
ABSTRACT
Transfer RNAs belong to the most abundant type of ribonucleic acid in the cell, and detailed investigations revealed correlations between alterations in the tRNA pool composition and certain diseases like breast cancer. However, currently available methods do not sample the entire tRNA pool or lack specificity for tRNAs. A specific disadvantage of such methods is that only full-length tRNAs are analysed, while tRNA fragments or incomplete cDNAs due to RT stops at modified nucleosides are lost. Another drawback in certain approaches is that the tRNA fraction has to be isolated and separated from high molecular weight RNA, resulting in considerable labour costs and loss of material. Based on a hairpin-shaped adapter oligonucleotide selective for tRNA transcripts, we developed a highly specific protocol for efficient and comprehensive high-throughput analysis of tRNAs that combines the benefits of existing methods and eliminates their disadvantages. Due to a 3ʹ-TGG overhang, the adapter is specifically ligated to the tRNA 3ʹ-CCA end. Reverse transcription prior to the ligation of a second adapter allows to include prematurely terminated cDNA products, increasing the number of tRNA reads. This strategy renders this approach a powerful and universal tool to analyse the tRNA pool of cells and organisms under different conditions in health and disease.
Introduction
tRNAs are important molecules in the biosynthesis of proteins, where they function as adapters between mRNA codons and the corresponding amino acids[Citation1,Citation2]. Correct expression and processing of tRNA primary transcripts are regulated by many different enzymes, ensuring that the final tRNA gains its functional structure [Citation3]. If not encoded in the genome, a CCA-triplet, essential for the subsequent aminoacylation, is added to the 3ʹ-end of the tRNA by the CCA-adding enzyme [Citation4–Citation6]. As all mature tRNAs carry this sequence, the CCA triplet allows for specific isolation of tRNAs.
Due to the degenerated genetic code, the 20 amino acids used in protein biosynthesis are encoded by more than 20 codons in the mRNA. As a consequence, there are many tRNA isoacceptors in a cell that are charged with the same amino acid but address different, yet amino acid-specific codons [Citation7]. Similarly, and especially in eukaryotes, isodecoders exist – tRNAs that recognize the same codon triplet but can differ at certain positions within their sequence [Citation8]. This variability and different frequency in synonymous codons and, consequently, also in the tRNAs used by a cell [Citation9], results in a codon usage bias [Citation10,Citation11]. This bias can be discriminative for different species [Citation12] and different cell types within an organism [Citation13–Citation15]. Additionally, codon usage biases and diverse tRNA abundances were detected in cells of different growth states [Citation16], and differences in the tRNA pool composition have been observed in healthy and tumorigenic cells [Citation2,Citation16–Citation18]. Hence, this feature can be used as a signature for the identification of specific cell types or the detection of tRNA pool aberrations linked to certain diseases.
Furthermore, mutations in the tRNA sequence as well as in the different factors involved in maturation, aminoacylation and translation can cause tRNA malfunction and lead to phenotypic manifestation including severe diseases [Citation19–Citation23]. Beyond their important role in protein synthesis, there is growing evidence that tRNA-derived fragments play important roles in diverse cellular processes like gene silencing or control of protein translation [Citation24,Citation25]. Accordingly, substantial efforts have been made to establish various methods for efficient monitoring of tRNA levels or identification of tRNA sequence aberrations [Citation26,Citation27] that can be used to investigate the roles of tRNAs in disease [Citation28]. However, the lack of preciseness and robustness limits the deep investigation of these molecules, and more efficient tools are required to utilize tRNAs as biomarkers.
Many methods for tRNA quantitation have been developed, highlighting the many challenges arising for high-throughput analysis of the tRNA pool within a cell. tRNAs are structurally very stable and often highly modified. Mature tRNAs carry a short, single-stranded 3ʹ-end and a double-stranded 5ʹ-end, resulting in difficult adapter ligation and reverse transcription – two essential steps in high-throughput RNA sequencing. Although tRNAs comprise a high amount of total RNA in the cell (10 – 15%) [Citation29], most approaches require a tRNA-specific enrichment step to reduce the tremendous background of ribosomal RNA (up to 80% in a cell) in the ligation reaction. Using a Y-shaped adapter that hybridizes to the 3ʹ-CCA end and is then ligated to the 5ʹ- and 3ʹ-ends, Shigematsu et al. developed the YAMAT-Seq method (Y-shaped Adapter-ligated MAture TRNA sequencing) to circumvent these problems, allowing for a selective tRNA amplification without prior enrichment [Citation30]. However, as adapter oligonucleotides are simultaneously fused to the tRNA 5ʹ- and 3ʹ-ends, only tRNAs that are fully reverse transcribed into cDNA are amplified, while prematurely terminated cDNAs, e.g. due to modifications and/or structural obstacles, are lost. Hence, no information concerning tRNA fragments or tRNAs carrying a substantial amount of nucleoside modifications are retrieved in the YAMAT procedure. Pang et al. overcame such problems by introducing a two-step adapter ligation, allowing to recover such cDNA fragments [Citation31]. This approach, however, requires an extensive purification of tRNA fractions out of a total RNA reparation, and the authors apply five consecutive HPLC preparation steps of the reaction intermediates, likely resulting in a considerable loss of valuable tRNA material. Thus, an urgent need remains for methods that are easy to handle and combine adapter ligation without prior tRNA isolation. For a comprehensive investigation of the tRNA pool, such a method should include also tRNA-derived cDNA fragments, as they contain valuable information concerning expression of tRNAs (that are otherwise not registered) and specific modification positions.
Here, we present the fast and powerful method LOTTE-seq (Long hairpin oligonucleotide based tRNA high-throughput sequencing) that combines the benefits of the approaches described above [Citation30,Citation31], while it simultaneously avoids their disadvantages. In LOTTE-seq, a CCA-specific 3ʹ-adapter is ligated to mature tRNAs without previous purification. The second adapter is ligated to the resulting cDNA 3ʹ-ends. As a result, both full-length and shorter cDNA fragments are included in the data analysis. Further, with the use of a selective adapter for mature tRNA 3ʹ-ends, no purification of a tRNA fraction is required, and a crude total RNA preparation can be used. We believe that this method is very useful for tRNA research and diagnostics in terms of tRNA pool composition and abundance of individual tRNAs in various cellular states such as proliferation, differentiation, stress or disease.
Results
Hairpin-adapter-based ligation
LOTTE-seq is a powerful method to analyse tRNA pools in a fast and easy way starting from crude RNA preparations (). The first step is the selective ligation of a 3ʹ-adapter to mature tRNAs carrying a 3ʹ-CCA end. To this end, total RNA can be used without prior laborious and possibly bias-introducing tRNA enrichment, where usually a considerable amount of material is lost. This specificity is achieved using a hairpin-shaped DNA oligonucleotide that contains a 3ʹ-TGGN (N = any nucleotide) overhang complementary to the mature tRNA 3ʹ-end, consisting of the discriminator position 73 (numbering according to Sprinzl et al.[Citation32]) and the CCA terminus. Such a hairpin oligonucleotide was first developed by Dittmar et al.[Citation14] for tRNA-microarray analysis and is distantly related to the Y-shaped adapter used in YAMAT-Seq[Citation30]. A significant difference in the design of our hairpin-shaped adapter is that it contains only deoxyribonucleotides. Accordingly, we use T4 DNA ligase to fuse the adapter to the tRNA 3ʹ-ends (Step A). The ligation product is reverse transcribed into a cDNA pool containing not only full-length cDNA products, but also incomplete molecules resulting from RT stops at nucleoside modifications (Step B). The single gel extraction step in the whole LOTTE-seq procedure separates the RT-products from unused hairpin adapter that could interfere within the second ligation step. The purified cDNA is then ligated to the second adapter (Step C). According to Pang et al.[Citation31] and our own experience[Citation33], ligating the second adapter to the cDNA 3ʹ-end and not to the tRNA 5ʹ-end leads to a considerable increase in sequence reads of the tRNA pool. Transfer RNAs contain many modified bases and have a highly stable secondary structure, leading to frequent premature cDNA terminations. If both adapters were ligated to the tRNA, only full-length cDNA products could be amplified, because prematurely terminated cDNA fragments lack the second adapter and, consequently, the PCR primer-binding site. In contrast, ligation of the second adapter to the cDNA 3ʹ-ends allows amplification not only of full-length cDNAs, but also of prematurely terminated cDNA products (Step D). As the 3ʹ-terminal 20 to 30 positions in a tRNA contain only very few modifications, these regions are readily reverse transcribed into cDNA and can be used to identify the individual tRNAs (Step E)[Citation31]. Hence, the tRNA reads in our approach contain much more information than those of standard procedures based on the adapter ligation to both tRNA ends.
Figure 1. Schematic workflow of the LOTTE-seq procedure. (A) A DNA hairpin-oligonucleotide (green) with a 3ʹ-TGGN overhang hybridizes to the tRNA 3ʹ-CCA end (tRNA in blue). T4 DNA ligase fuses the 3ʹ-end of the CCA terminus to the phosphorylated 5ʹ end of the adapter. (B) The tRNA is reverse transcribed with parts of the hairpin oligonucleotide serving as primer binding site. Secondary structure and modified bases can lead to premature RT stops and partial cDNA (yellow). (C) Using T4 RNA ligase I, a 5ʹ-phosphorylated and 3ʹ-blocked second adapter (red) is fused to the 3ʹ-end of the cDNA, leading to the generation of cDNA product with adapters on both sides (red and green). (D) This product is PCR-amplified with indexed primers binding to the adapter overhang sequences. (E) The cDNA library consisting of full-length as well as prematurely terminated tRNA sequences is analysed by high-throughput sequencing.
DNA hairpin ligation is highly selective
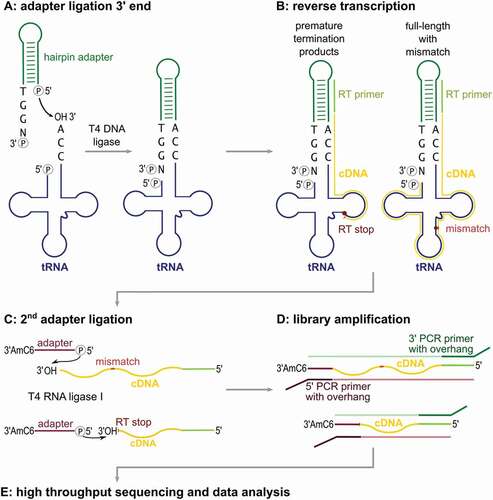
Without a selective adapter ligation to the tRNA 3ʹ-end, it is essential to efficiently separate tRNAs from other RNAs in the preparation, as these would be unavoidably included in the analysis. Apart from ribosomal RNA (rRNA) depletion based on commercially available kits, one possibility is to purify fractions containing small RNAs by gel electrophoresis, high salt precipitation or size exclusion chromatography. However, all these approaches result in a loss of tRNA material and, consequently, require an accordingly high amount of starting material [Citation34,Citation35]. In LOTTE-seq, we avoid such procedures and select tRNAs by a highly specific adapter ligation. Originally designed to introduce fluorescence labels in tRNA samples[Citation14], we use a DNA-only adapter with 3ʹ-TGGN overhang and an extended double-stranded region that introduces a primer binding site for reverse transcription and PCR (). Furthermore, as RNA-seq data can be prone to amplification or ligation biases [Citation31,Citation36], our hairpin design allows for the introduction of unique molecular identifier sequences (UMI) for the detection of such artefacts (Figure S1)[Citation37]. For optimal adapter ligation, we tested T4 DNA ligase, T4 RNA ligase 1 and T4 RNA ligase 2 (truncated KQ) in their specificity to fuse the adapter to in vitro transcribed tRNA with 3ʹ-CCA end (). T4 RNA ligase 1 requires single-stranded RNA molecules as substrates, while T4 RNA ligase 2 is more promiscuous and tolerates single- as well as double-stranded transcripts [Citation38–Citation40]. In contrast, T4 DNA ligase strictly requires double-stranded DNA or RNA and ligates either blunt ends or gapless nicks in a double-stranded region [Citation38,Citation41]. While both RNA ligase 1 and 2 fuse our hairpin adapter to the 3ʹ-end of a tRNA carrying the CCA-terminus, a considerable amount of ligation product is also observed when a tRNA transcript lacking the CCA-end was offered (). These side reaction products are the result of unspecific ligation of a single-stranded RNA 3ʹ-end to the 5ʹ-end of the adapter, without hybridizing to a CCA-sequence. T4 DNA ligase, however, does not accept single-stranded RNA 3ʹ-ends and only tolerates the nick region generated by the CCA-carrying tRNA hybridized to the adapter molecule. Hence, only T4 DNA ligase shows a specific and efficient ligation to the CCA-end-carrying tRNA (80 – 90% yield), while no unwanted side-reaction products with tRNA lacking the CCA end were observed, as they occurred with RNA ligases 1 and 2 ().
Figure 2. DNA hairpin adapter ligation. (A) DNA hairpin adapter for LOTTE-seq. The 5ʹ-end of the TGGN overhang is phosphorylated for ligation, the base-paired 3ʹ-end for blocking unwanted side reactions. RT primer binding site is indicated. (B) Adapter ligation catalysed by T4 DNA ligase, T4 RNA ligase 1 and T4 RNA ligase 2 (truncated KQ). Hairpin adapter was incubated with radioactively labelled in vitro transcribed yeast tRNAPhe with and without CCA-end. Only T4 DNA ligase fuses the tRNA with 3ʹ-CCA-end to the adapter hairpin at high selectivity, while RNA ligases 1 and 2 show considerable amounts of side reaction products with the transcript lacking a CCA-end (indicated by asterisks *). T4 RNA ligase 1 shows an additional high molecular weight product migrating in the upper part of the gel, probably resulting from the ligation of two tRNA molecules (**). The panel shows a prolonged exposure of the gels in order to visualize any unspecific ligation side reaction products. In a subsequent PCR-based amplification, such products will represent a considerable unwanted part of the sequence reads. (C) T4 DNA ligase-catalysed hairpin adapter ligation on tRNA transcripts with different 3ʹ-ends. Only the tRNA with a complete 3ʹ-CCA end was accepted for ligation, indicating a high specificity of the adapter ligation for mature tRNA 3ʹ-ends. When the tRNAs with different 3ʹ-ends were pooled, T4 DNA ligase exclusively selects the mature tRNA with CCA-end for ligation.
To further demonstrate the nick specificity of the T4 DNA ligase reaction, we investigated the ligation on tRNAs with partial CCA ends. In a mixture of in vitro transcribed tRNAs with such various 3ʹ-ends, only transcripts with complete 3ʹ-CCA end were ligated (), indicating that a complete CCA terminus is an absolute requirement for fusion to the adapter, while tRNAs ending with CC, C, or even completely lack the CCA end are not ligated at all (). Identical results were obtained when the UMI-containing hairpin adapter was used (Figure S1). Taken together, only T4 DNA ligase is highly specific for the adapter fusion to the tRNA CCA-end, while both RNA ligase reactions lead to unwanted side reaction products that reduce the sequence output of mature tRNAs in the preparation.
Hairpin adapter ligation to tRNA preparations
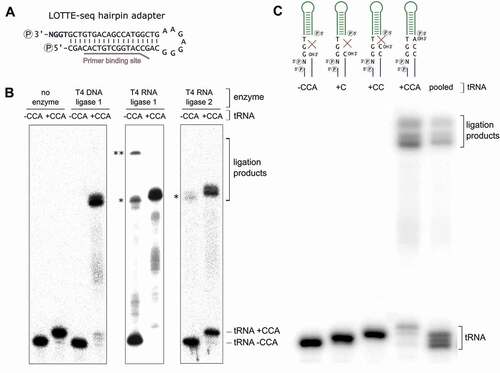
We also investigated the specificity of the hairpin adapter ligation on tRNAs isolated from total RNA. In contrast to the in vitro transcribed tRNA, the enriched tRNA pool of an organism is diverse in length and contains modified bases. We isolated the tRNA fraction of a Dictyostelium discoideum culture and performed the hairpin adapter ligation under the determined optimal conditions. In a control experiment, we incubated the tRNA preparation with snake venom phosphodiesterase (SVPD I) to remove the 3ʹ-CCA ends. This enzyme catalyzes the hydrolysis of 3ʹ-nucleotide phosphates and can easily be adjusted to only remove three nucleotides of the tRNA 3ʹ-end [Citation42,Citation43]. To restore and visualize the CCA end in a part of the preparation, the tRNA was incubated with the recombinant D. discoideum CCA-adding enzyme in the presence of NTPs spiked with[Citation32]P-ATP[Citation44]. This label allowed to monitor the efficiency of the ligation reaction ().
Figure 3. Hairpin adapter ligation to a tRNA pool preparation. (A) To introduce a radioactive label for visualization, CCA ends of the D. discoideum tRNA pool were removed by snake venom phosphodiesterase and restored by CCA-adding enzyme (D. discoideum) and NTPs spiked with α-32P-ATP. T4 DNA ligase fused the labelled tRNA pool to the hairpin adapter at high efficiency. (B) After reverse transcription and ligation of the second adapter, cDNA was amplified with indexed primers for Illumina deep sequencing. The original tRNA pool of D. discoideum as well as pool samples without or with restored CCA ends were analysed. Only samples with CCA ends (original or restored) gave rise to amplification products. The product length of 150 to 200 bp corresponds to the expected size of PCR products consisting of adapters and complete or partial tRNA sequences.
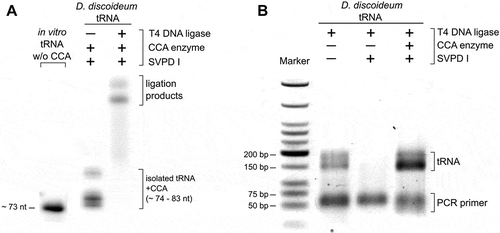
Next, the efficiency of the whole procedure including RT/PCR was tested. A SVPD I-treated tRNA sample was included as a negative control. Only the tRNA preparation with CCA ends (original or restored by CCA-addition) gave rise to PCR products with a size of 150–200 bp, as expected for tRNA-adapter ligation products (). The negative control, in contrast, did not produce any amplification products. The correct adapter ligation to the tRNA CCA ends was verified by sequence analysis of 10 individual reaction products after standard cloning (data not shown).
The prior isolation of tRNA usually results in an increased amount of specific ligation products and, consequently, tRNA-derived sequence data. Yet, it requires additional time-consuming treatment of the total RNA sample, lowering the overall yield. Further, extensive treatment of the RNA may distort the composition of the library so that also the relative abundance of tRNAs is affected. Hence, it is desirable to avoid such additional treatments and to use a total RNA preparation for tRNA analysis, and we applied our strategy to different amounts of total E. coli RNA, where we observed specific amplification of ligation products from starting material as low as 0.25 µg total RNA (Figure S2).
LOTTE-seq works for organism from all domains of life
To analyse the performance of our procedure, we subjected total RNA preparations of six model organisms to LOTTE-seq. Total RNA preparations of HEK293T cells (human), Spinacia oleracea (plant), Saccharomyces cerevisiae (fungi), Dictyostelium discoideum (Amoeba), Escherichia coli (Gram-negative bacteria) and Geobacillus stearothermophilus (Gram-positive bacteria) were investigated in two independent experiments and analysed on an Illumina MiSeq device with read numbers ranging from 1.4 to 3.2 million. In all approaches, LOTTE-seq specifically selects tRNA with a 3ʹ-CCA-end, including prematurely terminated cDNA fragments that represent the tRNA 3ʹ- part. Reads from non-tRNA sequences were found at a very low abundance with an average value of 3% (). tRNA sequences lacking the mature CCA-end were found only in 0.6% of all reads, indicating the high selectivity of our approach.
Table 1. Non-specific reads in tRNA-seq approaches.
To retrieve also information about tRNAs that cannot be fully reverse transcribed into cDNA, a two-step adapter ligation procedure was implemented, where the second adapter was fused to the 3ʹ-end of the generated cDNA[Citation31]. This strategy also includes cDNA fragments due to premature RT stops, so that a much higher number of (partial) tRNA sequences is represented in the sequence reads. Induced by robust RNA structures as well as base modifications (that are frequently found in tRNAs), such RT stops can affect the efficiency of reverse transcription [Citation45–Citation48]. While this decreases the number of reads spanning certain modifications, the corresponding pileup of RT stops in LOTTE-seq is an excellent indicator for these base modifications.
When performing LOTTE-seq for different organisms, we found that the cDNA yield differed dramatically from species to species. For bacterial samples, higher amounts of cDNA were obtained. This might be due to a less complex pattern of base modifications in these organisms [Citation49,Citation50]. In contrast, the relative amount of full-length tRNA was smaller in the human and plant samples, where tRNAs are usually modified to a greater extent. Many reads show termination sites corresponding to positions 9, 26, 37 and 58 in eukaryotic tRNAs and positions 22, 37 and 46 in bacterial tRNAs (Figure S3). Position 58 is represented by a highly conserved A residue frequently modified as m1A [Citation51,Citation52]. As base methylations can cause RT termination, it is highly likely that the observed read stops at this position represent the presence of this base modification. We further found read termination positions at A9 in eukaryotes and A22 in bacteria – positions that are also known to be modified as m1A[Citation52]. Read terminations at positions G26 (eukaryotes) and G46 (E. coli) might be caused by methylated guanosines (m1G, m22G or m7G). Additional termination signals were observed at positions 20 (D. discoideum) and 47 (D. discoideum, S. oleracea), where we cannot assign a specific modification to these positions, since there are no data available about tRNA modifications in these species. Compared to the eukaryotic samples, we observed less RT terminations in the bacterial tRNAs. Here, predominantly position 37, located downstream of the anticodon, is affected. This position carries a highly conserved purine residue that is frequently modified to keep the anticodon loop in an open conformation, as it avoids a detrimental base-pairing of positions 33 and 37[Citation50]. In conclusion, LOTTE-seq allows for the identification of certain nucleoside modifications in tRNAs. Depending on the reverse transcriptase, it is probably also possible to identify nucleotide misincorporations as RT signatures, as it was described for several of these enzymes [Citation47,Citation53–Citation56].
Discussion
In recent years, the investigation of tRNAs or tRNA pools and their correlation to translation efficacy and regulation, stress conditions and diseases developed into an important area of research [Citation2,Citation13,Citation57–Citation59]. There are many indications that tRNA abundance is associated with certain diseases [Citation60–Citation63]. However, the peculiar features of tRNA molecules render library preparation quite complicated and error-prone, and standard Illumina approaches are not very practical for their analysis, as can be seen in Fig. S4, where we compare LOTTE-seq with a standard sRNA TruSeq approach (5ʹ- and 3ʹ-adapter ligation followed by reverse transcription) and an optimized sRNA TruSeq protocol (3ʹ-adapter ligation followed by cDNA synthesis and subsequent cDNA adapter ligation). A reason for these difficulties is the high amount of modified bases [Citation64–Citation66] as well as stable secondary and tertiary structures of tRNAs [Citation45,Citation67]. Here, we present an improved method for efficient capturing of tRNAs for deep sequencing analysis that combines the advantages of two other valuable approaches, while avoiding their disadvantages. The usage of a DNA hairpin adapter that specifically hybridizes to the tRNA 3ʹ-CCA end ensures that exclusively mature tRNA transcripts are investigated. In the reaction catalysed by T4 DNA ligase[Citation14], only full-length CCA ends are accepted for ligation, in contrast to T4 RNA ligase 1 and T4 RNA ligase 2 (truncated KQ). tRNAs with partial or no CCA end are efficiently excluded, as T4 DNA ligase does not tolerate single-stranded nucleic acids or double strands that carry a gap in the hybrid region between CCA end and adapter overhang ().
Interestingly, the analysis of the tRNA sequence reads identified a series of stop signals resulting from reverse transcription termination at methylated base positions. Several experimental approaches exist that use such induced RT stop signals to specifically investigate the presence of individual types of modifications. ARM-seq[Citation68] as well as DM-seq[Citation69] compare untreated with enzymatically demethylated samples to identify certain base methylation positions in transcriptome data. With these methods, m1A was found at positions 9 and 58 in eukaryotes [Citation68,Citation69] and position 22 in some bacterial tRNAs[Citation70]. m1G is also found at position 9 in eukaryotes and at position 37 in both eukaryotic and bacterial tRNAs. As described above, LOTTE-seq reveals strong RT stop signals at these positions (Figure S3). The m1A22 RT stop is only visible in G. stearothermophilus tRNA but not in E. coli – an observation that was also made by Schwartz et al., who identified m1A22 in B. subtilis and S. aureus, but not in E. coli, where it is obviously absent[Citation70]. Furthermore, DM-tRNA-seq identified m2C32 in only five human tRNAs[Citation69], which is also in agreement with a weak RT stop signal at the corresponding position in LOTTE-seq. In all analysed eukaryotic species, we obtained a strong stop signal at position 26, corresponding to the m22G modification identified by DM-tRNA-seq at this position[Citation69]. Similarly, LOTTE-seq showed RT signals at position 46 in E. coli, where DM-tRNA-seq located m7G.
Taken together, this comparison shows that LOTTE-seq produces modification-specific RT stop signatures, demonstrating that this method can be used for the identification of certain base modifications. A combination of our approach with ARM-seq[Citation68], DM-tRNA-seq[Citation69] or AlkAniline-seq[Citation71], where enzymatic treatment of specific modifications is applied, would represent a promising strategy for the investigation of tRNA-based modifications. Such a combination – which is not possible with YAMAT – should dramatically increase the number of useable tRNA reads and thus facilitate the accurate identification of position-specific modifications.
When analysing human and plant samples, we were faced with the problem of the accurate mapping of tRNA reads as described by Hoffmann et al. [Citation72]. Human and plant genomes comprise 400–1000 tRNA genes (Supplementary Table S1) with many isodecoders, differing in only a few nucleotides [Citation8,Citation73–Citation76]. This complicates the allocation of reads to the corresponding gene, even for full-length tRNAs. Together with a relatively short read length, this leads to an inaccuracy of mapping. In order to discard reads that map to sequences other than tRNA genes, the reads were mapped against an artificial genome with masked tRNAs[Citation72]. For LOTTE-seq, however, we only select mature tRNAs with posttranscriptionally added 3ʹ-CCA termini representing an identification signature for tRNA reads.
Compared to the procedures described by Shigematsu[Citation30] and Pang[Citation31] in terms of specificity, LOTTE-seq shows a selectivity for tRNAs similar to YAMAT (). However, the use of T4 DNA ligase leads to an increased specificity for complete CCA-ends (only 0.6% non-CCA ends), while T4 RNA ligase 2 that was used in YAMAT also accepts unpaired single-stranded 3ʹ-ends, leading to 6.3% non-CCA-ends. A direct comparison of the tRNA pool composition identified by YAMAT and LOTTE-Seq, however, is not reasonable, as Shigematsu et al. used breast cancer cell lines (BT-474, SK-BR-3, MCF-7) in their analysis[Citation30], while we used human embryonic kidney cells (HEK293T). There is growing evidence that the cellular tRNA pool composition is not stable, but is actively adjusted to individual growing conditions or cell-type requirements, resulting in specific tRNA pools in different cells or organs [Citation2,Citation16,Citation60,Citation63,Citation77–Citation82]. As a result, these cell-type-specific differences render a comparison of the data obtained by YAMAT and LOTTE-Seq impossible. Also, a comparison of the data of Pang[Citation31] is not feasible, since neither the fraction of non-tRNA reads nor the fraction of non-CCA ends was reported. Therefore, we used our own sequencing data on D. discoideum and G. stearothermophilus that were generated by a procedure highly similar to the Pang approach[Citation33]. Instead of HPLC separation, the tRNA-containing small RNA fraction was isolated by high salt precipitation [Citation83,Citation84]. The subsequent steps were identical to the Pang strategy. A 3ʹ-adapter was fused to the small RNA preparation by truncated T4 RNA ligase 2. After cDNA synthesis, a second adapter was ligated to the cDNA 3ʹ-ends, the reaction products were amplified by PCR and sequenced on a MiSeq device. While this procedure also led to a considerable amount of tRNA reads (56.5%), the number of non-tRNA reads is much higher compared to YAMAT or LOTTE-seq (). Furthermore, the use of T4 RNA ligase leads to 42% of sequences ending with sequences other than CCA – a further indication that CCA-specific 3ʹ-adapters are highly selective, and – when combined with T4 DNA ligase reaction in LOTTE-seq – result in the highest number of reads with mature tRNA 3ʹ-ends. Furthermore, in all tRNA pools analysed by LOTTE-seq, we did not observe the appearance of jackpot tRNAs – high sequence reads resulting from ligation or amplification biases as described by Pang et al.[Citation31].
Taken together, LOTTE-seq is a highly robust and versatile approach that combines the pros of two – also very valuable – alternative procedures, while avoiding their cons. Combined with unique molecular identifiers, LOTTE-seq is a useful method to investigate the tRNA pools of different sources in a fast, convenient and reliable way.
Conclusions
As tRNAs are recognized as important contributors to regulatory mechanisms, the development of improved methods for the analysis of tRNAs in various cell states and organisms is an urgent need. LOTTE-seq renders the analysis of tRNA pools or individual transcripts (including some modification) more efficient and accurate. We hope that this not only improves the statistical relevance of tRNA expression data but also sets the stage to implementing tRNAs as a powerful biomarker for the detection of various cellular states.
Materials and methods
Total RNA isolation of different cells
Human embryonic kidney cells (HEK293T) were cultured under standard conditions. Cells were lysed with TRIzol® (Thermo Scientific) and used for total RNA isolation. Four leaves of Spinacia oleracea were disrupted in a CellCrusher® with liquid nitrogen and mixed with TRIzol®. Saccharomyces cerevisiae cells (BY4716) were grown as a liquid culture in YPAD medium overnight at 30°C. Cells were harvested by centrifugation, mixed with TRIzol® and disrupted in a FastPrep® cell homogenizer (MP Biomedicals) with 1 mm silica beads. Dictyostelium discoideum cells (gift of C. Hammann, Bremen) were taken up in TRIzol® (3 x 108 cells). Geobacillus stearothermophilus cells were cultivated in medium 220 at 50°C, harvested by centrifugation and treated with a FastPrep® cell homogenizer (MP-Biomedicals) and 1 mm silica beads. TRIzol® was added to the disrupted cells. Escherichia coli Top10 cells were cultured at 37°C in LB-medium, harvested by centrifugation and lysed with TRIzol®.
For the preparation of tRNAs, 0.5 M NaCl and 5% (v/v) PEG8000 were added to the total RNA preparation. After incubation for 30 min at −20°C, the sample was centrifuged twice for 30 min at 4°C and 10.000 x g. Small RNAs in the supernatant were precipitated with 100% ethanol[Citation84]. The precipitated RNAs were redissolved and purified by polyacrylamide gel electrophoresis. Gel extraction of the tRNA-containing bands was performed as described[Citation85].
Adapter ligation
A 100 pmol of the hairpin-shaped DNA adapter with 3ʹ-TGGN overhang (5ʹ-pCGACACTGTCGGTACCGACGGGAGAAGTCGGTACCGACAGTGTCGTGGNp-3‘) was incubated with 2–4 µg total RNA and 30 units T4 DNA ligase (NEB) in 66 mM Tris-HCl pH 7.6, 6.6 mM MgCl2, 10 mM DTT, 66 µM ATP and 25% (v/v) DMSO [Citation14] for 8 h at 32°C. The enzyme was heat-inactivated for 10 min at 65° and the ligation product was purified by ethanol precipitation.
For ligation with T4 RNA ligase 1, 50 pmol hairpin adapter and 10 pmol in vitro transcribed tRNAs with defined homogeneous 3ʹ-end (produced as described [Citation86,Citation87]) were incubated 50 mM Tris/HCl pH 8.0, 10 mM MgCl2, 0.2 mg/ml BSA, 1 mM hexamine cobalt(III) chloride, 12.5% PEG, 1 mM ATP and 30 units RNA ligase 1 (NEB) for 16 h at 16°C. The reaction was stopped by incubation at 65°C for 15 min.
For ligation with T4 RNA ligase 2, 10 pmol of the tRNA in vitro transcripts was incubated with 50 pmol hairpin adapter, 1 x T4 RNA ligase buffer (NEB) and 10 units RNA ligase 2 (NEB) for 1 h at 37°C following an incubation overnight at 4°C.
Reverse transcription
tRNA ligated at the 3ʹ-end to the hairpin adapter was incubated with 100 pmol32P-labelled RT primer (5‘-CAAGC TCGGTACCGACAGTG-3‘; underlined sequence represents primer binding site for subsequent PCR) and 2 mM dNTPs for 5 min at 65°C and cooled down to room temperature. Reaction buffer (Thermo Scientific), 5 mM DTT, RNase inhibitor and 15 units SuperScript IV RT (Thermo Scientific) were added, and the reaction was incubated at 55°C for 30 min. The enzyme was heat-inactivated for 10 min at 80°C. cDNA was separated on a poly-acrylamide gel and visualized by autoradiography before purification by gel extraction.
Ligation of cDNA adapter
The gel-purified cDNA and 100 pmol of a DNA-only version of the Illumina TruSeq small RNA kit adapter (5‘-pGATCGTCGGACTGTAGAACTCTGAAC-AminoC6–3ʹ) were incubated in 50% (v/v) PEG8000, 1 x T4 RNA ligase buffer (NEB), 1 mM ATP, 1 mM cobalt hexamine chloride and 10 units T4 RNA ligase 1 (NEB) for 16 h at 16°C. The enzyme was heat-inactivated for 10 min at 65°C.
Amplification of the cDNA library
cDNA carrying 5ʹ and 3ʹ adapter sequences was incubated in 1 x Phusion HF buffer, 0.2 µM dNTPs, 0.5 µM forward primer (5ʹ- AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGA-3ʹ; Illumina 5ʹ-PCR primer TruSeq small RNA library preparation kit), 0.5 µM index primer (5ʹ-CAAGCAGAAGACGGCATACGAGATNNNNNNTAGGACTCCATGCAAGCTCGGTACCGACAGTG-3ʹ; custom primer for Illumina TruSeq small RNA library preparation kit, NNNNNN – index) and 0.5 units Phusion HF DNA polymerase (Thermo Scientific) for 30 s at 98°C and amplified in 14 cycles (10 s at 98°C, 20 s at 60°C, 15 s at 72°C). A final elongation step for 2 min at 72°C completed the amplification. The PCR product was purified using the QiaQuick PCR purification kit (Qiagen) to remove unused PCR primers.
Optimized TruSeq small RNA protocol for tRNA library preparation
2–4 µg total RNA and 100 pmol 3ʹ-adapter (5ʹ-pUGGAATTCTCGGGTGCCAAGG-amino-C7-3ʹ) were incubated with 50 mM Tris-HCl (pH 8.0), 10 mM MgCl2, 0.2 mg/ml BSA, 1 mM cobalt hexamine chloride, 12.5% (v/v) PEG8000, 30 µM ATP and 10 units T4 RNA ligase 1 (NEB) for 16 h at 25°C. The reaction was purified with the GeneJET RNA Cleanup and Concentration Micro Kit (Thermo Scientific, #K0841) and used for reverse transcription under conditions described above.
Standard truseq small RNA protocol for library preparation
Total RNA library preparation was carried out according to the Illumina TruSeq small RNA library preparation protocol. A pre-adenylated 3ʹ-adapter was incubated with 1 µg of total RNA in the presence of T4 RNA ligase 2 (truncated KQ), while the 5ʹ-ligation was carried out with T4 RNA ligase 1 and a 5ʹ-RNA adapter. Reverse transcription and cDNA amplification were carried out according to the manufacturer's protocol (Illumina).
High-throughput RNA sequencing
Quality and concentration of the purified library constructs were determined on a 2100 Bioanalyzer (Agilent). High-throughput analysis of the libraries was done as single end run (150 nt) on a MiSeq System (Illumina®) with a custom primer designed for Illumina MiSeq analysis (5ʹ-CACTGTCGGTACCGAGCTTGCATGGAGTCCTA-3ʹ).
Bioinformatics analysis pipeline
For a detailed description, see the Supplementary Data.
Author Contributions
HB, PFS and MM conceived the project; HB, MM and LE designed the experiments; LE, AH and JF performed the experiments; HB, PFS, MM, LE, AH and JF analysed the tRNA-seq data; PFS and MM supervised the project; all authors wrote the MS.
Data Availability
All data of this study are included in this published article and its supplementary information files. RNA-seq data are available at NCBI BioProject (ID: PRJNA541863).
RNA-seq data are available at: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA541863
Supplemental Material
Download MS Word (847.2 KB)Acknowledgments
We thank Irene Coin, Leipzig University, for providing HEK293T cells. We thank Christian Hammann, Jacobs University Bremen, for providing Dictyostelium discoideum AX2 cells. Special thanks go to Christina Weinberg for scientific discussion.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Balakrishnan R, Park J, Karra K, et al. YeastMine--an integrated data warehouse for Saccharomyces cerevisiae data as a multipurpose tool-kit. Database (Oxford). 2012;2012:bar062.
- Goodarzi H, Nguyen HCB, Zhang S, et al. Modulated expression of specific tRNAs drives gene expression and cancer progression. Cell. 2016;165:1416–1427.
- Phizicky EM, Hopper AK. tRNA biology charges to the front. Genes Dev. 2010;24:1832–1860.
- Sprinzl M, Cramer F. The -C-C-A end of tRNA and its role in protein biosynthesis. In: Cohn WE, editor. Progress in nucleic acid research and molecular biology, place unknown, Vol. 22. Cambridge (MA): Academic Press, 1979. p. 1–69.
- Xiong Y, Steitz TA. Mechanism of transfer RNA maturation by CCA-adding enzyme without using an oligonucleotide template. Nature. 2004;430:640–645.
- Betat H, Rammelt C, Mörl M. tRNA nucleotidyltransferases: ancient catalysts with an unusual mechanism of polymerization. Cell Mol Life Sci. 2010;67:1447–1463.
- Goodenbour JM, Pan T. Diversity of tRNA genes in eukaryotes. Nucleic Acids Res. 2006;34:6137–6146.
- Parisien M, Wang X, Pan T. Diversity of human tRNA genes from the 1000-genomes project. RNA Biol. 2013;10:1853–1867.
- Ikemura T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol Biol Evol. 1985;2:13–34.
- Grantham R, Gautier C, Gouy M, et al. Codon catalog usage and the genome hypothesis. Nucl Acids Res. 1980;8:197.
- Grantham R, Gautier C, Gouy M, et al. Codon catalog usage is a genome strategy modulated for gene expressivity. Nucl Acids Res. 1981;9:r43–74.
- Sharp PM, Li WH. The codon Adaptation Index--a measure of directional synonymous codon usage bias, and its potential applications. Nucl Acids Res. 1987;15:1281–1295.
- Plotkin JB, Robins H, Levine AJ. Tissue-specific codon usage and the expression of human genes. Proc Natl Acad Sci U S A. 2004;101:12588–12591.
- Dittmar KA, Goodenbour JM, Pan T. Tissue-specific differences in human transfer RNA expression. PLoS Genet. 2006;2:e221.
- Camiolo S, Farina L, Porceddu A. The relation of codon bias to tissue-specific gene expression in Arabidopsis thaliana. Genetics. 2012;192:641–649.
- Gingold H, Tehler D, Christoffersen NR, et al. A dual program for translation regulation in cellular proliferation and differentiation. Cell. 2014;158:1281–1292.
- Randerath K, Agrawal HP, Randerath E. tRNA alterations in cancer. In: Nass G, editor. Modified nucleosides and cancer. Berlin, Heidelberg: Springer Berlin Heidelberg; 1983. p. 103–120.
- Grewal SS. Why should cancer biologists care about tRNAs? tRNA synthesis, mRNA translation and the control of growth. Biochim Biophys Acta. 2015;1849:898–907.
- Guo M, Chong YE, Shapiro R, et al. Paradox of mistranslation of serine for alanine caused by AlaRS recognition dilemma. Nature. 2009;462:808–812.
- Suzuki T, Nagao A, Suzuki T. Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu Rev Genet. 2011;45:299–329.
- Wang S, Li R, Fettermann A, et al. Maternally inherited essential hypertension is associated with the novel 4263AG mutation in the mitochondrial tRNAIle gene in a large Han Chinese family. Circ Res. 2011;108:862–870.
- Karaca E, Weitzer S, Pehlivan D, et al. Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and central nervous system function. Cell. 2014;157:636–650.
- Ishimura R, Nagy G, Dotu I, et al. RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science. 2014;345:455–459.
- Lee YS, Shibata Y, Malhotra A, et al. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev. 2009;23:2639–2649.
- Liu S, Chen Y, Ren Y, et al. A tRNA-derived RNA fragment plays an important role in the mechanism of arsenite -induced cellular responses. Sci Rep. 2018;8:16838.
- Krishnan P, Ghosh S, Wang B, et al. Genome-wide profiling of transfer RNAs and their role as novel prognostic markers for breast cancer. Sci Rep. 2016;6:32843.
- Sun C, Yang F, Zhang Y, et al. tRNA-derived fragments as novel predictive biomarkers for trastuzumab-resistant breast cancer. Cell Physiol Biochem. 2018;49:419–431.
- Kirchner S, Ignatova Z. Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat Rev Genet. 2015;16:98–112.
- Westermann AJ, Gorski SA, Vogel J. Dual RNA-seq of pathogen and host. Nat Rev Microbiol. 2012;10:618–630.
- Shigematsu M, Honda S, Loher P, et al. YAMAT-seq: an efficient method for high-throughput sequencing of mature transfer RNAs. Nucleic Acids Res. 2017;45:e70.
- Pang YLJ, Abo R, Levine SS, et al. Diverse cell stresses induce unique patterns of tRNA up- and down-regulation: TRNA-seq for quantifying changes in tRNA copy number. Nucleic Acids Res. 2014;42:e170.
- Sprinzl M, Horn C, Brown M, et al. Compilation of tRNA sequences and sequences of tRNA genes. Nucl Acids Res. 1998;26:148–153.
- Ernst FGM, Erber L, Sammler J, et al. Cold adaptation of tRNA nucleotidyltransferases: A tradeoff in activity, stability and fidelity. RNA Biol. 2018;15:144–155.
- Lukavsky PJ, Puglisi JD. Large-scale preparation and purification of polyacrylamide-free RNA oligonucleotides. RNA. 2004;10:889–893.
- Chionh YH, Ho C-H, Pruksakorn D, et al. A multidimensional platform for the purification of non-coding RNA species. Nucleic Acids Res. 2013;41:e168.
- Fu Y, Wu P-H, Beane T, et al. Elimination of PCR duplicates in RNA-seq and small RNA-seq using unique molecular identifiers. BMC Genomics. 2018;19:531.
- Kivioja T, Vähärautio A, Karlsson K, et al. Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods. 2011;9:72–74.
- Bullard DR, Bowater RP. Direct comparison of nick-joining activity of the nucleic acid ligases from bacteriophage T4. Biochem J. 2006;398:135–144.
- Nichols NM, Tabor S, McReynolds LA. RNA ligases. Curr Protoc Mol Biol. 2008;Chapter 3:Unit3.15.
- Munafó DB, Robb GB. Optimization of enzymatic reaction conditions for generating representative pools of cDNA from small RNA. RNA. 2010;16:2537–2552.
- Moore MJ, Sharp PA. Site-specific modification of pre-mRNA: the 2’-hydroxyl groups at the splice sites. Science. 1992;256:992–997.
- Deutscher MP. Reactions at the 3’ terminus of transfer ribonucleic acid. 3. Catalytic properties of two purified rabbit liver transfer ribonucleic acid nucleotidyl transferases. J Biol Chem. 1972;247:459–468.
- Reichert AS, Thurlow DL, Mörl M. A eubacterial origin for the human tRNA nucleotidyltransferase? Biol Chem. 2001;382:1431–1438.
- Levinger L, Oestreich I, Florentz C, et al. A pathogenesis-associated mutation in human mitochondrial tRNALeu(UUR) leads to reduced 3’-end processing and CCA addition. J Mol Biol. 2004;337:535–544.
- Wittig B, Wittig S. Reverse transcription of tRNA. Nucl Acids Res. 1978;5:1165–1178.
- Harrison GP, Mayo MS, Hunter E, et al. Pausing of reverse transcriptase on retroviral RNA templates is influenced by secondary structures both 5’ and 3’ of the catalytic site. Nucl Acids Res. 1998;26:3433–3442.
- Motorin Y, Muller S, Behm‐Ansmant I, et al. Identification of modified residues in RNAs by reverse transcription‐based methods. In: Gott JM, editor. RNA modification. San Diego, Calif.: Academic Press/Elsevier; 2007. p. 21–53.
- Wilusz JE. Removing roadblocks to deep sequencing of modified RNAs. Nat Methods. 2015;12:821–822.
- Grosjean H. DNA and RNA modification enzymes: structure, mechanism, function and evolution: nucleic acids are not boring long polymers of only four types of nucleotides: a guided tour. Austin, TX, USA: Landes Bioscience; 2009.
- Lorenz C, Lünse CE, Mörl M. tRNA modifications: impact on structure and thermal adaptation. Biomolecules. 2017;7. DOI:10.3390/biom7020035
- Hori H. Methylated nucleosides in tRNA and tRNA methyltransferases. Front Genet. 2014;5:144.
- Oerum S, Dégut C, Barraud P, et al. m1A Post-transcriptional modification in tRNAs. Biomolecules. 2017;7. DOI:10.3390/biom7010020
- Findeiss S, Langenberger D, Stadler PF, et al. Traces of post-transcriptional RNA modifications in deep sequencing data. Biol Chem. 2011;392:305–313.
- Gogakos T, Brown M, Garzia A, et al. Characterizing expression and processing of precursor and mature human tRNAs by hydro-tRNAseq and PAR-CLIP. Cell Rep. 2017;20:1463–1475.
- Helm M, Motorin Y. Detecting RNA modifications in the epitranscriptome: predict and validate. Nat Rev Genet. 2017;18:275–291.
- Kietrys AM, Velema WA, Kool ET. Fingerprints of modified RNA bases from deep sequencing profiles. J Am Chem Soc. 2017;139:17074–17081.
- Zhou Y, Goodenbour JM, Godley LA, et al. High levels of tRNA abundance and alteration of tRNA charging by bortezomib in multiple myeloma. Biochem Biophys Res Commun. 2009;385:160–164.
- Zhong J, Xiao C, Gu W, et al. Transfer RNAs mediate the rapid adaptation of escherichia coli to oxidative stress. PLoS Genet. 2015;11:e1005302.
- Torrent M, Chalancon G, Groot de NS, et al. Cells alter their tRNA abundance to selectively regulate protein synthesis during stress conditions. Sci Signal. 2018;11. DOI:10.1126/scisignal.aat6409.
- Pavon-Eternod M, Gomes S, Geslain R, et al. tRNA over-expression in breast cancer and functional consequences. Nucleic Acids Res. 2009;37:7268–7280.
- Hanada T, Weitzer S, Mair B, et al. CLP1 links tRNA metabolism to progressive motor-neuron loss. Nature. 2013;495:474–480.
- Clarke CJ, Berg TJ, Birch J, et al. The initiator methionine tRNA drives secretion of type II collagen from stromal fibroblasts to promote tumor growth and angiogenesis. Curr Biol. 2016;26:755–765.
- Orioli A. tRNA biology in the omics era: stress signalling dynamics and cancer progression. Bioessays. 2017;39. DOI:10.1002/bies.201600158
- Bjork G. Transfer RNA modification. Annu Rev Biochem. 1987;56:263–287.
- Agris PF, Vendeix FAP, Graham WD. tRNA’s wobble decoding of the genome: 40 years of modification. J Mol Biol. 2007;366:1–13.
- Pan T. Modifications and functional genomics of human transfer RNA. Cell Res. 2018;28:395–404.
- Verma IM. 6 Reverse Transcriptase. In: Sumner JB, editor. The enzymes: chemistry and mechanism of action. New York: Acad. Pr; 1951. p. 87–103.
- Cozen AE, Quartley E, Holmes AD, et al. ARM-seq: alkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat Methods. 2015;12:879–884.
- Zheng G, Qin Y, Clark WC, et al. Efficient and quantitative high-throughput tRNA sequencing. Nat Methods. 2015;12:835–837.
- Schwartz MH, Wang H, Pan JN, et al. Microbiome characterization by high-throughput transfer RNA sequencing and modification analysis. Nat Commun. 2018;9:5353.
- Marchand V, Ayadi L, Ernst FGM, et al. AlkAniline-Seq: profiling of m7 G and m3 C RNA modifications at single nucleotide resolution. Angew Chem Int Ed Engl. 2018;57:16785–16790.
- Hoffmann A, Fallmann J, Vilardo E, et al. Accurate mapping of tRNA reads. Bioinformatics. 2018;34:1116–1124.
- Chan PP, Lowe TM. GtRNAdb: A database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 2009;37:D93–7.
- Jühling F, Mörl M, Hartmann RK, et al. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009;37:D159–62.
- Geslain R, Pan T. Functional analysis of human tRNA isodecoders. J Mol Biol. 2010;396:821–831.
- Michaud M, Cognat V, Duchêne A-M, et al. A global picture of tRNA genes in plant genomes. Plant J. 2011;66:80–93.
- Topisirovic I, Sonenberg N. Distinctive tRNA repertoires in proliferating versus differentiating cells. Cell. 2014;158:1238–1239.
- Orioli A, Praz V, Lhôte P, et al. Human MAF1 targets and represses active RNA polymerase III genes by preventing recruitment rather than inducing long-term transcriptional arrest. Genome Res. 2016;26:624–635.
- Rapino F, Delaunay S, Rambow F, et al. Codon-specific translation reprogramming promotes resistance to targeted therapy. Nature. 2018;558:605–609.
- Huang S-Q, Sun B, Xiong Z-P, et al. The dysregulation of tRNAs and tRNA derivatives in cancer. J Exp Clin Cancer Res. 2018;37:101.
- Zhang Z, Ye Y, Gong J, et al. Global analysis of tRNA and translation factor expression reveals a dynamic landscape of translational regulation in human cancers. Commun Biol. 2018;1:234.
- Santos M, Fidalgo A, Varanda AS, et al. tRNA deregulation and its consequences in cancer. Trends Mol Med. 2019. DOI:10.1016/j.molmed.2019.05.011
- Cathala G, Savouret JF, Mendez B, et al. A method for isolation of intact, translationally active ribonucleic acid. DNA. 1983;2:329–335.
- Eichinger L, Rivero-Crespo F. Dictyostelium discoideum protocols. Humana Press; 2006. p. 221–222.
- Sambrook J, Russell DW. Isolation of DNA fragments from polyacrylamide gels by the crush and soak method. CSH Protoc. 2006;2006. DOI:10.1101/pdb.prot2936
- Schürer H, Lang K, Schuster J, et al. A universal method to produce in vitro transcripts with homogeneous 3’ ends. Nucleic Acids Res. 2002;30:e56.
- Mörl M, Hartmann RK. Production of RNAs with homogeneous 5′- and 3′-ends. In: Hartmann RK, Bindereif A, Schön A, et al., editors. Handbook of RNA biochemistry. Weinheim, Germany: Wiley-VCH; 2014. p. 29–44.