
ABSTRACT
Obesity is becoming a global problem. Research into the detailed mechanism of adipocyte development is crucial for the treatment of excess fat. Zinc finger protein 217 plays roles in adipogenesis. However, the underlying mechanism remains unclear. Here, we demonstrated that ZFP217 knockdown prevented the mitotic clonal expansion process and caused adipogenesis inhibition. Depletion of ZFP217 increased the expression of the m6A methyltransferase METTL3, which upregulated the m6A level of cyclin D1 mRNA. METTL3 knockdown rescued the siZFP217-inhibited MCE and promoted CCND1 expression. YTH domain family 2 recognized and degraded the methylated CCND1 mRNA, leading to the downregulation of CCND1. Consequently, cell-cycle progression was blocked, and adipogenesis was inhibited. YTHDF2 knockdown relieved siZFP217-inhibited adipocyte differentiation. These findings reveal that ZFP217 knockdown–induced adipogenesis inhibition was caused by CCND1, which was mediated by METTL3 and YTHDF2 in an m6A-dependent manner. We have provided novel insight into the underlying molecular mechanisms by which m6A methylation is involved in the ZFP217 regulation of adipogenesis.
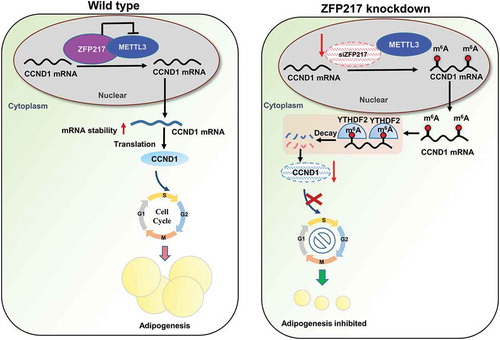
Graphical abstract
Introduction
Obesity, defined as excess body fat, is becoming a major global health problem. When more energy is taken in and less energy consumed in the organism, extra energy is stored as triacylglycerol, which makes the organism become fat [Citation1]. Excess fat is related to cardiovascular disease and type 2 diabetes [Citation2]. However, no effective treatment options are currently suitable for obesity. Understanding the mechanism of adipocyte development is important for future therapeutic strategies.
Adipocytes are the characteristic cells of adipose tissue. They can transform from preadipocytes into mature adipocytes with lipid accumulation [Citation3]. The progression of preadipocyte differentiation into adipocytes is accomplished with increased adipocyte numbers as well as increased adipocyte size. Adipocyte differentiation happens over several phases. Many transcription factors, such as peroxisome proliferator-activated receptor-gamma (PPARγ) and CCAAT/enhancer-binding proteins (C/EBPs), are the major regulators of adipogenic differentiation [Citation4]. Preadipocytes cultured to confluence in vitro became growth-arrested during the transition from G0 to G1 of the cell cycle. Mitotic clonal expansion (MCE) is obligatory for the 3T3-L1 preadipocyte differentiation into adipocytes. Growth-arrested preadipocytes concurrently reenter the MCE process and experience many cycles of cell division [Citation5]. In our recent study, we found that epigallocatechingallate (EGCG) could decrease the protein and gene levels of the MCE regulators and block the MCE process at the same time, thereby preventing the events of adipocyte differentiation [Citation6].
Cell differentiation is regulated by various transcription factors. In eukaryotes, many transcriptional regulators are zinc finger proteins. These elements participate in multiple physiological processes, including cell development and differentiation [Citation7]. For example, ZNF395 together with PPARG2 plays an important role in inducing dermal fibroblasts to undergo adipogenesis [Citation8]. ZNF638 has a mutual effect and transcriptionally coordinates with C/EBPs, leading to the expression of PPARγ [Citation9]. ZFP217 is a member of the Krüppel-type zinc finger protein family, plays important roles in tissue development and cell differentiation. Recently, a novel role for ZFP217 in adipogenesis has been reported, but the underlying mechanism remains largely unclear [Citation10].
RNA chemical modification is a vital posttranscriptional regulator. Like other RNA modifications, methylation of N6-adenosine (m6A), the most common intrinsic eukaryotic mRNA modification, has a close relationship with RNA structure, localization, and function [Citation11]. The installation of m6A on mRNA is controlled by a methyltransferase synthetic composed of the cofactors Wilms tumour 1-associated protein (WTAP) and the methyltransferase-like (METTL) enzymes METTL3 and METTL14. The fat mass and obesity-associated protein (FTO) enzyme or ALKB homolog 5 (ALKBH5) catalyzes m6A removal [Citation12]. The cytoplasmic YTH-domain family proteins can bind to m6A. YTHDF1 can promote mRNA translation efficiency, while YTHDF2 identifies mRNA m6A sites to mediate mRNA degradation [Citation13]. YTHDF3 synergizes with YTHDF1 to promote protein synthesis and influences methylated mRNA decay mediated through YTHDF2 [Citation14].
Our research shows that loss of ZFP217 increased METTL3 expression, leading to increased m6A level of the key cell-cycle regulator cyclin D1 (CCND1), which is proved to be a target of YTHDF2. CCND1 mRNA with higher m6A was recognized and bound by YTHDF2. The decreased mRNA stability and reduced CCND1 protein expression were mediated by YTHDF2. Therefore, MCE was partially arrested and adipogenesis was suppressed, implying a significant relationship between ZFP217 and cell-cycle progression mediated by m6A-modification in 3T3-L1 cell adipogenic mechanism.
Result
Loss of ZFP217 inhibited adipogenesis of 3T3-L1 cells
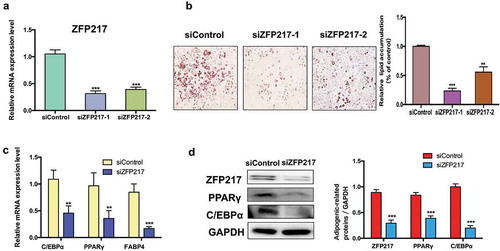
To investigate the effects of ZFP217 on adipogenesis, we conducted loss-of-function assays by using two distinct siRNAs to study the effect of ZFP217 on the adipogenic differentiation of 3T3-L1 cells. We analysed the expression of ZFP217 in 3T3-L1 cells by quantitative PCR with reverse transcription. Results exhibited that endogenous ZFP217 has been knockdown at least 70% at the RNA level in 3T3-L1 cells ()). We have chosen the better performance siRNA to carry out the following experiments. Adipocyte differentiation was evaluated by Oil Red O staining. Results showed that lipid deposition was impaired in ZFP217-knockdown cells in contrast with the control group, and caused an obvious decrease in the number of lipid droplets ()). The adipocyte marker genes and key adipogenic transcription factors PPARγ, fatty acid-binding protein 4 (FABP4) and CCAAT/enhancer-binding protein, alpha (C/EBPα) mRNA expression were markedly suppressed after ZFP217 knockdown ()). Consistent with this observation, the protein expression of PPARγ and C/EBPα were also significantly decreased due to the lack of ZFP217 in 3T3-L1 cells. Taken together, these results suggest that ZFP217 is essential to keep 3T3-L1 cells adipogenic.
Figure 1. ZFP217 knockdown inhibit 3T3-L1 adipogenesis.
(a) ZFP217 knockdown efficiency in 3T3-L1 cell lines. (b) Oil red O staining of control and ZFP-depleted cells after two-day post-confluence 3T3-L1 preadipocytes incubated with the hormonal induces (IBMX, dexamethasoneand insulin). Relative lipid accumulation was quantified with a microplate spectrophotometer at 500 nm. (c) The adipogenesis marker genes mRNA expression was analysed using qPCR. (d) Cells were collected by RIPA lysate after 24 h of induced and subjected to Western blot analyses using the corresponding antibodies. The right panel shows the relative protein levels quantified by densitometry and normalized to GAPDH. The data are presented as the mean ± SD of triplicate tests. *P< 0.05, **P< 0.01, ***P< 0.001 compared to the control group.
ZFP217-knockdown blocks MCE by inhibiting CCND1 expression
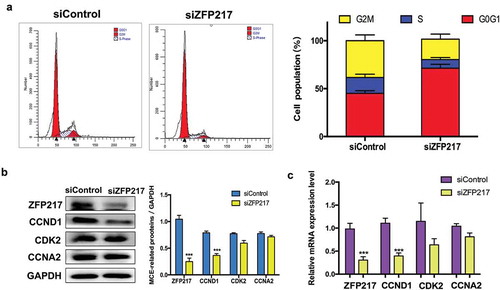
MCE is a vital prerequisite for adipogenesis that occurs within 48 h of adipogenic stimulation. Previous publications have reported that Zinc finger protein family play critical roles in cell cycle [Citation15–Citation17], we speculated that ZFP217 may involve in adipogenesis by regulating cell-cycle progress. To test the function of ZFP217 on MCE, the cell-cycle distribution of ZFP217-depleted cells cultured with MDI for 24 h was analysed by flow cytometry. Compared with the control cells, ZFP217 knockdown triggered more cells to accumulate at G1 stage, whereas the cell numbers in S-phase and G2/M-phase decreased ()). Therefore, cell-cycle progression was prolonged upon the deletion of ZFP217. Cyclins and cyclin-dependent kinases (CDKs) are two major kinds of regulator that determine the cell-cycle progression. Cyclin D1 (CCND1), cyclin-dependent kinase 2 (CDK2) and cyclin A2 (CCNA2) are required for G1 and S phase. To further assess the effects of ZFP217 on MCE, the expression of CCND1, CDK2 and CCNA2 were determined. The protein expression of CCND1 were significantly decreased upon ZFP217 deletion, while the other two factors showed no significant changes compared with the control ()) and the similar results have also been found in mRNA expression ()). These results suggest that decreased CCND1 expression contributed to siZFP217-induced prolonged G1/S transition.
Figure 2. ZFP217 knockdown induced cell-cycle arrest in G0G1 phase.
(a) Flow cytometry analysis of cell-cycle progression of control and ZFP217-depleted cells. Cells were harvested at 24 h after MDI initiated and stained. The percentages of cells in the G1, S and G2 phase were calculated using ModFit LT software. (b) Cell lysates were obtained after 24 h of induction and subjected to Western blot analyses using the corresponding antibodies. The right panel shows the relative protein levels quantified by densitometry and normalized to GAPDH. (c) The MCE-related genes mRNA expression was analysed using qPCR. The data are presented as the mean ± SD of triplicate tests. *P< 0.05, **P< 0.01, ***P< 0.001 compared to the control group.
METTL3 mediates siZFP217-induced decrease in CCND1 protein level
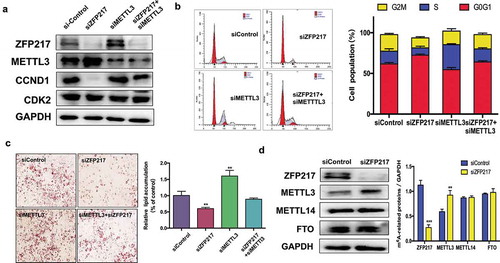
ZFP217 interacts with METTL3 to restrict mRNA transcript m6A deposition [Citation18], which raises the question whether ZFP217 influences CCND1 through METTL3 regulation. First, we detected the effect of ZFP217 on METTL3 expression. We found that ZFP217 knockdown led to increased METTL3 and decreased CCND1 protein expression ()). siMETTL3 also rescued the siZFP217-inhibited CCND1 protein level while CDK2 was not influenced by siZFP217 or siMETTL3. Which imply that ZFP217 participate in adipogenesis through CCND1. The flow cytometry further demonstrated that the METTL3 knockdown group had more cells in G2/M and S phases, and siMETTL3 rescued ZFP217 knockdown-inhibited cell proliferation ()), suggesting METTL3 plays a role in the siZFP217 caused impairment of the cell cycle. A specific and important association between ZFP217 and METTL3 maybe existed in 3T3-L1 cells. ORO staining analysis showed that knockdown of METTL3 partially rescued loss of ZFP217 caused phenomenon of adipocyte differentiation inhibited ()). These results led us to hypothesize that METTL3 is involved in the regulation of adipogenesis by ZFP217. METTL3 is a methyltransferase and always exerts its function in cooperation with other proteins, such as METTL14 and FTO. The RNA methyltransferase complex of WTAP, METTL3, and METTL14 can regulate adipogenesis through mitotic clonal expansion [Citation19]. To rule out the role of other proteins in our system, the effects of ZPF217 depletion on METTL14 and FTO expression were also examined. The immunoblot assays showed no significant distinction between control and ZFP217-knockdown groups except for METTL3 protein ()). Collectively, these findings indicate that ZFP217 may take part in adipogenesis by interacting with METTL3 to regulate adipogenesis.
Figure 3. ZFP217 regulate m6A methyltransferase METTL3 expression.
(a) The MCE and adipogenesis related protein expression. Cell lysates were obtained after 24 h of induction and subjected to Western blot to analysis MCE-related proteins. (b) Flow cytometry analysis of cell-cycle progression of several groups. The percentages of cells in the G1, S and G2 phase were calculated using ModFit LT software. (c) Oil Red O staining of control, ZFP217 knockdown, METTL3 knockdown and ZFP217+ METTL3 knockdown cells after MDI-induced for 8days. (d) ZFP217 knockdown increased METTL3 expression in 3T3-L1 preadipocytes during MCE. Total cell lysates were prepared and analysed by Western blot. Western blot analysis of each total protein was done in parallel. Immunoblots are a representative image of three independent experiments. The data are presented as the mean ± SD of triplicate tests. *P< 0.05, **P< 0.01, ***P< 0.001 compared to the control group.
CCND1 protein expression is mediated by METTL3 in an m6A-dependent manner
The plenty of m6A modification in RNA effectively influences all aspects of RNA metabolism, including cell differentiation and stress responses, resulting in alterations in an array of cellular processes [Citation20]. METTL3 is a recognized methyltransferase and regulates m6A modification dynamically, which suggests the hypothesis that METTL3 influences CCND1 expression through RNA m6A methylation.
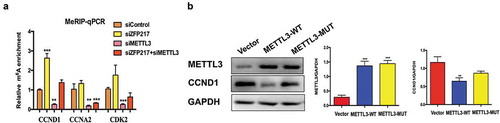
Based on the published m6A-Seq/RNA-Seq datasets [Citation21], many regulatory genes participate in the MCE process, including CCND1, CDK2 and CCNA2, which contain more than one m6A site in 3T3-L1 cells. To investigate the underlying mechanisms, we used methylated RNA immunoprecipitation combined with qPCR (MeRIP-qPCR) [Citation22] to examine m6A methylation levels in the CCND1, CDK2 and CCNA2 transcripts influenced by METTL3 and ZFP217 knockdown. Results showed that ZFP217 knockdown significantly increased while METTL3 knockdown significantly decreased m6A level in CCND1 mRNA. Importantly, CCND1 m6A level dramatically increased upon siZFP217 treatment and combining siZFP217 with siMETTL3 rescued the m6A level, leading to a similar m6A level to control cells ()). These results show that CCND1 protein expression depended on m6A. However, the m6A levels of CDK2 and CCNA2 were also affected by METTL3 but not influenced by ZFP217, suggesting ZFP217 inhibits adipogenesis not through a METTL3-mediated CDK2 or CCNA2 pathway. To further confirm CCND1 protein expression was mediated by METTL3 in an m6A-dependent manner, METTL3-WT and METTL3-MUT plasmids were generated. Overexpression of METTL3-WT significantly decreased CCND1 protein expression ()), while METTL3-MUT had little effect on CCND1, implying that METTL3 regulated CCND1 expression in a methylation-dependent way.
Figure 4. METTL3mediates gene and protein expression of CCND1 in an m6A-dependent manner.
(a) Methylated RNA immunoprecipitation (MeRIP)-qPCR analysis of m6A levels of CCND1 mRNA in each group after MDI-induced for 24 h. (b) Western blot analysis of METTL3 and CCND1 in cells after transfected with control, wild-type (WT) and mutant (MUT) METTL3 plasmid and MDI-induced for 24 h. The data are presented as the mean ± SD of triplicate tests. *P< 0.05, **P< 0.01, ***P< 0.001 compared to the control group.
CCND1 is bounded by YTHDF2, which decreases CCND1 mRNA stability, leading to its lower protein level
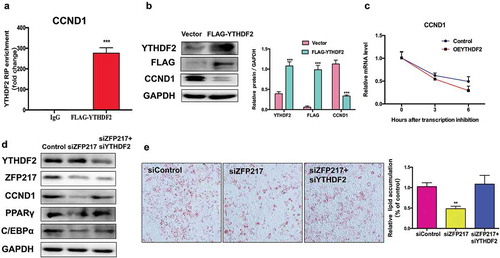
YTHDF2 selectively recognizes the m6A modification and promotes m6A-containing mRNA degradation [Citation23]. This raises the hypothesis that YTHDF2 binds to and degrades the m6A-containing CCND1 mRNA, further decreasing CCND1 protein level. To tested this hypothesis, we generated FLAG-tagged YTHDF2 plasmids to test whether CCND1 was bounded by YTHDF2. RNA immunoprecipitation combined with qPCR analysis confirmed that CCND1 was highly enriched in YTHDF2-RIP lysate ()). Next, we found that CCND1 protein level was dramatically reduced upon YTHDF2 overexpression ()). To further investigate whether CCND1 mRNA degradation happened because of YTHDF2, we used the transcription inhibitor actinomycin D to examine CCND1 mRNA lifetime in cells. Compared to the control group, CCND1 mRNA levels were significantly decreased in YTHDF2 overexpression cells ()), suggesting that YTHDF2 shortens the lifespan of CCND1 mRNA and thereby decreases its protein expression.
Figure 5. CCND1 is a target of YTHDF2 which could influence mRNA stability.
(a) RIP analysis of the interaction of CCND1 with FLAG in 3T3-L1 cells transfected with control or YTHDF2-FLAG. Enrichment of CCND1 with FLAG was measured by qPCR and normalized to input. (b) Western blot analysis of FLAG and YTHDF2 in cells transfected with control and YTHDF2-FLAG plasmid. The right panel shows the relative protein levels quantified by densitometry and normalized to GAPDH. (c) Lifespans of CCND1 expression in cells transfected with the YTHDF2 overexpression plasmid compared with those of cells transfected with empty vector. Relative mRNA levels quantified by qPCR. (d) Western blot analysis of CCND1 in control, ZFP217 knockdown or YTHDF2 knockdown cells after MDI-induced for 24 h. The right panel shows the relative protein levels quantified by densitometry and normalized to GAPDH. (e) ORO staining of control, ZFP217 knockdown and ZFP217+ YTHDF2 knockdown cells after MDI-induced for 8 days. The data are presented as the mean ± SD of triplicate tests. *P< 0.05, **P< 0.01, ***P< 0.001 compared to the control group.
Knockdown of YTHDF2 rescues CCND1 expression and adipogenesis of ZFP217-deficient cells
We next determined whether YTHDF2 plays a main role in the function of ZFP217 in adipogenesis. We found that YTHDF2 deletion rescued CCND1 protein expression in ZPF217-knockdown cells, and no interaction between YTHDF2 and ZPF217 was observed ()). These findings suggest YTHDF2 affects CCND1 protein expression not by directly influencing ZPF217. ORO staining analysis further revealed that decreased YTHDF2 expression partially rescued the impaired adipogenesis caused by loss of ZPF217 ()). Consistent with phenotype, YTHDF2 knockdown partially rescued the effect of ZPF217 knockdown of downregulating the adipocyte markers PPARγ and C/EBPα ()). In short, these results show that ZPF217, METTL3, and YTHDF2 act in synergy to mediate the expression of CCND1 through the m6A modification, affecting the MCE process and furthermore influencing adipogenesis.
Discussion
Adipocyte differentiation involves a series of gene expression events, and there are more than 100 transcription factors that participate in different levels of regulatory mechanism [Citation24]. Understanding the transcriptional network and the mechanism controlling adipogenesis is of fundamental importance. ZFP217 is known as a core component of transcriptional repressor protein complex with histone modification and involves in many biological processes, including cell differentiation and proliferation [Citation25]. ZFP217 has been reported to cooperate with Ezh2 in promoting adipogenesis, which imply that ZFP217 have potential effect on obesity [Citation10]. Here, we conducted a deeper study and found knockdown of ZFP217 can reduce the expression of key adipogenic-related factors and further inhibit adipogenesis, which is consistent with previous results.
ZFP217 was shown to positively regulate adipogenesis by repressing Wnt signalling [Citation10]. While in this study, we uncovered that ZFP217 knockdown significantly decreased the cell amount at G1 stage. Further study showed siZFP217 led to cell proliferation impairment by blocking the mitotic clonal expansion (MCE) in 3T3-L1 cells. MCE is the earliest event in adipogenesis and D-type cyclins are necessary for the differentiation of 3T3-L1 cells [Citation5]. CCND1 and CDK2 are two major factors that act in early G1 phase in response to external stimuli [Citation26]. ZFP217 knockdown decreased CCND1 gene and protein expression, thereby impairing cell-cycle progression and adipogenesis.
To further explore the mechanisms that ZFP217 influences CCND1 expression, we noticed ZFP217 has mutual effect with several RBPs and exert its function in the aspect of RNA metabolism. Study has shown the interaction between ZFP217 and METTL3 and METTL3-ZFP217 is maintained in an inactive complex. METTL3 is a member of the m6A methyltransferase complex. ZFP217 sequesters METTL3, diminishing METTL3 binding to RNAs [Citation18]. Our results found that ZFP217 knockdown increased METTL3 protein expression while other m6A-related proteins were not affected, which raises the question whether ZFP217 regulate adipogenesis via an METTL3-m6A dependent way? In the current study, we found that METTL3 knockdown increased the protein expression of CCND1 and promoted lipid droplet accumulation. METTL3 knockdown rescued the cell-cycle inhibition of siZFP217-inhibited cells, allowing them to transit into G2/M phase. We performed m6A-specific-methylated RNA immunoprecipitation (MeRIP) with next-generation sequencing (m6A-seq) in 3T3-L1 cells. By analysing m6A-seq data, we found an exact m6A site (2864 nt) in 3ʹUTR of CCND1 as shown in Figure S1. Based on this exact m6A site, we designed specific primers to amplify CCND1 gene to perform meRIP-qPCR. As expected, METTL3 has direct effect in reducing CCND1 m6A mRNA level. By expressing a catalytically inactive mutant METTL3, we found that METTL3-mediated CCND1 protein expression was m6A-dependent. Overall, METTL3 interacted with ZFP217 to regulate cell-cycle progression and influence adipogenesis. Here, we cannot exclude other factors that are affected by METTL3 activity can also contribute to the adipogenesis phenotype observed here. Since CCND1 protein was observed to play critical role in the current experiment, we mainly focused on CCND1 in this work. Whether other factors regulate adipogenesis through m6A methylation needs further study.
The m6A writer complex was mainly composed of METTL3, METTL14 and WTAP, whereas METTL14 is not enzymatically active and is therefore not a methyltransferase [Citation27–Citation30]. METTL14 acts as an adaptor to bind the RNA substrate and to promote the activity of METTL3, which is known as the only mRNA m6A methyltransferase so far. Also, we cannot rule out the possibility that other unknown methyltransferases regulate m6A level in the current study. In this study, we found knockdown of METTL3 also decreased m6A level of CDK2 mRNA ()), while the protein abundance of CDK2 was unchanged ()). In our previous work, FTO increased m6A level of CDK2 mRNA and promote CDK2 protein expression [Citation6]. The discrepancy in the correlation between m6A level and protein expression may be due to the different m6A sites targeted by FTO and METTL3. In fact, m6A writers and erasers could preferentially target to distinct regions of the same mRNA transcripts and lead to different fates of the target transcripts. For example, FTO preferentially erases m6A modifications on the 5′-terminal and middle exons of MYC mRNA and promotes the stability of MYC mRNA through inhibition of YTHDF2-mediated RNA decay [Citation31]. While loss of METTL14 decreases m6A abundance on the 3′-region of MYC mRNA and reduces the stability and translation of MYC mRNA [Citation32]. The different m6A sites in same transcripts could lead to different downstream process, like the recognition and bind by readers (m6A binding proteins), eventually result in the difference of protein abundance. Whether FTO and METTL3 targets different m6A sites of CDK2 mRNA need to be further investigated. Since this is not the main problem we focused on in this paper, we did not deeply study on this part. Nevertheless, it is an interesting problem, which deserves further study.
The YTHDF family comprises three paralogs that share high amino acid identity over their entire length, YTHDF1, YTHDF2, and YTHDF3. YTHDF2 is implicated in the control of m6A-associated mRNA stability, and knockdown of YTHDF2 stabilizes the mRNAs with YTHDF2-binding sites [Citation23]. Several researches have elucidated that YTHDF2 recognize m6A and recruiting the machinery of mRNA decay and further lead to mRNA degradation [Citation33–Citation35]. In our study, CCND1 protein expression is negatively correlate with the level of m6A-modified mRNA, suggesting that YTHDF2 participates in the control of CCND1 expression. RIP experiment proved CCND1 was bounded by YTHDF2. To understand further the molecular mechanism of its function, we have performed further experiments. Knockdown both YTHDF2 and ZFP217 can revitalize adipogenesis in 3T3-L1 cells. The interaction between ZFP217 and YTHDF2 happened in cell differentiation progress, it seems that ZFP217 can suppress adipogenesis through interact with YTHDF2.
Taken together, we identified ZFP217 to be pre-requisite for adipocyte differentiation by tightly coupling MCE regulation and m6A RNA modification. Our research is the first time to demonstrate that ZFP217 regulates adipogenesis in an m6A-dependent and YTHDF2-mediated way. These findings shed light on the regulation of ZFP217 is a valuable tool for excess-fat treatments, though further research is required.
Material and methods
Cell culture and adipocyte differentiation
Mouse 3T3-L1 pre-adipocytes were cultured in DMEM medium (DMEM, Gibco, USA) supplemented with 10% foetal bovine serum (FBS, Gibco, USA) and 1% penicillin-streptomycin (Gibco, USA). Two days’s post-confluence, cell differentiation was induced with the MDI differentiation medium containing 1 M dexamethasone (DEX, Sigma-Aldrich, USA), 0.5 mM 3-isobutyl-1- methylxanthine (IBMX, Sigma-Aldrich, USA), 1g/mL insulin (INS, Sigma-Aldrich, USA) and DMEM supplemented with 10% FBS. After 48 h (day 2), the culture medium was replaced with DMEM supplemented with 10% FBS, 1g/mL insulin, and replenished every other day until day 8. Cells were transfected with siZFP217 from day 0 to 8, and untreated cells were used as a control.
Oil Red O staining
After transfection with siZFP217, cells were growth-arrested for 2 days and treated with an adipogenic induction cocktail containing dexamethasone, isobutylmethylxanthine and insulin (the MDI complex). After 6 days of differentiation the fully cells were rinsed twice with PBS (Nanjing KeyGen Biotech, Nanjing, China), and then fixed with 10% methanol for 30 min at room temperature. Then, the cells were rinsed with PBS twice, 60% isopropanol twice, after airing stained with filtered Oil Red O (0.35 g, 60% isopropanol; Sigma, USA) for 30 min, followed by washing twice in water. Stained cells were visualized by using a SpectraMax Microplate Luminometer (Molecular Devices, San Jose, CA, USA). The cell differentiation was quantified by elution of stain with isopropanol and measurement of the absorbance at 490 nm.
Quantitative real-time PCR (qPCR)
Total RNA was extracted from 3T3-L1 cells using TRIzol (Invitrogen, USA) in accordance with a manufacturer’s protocol. Then, 2 μg of total RNA was reverse transcribed into cDNA by M-MLV reverse transcriptase (Invitrogen, USA). Real-time quantitative PCR analysis was performed using SYBR Green PCR Master Mix (Roche, China) with the ABI Step-One PlusTM Real-Time PCR System (Applied Biosystems, USA). The primer sequences are shown in Supplemental Table S2. The data were analysed following the 2−ΔΔCt method and calculated using GAPDH as an internal control.
Western blot analysis
Proteins were isolated using RIPA Lysis Buffer (Beyotime, China) in accordance to the manufacture’s protocol. The protein concentrations were measured using a BCA protein assay kit (Pierce, China). Twenty micrograms of nuclear proteins were loaded in each lane in SDS-PAGE for Western blotting. Equal amounts of protein were heated for 10 min in SDS/PAGE sample buffer (50 mMTris (pH 6.8), 100 mMdithiothreitol, 10% glycerol, 2% SDS, and 0.1% bromophenol blue). Proteins were separated by SDS-PAGE and then transferred to polyvinyl ident fluoride (PVDF) membranes (Millipore). The membranes were blocked with 5% non-fat milk and 0.1% Tween-20 at room temperature for 1 h and then incubated with primary antibodies overnight at 4°C. After the membranes were washed, they were incubated with HRP-conjugated secondary antibodies (Santa Cruz Biotechnology) at room temperature for 1 h. The immunoblots were visualized using chemiluminescence (ECL Plus detection system) and quantified using ImageJ software.
Plasmids and transfection
The wild-type METTL3-CDS expression plasmid was generated by cloning the full-length ORF of mouse METTL3 gene (NM_019721) into pcDNA3.1 mammalian expression vector. The mutant METTL3-CDS (D395A and W398A) was amplified by PCR and cloned into pcDNA3.1. The full-length cDNA of the mouse YTHDF2 (NM_145393) was generated by PCR amplification and cloned into pCDNA3.1 at Xhol/BamHI restriction sites (Thermofisher). Synthetic siRNA oligonucleotides specific for regions in the mouse ZFP217, METTL3 and YTHDF2 mRNA were designed and synthesized by GnenPharma (Shanghai, China). The sequence that gave successful knockdown were listed in Supplementary Table S1. To silence ZFP217, METTL3 or YTHDF2, cells were transfected with specific siRNA (50 nM). Cell transfection was achieved by using Lipofectamine 2000 (Invitrogen, USA) for plasmid and Lipofectamine RNAiMAX (Invitrogen, USA) for siRNA following the manufacturer’s protocols.
meRIP-qPCR
m6A immunoprecipitation experiments were performed as previously described [Citation19]. After 24 h transfection, total RNA was isolated from 3T3-L1 cells using TRIzol reagent. Two hundred μg of total RNA was subjected to immunoprecipitate with anti-m6A antibody or IgG coupled to Dynabeads (Invitrogen, USA) in immunoprecipitation buffer (10 mM sodium phosphate, 0.05% Triton X, and 140 mMNaCl) at 4°C for 2 h. The samples were washed three times with immunoprecipitation buffer, and RNA was eluted from the beads by incubating with elution buffer containing 4.2 μl of 20 mg/ml proteinase K for 1.5 h at 50°C. Following ethanol precipitation, the input RNA and eluted poly(A) RNA were reverse transcribed with random hexamers, and the enrichment was determined by qPCR. The Ct difference between the 1% input and the immunoprecipitated (IP) RNA was determined, and the relative enrichment was calculated as 2−dCT as described above. The primer sequences used to amplify the m6A peak region were as follows: mCCND1-m6A-F, 5ʹ-TGCCCCCAAGTTAACAACAGT −3ʹ and mCCND1-m6A-R, 5ʹ-CTGGGAGTCATCGGTAGCAG −3ʹ. mCDK2-m6A-F, 5ʹ-GCCATTCTCACCGT GTCCTT-3ʹ and mCDK2-m6A-R, 5ʹ-GTTCGGACAGGGA CTCCAAA-3ʹ. mCCN A2-m6A-F, 5ʹ-ACCTGCCTTCACTCATTGCT-3ʹ and mCCNA2-m6A-R, 5ʹ-TCC CGTATTGACTGTTGGGC-3ʹ.
RNA Immunoprecipitation (RIP)
RNA Immunoprecipitation was performed as previously described [Citation13]. 3T3-L1 cells overexpressing the FLAG-YTHDF2 peptide were collected with a cell lifter (two 15-cm plates for each) and were concentrated via centrifugation at 400 g for 5 min. The cell pellet was resuspended in 2 V of lysis buffer (150 mMKCl, 10 mM HEPES, 2 mM EDTA, 0.5% Nonidet P-40, 0.5 mM DTT, 1:100 protease inhibitor cocktail, and 400 U/ml RNase inhibitor) and incubated on ice for 10 min. Cell debris was removed via centrifugation at 15,000 g for 15 min at 4°C, and the supernatant was further cleared by passage through a 0.45 mm membrane syringe filter. A 50 mL aliquot of cell lysate was saved as input, and the remaining sample was incubated with anti-FLAG magnetic beads (MilliporeSigma) in ice-cold NT2 buffer [200 mM NaCl, 50 mM HEPES (pH 7.6), 2 mM EDTA, 0.05% Nonidet P-40, 0.5 mM DTT, and 200 U/ml RNase inhibitor] for 4 h at 4°C. The beads were washed 8 times and were saved as the immunoprecipitated fraction, 2 mg of which was loaded on a gel along with 10 mg of the input (corresponding to ~1% of overall input).
mRNA stability
Cells were transfected with either the vehicle or YTHDF2 plasmid for 24 h and then treated with 5 μg/ml actinomycin D (Sigma, USA) to inhibit global mRNA transcription. Samples were collected at 0, 3 and 6 h to assess degradation. RNA was extracted by using TRIzol reagent and then reverse transcribed as described above. The mRNA transcript levels of interest were detected by qPCR.
Flow cytometry
Cells transfected with control, siMETTL3 or siZFP217 were cultured in MDI medium for 24 h. The cells were harvested and washed with PBS, then fixed in 70% ice-cold ethanol at −20°C overnight. After washed with PBS, the cells were centrifuged and resuspended in 1 mL FxCycleTM PI/RNase Solution (Invitrogen, USA). Samples were incubated for 15 min at room temperature in the dark and subjected to flow cytometer (Becton Dickinson, USA). The analysis of cell-cycle distribution was performed with ModFit LT 5.0 Software.
Statistical analysis
Data values were presented as mean ± SEM. Statistical analyses were performed using two-tailed Student’s t-tests. P< 0.05 was considered as statistically significant.
Author contributions
Q.L. and Y.Z. performed the experiments, analysed data, and wrote the manuscript. R.W., Q.J. and M.C. performed experiments, analysed data, and edited the manuscript. Z.B., Y.L. and Y.Y. performed data analyses. J.F. and Y.W. revised the manuscript. X.W. conceived the project and revised the manuscript critically.
Disclosure of potential conflicts of interest
No potential conflict of interest was reported by the authors.
Supplemental Material
Download Zip (789.9 KB)Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Lee SW, Son JY, Kim JM, et al. Body fat distribution is more predictive of all-cause mortality than overall adiposity. Diabetes Obesity Metab. 2018;20:141–147.
- Buzaglo-Azriel L, Kuperman Y, Tsoory M, et al. Loss of muscle MTCH2 increases whole-body energy utilization and protects from diet-induced obesity. Cell Rep. 2017;18:1335–1336.
- Esteve RM. Adipose tissue: cell heterogeneity and functional diversity. Endocrinol Nutr. 2014;61:100–112.
- Tang QQ, Otto TC, Lane MD. CCAAT/enhancer-binding protein beta is required for mitotic clonal expansion during adipogenesis. Proc Natl Acad Sci U S A. 2003;100:850–855.
- Tang QQ, Otto TC, Lane MD. Mitotic clonal expansion: a synchronous process required for adipogenesis. Proc Natl Acad Sci U S A. 2003;100:44–49.
- Wu R, Yao Y, Jiang Q, et al. Epigallocatechin gallate targets FTO and inhibits adipogenesis in an mRNA m(6)A-YTHDF2-dependent manner. Int J Obes (Lond). 2018;42:1378–1388.
- Wei S, Zhang L, Zhou X, et al. Emerging roles of zinc finger proteins in regulating adipogenesis. Cell Mol Life Sci. 2013;70:4569–4584.
- Hasegawa R, Tomaru Y, de Hoon M, et al. Identification of ZNF395 as a novel modulator of adipogenesis. Exp Cell Res. 2013;319:68–76.
- Meruvu S, Hugendubler L, Mueller E. Regulation of adipocyte differentiation by the zinc finger protein ZNF638. J Biol Chem. 2011;286:26516–26523.
- Xiang H, Zhong ZX, Peng YD, et al. The emerging role of Zfp217 in ADIPOGENESIS. Int J Mol Sci. 2017;18:1367.
- Kate D, DPPJ M. 5′ UTR m6A promotes cap-independent translation. Cell. 2015;163:999–1010.
- Batista PJ, Molinie B, Wang J, et al. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. 2014;15:707–719.
- Wang X, Lu Z, Gomez A, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. NATURE. 2014;505:117–120.
- Shi H, Wang X, Lu Z, et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017;27:315–328.
- Ho T, Guilbaud G, Blow JJ, et al. The KRAB zinc finger protein Roma/Zfp157 Is a critical regulator of cell-cycle progression and genomic stability. Cell Rep. 2016;15:724–734.
- Tili E, Michaille JJ, Liu CG, et al. GAM/ZFp/ZNF512B is central to a gene sensor circuitry involving cell-cycle regulators, TGF{beta} effectors, Drosha and microRNAs with opposite oncogenic potentials. Nucleic Acids Res. 2010;38:7673–7688.
- Noguchi A, Adachi S, Yokota N, et al. ZFP36L2 is a cell cycle-regulated CCCH protein necessary for DNA lesion-induced S-phase arrest. Biol Open. 2018;7:bio.031575.
- Aguilo F, Zhang F, Sancho A, et al. Coordination of m(6)A mRNA methylation and gene transcription by ZFP217 regulates pluripotency and reprogramming. Cell Stem Cell. 2015;17:689–704.
- Kobayashi M, Ohsugi M, Sasako T, et al. The RNA methyltransferase complex of WTAP, METTL3, and METTL14 regulates mitotic clonal expansion in adipogenesis. Mol Cell Biol. 2018;38.
- Du Y, Hou G, Zhang H, et al. SUMOylation of the m6A-RNA methyltransferase METTL3 modulates its function. Nucleic Acids Res. 2018;46:5195–5208.
- Zhao X, Yang Y, Sun BF, et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014;24:1403–1419.
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. NATURE. 2012;485:201–206.
- Ivanova I, Much C, Di Giacomo M, et al. The RNA m(6)A reader YTHDF2 Is essential for the post-transcriptional regulation of the maternal transcriptome and oocyte competence. Mol Cell. 2017;67:1059–1067.
- Nakagami H. The mechanism of white and brown adipocyte differentiation. Diabetes Metab J. 2013;37:85–90.
- Quinlan KGR, Nardini M, Verger A, et al. Specific recognition of ZNF217 and other zinc finger proteins at a surface groove of C-terminal binding proteins. Mol Cell Biol. 2006;26:8159–8172.
- Fu M, Rao M, Bouras T, et al. Cyclin D1 inhibits peroxisome proliferator-activated receptor gamma-mediated adipogenesis through histone deacetylase recruitment. J Biol Chem. 2005;280:16934–16941.
- Liu J, Yue Y, Han D, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N-6-adenosine methylation. Nat Chem Biol. 2014;10:93–95.
- Sledz P, Jinek M. Structural insights into the molecular mechanism of the m(6)A writer complex. ELIFE. 2016;5.
- Wang P, Doxtader KA, Nam Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell. 2016;63:306–317.
- Wang X, Feng J, Xue Y, et al. Corrigendum: structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. NATURE. 2017;542: 260.
- Su R, Dong L, Li C, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. CELL. 2018;172: 90.
- Weng H, Huang H, Wu H, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell. 2018;22: 191.
- Chen M, Wei L, Law CT, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. HEPATOLOGY. 2018;67:2254–2270.
- Li M, Zhao X, Wang W, et al. Ythdf2-mediated m(6)A mRNA clearance modulates neural development in mice. Genome Biol. 2018;19: 69.
- Patil DP, Chen C, Pickering BF, et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. NATURE. 2016;537: 369.