
ABSTRACT
The de-regulation of microRNAs (miRNAs) is associated with multiple human diseases, yet cellular mechanisms governing miRNA abundance remain largely elusive. Human miR-122 is required for Hepatitis C proliferation, and low miR-122 abundance is associated with hepatic cancer. The adenylyltransferase Gld2 catalyses the post-transcriptional addition of a single adenine residue (A + 1) to the 3ʹ-end of miR-122, enhancing its stability. Gld2 activity is inhibited by binding to the Hepatitis C virus core protein during HepC infection, but no other mechanisms of Gld2 regulation are known. We found that Gld2 activity is regulated by site-specific phosphorylation in its disordered N-terminal domain. We identified two phosphorylation sites (S62, S110) where phosphomimetic substitutions increased Gld2 activity and one site (S116) that markedly reduced activity. Using mass spectrometry, we confirmed that HEK 293 cells readily phosphorylate the N-terminus of Gld2. We identified protein kinase A (PKA) and protein kinase B (Akt1) as the kinases that site-specifically phosphorylate Gld2 at S116, abolishing Gld2-mediated nucleotide addition. The data demonstrate a novel phosphorylation-dependent mechanism to regulate Gld2 activity, revealing tumour suppressor miRNAs as a previously unknown target of Akt1-dependent signalling.
Introduction
MicroRNAs (miRNAs) are critical regulators of gene expression that are essential to human life, normal cellular function, and development. De-regulation of miRNAs is, perhaps not surprisingly, associated with a number of human diseases[Citation1]. MiRNAs regulate the expression of many genes, including oncogenes, by complementary base pairing with the 3ʹ-untranslated regions (UTRs) of mRNAs, which normally inhibits protein synthesis[Citation1].
MiRNAs themselves are post-transcriptionally regulated by the addition of single or multiple adenine (A) or uridine (U) residues to their 3ʹ-ends. This untemplated RNA editing is now recognized as an important mechanism regulating cellular miRNA homeostasis [Citation2,Citation3]. The addition of a single A to the 3ʹ-end on certain miRNAs leads to increased stability [Citation4]. Conversely, the activity of mature miRNAs is reduced by the addition of a single 3ʹ-U residue [Citation5,Citation6]. The addition of multiple U residues to precursor miRNAs (pre-miRNAs) triggers subsequent degradation by the U-specific exonuclease Dis3L2 (Dis3-like exonuclease 2) [Citation3]. Although uridylation is commonly associated with silencing and degradation of RNAs, monouridylation of Group II pre-miRNAs lacking a critical 3ʹ-end overhang nucleotide is required for miRNA maturation and processing by Dicer [Citation7]. Cellular mechanisms that regulate miRNAs through 3ʹ-terminal nucleotide additions are of fundamental relevance to the molecular basis of diseases characterized by de-regulated miRNA metabolism [Citation3,Citation8].
A diverse family of terminal nucleotidyltransferases (TENTs) catalyses 3ʹ-A and U additions to RNAs in human cells. The nucleotidyltransferase Gld2 (germline development 2, TENT2) was first identified as a regulator of meiosis in Caenorhabditis elegans [Citation9] and was later shown to extend the poly(A) tails of mRNAs (), leading to enhanced mRNA stability and increased abundance of the encoded protein [Citation10]. In humans, Gld2 stabilizes miR-122 in the liver and fibroblasts through mono-adenylation [Citation4,Citation11] and mRNAs via poly-adenylation [Citation12] ().
Figure 1. Pathways regulated by Gld2 and domain architecture. (a) Known functions of Gld2. Gld2 stabilizes mature miRNA and mRNA through monoadenylation or polyadenylation of the 3ʹ-end. Mononucleotide addition of Group II pre-miRNAs on the 3ʹ-end by Gld2 allows recognition by Dicer to be processed to mature miRNAs. This is followed by strand selection by Argonaute (AGO) and incorporation into the RNA-induced silencing complex (RISC). The different pathways are represented by solid or dashed lines. (b) Schematic of Gld2 showing the nucleotidyltransferase domain (NTR) and poly(A) polymerase-like domain (PAP).
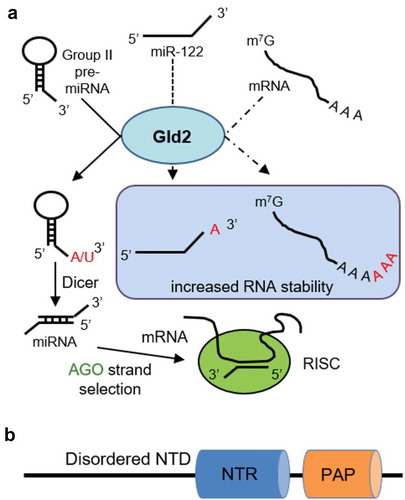
Gld2 is thought to be part of a larger protein complex involved in RNA modification and germ cell formation [Citation13]. Although some reports [Citation7] suggested that Gld2 may function as a uridylyltransferase, we recently characterized human Gld2 as a bona fide adenylyltransferase [Citation14]. Our data confirmed a basal activity of Gld2 with U, but the 80-fold higher catalytic efficiency for ATP makes the enzyme strongly selective for A additions [Citation14]. Gld2 encodes a nucleotidyltransferase domain and a poly(A) polymerase-associated domain that are required for catalytic activity as well as a disordered N-terminal domain of unknown function [Citation10] (), yet lacks identifiable RNA binding motifs. The crystal structure of a truncated C. elegans Gld2 in complex with the interacting protein Gld3 shows that the two essential Gld2 catalytic domains share the same fold as other nucleotidyltransferases [Citation15].
Cellular mechanisms that regulate miRNAs through 3ʹ-end nucleotide additions are of fundamental relevance to the molecular basis of diseases characterized by de-regulated miRNA metabolism [Citation3,Citation8]. Gld2 and its substrate miR-122 play a role in Hepatitis C virus (HCV) infection and in hepatic cancer [Citation16]. MiR-122 is one of the most abundant miRNAs in the liver, with an essential role in maintaining liver homeostasis and differentiation [Citation16]. During HCV infection, miR-122 binds to two sites in the viral 5ʹ-UTR of the Hepatitis C viral RNA and is required for HCV infection [Citation16,Citation17]. The miR-122 interaction with the 5ʹ-UTR enhances viral replication by increasing the formation of ribosome complexes to increase viral protein production. The binding of miR-122 to protein argonaute-2 (Ago2) in the RNA-induced silencing complex (RISC) also protects viral RNA from exonucleases [Citation16]. Interestingly, the HCV core protein was shown to bind to Gld2 in the cytoplasm and inhibit its nucleotide addition activity. The subsequent reduction in miR-122 abundance allows HCV to maintain low levels of viral protein production to facilitate continuous viral replication and infection of host cells [Citation18]. Consequently, inhibition of Gld2 by the HCV core protein decreases miR-122 stability and abundance. Low miR-122 levels, in turn, are associated with hepatic cancer, linking HCV infection to the development of hepatocellular carcinoma (HCC) [Citation18,Citation19]. Hepatitis B virus X-protein (HBx) was also shown to reduce Gld2 protein levels and cause an increase in cationic amino acid transporter 1 (CAT-1), a target of miR-122 [Citation20–Citation22]. CAT-1 is involved in the tumorigenesis of the Hepatitis B virus (HBV) [Citation20]. Miravirsen, an anti-miR-122 oligonucleotide, is in Phase II trials to treat Hepatitis C and has been shown to decrease levels of miR-122 for a prolonged period of time, resulting in decreased HCV RNA levels in patients [Citation23–Citation25]. As high levels of miR-122 have been observed in colorectal liver metastasis, Miravirsen has been suggested as a potential anti-cancer drug as well [Citation26].
While it is clear that Gld2 plays a role in promoting miRNA stability [Citation3,Citation14,Citation16], cellular mechanisms that regulate Gld2 activity were previously unknown. In HCC cells, miR-122 is destabilized despite no observed changes in Gld2 protein levels [Citation18,Citation19]. These data suggest the existence of a clinically relevant mechanism that regulates Gld2 activity via post-translational modification. We demonstrate that Gld2 activity is indeed regulated by phosphorylation. We found that Gld2 is phosphorylated at specific serine residues in the disordered N-terminal domain in vivo, which dramatically impact catalytic activity and substrate specificity. We found protein kinases A (PKA) and B (Akt1) site-specifically phosphorylate Gld2 at S116, which abolishes 3ʹ-nucleotide addition activity. The data reveal tumour suppressor miRNAs as a previously unrecognized target of oncogenic protein kinases.
Results
Gld2 plays an important role in miRNA stability, but the regulation of Gld2 activity or substrate specificity is unknown. Studies in other nucleotide polymerases [Citation27,Citation28] found that phosphorylation of serine and threonine residues can increase activity or processivity. For example, serine phosphorylation in the disordered C-terminal domain of RNA polymerase II is required for transcription initiation and elongation [Citation27]. Phosphorylation of the terminal uridylyltransferase Tut1 at S6 is required for Tut1 nuclear retention and regulation of specific mRNAs [Citation28]. Multiple independent proteome-level mass spectrometry studies of human cells revealed phosphorylated residues in Gld2 [Citation29–Citation31], including five conserved serine residues (S62, S69, S95, S110, S116) in the disordered N-terminal domain (). Despite these observations, the putative Gld2 kinase(s) and the impact of phosphorylation on Gld2 activity was unknown.
Figure 2. Multiple sequence alignment of mammalian Gld2 sequences. Sequences were downloaded from NCBI and the alignment and editing were performed with Muscle [Citation68], MultiSeq from VMD 1.8.7 [Citation69], and Wasabi [Citation70]. Numbers above the alignment indicate the position in H. sapiens Gld2.
![Figure 2. Multiple sequence alignment of mammalian Gld2 sequences. Sequences were downloaded from NCBI and the alignment and editing were performed with Muscle [Citation68], MultiSeq from VMD 1.8.7 [Citation69], and Wasabi [Citation70]. Numbers above the alignment indicate the position in H. sapiens Gld2.](/cms/asset/225d6cb7-6bec-442c-90c2-69f365cc0104/krnb_a_1608754_f0002_oc.jpg)
Phosphorylation of the Gld2 N-terminal domain in HEK 293 cells
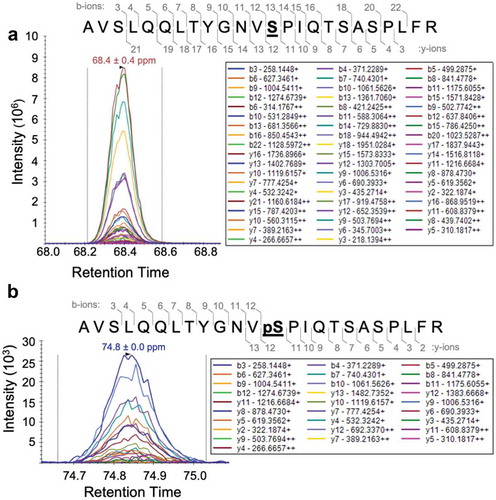
Although the above studies suggest the existence of a Gld2 kinase in human cells, we analysed Gld2 following incubation with HEK 293 cell lysates to confirm phosphorylation activity towards Gld2. The HEK 293 cells were stimulated with epidermal growth factor (EGF) to activate cellular kinases. Purified Gld2 (Figure S1) was incubated with cell extracts from HEK 293 cells after EGF stimulation of signalling pathways. The phosphorylation status of Gld2 was subsequently analysed by mass spectrometry, and we unambiguously identified phosphorylation at S62 in the sample incubated with EGF-stimulated cell lysate (). We did not identify pS62 in unstimulated cells or in recombinantly produced Gld2. As Gld2 has been previously shown to be involved in miRNA metabolism [Citation4,Citation7], this indicates the existence of physiologically relevant signalling pathways connecting EGF-stimulated protein kinases to miRNA metabolism via phosphorylation-dependent regulation of Gld2. EGF activates many cellular pathways involved in regulating growth, proliferation, differentiation, and survival. EGF binds to receptor tyrosine kinases, leading to their activation. This in turn activates cascades of cellular kinases. Within 10 min of EGF stimulation, mammalian cells display 100s of new phosphorylation events [Citation32]. Data from this study also indicate that following EGF stimulation 30 tyrosine and more than 100 serine/threonine kinases are activated by phosphorylation, which may be responsible for S62 phosphorylation. Any of these serine/threonine kinases are potential candidates for catalysing S62 phosphorylation.
Figure 3. Gld2 is phosphorylated at S62 when incubated with EGF-stimulated HEK 293 cell extract. Mass spectra of Gld2 after incubation with cell extracts from EGF stimulated HEK 293 cells showing (a) unphosphorylated S62 and (b) phosphorylation at S62 (bolded and underlined). The intensity for y and b-ions resulting from fragmentation of the peptide containing S62 is shown; these intensities are overlaid on the retention time position of the full peptide mass. M/z values for each y and b ion are shown. Gld2 was isolated using a GST Spintrap column before mass spectrometry. Trypsin was used to generate the Gld2 peptides and the ions from the peptides are shown in the mass spectra. A full scanning was performed to obtain all possible modifications and was followed by parallel reaction monitoring (PRM) to verify the modification at S62.
Gld2 N-terminal domain phosphomimetic variants regulate catalytic activity
To rapidly assess the effects of phosphorylation at positions in the Gld2 N-terminal domain, human Gld2 variants were produced in Escherichia coli with respective serine phosphorylation sites mutated to the phosphomimetic glutamic acid [Citation33]. Wild type and phosphomimetic Gld2 variants were produced and purified to homogeneity (Figure S1). Kinetic parameters for specific activity and binding affinity were determined for wild-type Gld2 and phosphomimetic variants with glutamate substitutions at the phosphorylation sites S62, S69, S95, S110, and S116.
Nucleotide addition activity was measured by incubating each enzyme variant with an RNA substrate and [α-32P]-ATP (, and S2A, S2B). Enzymatic and binding assays were conducted with RNA substrates miR-122 and a mRNA poly(A) tail mimic of 15 adenine residues (15A). MiR-122 and 15A RNA were used based on previous studies demonstrating their competence as Gld2 substrates [Citation4,Citation14].
Figure 4. Phosphomimetic Gld2 variants modulate catalytic activity and RNA binding. (a) Activity assay gels of wild type and mutant Gld2. Wild type (WT) Gld2 and glutamic acid mutants were incubated with [α-32P]-ATP and miR-122 (22 nts) or 15A RNA (15 nts) at 37°C with samples taken every 2 min for 8 min. Reactions were repeated in triplicate (R1-3) and analysed via gel electrophoresis and phosphorimaging. A no enzyme control was performed for each RNA substrate in triplicate and the average of the no enzyme triplicates for each RNA was calculated for the 0-min time point. Reaction products were quantified by exposing a Whatman filter strip dotted with different known concentrations of [α-32P]-ATP to the same phosphorscreen as the gel. R, replicate. (b) and (c) Bar graphs showing the fold change in specific activity (SA) at 1 µM ATP and binding affinity (KD) between wild-type Gld2 and Gld2 glutamic acid mutants with (b) miR-122 (22nt) or (c) oligo(A) tail mimic 15A RNA (15nt). Each Gld2 variant was incubated with 1 µM unlabelled and [α-32P]-labelled ATP and 2 µM RNA substrate. Samples were taken every 2 min and stopped with the addition of 2x RNA loading dye. Reactions were analysed via electrophoretic separation and subsequent phosphorimaging. Specific activity is the activity of an enzyme per milligram of purified enzyme and was calculated from the linear slope of the curve. Fluorescence anisotropy was used to determine the KD. Each Gld2 enzyme was incubated with a RNA substrate fluorescently labelled on the 5ʹ-end with 6-FAM and incubated at room temperature for 20 min. Fluorescence polarization was measured at Ex. 492 nm and Em. 535/20 nm and the KD was calculated using SigmaPlot. Error bars represent one standard error calculated from triplicate reactions. Significant changes calculated using a two-tailed t-test are indicted by asterisks. p ≤ 0.05 (*); p ≤ 0.01 (**); p ≤ 0.001 (***). Fold changes were calculated using data from and Figure S2.
![Figure 4. Phosphomimetic Gld2 variants modulate catalytic activity and RNA binding. (a) Activity assay gels of wild type and mutant Gld2. Wild type (WT) Gld2 and glutamic acid mutants were incubated with [α-32P]-ATP and miR-122 (22 nts) or 15A RNA (15 nts) at 37°C with samples taken every 2 min for 8 min. Reactions were repeated in triplicate (R1-3) and analysed via gel electrophoresis and phosphorimaging. A no enzyme control was performed for each RNA substrate in triplicate and the average of the no enzyme triplicates for each RNA was calculated for the 0-min time point. Reaction products were quantified by exposing a Whatman filter strip dotted with different known concentrations of [α-32P]-ATP to the same phosphorscreen as the gel. R, replicate. (b) and (c) Bar graphs showing the fold change in specific activity (SA) at 1 µM ATP and binding affinity (KD) between wild-type Gld2 and Gld2 glutamic acid mutants with (b) miR-122 (22nt) or (c) oligo(A) tail mimic 15A RNA (15nt). Each Gld2 variant was incubated with 1 µM unlabelled and [α-32P]-labelled ATP and 2 µM RNA substrate. Samples were taken every 2 min and stopped with the addition of 2x RNA loading dye. Reactions were analysed via electrophoretic separation and subsequent phosphorimaging. Specific activity is the activity of an enzyme per milligram of purified enzyme and was calculated from the linear slope of the curve. Fluorescence anisotropy was used to determine the KD. Each Gld2 enzyme was incubated with a RNA substrate fluorescently labelled on the 5ʹ-end with 6-FAM and incubated at room temperature for 20 min. Fluorescence polarization was measured at Ex. 492 nm and Em. 535/20 nm and the KD was calculated using SigmaPlot. Error bars represent one standard error calculated from triplicate reactions. Significant changes calculated using a two-tailed t-test are indicted by asterisks. p ≤ 0.05 (*); p ≤ 0.01 (**); p ≤ 0.001 (***). Fold changes were calculated using data from Table 1 and Figure S2.](/cms/asset/11e6e5f4-aab0-4e66-8e0d-53124c41dee6/krnb_a_1608754_f0004_oc.jpg)
Depending on the residue location, phosphomimetic substitutions had distinct effects on enzyme activity (, ). S62E markedly increased activity with both RNAs compared to wild-type Gld2, indicating an overall activating effect. In contrast, an S116E mutation severely decreased Gld2 activity with miR-122 and 15A RNA. Interestingly, S110E increased activity for miR-122 but decreased activity for 15A RNA. Gld2 S69E showed no significant changes in activity for either RNA while S95E was 1.6-fold more active with 15A RNA.
Table 1. Activity and RNA-binding of wild type (WT) and phosphomimetic Gld2 variants.
Based on a comparison of the specific activities (, ), only S62E and S116E displayed statistically significant changes in activity with both RNA substrates. S62E enhanced the nucleotide addition activity by ~5-fold. S116E exhibited the opposite effect, decreasing Gld2 activity by 111-fold with miR-122 and 16-fold with 15A RNA. As the S62E and S116E mutants displayed opposite effects on Gld2 activity, a double mutant (S62E, S116E) was generated to investigate cumulative effects. Interestingly, the inhibitory effect of the S116E mutation overpowered the activating effect of the S62E mutation and decreased the activity of Gld2 74-fold with miR-122 and 5-fold with 15A RNA compared to the wild type enzyme. The double mutant counteracted the silencing effect of S116E alone by 3.1-fold with 15A RNA and 1.5-fold with miR-122.
The ability of the mutants to alter the nucleotide addition activity varied between RNA substrates. The molecular basis for the higher specific activity of Gld2 with miR-122 compared to 15A RNA remains to be elucidated to discern whether Gld2 recognizes a specific RNA sequence and/or discriminates substrates based on the RNA length.
Gld2 phosphomimetic substitutions impact RNA substrate affinity
As changes in catalytic activity were RNA-dependent, the RNA binding affinities of all Gld2 phosphomimetic variants were quantified using fluorescence anisotropy (Figures S2C, S2D). The binding affinities (KD) for all enzyme variants were in the nanomolar range (, ). Changes in RNA binding affinity were substrate dependent. The binding affinity to miR-122 was unchanged for most mutants, except for S62E, which showed a 5.5-fold increase in RNA binding compared to wild-type Gld2 with miR-122. The same mutant showed no change in binding to a 15A substrate. For the 15A RNA, two mutants (S69E and S95E) showed a decrease in affinity, while all other mutants showed no change. With a 6.6-fold reduced KD compared to wild type, Gld2 S95E showed the most dramatic impact on 15A RNA binding; S69E was 3.3-fold decreased in binding affinity to 15A.
Overall, our phosphomimetic analysis suggests that each phosphorylation site has a distinct role in regulating Gld2 activity or substrate selectivity. We found that S62E increases activity with either no change (15A) or with increased binding affinity (miR-122), which may favour miRNA over mRNA stabilization. S69E and S95E caused no significant change in activity, but decreased affinity towards 15A RNA, indicating a reduced preference for mRNA adenylation. S110E appears to have an insignificant impact on activity and binding on the tested substrates. Finally, S116E and the S62E/S116E double mutant markedly reduced activity without significantly impacting RNA binding. The data indicate that Gld2 S116E and the double mutant are able to bind to the RNA target, but perhaps not in a catalytically competent conformation.
PKA and Akt1 site-specifically phosphorylate Gld2 at S116
As the phosphomimetic mutants displayed significant changes in activity and RNA substrate binding compared to wild-type Gld2, we next identified kinases that phosphorylate the Gld2 N-terminus. PhosphoMotif Finder [Citation34] and GPS 3.0 [Citation35] were used to generate a list of potential kinases (Table S2). Using the kinase assay detailed in Materials and Methods, wild-type Gld2 was incubated with recombinant and active human kinases (CK2α, CK2 holoenzyme, CDK5, PKA, and Akt1) predicted to have a recognition motif in Gld2 (). The kinase Abl, which was not identified as a potential Gld2 kinase, was used as a negative control. We used our recently developed approach [Citation36] combining genetic code expansion with in vivo enzymatic phosphorylation to prepare fully activated and purified recombinant Akt1 with programmed phosphorylation at both activating sites (ppAkt1T308,S473). This method involves protein production in an E. coli strain that co-expresses the kinase PDK1 to phosphorylate Akt1 at T308. The strain also genetically encodes phosphoserine (pSer) at UAG codons. The serine codon at position 473 was replaced with a UAG codon to direct pSer incorporation in Akt1.
Figure 5. Akt1 and PKA phosphorylate Gld2 at S116. (a) Gld2 or Gld2 S116A were incubated with [γ-32P]-ATP and the indicated kinases. Formation of phosphorylated Gld2 (75 kDa) was monitored by electrophoretic separation and subsequent phosphorimaging. R, replicate. (b) Quantification of phosphorylated product formation from a kinase reaction over 15 min. Doubly phosphorylated Akt1 displays a low level of autophosphorylation, which we measured in the absence of a substrate peptide. (c) Mass spectra of unphosphorylated Gld2 or Gld2 phosphorylated by CK2α, ppAkt1T308,S473 (ppAkt1), or PKA. Unphosphorylated peptide is indicated by the light pink peak and the phosphorylated peptide by the dark pink peak. Position 116 is bolded and underlined. (d) Bar graphs showing the fold change in specific activity (SA) at 1 µM ATP between treated WT (tWT) and Gld2 phosphorylated by PKA with miR-122 (22nt) or oligo(A) tail mimic 15A RNA (15nt). The inset shows the fold change in activity of Gld2 phosphorylated by PKA compared to tWT. Error bars show one standard error calculated from triplicate reactions. Significant changes calculated using a two-tailed t-test are indicted by asterisks. p ≤ 0.001 (***).
![Figure 5. Akt1 and PKA phosphorylate Gld2 at S116. (a) Gld2 or Gld2 S116A were incubated with [γ-32P]-ATP and the indicated kinases. Formation of phosphorylated Gld2 (75 kDa) was monitored by electrophoretic separation and subsequent phosphorimaging. R, replicate. (b) Quantification of phosphorylated product formation from a kinase reaction over 15 min. Doubly phosphorylated Akt1 displays a low level of autophosphorylation, which we measured in the absence of a substrate peptide. (c) Mass spectra of unphosphorylated Gld2 or Gld2 phosphorylated by CK2α, ppAkt1T308,S473 (ppAkt1), or PKA. Unphosphorylated peptide is indicated by the light pink peak and the phosphorylated peptide by the dark pink peak. Position 116 is bolded and underlined. (d) Bar graphs showing the fold change in specific activity (SA) at 1 µM ATP between treated WT (tWT) and Gld2 phosphorylated by PKA with miR-122 (22nt) or oligo(A) tail mimic 15A RNA (15nt). The inset shows the fold change in activity of Gld2 phosphorylated by PKA compared to tWT. Error bars show one standard error calculated from triplicate reactions. Significant changes calculated using a two-tailed t-test are indicted by asterisks. p ≤ 0.001 (***).](/cms/asset/35029c31-cf1c-4376-a552-d9fcb6e0c41e/krnb_a_1608754_f0005_oc.jpg)
Following incubation of Gld2 and [γ-32P]-ATP with each kinase in separate reactions, the radio-labelled phosphorylated Gld2 (pGld2) product was only observed when Gld2 was incubated with PKA or ppAkt1T308,S473 (Figure S3, ). Quantification of the Akt1-dependent reaction showed a rapid increase in phosphorylated Gld2 over a 15-min time course (). Independent pGld2 preparations resulting from incubation with PKA or ppAkt1T308,S473 were analysed by mass spectrometry to determine the site(s) of phosphorylation. Both un-phosphorylated Gld2 and Gld2 incubated with CK2α, which was inactive in phosphorylating Gld2, were also analysed by mass spectrometry as controls (). S116 was unambiguously identified as the site of specific phosphorylation by Akt1 and PKA. Phosphorylation at S116 was not observed in un-phosphorylated Gld2 or in the preparation incubated with CK2α. Next, S116 was mutated to an alanine residue (Figure S1) to determine if S116 represents the sole Akt1 and PKA phosphorylation site on Gld2. The kinase assay was repeated with Gld2 S116A incubated with PKA or ppAkt1T308,S473 (). For both kinases, no phosphorylated Gld2 S116A product was observed, confirming that PKA and Akt1 phosphorylate Gld2 specifically and exclusively at S116.
Phosphorylation of Gld2 at S116 abolishes nucleotide addition activity
The phosphomimetic mutant (S116E) was not competent in nucleotide addition, yet the variant retained RNA binding affinity (, ). As Akt1 and PKA both phosphorylate S116, we produced pGld2S116 following incubation with PKA as noted above to investigate the nucleotide addition activity of Gld2 with phosphate on S116. We performed nucleotide addition activity assays with pGld2S116 and with an un-phosphorylated Gld2 control without PKA addition.
PKA-dependent phosphorylation of Gld2 decreased nucleotide addition activity by two orders of magnitude (, S4). Significant reductions in activity were observed for both 15A RNA (~45 fold) and miR-122 (~400 fold) substrates. Although both pGld2S116 and the S116E phosphomimetic variant reduced nucleotide addition activity, as we anticipated, the phosphate at S116 had a significantly stronger inhibitory effect compared to acidic amino acid substitutions (~3 fold). This observation is even more striking in light of the fact that our pGld2S116 preparations are only partially phosphorylated (), suggesting phosphorylation of the Gld2 N-terminal domain is a potent mechanism for the cell to control nucleotide addition activity.
Nucleotide addition activity assays and binding assays were also performed for the S116A mutant (, Figure S5). Alanine substitutions are often used as phospho-ablated enzyme models. Although the mutant was expected to act similarly to the wild type enzyme, the alanine substitution in fact reduced the activity by 23-fold with miR-122. As described above, the S116E mutation reduced activity by 111-fold with miR-122, indicating that a serine residue is crucial at this position and cannot be replaced by alanine. Thus, S116A is not an appropriate model for an un-phosphorylated Gld2 and indicates that a serine in position S116 is required for full activity. We previously showed that alanine is not necessarily a good model for a non-phosphorylatable residue [Citation36]. Overall, these data indicate that ppAkt1T308,S473 dependent phosphorylation of Gld2 inactivates Gld2. Our data reveals a novel molecular pathway linking Akt1 activity to miR-122 stability and activity in vivo.
Discussion
Gld2 activity is regulated by phosphorylation
Gld2 is a key regulator of the stabilization and maturation of tumour suppressors miR-122 and let-7 [Citation4,Citation7,Citation11]. Cellular mechanisms that regulate Gld2 mediated nucleotide addition were previously unknown. Data from HCC cells [Citation18] and proteomic analysis [Citation29,Citation31] implicated post-translational modification as a potential mechanism regulating Gld2 activity. Here, we presented the first evidence that serine phosphorylation of Gld2 has a profound impact on catalytic activity and RNA binding.
We identified phosphorylation sites in the N-terminal domain of Gld2 that positively or negatively regulate nucleotide addition activity. Our findings are reminiscent of regulation identified in other polymerases. Phosphorylation of specific serine residues in the C-terminal domain of RNA polymerase II and in the terminal uridylyltransferase Tut1 regulates their activity and substrate recognition [Citation27,Citation28]. For RNA polymerase II, serine phosphorylation in the disordered C-terminal domain is required for promoter clearance [Citation27]. Phosphorylation of the uridylyltransferase Tut1 at position S6 plays a role in its regulation of specific mRNAs and in its nuclear retention, possibly by facilitating interactions between Tut1 and nuclear proteins [Citation28]. Similarly, we found that both phosphomimetic mutations to acidic residues or true phosphorylation of Gld2 at different sites in the N-terminal domain substantially altered substrate specificity, enhanced, or abolished enzyme activity. Although these phosphorylation sites are conserved among mammalian Gld2 proteins, they are not conserved in the human terminal uridylyltransferases Tut4 and Tut7, as these enzymes lack the large disordered N-terminal region.
Undoubtedly, human cells possess a robust Gld2 phosphorylation activity (). We found that lysates from EGF-stimulated HEK 293 cells were active in phosphorylating Gld2 at S62. In this particular experiment, we were not able to identify additional phosphorylation sites in Gld2, suggesting that additional Gld2 kinases may not be activated or sufficiently active in our experimental conditions. While Akt1 and PKA are expressed in HEK 293 cells, Akt1 activity in these cells even upon EGF stimulation is low [Citation37] and likely not sufficient to yield quantitative phosphorylation of Gld2 S116 required for mass spectrometry. Alternatively, Gld2 phosphatases may be active at sites other than Ser62. The data, nevertheless, show that human cells are competent in phosphorylation of Gld2 in its N-terminal domain.
Our experiments with phosphomimetic mutants indicate that phosphorylation at S62 significantly increases Gld2 activity with miR-122 and the 15A RNA. Although Gld2 S62E showed a significant increase in activity with both RNAs, increased binding affinity was only observed with miR-122. It is interesting to note that wild-type Gld2 is ninefold more active with miR-122 than with the poly(A) tail mimic. Even the increase in activity with the poly(A) tail mimic by S62E does not reach the level of wild type activity with miR-122. In contrast, experiments with Gld2 S116E and pGld2S116 show that phosphorylation at this site almost abolishes nucleotide addition activity. Although enzyme activity is more than 100-fold reduced with miR-122, Gld2 S116E and pGld2S116 still retained very low levels of enzymatic activity. In comparison, a mutation in the active site, D215A, completely abolished enzyme activity on miRNA substrates [Citation4,Citation18]. D215A is part of the conserved catalytic triad responsible for activity [Citation3,Citation38,Citation39], while S116 is found in the disordered N-terminal domain, which was previously shown to be dispensable for catalytic activity in the related uridylyltransferase Cid1 [Citation40–Citation42], but we here show that its function lies in the regulation of enzyme activity. Using enzymatic assays and mass spectrometry, we identified and validated Akt1 and PKA as kinases with site-specific phosphorylation activity at S116 in Gld2. Although acidic amino acids are not always able to mimic the functional impact of phosphate [Citation36], the glutamate variant showed reduced activity similarly to the pGld2S116 enzyme. Phosphorylation at S116, however, led to a significantly greater reduction in Gld2 activity, which was two orders of magnitude below the activity of the un-phosphorylated enzyme. Thus, phosphorylation at S116 effectively controls Gld2 activity.
During HCV infection, the Hepatitis C core protein binds to Gld2 to inhibit its adenylation activity. Core protein binding is somewhat inefficient, with 13% binding at a 1:1 ratio. Nonetheless, this inhibition leads to a reduction of cellular miR-122 levels by 30%[Citation18]. Even partial phosphorylation of cellular Gld2 at S116 is expected to have a similar or greater effect on miR-122 levels, making posttranslational modification of Gld2 an efficient means to control cellular miRNA levels.
Although phosphorylation of S116 inhibits Gld2 catalytic activity, RNA binding affinity was unperturbed, suggesting Gld2 phosphorylated at S116 binds the RNA substrate in a non-productive conformation. The crystal structures of related nucleotidyltransferases from yeast [Citation43] and C. elegans [Citation15,Citation44] as well as the human terminal uridylyltransferases Tut1 and Tut7 [Citation45,Citation46] revealed a conserved positively charged surface that may facilitate RNA binding. Despite these efforts, no structural information is available on the N-terminal domain of Gld2. Our data suggest that Gld2 can assume different RNA binding modes. The wild-type Gld2 enzyme binds RNA with high affinity in a catalytically competent mode, which is perhaps stabilized yet further in the activating mutants S62E and S110E. Conversely, Gld2 variants with phosphomimetic substitution or phosphorylation at S116 appear to bind the RNA substrate in a non-catalytic mode that interferes with nucleotide addition. This is not unique to Gld2, as other cases of catalytically incompetent enzymes have been described in the literature. A small deletion in RNA polymerase (RNAP) leads to a catalytically incompetent RNAP that remains bound to the promoter complex [Citation47]. It is also well known that phosphorylation of the C-terminal domain of RNAP II is required for promoter clearance but not its activity [Citation27,Citation48]. While the structural basis for phosphorylation-dependent modulation of Gld2 activity is not yet defined, our experiments suggest an allosteric mechanism. We identified Gld2 variants that impact activity independently of RNA binding, indicating that allosteric or conformational changes in the Gld2 RNA complex may play an important role in Gld2 catalysed nucleotide addition.
Furthermore, in the cell, phosphorylation of Gld2 may affect interactions with proteins that are implicated in RNA substrate selectivity [Citation12,Citation49–Citation52]. In humans, Gld2 was recently shown to interact with the RNA-binding protein Quaking (QKI-7) to facilitate polyadenylation of target mRNAs. QKI-7 was shown to bind between residues 1–141 on Gld2 [Citation52], which correspond to the disordered N-terminal domain, including all of the phosphorylation sites investigated here. Future efforts will determine the effect of phosphorylation on the ability of RNA binding proteins to interact with and regulate Gld2 activity and substrate specificity.
Oncogenic protein kinases signal to miRNA regulation
We identified Gld2 as a previously unknown substrate of two oncogenic kinases. PKA and Akt1 belong to the evolutionarily conserved AGC family of protein kinases that are activated upon stimulation with growth factors such as EGF [Citation53]. PKA is the key kinase in the cyclic AMP signalling pathway that is activated upon hormone binding to G-protein coupled receptors (GPCR). PKA activation has been shown to modulate the expression of miR-122 through the extracellular signal-regulated kinase (ERK) [Citation54] and miRNA let-7b levels through protein phosphatase 2A (PP2A) activation but the underlying pathway remained unclear [Citation55].
Akt1 is a central hub of the PI3K/Akt signalling pathway, which is the most commonly activated signal transduction pathway in human cancers [Citation56]. Active Akt1 signals for cell survival and proliferation while also inhibiting apoptosis [Citation53]. Akt1 activity is dependent on phosphorylation at two key regulatory sites (T308, S473). Over-active and hyper-phosphorylated Akt1 is a hallmark of diverse human malignancies [Citation56,Citation57], while the unmodified Akt1 protein is inactive and rapidly degraded in cells [Citation58]. Several reports show that Akt1 expression is regulated by miRNAs. The miRNAs miR-564 and miR-215 directly negatively regulate Akt1 mRNA stability [Citation59,Citation60], while miRNA let-7 inhibits cyclin D1 expression, leading to reduced Akt1 phosphorylation at S473 [Citation61]. Conversely, miRNA-122 overexpression is associated with increased Akt1 phosphorylation in T-cell lymphoma and renal cell carcinoma cells [Citation62], enhancing cancer progression. These reports correlate PKA or Akt1 activity with miRNA levels yet fail to identify the factor linking PKA or Akt1 activity to miRNA levels, exposing gaps in our understanding of the signal transduction network. In addition, neither PKA nor Akt1 were previously shown to directly phosphorylate any regulator of RNA.
Although miRNAs are known to regulate Akt1, the ability of Akt1 activity to control an enzyme involved in the regulation of miRNA metabolism has not been documented previously. The Akt isozyme Akt2 is known to phosphorylate the single-stranded RNA binding protein KH-type splicing regulatory protein (KSRP), allowing it to switch its RNA preference from mRNA to primary miRNA (pri-miRNA) and facilitate Drosha processing to pre-miRNA [Citation63]. Although KSRP binds RNA, no enzyme that directly regulates RNA stability has been previously identified as a substrate of Akt1. Using precise biochemical experiments, we found that both Akt1 and PKA site-specifically phosphorylate Gld2 at S116, near abolishing Gld2 activity with miRNA and mRNA substrates.
Relevance of Gld2 phosphorylation to disease
Hyperactivity of Akt1 and PKA is common in many cancers [Citation64,Citation65]. Our data suggest that phosphorylation of Gld2 by these kinases would further promote carcinogenesis by destabilizing tumor suppressor miRNAs, thus, further inducing tumorigenesis. Abolishing Gld2 activity leads to a decrease in levels of tumor suppressor miRNAs including miR-122 and let-7 [Citation7,Citation16] (). Decreased miR-122 and let-7 levels and activity enable over- or un-regulated expression of their target genes, including oncogenes with roles in cell growth, metastasis, and apoptosis [Citation16,Citation66].
Figure 6. Model of Akt1-mediated regulation of Gld2. Gld2 monoadenylates miRNAs to increase stability and adds a single nucleotide to pre-miRNAs to enable recognition by Dicer and miRNA maturation. Decreased miRNA levels (e.g. let-7b [Citation55]) are associated with decreased Akt1 phosphorylation status and activity. Conversely, fully activated Akt1 (ppAkt1T308,S473) phosphorylates Gld2 at S116 and silences the stabilizing/maturing effect of Gld2 on miRNA. Thus, through phosphorylation of Gld2, Akt1 activity is expected to reduce miRNA levels. Phosphorylation is indicated by the orange circles.
![Figure 6. Model of Akt1-mediated regulation of Gld2. Gld2 monoadenylates miRNAs to increase stability and adds a single nucleotide to pre-miRNAs to enable recognition by Dicer and miRNA maturation. Decreased miRNA levels (e.g. let-7b [Citation55]) are associated with decreased Akt1 phosphorylation status and activity. Conversely, fully activated Akt1 (ppAkt1T308,S473) phosphorylates Gld2 at S116 and silences the stabilizing/maturing effect of Gld2 on miRNA. Thus, through phosphorylation of Gld2, Akt1 activity is expected to reduce miRNA levels. Phosphorylation is indicated by the orange circles.](/cms/asset/f8ef57a1-c7f4-4f1a-9b30-17f55ccd93e8/krnb_a_1608754_f0006_oc.jpg)
In a related disease context, Gld2 is downregulated in Hepatitis B and inhibited in Hepatitis C infections [Citation16,Citation18]. Both HBV and HCV are contributing factors in the development of HCC and other liver diseases due to the dysregulation in miR-122 levels and the resulting expression of miR-122 regulated oncogenes [Citation16,Citation19]. The extent of Gld2 catalysed mRNA adenylation, and the effect of specific phosphorylations on the transcriptome and miRnome remain to be investigated and may reveal additional contributions of Gld2 regulation to pathogenesis.
Conclusion
While hundreds of Akt1 substrates have been validated and/or predicted in human cells [Citation67], miRNA editing enzymes were not previously known to be part of the Akt1 signalling network. Similarly, it was unclear how Gld2 activity may be regulated to respond to external stimuli and signaling pathways, in turn controlling miRNAs and an even larger number of downstream mRNA substrates. We here revealed the first link between the activity of oncogenic kinases Akt1 and PKA and the regulation of Gld2. We found that HEK 293 cells contain N-terminal Gld2 kinase activity and that phosphorylation sites in the N-terminal domain of Gld2 can either positively or negatively regulate nucleotide addition activity. We identified Gld2 as a bona fide substrate of PKA and Akt1, and the site-specific phosphorylation catalysed by either kinase at Gld2 S116 abolishes nucleotide addition activity. While the overall impact of Akt1/PKA signalling on miRNA metabolism remains to be investigated in a cellular context, these data significantly enhance our knowledge on miRNA regulation and reveal a previously unrecognized link between oncogenic signal transduction and the regulation of tumor suppressor miRNAs.
Materials and methods
Multiple sequence alignment
Alignments were performed as previously described [Citation14]. Briefly, 250 mammalian Gld2 sequences were downloaded from NCBI. Sequence alignment and alignment editing were performed with Muscle [Citation68], MultiSeq from VMD 1.8.7 [Citation69], and Wasabi [Citation70].
Plasmids, protein purification, and western blotting
Homo sapiens Gld2 was codon-optimized (Genewiz, South Plainfield, NJ, USA) for expression in Escherichia coli. The gene was cloned into pGEX-6P-2 with a N-terminal TEV cleavage site using BamHI and XhoI restriction sites. Mutants were generated through site-directed mutagenesis [Citation33]. All primers are listed in Table S1. Successful cloning was verified by DNA sequencing at the London Regional Genomics Centre, London, ON, Canada. Gld2 protein production, purification, and Western blotting details are in the Supplementary Data. Cloning of Akt1 and PDK1 and Akt1 production and purification were previously described [Citation36].
Nucleotide addition assay
Gld2-catalysed reactions and product quantification were carried out as described previously [Citation14]. Specific details can be found in the Supplementary Data.
Fluorescence anisotropy
Various concentrations of Gld2 were incubated with 20 nM 5ʹ-end labelled miR-122 (22 nt) or 15A RNA (15 nt) room temperature in the dark. The RNAs were labelled on the 5ʹ-end with 6-carboxyfluorescein (6-FAM) (Sigma-Aldrich). Fluorescence polarization was measured on a Victor3V (PerkinElmer) with an excitation of 492 nm and emission of 535/20 nm and a binding curve was generated to calculate the dissociation constants (KD). More details can be found in the Supplementary Data.
Identification of potential kinases
Two online tools were used to generate a list of potential Gld2 kinases. PhosphoMotif Finder identifies putative kinase binding sequences in a query sequence based on the binding motifs of kinases as well as their substrate sequences identified in the literature [Citation34]. GPS 3.0 predicts kinase phosphorylation sites in the query sequence using a computational prediction program [Citation35].
Dot plot kinase activity assays
Kinase assays were performed as previously described [Citation36]. Briefly, reactions containing a kinase, wild-type Gld2, and [γ-32P]-ATP (Perkin Elmer) were carried out at 37°C Samples were taken at various timepoints and stopped by spotting on P81 paper. The P81 paper was washed, air-dried, exposed to a phosphor screen, and visualized with a phosphorimager (Storm 860 Molecular Imager).
Kinase activity assays using SDS gels
In the following assays, Gld2 was tested as a protein substrate for several human kinases. Kinase assays were performed in 60 µL reactions containing 900 nM Gld2, kinase buffer (20 mM MOPS [pH 7.0], 25 mM β-glyceraldehydephosphate, 25 mM MgCl2, 5 mM EGTA [pH 8.0], 1 mM Na2VO4, 0.1 mM ATP, 13.2 nM [γ-32P]-ATP (Perkin Elmer)). Reactions were initiated with the addition of the specified kinase. Since kinases varied in activity, final concentrations were adjusted according to published values using 25 nM CK2α, 1.43 nM CK2 holoenzyme, 33 nM fully activated ppAkt1T308,S473, 30 nM Abl, 0.45 nM PKA, or 12 nM CDK5 in a kinase dilution buffer (0.1 mg/mL BSA, 5 mM MOPS [pH 7.2], 25 mM β-glyceraldehydephosphate, 5 mM MgCl2, 1 mM EGTA [pH 8.0], 1 mM Na2VO4, 100 mM NaCl). Reactions were incubated at 37°C for 15 min on a microcentrifuge shaker. Samples (20 µL) were taken every 5 min and the reaction was stopped with the addition of 2 x SDS loading dye. Purified recombinant kinases CK2α, CK2 holoenzyme, Abl, PKA, and CDK5 were a generous gift from Dr David W. Litchfield (The University of Western Ontario, Canada). Reaction products at each time point were separated on a 10% polyacrylamide SDS gel. The gel was exposed to a storage phosphor screen overnight at −80°C and visualized with a phosphorimager (Storm 860 Molecular Imager). Kinase assays and quantification were previously described [Citation36,Citation71]. Kinase assays using unlabelled ATP for further downstream assays are described in the Supplementary Data.
Phosphorylation of Gld2 using HEK 293 cell extract
HEK 293 cells in 100 mm plates were grown to approximately 90% confluency in Dulbecco’s Modified Eagle’s Medium (DMEM) (319–005-3L, Wisent) with 10% fetal bovine serum (FBS) (098150, Lot 185,700, Wisent) and 1% penicillin-streptomycin (450–201-EL, Wisent) as described previously [Citation72]. Epidermal growth factor (EGF) was added to each plate to a final concentration of 50 ng/mL and incubated at 37°C for 1 hr. Cells were harvested and resuspended in 5 x kinase buffer with 5 mM phenylmethylsulfonyl fluoride (PMSF). Cells were broken with a Q125 Sonicator (Qsonica) six times at 20% amplitude and 1 sec on, 1 sec off. Using the cell extract as a source of active kinases, we performed a kinase reaction. This was repeated with unstimulated HEK 293 cell extract. Gld2 was isolated using a GST Spintrap column, and possible Gld2 phosphorylation sites were analysed by mass spectrometry.
Mass spectrometry
Mass spectrometry (MS) analysis of tryptic digested peptides was carried out on the EasyLC1000-QExactive tandem LC-MS system (ThermoFisher Scientific). A full scanning (full MS/dd-MS2 TopN, data-dependent acquisition mode in a Q-Exactive) was performed to obtain an overview of all possible protein modifications within Gld2. Parallel-reaction monitoring (PRM) was then carried out to further verify the phosphorylation at S62 or S116 in Gld2. We analysed Gld2 and Gld2 phosphorylated by purified recombinant kinases and by HEK 293 cell extract. Gld2 or pGld2 were precipitated in ice-cold acetone/ethanol/acetic acid (50/50/0.1 v/v/v). The protein precipitate was re-suspended in 8 M urea then reduced in 5 mM DTT at 37°C for 1 hr and alkylated in 14 mM iodoacetamide (IAA) in darkness at RT for 1 hr. Unreacted IAA was neutralized by adding 5 mM DTT and final protein concentration was determined by Bradford assay. Trypsin digestion was performed at 37°C overnight with a protein: trypsin ratio of 20:1 w/w. The digest was desalted in a C18 column (Phenomenex, Torrance, CA, USA) according to the manufacturer’s protocol and re-suspended in MS-grade water for MS injection. Data were analysed using Skyline software [Citation73].
Supplemental Material
Download MS Word (363.6 KB)Acknowledgments
We are thankful to Dr David W. Litchfield, Dr Laszlo Gyenis, and Teresa Nuñez de Villavicencio-Diaz for their advice on kinases and their gift of CK2α, CK2 holoenzyme, Abl, PKA, and CDK5 kinases and to Matthew Turk and Anish Engineer for advice and discussion.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Data
Supplementary data for this article can be accessed on publisher’s website.
Additional information
Funding
References
- Paul P, Chakraborty A, Sarkar D, et al. Interplay between miRNAs and human diseases. J Cell Physiol. 2018;233:2007–2018.
- De Almeida C, Scheer H, Zuber H, et al. RNA uridylation: a key posttranscriptional modification shaping the coding and noncoding transcriptome. Wiley Interdiscip Rev RNA. 2018;9:e1440.
- Chung CZ, Seidl LE, Mann MR, et al. Tipping the balance of RNA stability by 3ʹ editing of the transcriptome. Biochim Biophys Acta. 2017;1861:2971–2979.
- D’Ambrogio A, Gu W, Udagawa T, et al. Specific miRNA Stabilization by Gld2-catalyzed Monoadenylation. Cell Rep. 2012;2:1537–1545.
- Jones MR, Blahna MT, Kozlowski E, et al. Zcchc11 uridylates mature miRNAs to enhance neonatal IGF-1 expression, growth, and survival. PLoS Genet. 2012;8:e1003105.
- Jones MR, Quinton LJ, Blahna MT, et al. Zcchc11-dependent uridylation of microRNA directs cytokine expression. Nat Cell Biol. 2009;11:1157–1163.
- Heo I, Ha M, Lim J, et al. Mono-uridylation of pre-microRNA as a key step in the biogenesis of group II let-7 microRNAs. Cell. 2012;151:521–532.
- Winter J, Jung S, Keller S, et al. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009;11:228–234.
- Kadyk LC, Kimble J. Genetic regulation of entry into meiosis in Caenorhabditis elegans. Dev Camb Engl. 1998;125:1803–1813.
- Wang L, Eckmann CR, Kadyk LC, et al. A regulatory cytoplasmic poly(A) polymerase in Caenorhabditis elegans. Nature. 2002;419:312–316.
- Katoh T, Sakaguchi Y, Miyauchi K, et al. Selective stabilization of mammalian microRNAs by 3ʹ adenylation mediated by the cytoplasmic poly(A) polymerase GLD-2. Genes Dev. 2009;23:433–438.
- Glahder JA, Norrild B. Involvement of hGLD-2 in cytoplasmic polyadenylation of human p53 mRNA. APMIS. 2011;119:769–775.
- Nousch M, Minasaki R, Eckmann CR. Polyadenylation is the key aspect of GLD-2 function in C. elegans. Rna N Y N. 2017;23:1180–1187.
- Chung CZ, Jo DHS, Heinemann IU. Nucleotide specificity of the human terminal nucleotidyltransferase Gld2 (TUT2). Rna N Y N. 2016;22:1239–1249.
- Nakel K, Bonneau F, Eckmann CR, et al. Structural basis for the activation of the C. elegans noncanonical cytoplasmic poly(A)-polymerase GLD-2 by GLD-3. Proc Natl Acad Sci U S A. 2015;112:8614–8619.
- Bandiera S, Pfeffer S, Baumert TF, et al. miR-122–a key factor and therapeutic target in liver disease. J Hepatol. 2015;62:448–457.
- Masaki T, Arend KC, Li Y, et al. miR-122 stimulates hepatitis C virus RNA synthesis by altering the balance of viral RNAs engaged in replication versus translation. Cell Host Microbe. 2015;17:217–228.
- Kim G-W, Lee S-H, Cho H, et al. Hepatitis C Virus Core Protein Promotes miR-122 Destabilization by Inhibiting GLD-2. PLoS Pathog. 2016;12:e1005714.
- Moriya K, Fujie H, Shintani Y, et al. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med. 1998;4:1065–1067.
- Dai R, Peng F, Xiao X, et al. Hepatitis B virus X protein-induced upregulation of CAT-1 stimulates proliferation and inhibits apoptosis in hepatocellular carcinoma cells. Oncotarget. 2017. DOI:10.18632/oncotarget.17631.
- Chang J, Nicolas E, Marks D, et al. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004;1:106–113.
- Peng F, Xiao X, Jiang Y, et al. HBx down-regulated Gld2 plays a critical role in HBV-related dysregulation of miR-122. PloS One. 2014;9:e92998.
- Gebert LFR, Rebhan MAE, Crivelli SEM, et al. Miravirsen (SPC3649) can inhibit the biogenesis of miR-122. Nucleic Acids Res. 2014;42:609–621.
- Van der Ree MH, van der Meer AJ, van Nuenen AC, et al. Miravirsen dosing in chronic hepatitis C patients results in decreased microRNA-122 levels without affecting other microRNAs in plasma. Aliment Pharmacol Ther. 2016;43:102–113.
- Janssen HLA, Reesink HW, Lawitz EJ, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368:1685–1694.
- Iino I, Kikuchi H, Miyazaki S, et al. Effect of miR-122 and its target gene cationic amino acid transporter 1 on colorectal liver metastasis. Cancer Sci. 2013;104:624–630.
- Hirose Y, Ohkuma Y. Phosphorylation of the C-terminal domain of RNA polymerase II plays central roles in the integrated events of eucaryotic gene expression. J Biochem (Tokyo). 2007;141:601–608.
- Mohan N, Ap S, Francis N, et al. Phosphorylation regulates the Star-PAP-PIPKIα interaction and directs specificity toward mRNA targets. Nucleic Acids Res. 2015;43:7005–7020.
- Mertins P, Mani DR, Ruggles KV, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534:55–62.
- Mertins P, Yang F, Liu T, et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol Cell Proteomics MCP. 2014;13:1690–1704.
- Kettenbach AN, Schweppe DK, Faherty BK, et al. Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci Signal. 2011;4:rs5.
- Francavilla C, Papetti M, Rigbolt KTG, et al. Multilayered proteomics reveals molecular switches dictating ligand-dependent EGFR trafficking. Nat Struct Mol Biol. 2016;23:608–618.
- Edelheit O, Hanukoglu A, Hanukoglu I. Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure-function studies. BMC Biotechnol. 2009;9:61.
- Amanchy R, Tian XC, Kubota C, et al. A curated compendium of phosphorylation motifs. Nat Biotechnol. 2007;25:285–286.
- Xue Y, Ren J, Gao X, et al. GPS 2.0, a Tool to Predict Kinase-specific Phosphorylation Sites in Hierarchy. Mol Cell Proteomics MCP. 2008;7:1598–1608.
- Balasuriya N, Kunkel MT, Liu X, et al. Genetic code expansion and live cell imaging reveal that Thr308 phosphorylation is irreplaceable and sufficient for Akt1 activity. J Biol Chem. 2018;293:10744–10756.
- Lee RS, House CM, Cristiano BE, et al. Relative Expression Levels Rather Than Specific Activity Plays the Major Role in Determining In Vivo AKT Isoform Substrate Specificity. Enzyme Res. 2011;2011:720985.
- Schmidt M-J, Norbury CJ. Polyadenylation and beyond: emerging roles for noncanonical poly(A) polymerases. Wiley Interdiscip Rev RNA. 2010;1:142–151.
- Munoz-Tello P, Gabus C, Thore S. Functional implications from the Cid1 poly(U) polymerase crystal structure. Struct Lond Engl 1993. 2012;20:977–986.
- Chung CZ, Jaramillo JE, Ellis MJ, et al. RNA surveillance by uridylation dependent RNA decay in Schizosaccharomyces pombe. Nucleic Acids Res. 2019;47:3045–3057.
- Rissland OS, Mikulasova A, Norbury CJ. Efficient RNA polyuridylation by noncanonical poly(A) polymerases. Mol Cell Biol. 2007;27:3612–3624.
- Yates LA, Amode MR, Barrell D, et al. Structural plasticity of Cid1 provides a basis for its distributive RNA terminal uridylyl transferase activity. Nucleic Acids Res. 2015;43:2968–2979.
- Lunde BM, Magler I, Meinhart A. Crystal structures of the Cid1 poly (U) polymerase reveal the mechanism for UTP selectivity. Nucleic Acids Res. 2012;40:9815–9824.
- Nakel K, Bonneau F, Basquin C, et al. Structural basis for the antagonistic roles of RNP-8 and GLD-3 in GLD-2 poly(A)-polymerase activity. Rna N Y N. 2016;22:1139–1145.
- Faehnle CR, Walleshauser J, Joshua-Tor L. Multi-domain utilization by TUT4 and TUT7 in control of let-7 biogenesis. Nat Struct Mol Biol. 2017;24:658–665.
- Yamashita S, Takagi Y, Nagaike T, et al. Crystal structures of U6 snRNA-specific terminal uridylyltransferase. Nat Commun. 2017;8:15788.
- Maitra A, Shulgina I, Hernandez VJ. Conversion of active promoter-RNA polymerase complexes into inactive promoter bound complexes in E. coli by the transcription effector, ppGpp. Mol Cell. 2005;17:817–829.
- Phatnani HP, Greenleaf AL. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 2006;20:2922–2936.
- Barnard DC, Ryan K, Manley JL, et al. Symplekin and xGLD-2 are required for CPEB-mediated cytoplasmic polyadenylation. Cell. 2004;119:641–651.
- Kim JH, Richter JD. Opposing polymerase-deadenylase activities regulate cytoplasmic polyadenylation. Mol Cell. 2006;24:173–183.
- Rouhana L, Wang L, Buter N, et al. Vertebrate GLD2 poly(A) polymerases in the germline and the brain. Rna N Y N. 2005;11:1117–1130.
- Yamagishi R, Tsusaka T, Mitsunaga H, et al. The STAR protein QKI-7 recruits PAPD4 to regulate post-transcriptional polyadenylation of target mRNAs. Nucleic Acids Res. 2016;44:2475–2490.
- Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11:9–22.
- Menon B, Sinden J, Franzo-Romain M, et al. Regulation of LH receptor mRNA binding protein by miR-122 in rat ovaries. Endocrinology. 2013;154:4826–4834.
- Yamada S, Tsukamoto S, Huang Y, et al. Epigallocatechin-3-O-gallate up-regulates microRNA-let-7b expression by activating 67-kDa laminin receptor signaling in melanoma cells. Sci Rep. 2016;6:19225.
- Agarwal E, Brattain MG, Chowdhury S. Cell Survival and Metastasis Regulation by Akt Signalling in Colorectal Cancer. Cell Signal. 2013;25:1711–1719.
- Spencer A, Yoon SS, Harrison SJ, et al. The novel AKT inhibitor afuresertib shows favorable safety, pharmacokinetics, and clinical activity in multiple myeloma. Blood. 2014;124:2190–2195.
- Oh WJ, Wu CC, Kim SJ, et al. mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. Embo J. 2010;29:3939–3951.
- Yao J, Zhang P, Li J, et al. MicroRNA-215 acts as a tumor suppressor in breast cancer by targeting AKT serine/threonine kinase 1. Oncol Lett. 2017;14:1097–1104.
- Ru N, Zhang F, Liang J, et al. MiR-564 is down-regulated in osteosarcoma and inhibits the proliferation of osteosarcoma cells via targeting Akt. Gene. 2018;645:163–169.
- Sun H, Ding C, Zhang H, et al. Let‑7 miRNAs sensitize breast cancer stem cells to radiation‑induced repression through inhibition of the cyclin D1/Akt1/Wnt1 signaling pathway. Mol Med Rep. 2016;14:3285–3292.
- Lian J-H, Wang W-H, Wang J-Q, et al. MicroRNA-122 promotes proliferation, invasion and migration of renal cell carcinoma cells through the PI3K/Akt signaling pathway. Asian Pac J Cancer Prev APJCP. 2013;14:5017–5021.
- Blahna MT, Hata A. Regulation of miRNA biogenesis as an integrated component of growth factor signaling. Curr Opin Cell Biol. 2013;25:233–240.
- Manning BD, Toker A. AKT/PKB Signaling: navigating the Network. Cell. 2017;169:381–405.
- Caretta A, Mucignat-Caretta C. Protein Kinase A in Cancer. Cancers (Basel). 2011;3:913–926.
- Johnson SM, Grosshans H, Shingara J, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647.
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274.
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797.
- Roberts E, Eargle J, Wright D, et al. MultiSeq: unifying sequence and structure data for evolutionary analysis. BMC Bioinformatics. 2006;7:382.
- Veidenberg A, Medlar A, Löytynoja A. Wasabi: an Integrated Platform for Evolutionary Sequence Analysis and Data Visualization. Mol Biol Evol. 2016;33:1126–1130.
- Balasuriya N, McKenna M, Liu X, et al. Phosphorylation-Dependent Inhibition of Akt1. Genes (Basel). 2018;9:450.
- Turk MA, Chung CZ, Manni E, et al. MiRAR-miRNA Activity Reporter for Living Cells. Genes (Basel). 2018;9:305.
- MacLean B, Tomazela DM, Shulman N, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinforma Oxf Engl. 2010;26:966–968.