth monitored over 8 h. (b) Investigation of the effects of mutated CsrA binding sites in SR1 (pGKSR1mut2) and ahrC mRNA (ahrCmut1/3) on growth in CSEA. The indicated strains were used. Growth curves in CSE and CSEG are identical and not shown. Data shown with standard deviations (error bars are very small) are the results of four biological replicates.
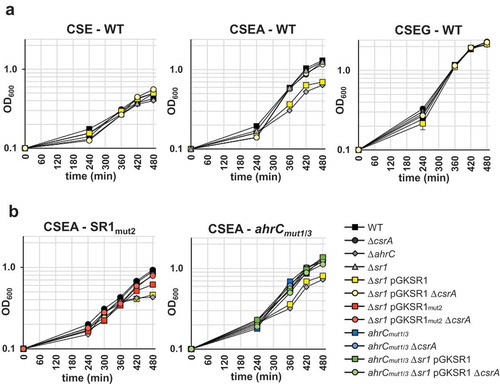
To corroborate an effect of CsrA on SR1, we compared the growth of ∆sr1 strains containing pGKSR1mut2 or pGKSR1 in the presence and absence of CsrA. The growth of all strains in CSE and CSEG was similar (not shown). In CSEA mutation of the decisive GGA2 motif of SR1 had an intermediate effect (slight growth impairment) between wild-type, overexpression (pGKSR1) and knockout (∆sr1) strains in the presence of CsrA compared to its absence ( left). This indicates that either SR1mut2 bound less efficiently to ahrC mRNA or that binding of CsrA to SR1 plays at least a minor role.
To investigate an effect of mutated GGA1* and GGA3* in ahrC mRNA, DB104 (ahrCmut1/3) was analysed in the presence and absence of CsrA as well as pGKSR1 for growth in CSE, CSEG and CSEA as above. Again, all strains grew identically in CSE and CSEG (not shown). In CSEA, the ∆ahrC strain showed impaired growth in the presence of CsrA as above ( right). In the absence of CsrA, all strains grew identically. If binding of CsrA to GGA1* and GGA3* in ahrC mRNA is required for initial contact with SR1, sr1 overexpression should not compensate for the ahrCmut1/3 mutations in the presence of CsrA. Whereas overexpression of SR1 decreased the growth of the wild-type strain DB104, it did not affect the growth of the ahrCmut1/3 mutant strain confirming our hypothesis. A comparison between ahrCmut1/3 in the presence and absence of CsrA showed no growth difference, which agrees with the finding that CsrA cannot bind ahrCmut1/3 mRNA in vivo.
In conclusion, CsrA is an integral part of the sr1/ahrC regulatory system in vivo as demonstrated in .
EMSAs with mutated SR1 and ahrC mRNA species suggest a mechanism of action for CsrA
To unravel how CsrA promotes complex formation between SR1 and ahrC mRNA, we performed EMSAs with combinations of internally labelled wild-type and mutated SR1 and unlabelled wild-type and mutated ahrC mRNA species.
Our previous structure probing analysis of SR1, ahrC mRNA and the SR1/ahrC RNA complex revealed that both RNAs fold in a modular way whereby the structure of the 5ʹ proximal region of either RNA was the same as in longer species [Citation7]. Although binding factors like CsrA immediately associate upon RNA synthesis, we believe that the above finding allows to use preformed RNA in the EMSA described below.
As shown in (a repetition of the experiment in ) CsrA enhances complex formation about two-fold between wild-type SR1 and ahrC mRNA. This is in good agreement with the ≈4.5-fold effect of CsrA on the proc activities (). Surprisingly, the same result was obtained when SR1mut2 was paired with wild-type ahrC mRNA. Complex formation with and without CsrA was 30% less efficient than in the case of SR1WT, because SR1 GGA2 overlaps region C, which is complementary with region C’ in ahrC (). However, despite the interruption of complete base-pairing by the GGA2 mutation, CsrA was able to overcome this defect at 400 nM ahrC mRNA with the amount of complex increasing from 24 to 42% (). This suggests that CsrA enhances SR1/ahrC mRNA complex formation without the requirement to bind to SR1. As expected from our data in , no SR1-CsrA complex was visible. GGA1* in ahrC is located outside of region G’ that is required for the initial contact with SR1. Therefore, ahrCmut1 RNA was as efficient as wild-type ahrC mRNA in binding SR1WT in the absence of CsrA (). However, CsrA was no longer capable of enhancing complex formation, suggesting that GGA1* in ahrC mRNA is crucial for promotion of complex formation by CsrA. Binding of ahrCmut2+3 RNA to SR1WT () was significantly impaired in the absence of CsrA (10% compared to 38%), as GGA2* and GGA3* are adjacent to complementary region G’ (see ). CsrA could not overcome this pairing deficiency, and, thus, GGA2* and GGA3* are also important for CsrA enhancing ahrC mRNA/SR1 complex formation. Furthermore, both in the presence and the absence of CsrA, complex formation between SR1 and ahrCmut1/2/3 RNA was significantly three- to four-fold impaired, as both complementary region G’ and the essential CsrA binding motifs were affected (Fig. S6). As a negative control, ahrCmut6 RNA was employed (Fig. S6). This mutant still contains the important motifs GGA1*-3* and all complementary regions to SR1. Therefore, the SR1/ahrC mRNA complex formation is enhanced ≈1.7-fold in the presence of CsrA. In all instances except SR1mut2, CsrA bound SR1 as expected (, Fig. S6). Since pGKSR1mut2 behaved similarly to pGKSR1 in the presence or absence of CsrA in regulating the procABC activity (), the combination of in vitro and in vivo data suggests the following mechanism presented in : CsrA first binds GGA1* in ahrC mRNA. This facilitates binding of GGA3* in ahrC RNA, which had blocked the stem-loop of the decisive G’ region. Binding of CsrA to motifs 1 and 3 induces slight structural alterations in ahrC mRNA that make region G’ accessible to binding of complementary SR1 region G (see ). Subsequently, the other complementary regions A to F in SR1 and A’ to F’ in ahrC mRNA interact with each other. We hypothesize that even though CsrA binding motif 2 in SR1 is not needed for the SR1/ahrC mRNA contact it might be involved in forming a bridge to one of the downstream motifs in ahrC mRNA to promote this later interaction. summarizes the SR1/ahrC/CsrA regulatory cascade.
Figure 9. Complex formation between wild-type and mutant SR1 and ahrC mRNA species in the presence and absence of CsrA.
1 µM CsrA was employed if indicated and complex formation monitored as described in Materials and Methods. The internally α 32[P]-[UTP] labelled SR1 species (0.15 fmol per reaction) were used in at least 100-fold lower equimolar amounts compared to the unlabelled ahrC mRNA species. Used ahrC mRNA concentrations are indicated. (a) Complex formation between SR1WT and ahrCWT mRNA. (b) Complex formation between SR1mut2 and ahrCWT mRNA. (c) Complex formation between SR1WT and ahrCmut1 mRNA (d) Complex formation between SR1WT and ahrCmut2+3 mRNA. Autoradiograms of the gels are shown. Below the gels, the percentage of SR1/ahrC mRNA complexes calculated with the AIDA software (Raytest) is indicated.
![Figure 9. Complex formation between wild-type and mutant SR1 and ahrC mRNA species in the presence and absence of CsrA.1 µM CsrA was employed if indicated and complex formation monitored as described in Materials and Methods. The internally α 32[P]-[UTP] labelled SR1 species (0.15 fmol per reaction) were used in at least 100-fold lower equimolar amounts compared to the unlabelled ahrC mRNA species. Used ahrC mRNA concentrations are indicated. (a) Complex formation between SR1WT and ahrCWT mRNA. (b) Complex formation between SR1mut2 and ahrCWT mRNA. (c) Complex formation between SR1WT and ahrCmut1 mRNA (d) Complex formation between SR1WT and ahrCmut2+3 mRNA. Autoradiograms of the gels are shown. Below the gels, the percentage of SR1/ahrC mRNA complexes calculated with the AIDA software (Raytest) is indicated.](/cms/asset/87d30f48-8206-4cbb-beed-e7ef7f37f48e/krnb_a_1605811_f0009_b.gif)
Figure 10. Function of CsrA in the SR1/ahrC mRNA system.
(a) Working model on the mechanism of CsrA action. SR1 binds ahrC mRNA within 7 complementary regions (A-G). ahrC mRNA region G’ responsible for the initial contact is partially sequestered in a short stem loop structure. In the absence of CsrA breathing of the stem loop structure opens G’ and allows inefficient binding of SR1. The interaction with SR1 induces structural changes within ahrC mRNA which prevent translation initiation. The presence of CsrA increases the SR1 mediated repression of ahrC mRNA translation as it facilitates the SR1/ahrC mRNA interaction. Initial CsrA binding to GGA1* and 3* opens ahrC region G’ which allows a more efficient SR1 binding and, hence, a more efficient repression of ahrC translation. Binding of additional CsrA molecules to GGA2 and 3 in SR1 and other GGA*s in ahrC mRNA that could further promote complex formation or stabilize the SR1/ahrC mRNA complex is conceivable. Yellow boxes: GGA motifs (numbering according to ); orange box: initial SR1/ahrC interacting regions G and G’; grey boxes: additional SR1/ahrC interacting regions; green circle: CsrA. (b) Flow diagram representing the regulatory functions of CcpN, SR1, CsrA and AhrC. CcpN is a transcription factor that represses sr1 transcription under gluconeogenic conditions [Citation28–Citation30]. SR1 is a trans-encoded regulatory RNA that binds ahrC mRNA thus inhibiting translation [Citation7,Citation8]. AhrC is the transcriptional activator of the rocABC and rocDEF operons [Citation7,Citation27]. For its activity, it has to bind L-arginine [Citation27]. CsrA promotes the base-pairing interaction between SR1 and ahrC mRNA (see ).
![Figure 10. Function of CsrA in the SR1/ahrC mRNA system.(a) Working model on the mechanism of CsrA action. SR1 binds ahrC mRNA within 7 complementary regions (A-G). ahrC mRNA region G’ responsible for the initial contact is partially sequestered in a short stem loop structure. In the absence of CsrA breathing of the stem loop structure opens G’ and allows inefficient binding of SR1. The interaction with SR1 induces structural changes within ahrC mRNA which prevent translation initiation. The presence of CsrA increases the SR1 mediated repression of ahrC mRNA translation as it facilitates the SR1/ahrC mRNA interaction. Initial CsrA binding to GGA1* and 3* opens ahrC region G’ which allows a more efficient SR1 binding and, hence, a more efficient repression of ahrC translation. Binding of additional CsrA molecules to GGA2 and 3 in SR1 and other GGA*s in ahrC mRNA that could further promote complex formation or stabilize the SR1/ahrC mRNA complex is conceivable. Yellow boxes: GGA motifs (numbering according to Figure 1); orange box: initial SR1/ahrC interacting regions G and G’; grey boxes: additional SR1/ahrC interacting regions; green circle: CsrA. (b) Flow diagram representing the regulatory functions of CcpN, SR1, CsrA and AhrC. CcpN is a transcription factor that represses sr1 transcription under gluconeogenic conditions [Citation28–Citation30]. SR1 is a trans-encoded regulatory RNA that binds ahrC mRNA thus inhibiting translation [Citation7,Citation8]. AhrC is the transcriptional activator of the rocABC and rocDEF operons [Citation7,Citation27]. For its activity, it has to bind L-arginine [Citation27]. CsrA promotes the base-pairing interaction between SR1 and ahrC mRNA (see Fig. 10A).](/cms/asset/5634db70-9a91-4781-9ec2-b59acb5967bf/krnb_a_1605811_f0010_c.jpg)
Even high concentrations of hag mRNA do not interfere with the ability of CsrA to promote the SR1/ahrC mRNA interaction
To date, hag is the only published target of CsrA in B. subtilis [Citation22,Citation23,Citation31]. While hag mRNA is present under most conditions in high intracellular amounts [Citation32] , SR1 is only present in significant intracellular amounts under gluconeogenic conditions with 200–225 molecules/cell [Citation8] and under conditions of sporulation [Citation32].
To compare the binding affinity of CsrA to SR1 with that to hag RNA, experiments with equimolar amounts of two labelled RNAs in the same reaction were employed to investigate the binding preference of CsrA to SR1, ahrC mRNA and hag RNA (Fig. S7A). SR1 or ahrC mRNA in the presence of hag RNA, where all were labelled and adjusted to 400 nM, were mixed and incubated with increasing amounts of CsrA. In both cases, hag RNA was already bound by CsrA at much lower concentrations (0.1 µM CsrA) than SR1 (0.7 µM CsrA) or ahrC mRNA (0.4 µM CsrA), confirming a higher affinity of CsrA to hag RNA. This is in accordance with the previously determined Kd of 115 nM [Citation33] . In conclusion, hag RNA was significantly more efficient in binding CsrA than either SR1 or ahrC mRNA.
Since the higher affinity of CsrA to hag mRNA might interfere with CsrA binding to ahrC mRNA or SR1, we investigated whether CsrA can promote the interaction of SR1 and ahrC mRNA in the presence of an excess amount of hag RNA. As shown in Fig. S7B, there is no difference in the amount of SR1/ahrC mRNA complex formed even in the presence of 800 nM hag RNA. Thus, although hag RNA (800 nM) is present at a two-fold higher concentration than ahrC mRNA (400 nM), it does not interfere with the ability of CsrA to promote the SR1/ahrC mRNA interaction. In summary, the higher affinity of CsrA for hag mRNA does not impede CsrA in promoting complex formation between SR1 and ahrC mRNA.
CsrA does not bind to three other currently known trans-encoded sRNAs from B. subtilis
To investigate if SR1 is the only one of the four currently well-characterized trans-encoded sRNAs from B. subtilis that is bound by CsrA, we employed EMSA on FsrA [Citation34], RnaC [Citation35] and RoxS [Citation33] in parallel to SR1 (Fig. S8). Three CsrA concentrations between 0.04 and 1 µM were used. Whereas about 40% of SR1 were bound at 0.2 µM, and about 70% at 1 µM, no complex between CsrA and any of the other sRNAs was detectable. Therefore, we can conclude that CsrA does not seem to play a role in the function of these sRNAs.
Discussion
Whereas in Gram-negative bacteria, in particular enterobacteria, the abundant RNA chaperone Hfq plays an important role in sRNA-mediated gene regulation [Citation36,Citation37], in Gram-positive species, this was only observed in one instance, in Listeria monocytogenes [Citation38]. Neither we nor others found a role of Hfq in either stabilizing an sRNA or promoting an sRNA/target RNA interaction in B. subtilis or S. aureus [Citation8,Citation39]. Streptococcus species do not even encode Hfq. Furthermore, by testing >150 conditions, no effect of Hfq on posttranscriptional regulation in B. subtilis could be observed [Citation40] and only six out of 100 known or predicted sRNAs displayed an altered abundance in the absence of Hfq [Citation41]. Therefore, it can be assumed that another protein might fulfil the function of Hfq in Gram-positive bacteria.
CsrA is an RNA binding protein that has been predominantly investigated in Gram-negative bacteria as a regulator of mRNA translation or RNA processing. More recently discovered roles of CsrA reviewed by Vakulskas et al. [Citation14] include the protection of an mRNA against an RNase [Citation19] or revealing a rut site required for rho-dependent transcription termination [Citation20]. In B. subtilis, the flagellin encoding hag mRNA was so far the only RNA for which a role of CsrA had been discovered. It contains two CsrA binding sites, one upstream of and one overlapping the SD sequence [Citation42]. The location of these motifs corresponds to those in CsrA-regulated mRNAs in Gram-negative bacteria and allows translation inhibition of hag mRNA by CsrA [Citation42]. Here, we demonstrate that CsrA can promote the complex formation between a regulatory sRNA and its partially complementary target mRNA in vitro () which has profound consequences in vivo (, ). This is a completely novel role for CsrA not only in B. subtilis, but in bacteria in general. Interestingly, such a role has been anticipated recently based on the results of an integrated transcriptomics study [Citation21]. We show that CsrA can bind both the small regulatory RNA SR1 and its primary target, ahrC mRNA, at nanomolar range (, S1). In vitro, CsrA promotes the interaction of both RNAs even in the presence of an excess of hag mRNA (Fig. S7) although hag mRNA binds CsrA with a higher affinity than SR1 or ahrC mRNA [Citation42]. In B. subtilis, under most conditions, hag mRNA is much more abundant than ahrC mRNA or SR1 [Citation8,Citation34]. However, it is unlikely that hag mRNA significantly impairs the formation of the ternary SR1/ahrC mRNA/CsrA complex in vivo. First, under the conditions for sr1 expression, gluconeogenesis [Citation6,Citation8] and sporulation [Citation34], hag mRNA abundance is decreased to levels comparable to SR1 [Citation34]. Second, CsrA should not be a limiting component, since micromolar concentrations were measured in E. coli [Citation42] and estimated in B. subtilis (not shown).
How might CsrA promote the interaction between SR1 and ahrC mRNA? B. subtilis CsrA binds to SR1 GGA motif 2 in a single-stranded region. In ahrC mRNA, motifs 1 and 3 contain two single-stranded nt, and motif 2 only one, and, when all mutated, prevent CsrA binding of the ahrC136 mRNA 62–95 nt downstream of the AUG start codon (). Interestingly, binding of CsrA to ahrC136 mRNA induced structural changes upstream of GGA2* () that make nt C113 and U118 flanking region G’ more accessible to the initial contact with region G of SR1 [Citation7]. Analysis of the AhrC-regulated procABC promoter confirmed that GGA1* and GGA3* are important for CsrA binding to ahrC RNA in vivo (). Regulation by SR1 was impaired when these motifs were mutated although they reside outside of region G’ complementary to SR1 region G. Unexpectedly, the alteration of decisive CsrA binding motif 2 of SR1 (pGKSR1mut2) resulted in only a minor effect on procABC activity (). This was in line with CsrA still being able to promote complex formation despite a 30% reduced binding affinity of SR1mut2 to ahrCWT mRNA (). Apparently, initial binding of CsrA to SR1 motif 2 is not required to enhance SR1/ahrC mRNA complex formation, whereas binding to ahrC mRNA is. Instead, CsrA might restructure ahrC mRNA to allow SR1 to access its initial binding site G’.
The NMR solution structure of RsmE – the CsrA functional homologue from P. fluorescens – bound to its target sites in hcnA mRNA revealed that a single protein dimer clamps two target sites, one at the SD sequence and the second upstream of it [Citation43]. Indeed, in the majority of cases in Gram-negative bacteria, two binding sites in a distance between 10 and 63 nt have been found to be bridged by one CsrA or Rsm homodimer. This might also apply to binding sites 1 and 3 in ahrC mRNA that are located 23 nt apart. As we have shown previously, SR1 binding ≈90 nt downstream of the ahrC start codon induces structural alterations at the RBS, which eventually inhibit translation initiation [Citation8]. Now we demonstrate that binding of CsrA to at least two sites in ahrC RNA (between 72 and 100 nt downstream of the SD sequence) enhances SR1/ahrC mRNA base-pairing about two-fold and propose that this allows SR1 to induce the structural alterations that prevent 30S subunit binding to the ahrC RBS. Indeed, the 5ʹ CsrA binding motifs 1 to 3 in ahrC mRNA flank the G’ region, which is the initial interaction site between SR1 and ahrC mRNA [Citation7]. EMSAs with SR1WT and ahrCmut1 suggest that ahrC GGA1* might be bound first by CsrA, as CsrA was not able to facilitate pairing with SR1WT when this motif was mutated (). Based on the EMSAs with ahrCmut2+3 or ahrCmut1/2/3 (, , S6) we hypothesize that subsequently, CsrA interacts with GGA3* which opens up region G’ required for the initial contact with SR1 [Citation8]. GGA2 and GGA3 in SR1 are located between complementary regions C and E, which interact later with C’ and E’ in ahrC mRNA. However, C and E contribute substantially to the formation of the SR1/ahrC mRNA duplex, as combined mutations in C, E and F completely abolished duplex formation [Citation8]. The importance of these regions for target RNA binding was also corroborated by the 30% reduced ability of SR1mut2 to interact with ahrCWT RNA. As this point mutation did not impede CsrA to promote complex formation with ahrCWT RNA we suggest that CsrA might bind GGA2 later during interaction of SR1 regions A to F with ahrC regions A’ to F’, bridging it with one of the downstream GGA* motifs in ahrC mRNA, thus stabilizing the SR1/ahrC mRNA complex during its formation. presents a working model on the role of CsrA in the SR1/ahrC mRNA system. Alternatively, CsrA binding to SR1 might not be necessary at all for enhancing complex formation, but required for the regulation of other, still unidentified SR1 targets. On average, trans-encoded sRNAs have at least five targets, some of them even more than 40 [Citation1]. Since ahrC mRNA is currently the only known target of SR1, we cannot rule out this alternative.
Since the regulatory system SR1/ahrC/CsrA controls arginine catabolism, we investigated growth in minimal media with and without arginine (). As expected, overexpression of sr1 from pGKSR1 impaired the use of arginine as sole carbon source. This is consistent with SR1 inhibiting translation of ahrC mRNA encoding the transcription activator of the arginine catabolic operons [Citation8] (,). Deletion of csrA relieved growth impairment () confirming that CsrA is an integral part of the regulatory system. When CsrA binding motifs 1 and 3 of ahrC were mutated, growth was not impaired in the presence of CsrA (). This substantiates that CsrA cannot bind ahrCmut1/3 mRNA at these crucial motifs. Furthermore, whereas sr1 overexpression inhibited the growth of the wild-type, it did not affect the growth of the ahrCmut1/3 strain confirming again that CsrA binding is the prerequisite for efficient SR1 binding to ahrC mRNA (). These data together with the results shown in further prove the concerted action of CsrA and SR1 in regulating ahrC.
Recently, ProQ was discovered to be a new RNA chaperone in Gram-negative bacteria [Citation44,Citation45]. Chromosome-encoded ProQ belongs to the same family as FinO encoded on F and R1 plasmids (rev. in [Citation46]) that enhances – like Hfq – the interaction between an antisense RNA (FinP) and its target traJ mRNA [Citation47]. Recently, it was shown that ProQ promotes the interaction between the sRNA RaiZ and its target, hu-α mRNA, to inhibit its translation [Citation48]. However, ProQ is not encoded in the genomes of Gram-positive bacteria. As mentioned above, no broader role for Hfq could be established in sRNA/target RNA systems in Gram-positives either. Therefore, it cannot be excluded that CsrA which is broadly conserved and annotated in more than 1500 species (rev. in [Citation49]) might be a chaperone that fulfils the role of Hfq or ProQ in Gram-positive bacteria. Unfortunately, to date, only three other trans-encoded sRNAs, FsrA, RnaC and RoxS from B. subtilis, have been investigated in detail and their target mRNAs identified [Citation34–Citation34]. Only RnaC and FsrA contain one GGA motif but do not bind CsrA. In addition, for FsrA it has been speculated that one or more of three small basic proteins, FbpA, B and C, might be the required RNA chaperones [Citation34]. However, an RNA binding activity has not been verified experimentally for any of them. Genetic evidence indicates that at least the 48 aa FbpB is required for FsrA to translationally repress the lutABC mRNA [Citation50]. It is still unclear if the role of FbpA, B or C is confined to the FsrA regulon. Notwithstanding, it cannot be excluded that other sRNAs from B. subtilis or other Gram-positive bacteria, whose targets have not yet been identified, might use CsrA as a chaperone to facilitate the interaction with their target mRNAs.
In 2018 it was found that two sRNAs, SgrS and DicF play only an accessory role in regulating the mannose reporter gene manX by promoting Hfq binding near the manX RBS, so that Hfq itself can directly interfere with ribosome binding [Citation51]. In the SgrS/DicF-manX case, the RNA chaperone apparently swapped the role with the sRNAs SgrS and DicF. Such an accessory role can, however, be excluded for SR1, as CsrA is unable to repress ahrC translation in the absence of SR1 (,).
Lately, it was reported that in the E. coli enterocyte effacement locus (LEE) Hfq plays a repressive and CsrA an activating role on sepL mRNA [Citation52]. However, since the motif required for CsrA binding overlaps the second Hfq interaction site, binding of both chaperones is mutually exclusive. Interestingly, in the SR1/ahrC system, control seems to be the opposite way around: Hfq activates ahrC translation [Citation8], whereas CsrA helps to inhibit ahrC translation by promoting SR1/ahrC RNA base-pairing. Since Hfq binds nt 17 to 21 of ahrC mRNA [Citation8] and CsrA binding motifs 1 to 3 are located between nt 98 and 131, binding of both RNA chaperones can occur simultaneously. This is in accordance with the results of the recent extended transcriptome research report, where an effect of E. coli CsrA on 11 base-pairing sRNAs was detected by CLIP-Seq and/or RNA abundance studies. Similar to ahrC mRNA, for GadY and Spot42, the CsrA binding sites do not overlap with the binding sites for Hfq known to promote the interaction of these sRNAs with their target mRNAs [Citation21].
So far, only a few examples are known where both Hfq and CsrA are involved in posttranscriptional control of gene expression. Future research will reveal whether these are exceptions. Most probably, the new role of CsrA in the promotion of sRNA/target RNA pairing will not be restricted to the SR1/ahrC case. We aim to use a more global approach to investigate the role of CsrA in sRNA/target RNA systems in Bacillus subtilis.
Materials and methods
Enzymes and chemicals
Chemicals used were of the highest purity available. Q5 DNA polymerase, T7 RNA polymerase, CIP and polynucleotide kinase were purchased from NEB, Firepol Taq polymerase from Solis Biodyne, RNases T1 and T2 from Sigma Aldrich, S1 nuclease from Thermo Scientific and RNase A and RNasin from Promega.
Strains, media and growth conditions
E. coli strains DH5α [Citation53], TOP10 (∆csrA/∆pgaA) [Citation54] and JVS-00141 (Δhfq) were used for cloning and CsrA purification, respectively. B. subtilis strains DB104 [Citation55] and DB104 (∆csrA::cat) (this study) were used for in vivo experiments. Complex TY medium (16 g/l Tryptone, 10 g/l yeast extract, 5 g/l NaCl) and CSE minimal medium (70 mM K2HPO4, 30 mM KH2PO4, 25 mM (NH4)2SO4, 0.5 mM MgSO4, 10 µM MnSO4, 22 mg/l Fe ammonium citrate, 3 g/l Na succinate, 4 g/l K glutamate and 10 mg/l histidine) served as cultivation media. For growth experiments in CSE, precultures inoculated from a fresh TY plate were grown in 5 ml liquid TY medium for 2 h, centrifuged, washed and resuspended in 1 ml CSE medium without glucose. Aliquots were used to inoculate 10 ml of CSE, CSE with 0.5% glucose (CSEG) or CSE with 8.7 g/l L-arginine (CSEA) to OD600 = 0.1 and grown for 8 h. If necessary, antibiotics were added as follows: For E. coli 100 µg/ml ampicillin or 25 µg/ml kanamycin and for B. subtilis 100 µg/ml spectinomycin, 5 µg/ml chloramphenicol, 5 µg/ml erythromycin or 12 µg/ml kanamycin.
Protein purification
E. coli Δhfq or ΔcsrA/pgaA strains containing plasmid pGPPM1 were used for purification of C-terminally Strep-tagged B. subtilis CsrA (CsrAStrep). Purification was performed as described using the Strep-Tactin system from IBA (Göttingen) [Citation9]. CsrA preparations from both strains bound SR1 with the same affinity (Fig. S1).
In vitro transcription, preparation of total RNA and Northern blotting
In vitro transcription was performed as described [Citation8]. Two-step PCRs were employed to introduce mutations by exchanging nucleotides of the complementary inner primers (see Table S1). Preparation of total RNA and Northern blotting including the determination of RNA half-lives were carried out as described previously [Citation6], except that for qRT-PCR only 125 µl time samples were taken and directly added to 250 µl RNAprotect Bacteria Reagent (Qiagen), vortexed and incubated for 5 min at room temperature. After centrifugation, the pellets were flash-frozen in liquid nitrogen and stored until use at −20°C. The isolated RNA was treated for 1 h at 37°C with 4 µl DNase I (RNase-free, NEB), followed by one phenol and two chloroform extractions. After ethanol precipitation, the RNA was dissolved in 20 µl bidist (bidistilled water).
Analysis of RNA-RNA complex formation and structure probing
Both SR1 and ahrC mRNA were synthesized in vitro from PCR-generated template fragments with specific primer pairs (see Table S1 for all oligonucleotides) using T7 RNA polymerase, purified from 6% denaturing PAA gels and resolved in TMN buffer (20 mM Tris-acetate pH 7.5, 2 mM MgCl2, 100 mM NaCl). RNAs were incubated for 2 min at 95°C, 2 min on ice followed by 30 min at 37°C to allow for proper folding. For RNA-RNA complex formation, various concentrations of unlabelled target RNA (determined by UV spectrophotometry) were incubated with 2000 cpm of α-32P-[UTP] labelled SR1 and 0.1 g/l tRNA in TMN buffer for 15 min at 37°C. One volume of stop solution (0.5 x TBE, 10% glycerol, 0.05% bromophenol blue and 0.05% xylene cyanol) was added, the mixtures rapidly cooled on ice, immediately loaded onto a 4%, 6% or 8% native PAA gel containing 1 x TBE buffer and separated in 0.5-fold TBE with 20 mA at 4°C for 4 h. Dried gels were analysed by PhosphorImaging in a Biostep PhosphorImager using AIDA Image Analyzer 5.0 software (Raytest). Structure probing of 5ʹ labelled RNAs was performed as follows: After binding of CsrAStrep to SR1 or ahrC RNA (30 000 cpm) in TMN buffer (total volume 5 µl) for 10 min at 37°C, 5 µl 1 x TMN buffer containing the diluted RNase T1, T2, A or S1 nuclease and 0.4 µl tRNA were added and cleavage conducted for 5 min at 37°C. For S1 nuclease cleavage, 2 mM ZnCl2 were added. A negative control without RNases was used. The reaction was stopped by addition of one volume formamide loading dye (90% formamide, 15 mM EDTA, 0.05% bromophenol blue and 0.05% xylene cyanole) and incubation on ice. After heat denaturation for 5 min at 95°C, the reaction mix was separated alongside a T1 ladder on a denaturing 8% or 6% PAA gel at 25 mA.
Analysis of CsrA binding to RNA
Internally α-32P-[UTP] labelled SR1 or ahrC mRNA were synthesized by T7 RNA polymerase on PCR templates, gel-purified and incubated with different concentrations of CsrAStrep for 10 min at 37°C. Stop solution (10% glycerol, 0.5x TBE, 0.05% bromophenol blue and 0.05% xylene cyanol) was added and samples were separated in native 6% or 8% PAA gels run at 4°C. Dried gels were quantified by PhosphorImaging as above.
cDNA synthesis and qRT-PCR
10 µl DNase I treated RNA was used for the synthesis of cDNA with 50 U SuperScript II reverse transcriptase (Invitrogen) for 50 min at 42°C, followed by digestion with 1 µl RNase A for 10 min at 37°C. After one phenol and two chloroform extractions followed by ethanol precipitation, the pellet was dissolved in 20 µl bidist. Quantitative real-time PCR was performed with the Maxima SYBR Green/ROX qPCR Master Mix (ThermoFisher) and the Mx3005P system (Stratagene) in 96 well blocks. Each well contained 9 µl bidist, 1.5 µl of primer mix (5 pmol/ml each in bidist) and 2 µl undiluted template cDNA. The reaction was started after addition of 12.5 µl qPCR MasterMix 2x in a darkened room. Forty cycles with denaturation for 30 s at 95°C, annealing for 30 s at 48°C and extension for 30 s at 72°C were used. Only for 5S rRNA detection, the template cDNA was 1000-fold diluted. Immediately after qRT-PCR, a melting point analysis with MxPro software (Stratagene) was carried out to test for product specificity. For final validation, a modified ∆∆Ct method was employed defining the number of cycles at which the fluorescence exceeds a certain threshold as Ct value. For evaluation, MxProSoftware from Stratagene was used.
Construction of plasmids and strains
E. coli/B. subtilis shuttle vector pGPPM1 (see Table S2 for all plasmids) for constitutive expression and purification of C-terminally Strep-tagged B. subtilis CsrA (CsrAStrep) was constructed by cloning a BamHI/SphI fragment obtained by PCR with primer pair SB2497/SB2498 on chromosomal DNA of B. subtilis into the BamHI/SphI pGP382 vector. The insert sequence was confirmed by sequencing. B. subtilis strain DB104 (∆csrA::spec) was constructed by transformation of strain DB104 with chromosomal DNA isolated from B. subtilis strain GP469. B. subtilis strain DB104 (amyE::pGAB kan) was constructed by transformation of DB104 with linearized integration vector pGAB1. Plasmid pINT6E constructed by exchanging the chloramphenicol resistance gene of plasmid pINT6 [Citation7] by the erythromycin resistance gene of plasmid pE194 was integrated into the B. subtilis DB104 chromosome resulting in DB104 (∆ahrC::ery). Successful ahrC knockout was confirmed by PCR on chromosomal DNA. For the construction of the csrAC-Flag strain, long-flanking-homology (LFH) PCR was used. Twenty-five cycles of PCR with Q5 polymerase were employed to generate four PCR fragments. The csrA FRONT cassette was obtained with primers SB3063/SB3078, the chloramphenicol resistance gene with SB3079/SB3080, and the csrA BACK cassette with SB3081/SB3066. SB3078 and SB3079 contain the sequence for the FLAG-tag to be introduced at the C-terminus of the chromosomally encoded csrA gene. After purification from agarose gels, the four fragments were pooled and subjected to 10 cycles with Q5 polymerase without primers for annealing, followed by 25 cycles for amplification with primer pair SB3063/SB3066. The resulting long fragment was directly used to transform DB104 to allow homologous recombination, and selection was performed on TY plates with chloramphenicol. The successful strain construction and expression of csrAC-Flag were confirmed by PCR on chromosomal DNA and Western blotting with anti-FLAG antibodies, respectively.
Plasmid pGKSR1mut2 carrying a mutation in GGA2 of sr1 was constructed by a two-step-PCR as follows: Chromosomal DNA of B. subtilis DB104 was used as a template to obtain two PCR fragments using primer pairs SB3199/SB1580 and SB1579/SB317, respectively. These fragments were isolated from an agarose gel and subjected to another PCR with primers SB3199 and SB317. The resulting fragment was isolated, digested with BamHI and HindIII and inserted into the BamHI/HindIII vector of pGK14.
The chromosomal ahrC mutant altered in GGA1* (to AGA) and GGA3* (to GGG) was constructed by LFH PCR as follows: The 900 bp FRONT fragment was generated with primer pairs SB3298/SB3321 and the 400 bp INSERT fragment with SB3320 and SB3307. A 1 kb BACK fragment and a 900 bp kanamycin resistance cassette obtained with SB3317/SB3313 and SB3308/SB3316, respectively, were used. Twenty-five PCR cycles with Q5 polymerase were employed to generate all fragments. The final PCR joining the four fragments was performed with primer pair SB3298/SB3313 as described above for the csrAC-FLAG strain. DB104 was transformed directly with the resulting long fragment, and selection was for kanamycin resistance. The mutant was confirmed by sequencing. The LFH PCRs were performed with denaturation for 30 s at 98°C, annealing for 30 s at 42°C and elongation at 72°C for 1 min per expected 1000 bp.
Construction of transcriptional and translational lacZ fusions and determination of β-galactosidase activities
For the construction of transcriptional lacZ fusions, PCR fragments were obtained on chromosomal DNA as template with primer pairs SB2744/2745 and SB2746/2747, digested with BamHI and EcoRI and inserted into pMG16 [Citation56] cleaved with the same enzyme pair yielding pMGP21 (procABC) and pMGP22 (procDEF), respectively. For the construction of translational lacZ fusions, PCR fragments were generated with primer pairs SB2774/SB2775 (ccpN), SB2778/SB2779 (rocA) and SB2780/SB2781 (rocD), cleaved with BamHI and EcoRI and inserted into the BamHI/EcoRI pGAB1 vector resulting in pGABP1-3, respectively. In pGABP1-3, transcription is under control of the constitutive heterologous promoter pIII. All vectors carrying lacZ fusions were confirmed by sequencing, linearized with ScaI and integrated into the chromosomal amyE locus. β-galactosidase activities were measured as described [Citation9].
Construction of B. subtilis strains containing a translational ahrC-lacZ fusion in the amyE locus
For the construction of the ahrC483-lacZ fusion, LFH PCR was used. Four fragments, amyE FRONT cassette (SB3027/SB3028, chromosomal DNA as template), ahrC483 under control of promoter pI (SB3075/SB3062, chromosomal DNA), spectinomycin resistance gene (SB3032/SB3074, pMG8 [Citation27,Citation57]) and lacZ (SB3061/3030, pGAB1), were generated by 25 cycles of PCR with Q5 polymerase. After purification from agarose gels, the four fragments were pooled and subjected to 10 cycles with Q5 polymerase without primers for annealing, followed by 25 cycles for amplification with primer pair SB3027/SB3030. The resulting long fragment was directly used to transform DB104 (amyE::pGAB kan) to allow homologous recombination. Selection was performed on TY plates with spectinomycin and X-Gal. To enable later transformation of this strain with sr1 overexpression plasmid pWSR1 encoding kanamycin resistance (kanR), the remaining kanR gene was inactivated by insertion of an erythromycin resistance (eryR) cassette using LFH PCR as above. FRONT and the BACK cassettes were obtained with SB3391/SB3392 or SB3395/SB3396, respectively, on pGAB1 as template, and the eryR insert with SB3393/SB23394 on pMG10 [Citation57] as template. The resulting long fragment was used to transform DB104 (amyE::ahrC-lacZ spec kan) yielding DB104 (amyE::ahrC-lacZ spec ery). Selection was for erythromycin resistance and kanamycin sensitivity.
In vitro translation
The PURExpress® In Vitro Protein Synthesis Kit (NEB) was used for in vitro translation. A full-length in vitro transcribed ahrC RNA served as template. 10 µl reactions, each containing 2.5 µCi of [Citation34] 35S-labelled methionine (1000 Ci/mmol), were pipetted as described in the manufacturers’ instructions, mixed with 1 µl ahrC RNA (300 nM final concentration), either 6 µl CsrA (final concentration 800 nM) or buffer W (IBA) and 1 µl SR1start to stop (final concentration 3 µM) or bidist and incubated at 32°C for 20 h. Reaction products were separated on a 17.5% Tris/glycine/SDS PAA gel. Dried gels were quantified by PhosphorImaging as above.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental Material
Download PDF (2 MB)Acknowledgments
The authors are grateful to Jörg Stülke (Göttingen) for providing plasmid pPG382 and B. subtilis ∆csrA strain GP469. We thank Jörg Vogel and Cynthia Sharma (Würzburg) for sending us E. coli strains JVS-00141 (∆hfq) and TOP10 (∆csrA/∆pgA), respectively.
Supplementary materials
Supplementary data for this article can be accessed here.
Additional information
Funding
References
- Wagner EG, Romby P. Small RNAs in bacteria and archaea: who they are, what they do, and how they do it. Adv Genet. 2015;90:133–208.
- Brantl S, Brückner R. Small regulatory RNAs from low-GC Gram-positive bacteria. RNA Biol. 2014;11:443–456.
- Brantl S. Bacterial chromosome-encoded small regulatory RNAs. Future Microbiol. 2009;4:85–103.
- Brantl S. Acting antisense: plasmid- and chromosome-encoded sRNAs from Gram-positive bacteria. Future Microbiol. 2012;7:853–871.
- Gimpel M, Brantl S. Dual-function small regulatory RNAs in bacteria. Mol Microbiol. 2017;103:387–397.
- Licht A, Preis S, Brantl S. Implication of CcpN in the regulation of a novel untranslated RNA (SR1) in Bacillus subtilis. Mol Microbiol. 2005;58:189–206.
- Heidrich N, Chinali A, Gerth U, et al. The small untranslated RNA SR1 from the Bacillus subtilis genome is involved in the regulation of arginine metabilism. Mol Microbiol. 2006;62:520–536.
- Heidrich N, Moll I, Brantl S. In vitro analysis of the interaction between the small RNA SR1 and its primary target ahrC mRNA. Nucleic Acids Res. 2007;35:331–346.
- Gimpel M, Heidrich N, Mäder U, et al. A dual-function sRNA from B. subtilis: SR1 acts as peptide encoding mRNA on the gapA operon. Mol Microbiol. 2010;76:990–1009.
- Gimpel M, Brantl S. Dual-function sRNA-encoded peptide SR1P modulates moonlighting activity of B. subtilis GapA. RNA Biol. 2016;13:916–926.
- Gimpel M, Maiwald C, Wiedemann C, et al. Characterization of the interaction between the small RNA-encoded peptide SR1 and GapA from Bacillus subtilis. Microbiology. 2017;163:1248–1259.
- Gimpel M, Preis H, Barth E, et al. SR1 – a small RNA with two remarkably conserved functions. Nucleic Acids Res. 2012;40:11659–11672.
- Romeo T, Vakulskas CA and Babitzke P. Post-transcriptional regulation on a global scale: form and function of Csr/Rsm systems. Environ Microbiol. 2013;15:313–324.
- Vakulskas CA, Potts AH, Babitzke P, et al. Regulation of bacterial virulence by Csr (Rsm) systems. Microbiol Mol Biol Rev. 2015;79:193–224.
- Dubey AK, Baker CS, Romeo T, et al. RNA sequence and secondary structure participate in high-affinity CsrA-RNA interaction. RNA. 2005;11:1579–1587.
- Baker CS, Morozov I, Suzuki K, et al. CsrA regulates glycogen biosynthesis by preventing translation of glgC in Escherichia coli. Mol Microbiol. 2002;44:1599–1610.
- Park H, Yakhnin H, Connolly M, et al. CsrA participates in a PNPase autoregulatory mechanism by selectively repressing translation of pnp transcripts that have been previously processed by RNase III and PNPase. J Bacteriol. 2015;197:3751–3759.
- Patterson-Fortin LM, Vakulskas CA, Yakhnin H, et al. Dual posttranscriptional regulation via a cofactor-responsive mRNA leader. J Mol Biol. 2013;425:3662–3677.
- Yakhnin AV, Baker CS, Vakulskas CA, et al. CsrA activates flhDC expression by protecting flhDC mRNA from RNase E-mediated cleavage. Mol Microbiol. 2013;87:851–866.
- Figueroa-Bossi N, Schwartz A, Buillemardet B, et al. RNA remodeling by bacterial global regulator CsrA promotes Rho-dependent transcription termination. Genes Dev. 2014;28:1239–1251.
- Potts AH, Vakulskas CA, Pannuri A, et al. Global role of the bacterial post-transcriptional regulator CsrA revealed by integrated transcriptomics. Nat Comm. 2017;8:1596.
- Mukherjee S, Yakhnin H, Kysela D, et al. CsrA-FliW interaction governs flagellin homeostasis and a checkpoint on flagellar morphogenesis in Bacillus subtilis. Mol Microbiol. 2011;82:447–461.
- Mukherjee S, Oshiro RT, Yakhnin H, et al. FliW antagonizes CsrA RNA binding by a noncompetitive allosteric mechanism. Proc Natl Acad Sci USA. 2016;113:9870–9875.
- Brantl S, Nuez B, Behnke D. In vitro and in vivo analysis of transcription within the replication region of plasmid pIP501. Mol Gen Genet. 1992;234:105–112.
- Heidrich N, Brantl S. Antisense-RNA mediated transcriptional attenuation: importance of a U-turn loop structure in the target RNA of plasmid pIP501 for efficient inhibition by the antisense RNA. J Mol Biol. 2003;333:917–929.
- Brantl S. The copR gene product of plasmid pIP501 acts as a transcriptional repressor at the essential repR promoter. Mol Microbiol. 1994;14:473–483.
- Miller CM, Baumberg S, Stockley PG. Operator interactions by the Bacillus subtilis arginine repressor/activator, AhrC, novel positioning and DNA-mediated assembly of a transcriptional activator at catabolic sites. Mol Microbiol. 1997;26:37–48.
- Licht A, Brantl S. Transcriptional repressor CcpN from Bacillus subtilis compensates asymmetric contact distribution by cooperative binding. J Mol Biol. 2006;364:434–448.
- Licht A, Golbik R, Brantl S. Identification of ligands affecting the activity of the transcriptional repressor CcpN from Bacillus subtilis. J Mol Biol. 2008;380:17–30.
- Licht A, Brantl S. The transcriptional repressor CcpN from Bacillus subtilis uses different repression mechanisms at different promoters. J Biol Chem. 2009;284:30032–30038.
- Yakhnin H, Pandit P, Petty TJ, et al. CsrA of Bacillus subtilis regulates translation initiation of the gene encoding the flagellin protein (hag) by blocking ribosome binding. Mol Microbiol. 2007;64:1605–1620.
- Nicolas P, Mäder U, Dervyn E, Rochat T, Leduc A, Pigeonneau N, Bidnenko E., et al. Condition-dependent transcriptome reveals high-level regulatory architecture in bacillus subtilis. Science. 2012;335:1103-1106.
- Durand S, Braun F, Lioliou E, Romilly C, Helfer AC, Kuhn L, Quittot N, Nicolas P, Romby P,Condon C. A nitric oxide regulated small rna controls expression of genes involved in redox homeostasis in bacillus subtilis. Plos Genet. 2015;11:e1004957.
- Gaballa A, Antelmann H, Aguilar C, Khakh SK, Song KB, Smaldone GT,Helmann JD. The bacillus subtilis iron-sparing response is mediated by a fur-regulated small rna and three small, basic proteins. Proc Natl Acad Sci Usa. 2008;105:11927-11932. DOI:
- Mars RA, Nicolas P, Coccolini M, Reilman E, Reder A, Schaffer M, Mäder U, Völker U. Van dijl jm and denham el (2015) small regulatory rna-induced growth rate heterogeneity of bacillus subtilis. Plos Genet. 2015;11:e1005046.
- Vogel J, Luisi BF. Hfq and its constellation of RNA. Nat Rev Microbiol. 2011;9:578–589.
- Kavita K, de Mets F, Gottesman S. New aspects of RNA-based regulation by Hfq and its partner sRNAs. Curr Opin Microbiol. 2018;42:53–61.
- Nielsen JS, Lei LK, Ebersbach T, et al. Defining a role for Hfq in Gram-positive bacteria: evidence for Hfq-dependent antisense regulation in Listeria monocytogenes. Nucleic Acids Res. 2010;38:907–919.
- Bohn C, Rigoulay C, Bouloc P. No detectable effect of RNA-binding protein Hfq absence in Staphylococcus aureus. BMC Microbiol. 2007;7:10.
- Rochat T, Delumeau O, Figueroa-Bossi N, et al. Tracking the elusive function of Bacillus subtilis Hfq. PLoS One. 2015;10:e0124977.
- Hämmerle H, Amman F, Vecerek B, et al. Impact of Hfq on the Bacillus subtilis transcriptome. PLoS One. 2014;9:e98661.
- Yakhnin AV, Pandit P, Petty TJ, et al. CsrA of Bacillus subtilis regulates translation initiation of the gene encoding the flagellin protein (hag) by blocking ribosome binding. Mol Microbiol. 2007;64:1605–1620.
- Schubert M, Lapouge K, Duss O, et al. Molecular basis of messenger RNA recognition by the specific bacterial repressing clamp RsmA/CsrA. Nat Struct Mol Biol. 2007;14:807–813.
- Attaiech L, Boughammoura A, Brochier-Armanet C, et al. Silencing of natural transformation by an RNA chaperone and a multitarget small RNA. Proc Natl Acad Sci USA. 2016;113:8813–8818.
- Smirnov A, Förstner KU, Holmqvist E, et al. Grad-seq guides the discovery of ProQ as a major small RNA-binding protein. Proc Natl Acad Sci USA. 2016;113:11591–11596.
- Mark Glover JN, Chaulk SG, Edwards RA, et al. The FinO family of bacterial RNA chaperones. Plasmid. 2015;78:79–87.
- Jerome LJ, van Biesen T, Frost LS. Degradation of FinP antisense RNA from F-like plasmids: the RNA-binding protein, FinO, protects FinP from ribonuclease E. J Mol Biol. 1999;285:1457–1473.
- Smirnov A, Wang C, Drewry LL, et al. Molecular mechanism of mRNA repression in trans by a ProQ-dependent small RNA. EMBO J. 2017;36:1029–1045.
- Van Assche E, van Puyvelde S, Vanderleyden J, Steenackers HP. RNA-binding proteins involved in post-transcriptional regulation in bacteria. Front Microbiol. 2015;6:141.
- Smaldone GT, Antelmann H, Gaballa A, et al. The FsrA sRNA and FbpB protein mediate the iron-dependent interaction of the Bacillus subtilis lutABC iron-sulfur-containing oxidases. J Bacteriol. 2012;194:2586–2593.
- Azam MS, Vanderpol CK. Translational regulation by bacterial small RNAs via an unusual Hfq-dependent mechanism. Nucleic Acids Res. 2018;46:2585–2599.
- Wang D, McAteer SP, Wawszczyk AB, et al. An RNA-dependent mechanism for transient expression of bacterial translocation filaments. Nucleic Acids Res. 2018;46:3366–3381.
- Hanahan D. Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983;166:557–580.
- Dugar G, Svensson SL, Bischler T, et al. The CsrA-FliW network controls polar localization of the dual-function flagellin mRNA in Campylobacter jejuni. Nat Commun. 2016;7:11667.
- Kawamura F, Doi RH. Construction of a Bacillus subtilis double mutant deficient in extracellular alkaline and neutral proteases. J Bacteriol. 1984;160:442–444.
- Müller P, Jahn N, Ring C, et al. A multistress-responsive type I toxin-antitoxin system: bsrE/SR5 from the B. subtilis chromosome. RNA Biol. 2015;13:511–523.
- Gimpel M, Brantl S. Construction of a modular plasmid family for chromosomal integration in Bacillus subtilis. J Microbiol Methods. 2012;91:312–317.