
ABSTRACT
Neuronal microexons represent the most highly conserved class of alternative splicing events and their timed expression shapes neuronal biology, including neuronal commitment and differentiation. The six-nt microexon 34ʹ is included in the neuronal form of TAF1 mRNA, which encodes the largest subunit of the basal transcription factor TFIID. In this study, we investigate the tissue distribution of TAF1-34ʹ mRNA and protein and the mechanism responsible for its neuronal-specific splicing. Using isoform-specific RNA probes and antibodies, we observe that canonical TAF1 and TAF1-34ʹ have different distributions in the brain, which distinguish proliferating from post-mitotic neurons. Knockdown and ectopic expression experiments demonstrate that the neuronal-specific splicing factor SRRM4/nSR100 promotes the inclusion of microexon 34ʹ into TAF1 mRNA, through the recognition of UGC sequences in the poly-pyrimidine tract upstream of the regulated microexon. These results show that SRRM4 regulates temporal and spatial expression of alternative TAF1 mRNAs to generate a neuronal-specific TFIID complex.
Introduction
Alternative microexons of 3 to 27 nt represent the most highly conserved class of pre-mRNA splicing events in vertebrates [Citation1]. These exons are predominantly found in the nervous system and preserve the open reading frame inserting only a few amino acids in loop regions of the corresponding protein [Citation1]. A major regulator of this “exon microsurgery” is the neuronal-specific Ser/Arg repeat splicing factor of 100 kDa (nSR100), also known as Serine/Arginine Repetitive Matrix 4 (SRRM4) [Citation1], which recognizes UGC-containing intronic sequences of the pre-mRNA to stimulate microexon inclusion [Citation2]. Disruption of SRRM4 results in multiple neurodevelopmental defects and has been causally linked to neurological disorders [Citation3,Citation4]. One of the candidate substrates of SRRM4 is TAF1 microexon 34ʹ [Citation1]. The TAF1 gene encodes the largest subunit of basal transcription factor IID (TFIID), which is the primary core promoter recognition factor and nucleates RNA polymerase II (pol II) preinitiation complex assembly for transcription [Citation5]. While different TAF1 isoforms are ubiquitously expressed, neuronal tissues express the TAF1-34ʹ mRNA isoform (aka N-TAF1) [Citation6]. This isoform results from the inclusion of the 6-nt long microexon 34ʹ, encoding two additional amino acids (alanine and lysine), to the canonical TAF1 (cTAF1) protein of 1893 residues [Citation6]. The direct contributions of neuronal splicing factors such as SRRM4 to the formation of TAF1 mRNAs and their expression patterns in the brain have not yet been explored.
Mutations in the TAF1 gene have been associated with neurodevelopmental [Citation7] and neurodegenerative conditions [Citation6]. In particular, perturbations of TAF1 mRNA biosynthesis have been associated with X-linked dystonia-parkinsonism (XDP, MIM: 314250) [Citation8], an adult onset, neurodegenerative condition with progressive loss of voluntary motor control replaced by severe motor contractions (dystonia) combined with or replaced by parkinsonism features [Citation9,Citation10]. The neuropathology of XDP is characterized by decreased numbers of neural progenitors in the subventricular zone [Citation11] and prominent loss of medium spiny neurons within the striatum [Citation12], a forebrain region that controls voluntary movement. All XDP patients harbour the insertion of an SVA (SINE-VNTR-Alu) retrotransposon of the F-subclass into TAF1 intron 32 [Citation6], which has been proposed to affect expression and alternative splicing of TAF1 mRNAs [Citation6,Citation8].
Given the involvement of TAF1 mRNA processing in human neurological disorders, we investigated the relation of SRRM4 to the brain-specific distribution of cTAF1 and TAF1-34ʹ. Discrimination of microexon-containing mRNAs from canonical mRNAs by in situ methods is challenging due to the very small size of the micro-exon. We tested BaseScope™ probes in mouse brains to discriminate mRNA molecules that differ in only 6 nt. By employing this technique, we have found that Taf1-34ʹ mRNAs are enriched in post-mitotic neurons, whereas cTaf1 is more widely expressed in the brain, including cells undergoing division and post-mitotic neurons. BaseScope™ detection was validated at the protein level by using antibodies specific to TAF1 proteins that include or exclude microexon 34ʹ. Employing mouse and human cell systems we find that SRRM4 is required and sufficient to promote microexon 34ʹ inclusion in TAF1 mRNAs in neuronal and non-neuronal backgrounds. The splicing event is mediated by SRRM4 recognition of two UGC motifs located in the poly-pyrimidine tract upstream of microexon 34ʹ. Taken together, these results provide strong evidence that SRRM4 directs inclusion of microexon 34ʹ in TAF1 mRNAs to regulate the temporal and spatial expression of different TAF1 protein isoforms in mammalian brains.
Results
Analysis of cTaf1, Taf1-34ʹ and Srrm4 expression patterns in the mouse brain
To investigate the link between the mRNA expression of Taf1-34ʹ and the neuron-specific splicing factor Srrm4, we first explored their expression patterns in mouse brain sections at the level of the striatum. We employed in situ hybridization (ISH) with the BaseScope™ method, to discriminate between cTaf1 and Taf1-34ʹ mRNAs using specific probes against the 6-nt microexon 34ʹ or against the sequence spanning the flanking exons. In adult mouse brain sections, cTaf1 probes detected a broadly distributed expression in cerebral cortex, corpus callosum, striatum and septum (). Prominent cTaf1 expression was detected in cells along the ventricle wall and within the neurogenic sub-ventricular zone (SVZ) (’ and ). Comparison of cTaf1 and Taf1-34ʹ signals indicated clear differences in their distribution patterns. The Taf1-34ʹ signal was more prominent in the cerebral cortex compared to cTaf1 and Taf1-34ʹ mRNA expression was sparse in the glial-rich corpus callosum, the ventricle wall and the SVZ ( and ). Similarly, the expression of Srrm4, detected by an RNAscope® probe, which uses multiple probes to increase signal amplification [Citation13], was abundant in neuron-rich regions relative to the low levels in the glial-rich corpus callosum ( and ). Srrm4-positive cells were also present along the ventricle wall and SVZ ().
Figure 1. Differential expression of cTaf1, Taf1-34ʹ and Srrm4 mRNAs in the mouse brain.
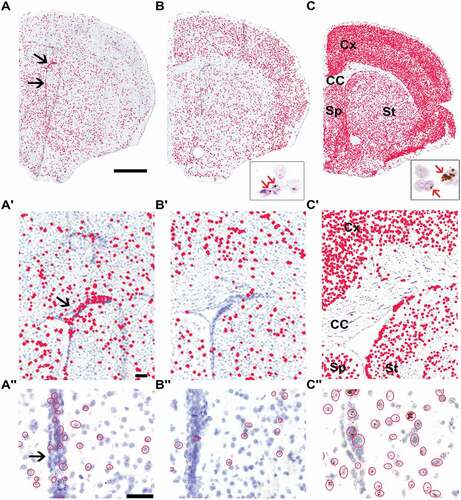
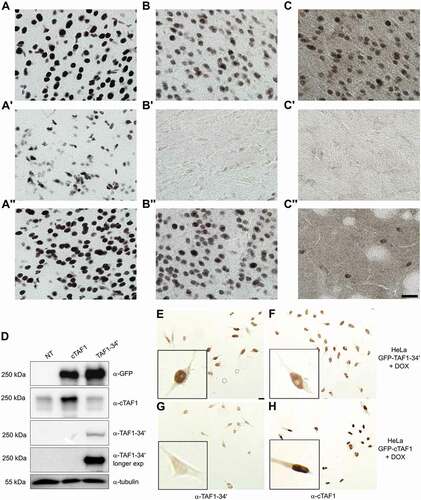
The mRNA ISH results were validated by immunohistochemical analyses (IHC) of different mouse brain regions by cTaf1-, Taf1-34ʹ- and Srrm4-specific antibodies (). The affinity-purified Taf1 antibodies developed in this study were directed against the region spanning microexon 34ʹ as the isoform-specific epitope, which is identical between mouse and human TAF1-34ʹ. The sera displayed high specificity in the detection by IHC and immunoblotting of both endogenous () and ectopically expressed proteins ( and Supplementary Fig. 1), with very limited, if any cross-reactivity when tested against the counterpart isoform ( and Supplementary Fig. 1). As additional validation, we performed IHC and confirmed that the antibodies against Taf1-34ʹ and Srrm4 were selectively immunoreactive with cell nuclei in neuron-rich regions of the brain, whereas the antibody against cTaf1 was ubiquitously immunoreactive across neuronal and non-neuronal cell nuclei (Supplementary Fig. 1). We next examined the distribution of cTaf1-, Taf1-34ʹ- and Srrm4 immunoreactivity within the brain. Consistent with the mRNA results, cTaf1 protein expression was detected in the nuclei of many cells throughout the brain, including in cells of the glial-rich corpus callosum and in post-mitotic neurons within the cerebral cortex and the striatum (-A’’). By contrast, Taf1-34ʹ expression was selectively enriched in neuronal nuclei ( and ) and was low within the corpus callosum (). The expression of Srrm4 also appeared enriched in neuronal nuclei and at low levels in the corpus callosum (). Magnification of sections at levels including the striatum showed that expression was high in the neocortex () but sparsely distributed in striatal cell nuclei ().
Figure 2. Overlapping expression of Taf1-34ʹ and Srrm4 proteins in the mouse brain.
The ISH and IHC performed on mouse brains indicated that cTaf1 and Taf1-34ʹ have different expression patterns within the forebrain. We therefore validated this differential distribution in the human brain by analysing RNA-seq data (Supplementary Fig. S2, panels A-B) and RT-PCR (Supplementary Fig. S2, panels C-D) from specimens from the striatum (caudate nucleus and putamen), neocortex, motor cortex, and thalamus. Consistent with the ISH data from the mouse brain, we confirmed that the inclusion of microexon 34ʹ is more prominent in the neocortex than within the striatum, while not detectable in the thalamus. The expression pattern of SRRM4 correlates with microexon 34ʹ inclusion, supporting their inter-dependency.
In conclusion, the mRNA ISH, RNA-seq and IHC analyses show that Taf1-34ʹ and Srrm4 are preferentially expressed in post-mitotic neurons relative to the more widespread expression of cTaf1. Importantly, the IHC analysis validated the sensitivity and accuracy of BaseScope™ technology to detect microexon 34ʹ-containing Taf1 mRNAs in situ () indicating that this is a versatile approach for the in situ detection of microexon sequences.
Inclusion of microexon 34ʹ into TAF1 mRNAs occurs in the later stages of neuronal differentiation
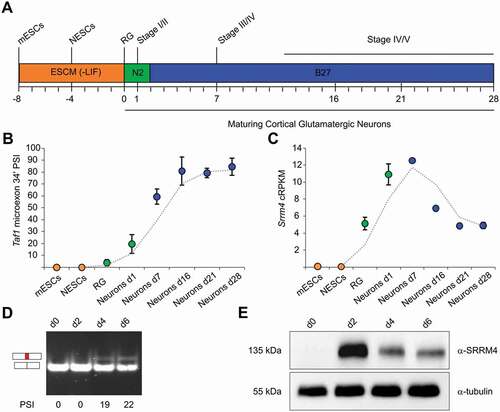
The analysis of cTaf1 and Taf1-34ʹ expression in the mouse brain suggested that these two isoforms display a temporal regulation. To investigate when during neural development Taf1-34ʹ first emerges, we examined the expression profile of this isoform using RNA-seq data collected at different time points during differentiation of mouse embryonic stem cells (mESCs) into cortical glutamatergic neurons [Citation14] (). Inclusion of microexon 34ʹ was evaluated using the Percentage-Spliced-In (PSI) metric, which determines the percentage of Taf1 mRNAs containing microexon 34ʹ. This study indicated that microexon 34ʹ is incorporated into Taf1 mRNA at the beginning of neuronal differentiation, reaching the highest value in mature post-mitotic neurons (). At earlier stages, when neuroepithelial cells and radial glia were undergoing divisions, microexon 34ʹ was not detectable. The expression of Srrm4 mRNA began in glial daughters and peaked in the early phase of neuronal maturation. As neuronal differentiation proceeded, Srrm4 expression decreased (). These results indicate a switch from cTaf1 to Taf1-34ʹ expression in the early phase of neuronal differentiation with a further increase in Taf1-34ʹ expression in mature neurons. Interestingly, Srrm4 expression peaks with the earliest detection of microexon 34ʹ, suggesting that Srrm4 initiates the transition of cTaf1 to Taf1-34ʹ mRNAs during fate determination. We next validated the temporal expression pattern of SRRM4 and the resulting shift between cTAF1 and TAF1-34ʹ in a human differentiation cell system, using the LUHMES cell line. LUHMES are human mesencephalic cells, which have been immortalized by v-myc. This oncogene is repressed in a tetracycline-dependent manner, which induces LUHMES cells to exit cell cycle and differentiate into morphologically and biochemically post-mitotic dopamine-like neurons [Citation15,Citation16]. Immunoblot analysis showed that endogenous SRRM4 was detectable at day 2 of differentiation and its expression was decreasing in later differentiation stages (). Consistent with SRRM4 expression, the incorporation of microexon 34ʹ into TAF1 mRNA was observed only in the latest differentiation points (day 4 and day 6) ().
Taken together, this data suggests that the neuronal SRRM4 splicing factor is the driver of the switch of cTAF1 to TAF1-34ʹ expression, which coincides with the neuronal maturation of mammalian brain cells.
Figure 3. Expression of cTAF1, TAF1-34ʹ and SRRM4 mRNAs during in vitro neuronal differentiation.
SRRM4 promotes the inclusion of the alternative microexon 34ʹ in TAF1 mRNA
To investigate the direct involvement of SRRM4 in TAF1 pre-mRNA splicing, we engineered HeLa cells to conditionally express an N-terminally GFP-tagged SRRM4. DOX-inducible expression of this transgene was confirmed by immunoblotting and immunofluorescence ( and ). Consistent with recent results [Citation17], the exogenous GFP-SRRM4 localized to the nuclear compartment () within the nuclear speckles, which was confirmed by its co-localization with the SRSF2/SC35 splicing factor ().
Figure 4. SRRM4 promotes the inclusion of the alternative microexon 34ʹ in TAF1 mRNA.
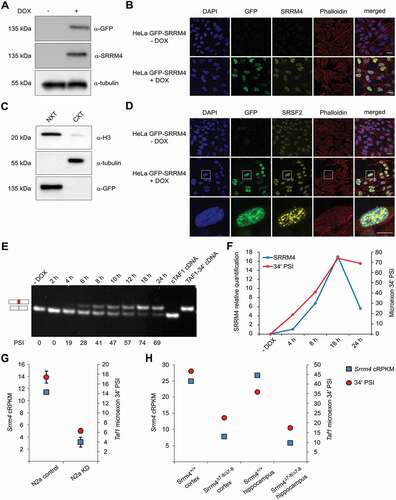
The contribution of SRRM4 in microexon 34ʹ incorporation in TAF1 mRNA was examined during a time-course induction of GFP-SRRM4 expression in HeLa cells (Supplementary Fig. S3A and S3B). For this analysis, we developed a polyacrylamide gel system to resolve RT-PCR products containing the 6-nt of microexon 34ʹ from cTAF1. With this system, we observed that the progressive SRRM4 expression coincided with increased microexon 34ʹ inclusion that reached a PSI of 74% after 18 h of DOX treatment (). Microexon 34ʹ inclusion lagged only shortly behind GFP-SRRM4 induction, which indicates a rapid turnover of the pool of TAF1 mRNAs (). We examined two other microexon splicing events, the incorporation of microexon 8A in KDM1A and microexon 16 in DAAM1 attributed to SRRM4 and observed inclusion of both microexons into their respective mRNAs upon SRMM4 induction in HeLa cells (Supplementary Fig. S3C and S3D).
To determine whether Srrm4 is required for inclusion of Taf1 microexon 34ʹ in neuronal cells, we examined RNA-seq data from mouse neuroblastoma N2a cells after siRNA-mediated knockdown of endogenous Srrm4 expression [Citation2]. The PSI for microexon 34ʹ inclusion in untreated N2a cells was ~18% and was reduced to 6% after Srrm4 knockdown (). The interdependency of microexon 34ʹ and Srrm4 was also confirmed using RNA-seq data from neocortical and hippocampal samples taken from a conditional Srrm4 knockout model [Citation3]. These results demonstrate that the reduced expression of Srrm4 correlates with reduced microexon 34ʹ incorporation in Taf1 mRNA in vivo (). Besides microexon 34ʹ, additional alternative splicing (AS) events have been described for the downstream exons of TAF1 [Citation6,Citation8,Citation18,Citation19]. To determine whether SRRM4 contributes to these splicing events, TAF1 mRNAs from GFP-SRRM4-expressing HeLa cells were scanned by RT-PCR using primer pairs spanning exon 30 to the final exon 38. The primers were designed to analyse the splicing events occurring across introns or exons of this region, to detect both alternative exon inclusion and exon skipping. This scanning confirmed that TAF1 mRNA undergoes several AS events and that only the inclusion of microexon 34ʹ is SRRM4-dependent (Supplementary Fig. S4). In addition, this analysis confirmed that the alternative splicing of microexon 34ʹ and exon 35ʹ are independent events and that the two alternative exons can be part of the same TAF1 isoform. To determine whether SRRM4 supports TAF1 microexon 34ʹ inclusion in different cell types, we generated GFP-SRRM4 expressing cells derived from human RPE1 retinal pigmented epithelial cells and human U-2 OS osteosarcoma cells ( to ). As with HeLa cells, DOX-induction of GFP-SRRM4 expression in these two non-neuronal backgrounds resulted in the inclusion of microexon 34ʹ in TAF1 mRNA (). The PSI values for microexon 34ʹ correlated with GFP-SRRM4 expression levels across different cell types. Compared to HeLa and U-2 OS cells, RPE1 cells expressed GFP-SRRM4 at lower levels and 50% of TAF1 mRNAs included microexon 34ʹ. By contrast, HeLa and U-2 OS cells expressed GFP-SRRM4 at similarly high levels and displayed a similar PSI (69% and 76% after 24 h of induction, respectively).
Figure 5. SRRM4 promotes the alternative splicing of TAF1-34ʹ in different non-neuronal cell lines.
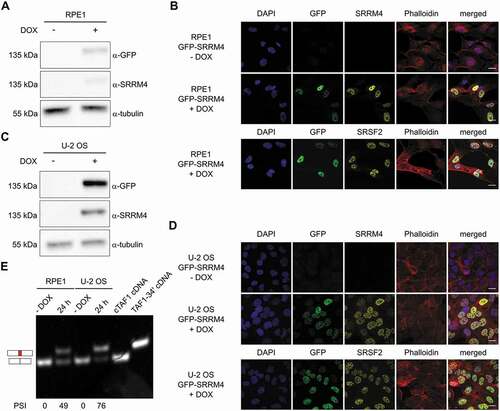
Taken together, these results show that exogenous expression of SRRM4 directs inclusion of microexon 34ʹ in TAF1 mRNAs. This alternative splicing event can be induced in non-neuronal cell systems, which underlines the powerful action of SRRM4 in driving microexon 34ʹ inclusion.
Interactome of ectopically expressed SRRM4 supports its splicing function
Several SRRM4 interactors have been described by mass spectrometry in 293T cells [Citation2] and N2a cells [Citation17]. We examined the hierarchy of SRRM4 interactors by iBAQ-based quantitative mass spectrometry (qMS) of GFP-SRRM4 purified from nuclear extracts of DOX-induced HeLa cells (Supplementary Fig. S3E). This procedure identified U2 snRNP auxiliary splicing factor, composed of the U2AF1 (U2AF35)/U2AF2 (U2AF65), as a major interactor of SRRM4. This complex, which binds to the 3ʹ AG dinucleotide and the polypyrimidine tract element, promotes the recruitment of U2 snRNP to adjacent branch sites [Citation20]. Stoichiometry values indicated that ~40% of GFP-SRRM4 protein forms a stable complex with U2AF1/U2AF2. Among the other GFP-SRRM4 interactors are the RNPS1 and SRSF11 splicing factors (relative stoichiometries of 0.25 and 0.01, respectively), which have been recently identified as co-regulators of SRRM4-dependent microexon splicing [Citation17]. Also consistent with recent results [Citation17], core components of the Exon Junction Complex (EIF4AIII, RBM8A, and MAGOHB) and its auxiliary proteins (PNN, ACIN1, SRRM2 and SAP18) were also identified, and these account for 10-20% of the recovered SRRM4 complexes. Our analysis further confirmed the interaction with FBXW11, which has been proposed recently as a regulator of SRRM4 proteolysis [Citation17]. The list of statistically significant interactors of GFP-SRRM4 in HeLa cell nuclear extract (with relative stoichiometry above 0.01) is provided in Supplementary Table S2.
UGC motifs upstream of TAF1 microexon 34ʹ are critical for SRRM4-mediated alternative splicing
A previous study identified UGC-containing motifs as critical SRRM4 binding sites to promote neuronal microexon inclusion [Citation2]. Sequence analysis of TAF1 intron 34ʹ sequences revealed the presence of two UGC motifs located in the proximity of the 3ʹ splice site of microexon 34ʹ (−18 and −29 nt). We investigated their involvement in microexon 34ʹ incorporation in a minigene reporter assay. The TAF1 minigene reporter included TAF1 sequences spanning from exon 33 to the final codon of exon 35. This embeds the sequence of the neuron-specific microexon 34ʹ with the flanking introns 33 and 34. The minigene reporter was designed as an in-frame fusion product with a GFP N-terminal tag to permit GFP-specific enrichment during RT-PCR analysis and to allow the discrimination from endogenous TAF1 mRNA splicing events (). TAF1 isoform-specific antibodies also allowed to examine the cTAF1 to TAF1-34ʹ switch at the protein level. Whereas transfection of the wild-type minigene reporter (MG) in 293T cells resulted only in cTAF1 products, co-transfection of the minigene with GFP-SRRM4 induced the incorporation of microexon 34ʹ sequence as determined by RT-PCR analysis of mRNAs () and by TAF1-34ʹ specific antibodies (). The two UGC motifs were subsequently mutated into UcC or UaC (). We found that mutagenesis of a single UGC reduced microexon 34ʹ incorporation by two-fold, whereas double UGC mutants displayed strongly reduced microexon 34ʹ incorporations (average PSI wild-type mini-gene: 58.5% vs average PSI double UGC mutants: 5%) (-).
Figure 6. Microexon 34ʹ incorporation is regulated by upstream UGC motifs.
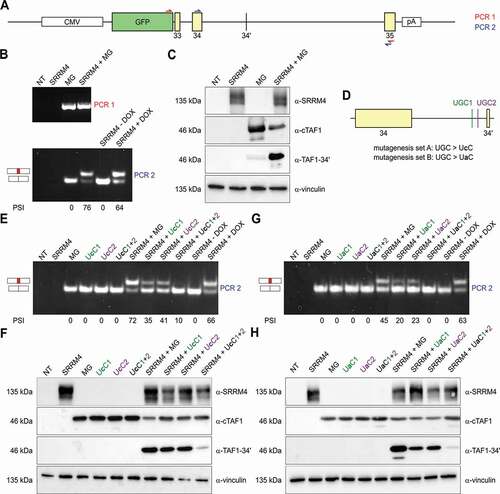
Taken together, the TAF1 minigene transfection experiments demonstrate that expression of SRRM4 promotes microexon 34ʹ inclusion in TAF1 mRNA, which is dependent on the two UGC motifs located in poly-pyrimidine tract just upstream the regulated microexon 34ʹ.
Discussion
Alternative splicing (AS) of mRNAs is one of the central mechanisms contributing to the diversification of the coding capacity of metazoan genomes. The AS regulatory networks are primarily directed by combinations of tissue-specific splicing factors, which enhance or silence specific splicing choices through their binding to cis-acting sequences in pre-mRNAs [Citation21]. Among vertebrate tissues, the nervous system shows the most extensive and conserved AS patterns [Citation22] required to generate protein diversity for specification and function of different cell types [Citation23,Citation24].
In this study, we investigated the spatial and temporal distribution of TAF1-34ʹ, the neuron-specific isoform of TAF1. The overlapping patterns of isoform-specific mRNA and protein validate the BaseScope™ method to differentiate mRNAs, which only differ by a 6-nt insertion. The specific staining patterns suggested that canonical and neuron-specific TAF1 isoforms have distinct distributions in specific regions of the brain in both mice and humans. One striking feature of these distributions is their opposing expression patterns in proliferating versus post-mitotic regions. A shift from cTAF1 to TAF1-34ʹ occurs upon neuronal maturation and this shift was confirmed by in vitro differentiation of mouse embryonic stem cells and human mesencephalic cells differentiation. The observation that TAF1 and TAF1-34ʹ are expressed in specific brain regions and at defined times of neuronal development suggest that these two isoforms exert distinct functions during development. TAF1 is part of the basal transcription factor TFIID, which represents a ~ 1.5 MDa complex composed of TBP (TATA-binding protein) and 13 TBP-associated factors (TAFs) [Citation25]. Within the complex, TAF1 contacts TBP [Citation26] and TAF7 [Citation27,Citation28] as well as downstream core promoter elements [Citation29–Citation31]. Although TFIID is regarded as a basal transcription factor with ubiquitous expression, tissue-specific TFIID variants have been reported [Citation32]. An example is TAF7L, the testis-specific paralog of TAF7. In late spermatogenesis, while overall TAF7 levels are reduced, TAF7L is expressed to associate in a testis-specific TFIID [Citation33]. An additional testis-specific TAF is TAF1L, the paralog of TAF1. TAF1L can also bind directly to TBP [Citation34] and can replace TAF1 within TFIID (unpublished observation). Tissue-specific functions of TAFs have not only been identified in germ lines, but also in neuronal cells. TAF9B, a paralog of TAF9, promotes alternative gene expression by specifically binding to both promoters and distal enhancers of neuronal genes [Citation35]. These observations indicate that tissue-specific forms of TFIID can display promoter-specificity driving tissue-specific transcription programmes. By analogy, we propose that the TAF1-34ʹ isoform could exert a specific neuronal function. We have found that TAF1-34ʹ efficiently incorporates into the TFIID complex (unpublished observations). This suggests that TAF1-34ʹ could regulate cell-type specific transcription through mediating cell-type specific protein-protein and/or protein-DNA interactions of TFIID. This proposal is in line with observations that neuron-specific microexons encode residues that overlap surface-accessible and looped regions of proteins that often coincide with modular domains involved in protein-protein interactions [Citation1]. This observation strengthens the concept that tissue-specific AS rewires protein-protein interaction networks [Citation36–Citation38]. Interestingly, the two amino acids encoded by microexon 34ʹ are located in the unstructured C-terminal region of TAF1. The addition of an alanine-lysine dipeptide in this region might create a new binding interface or an acceptor for post-translational protein modifications. Additional studies including TAF1-34ʹ-specific proteomics and ChIP-seq, combined with corresponding single-cell RNA-seq profiles for different brain regions, would test these hypotheses.
The alternative expression of TAF1 and TAF1-34ʹ isoforms is restricted to specific cell types in the brain and we show that the neuronal splicing factor SRRM4 directs this switch. Using both ISH and IHC, we could confirm the overlapping expression pattern between Srrm4 and Taf1-34ʹ. Interestingly, while the cortex showed very high correlation ( and ), the striatum sample challenged this finding, showing strong immunoreactivity for Taf1-34ʹ but a weak Srrm4 staining (B’’-C’’). This data could indicate that a low expression of Srrm4, not detectable by IHC, is sufficient to promote Taf1-34ʹ microexon inclusion. On the other hand, this could also suggest that an intricate network of AS regulation initiated by Srrm4 is sustained by other splicing factors that have functions overlapping with Srrm4. This second hypothesis could be of interest in regards of detection of low levels of Taf1-34ʹ in non-neuronal tissues, as striatal muscles (Fig. S1A), but additional experiments would be required to identify these factors. The involvement of SRRM4 in cTAF1-TAF1-34ʹ switch is demonstrated by the fact that the presence of SRRM4 is sufficient to direct microexon 34ʹ incorporation into TAF1 mRNA in several non-neuronal cell systems. Consistent with this, the incorporation of microexon 34ʹ into TAF1 mRNA is strikingly impaired when SRRM4 binding sites located just upstream of microexon 34ʹ are mutated. Reduction of microexon 34ʹ splicing has been linked to XDP [Citation6,Citation39,Citation40]. In order to identify the molecular mechanism linking the pathogenesis of the disease with a defect in TAF1 alternative splicing, it would be important to investigate the possible SVA-induced alteration in the SRRM4-dependent 34ʹ incorporation. In this context, the tools developed in this study will be used to investigate cTAF1, TAF1-34ʹ and SRRM4 expression, both at mRNA and protein levels, in XDP-derived material and XDP post-mortem brains.
Several neurodegenerative diseases that show tissue-specific cell loss are known to be caused by genetic alterations in ubiquitously expressed genes. This disconnection has been a mystery that has incurred numerous hypotheses to account for the cell type-specific vulnerability. Our study combined with others [Citation3,Citation41,Citation42] raises the possibility that microexons that were not previously annotated or evaluated will define tissue-specific isoforms of these ‘ubiquitous’ disease genes. This concept strongly applies to XDP as it affects TAF1, encoding a crucial component for pol II-mediated gene transcription. Our study demonstrates that the splicing factor SRRM4 directs neuron-specific TAF1 AS profiles, which provides a mechanism to account for the restricted tissue-specificity of XDP pathology. In addition, our results suggest that the neuronal splicing factor SRRM4 shapes TFIID to drive a neuronal-specific transcription programme that is important for mature brain function in vertebrates.
Materials and methods
Mice
For histological experiments we used 1 to 4 months old female and male mice of C57BL/6 and FVB genetic backgrounds. Mice were group-housed by sex and kept under standard laboratory conditions. All procedures were approved by the Animal Care and Use Committee at the Massachusetts Institute of Technology (MIT), which is AALAC accredited.
In situ hybridization on mouse tissue
Mice were deeply anesthetized with isoflurane prior to brain removal and its placement on ice. A thick block containing the striatum was dissected and placed into a cryomold containing cold Optimal Cutting Temperature (OCT) Compound (Tissue-Tek, Sakura Finetek Inc., Kyoto, Japan). The cryomold was then filled with OCT and further cooled, just until opaque, in a bath of methylbutane that was pre-cooled on dry ice. Cryomolds were sealed in plastic bags and stored at −80°C until cut into 10 μm sections on a cryostat. Sections were placed onto positively charged slides (Leica, Wetzlar, Germany) and stored at −80°C until use. For detection of Srrm4 mRNA, single-plex chromogenic in situ hybridization was done as described in the RNAscope® 2.5 manual from ACDBio (Silicon Valley, CA), with a probe directed to Srrm4 (cat. #529,068, ACDBio). For detection of microexon 34ʹ in Taf1-34ʹ, and the exon 34 to exon 35 junction site in cTaf1, we used BaseScope™ (ACDBio) probes (cat. #713,761 for Taf1-34ʹ and cat. #713,771 for cTaf1) and the provided protocol. Slides were scanned at 40X magnification with an Aperio slide scanner and images were processed in Aperio ImageScope (Leica).
Immunohistochemistry on mouse tissue and HeLa cells
Mice were deeply anesthetized with isoflurane and trans-cardially perfused with 0.9% saline solution followed by 4% (wt/vol) freshly depolymerized paraformaldehyde in 0.1 M NaKPO4 buffer as fixative. Dissected brains were post-fixed for 90 min and then were transferred to cryoprotective 25% (vol/vol) glycerol sinking solution for 12–24 hours. Brains were then frozen and cut at 30 μm on a sliding microtome, and sections were placed in 0.1% sodium azide in 0.1 M phosphate buffer solution (PBS) made from NaKPO4 for storage at 4°C. Sections were treated for antigen retrieval by submerging in 95°C, pH 6.0 unmasking solution (Vector laboratories, Burlingame, CA) for 10 min. See https://www.protocols.io/view/immunofluorescence-for-free-floating-brain-section-kracv2e dx.doi.org/10.17504 (2017) for extra details of the immunoreactivity reactions. Briefly, sections were rinsed and treated for 10 min with 3% H2O2 in PBS with 0.2% Triton X-100. Sections were incubated in TSA blocking solution (PerkinElmer, Waltham, MA) and then in primary antiserum overnight at 4°C. Sections were subsequently rinsed and incubated in a biotinylated secondary anti-rabbit antibody and followed by the Vectastain Peroxidase ABC System (Vector Laboratories) according to manufacturer’s instructions. Sections were mounted and coverslipped with Eukitt (Electron Microscopy Sciences, Hatfield, PA). Immunohistochemical images were obtained on an Olympus BX61 microscope with a 100 × 1.35 NA oil immersion objective. The same procedure was adopted to perform IHC on HeLa cells.
Preparation of human brain specimens
Flash frozen human hemispheres from unremarkable donors were acquired from the Netherlands Brain Bank. Frozen hemispheres were dissected into coronal slices in a − 20°C climate control room using a 30 cm cutting edge brain dissecting knife (Fine Scientific Tools, Foster City, CA). Further dissection of selected regions was performed using pre-cooled scalpel blades (Fine Scientific Tools) and 3 mm biopsy punches (Microtech, Fletcher, NC). Frozen tissue was pulverized using a mortar and pestle while submerged in liquid nitrogen.
Plasmid and cell lines generation
The pcDNA5/FRT/TO/GFP-SRRM4 expression plasmid was generated using the GATEWAY cloning system according to the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, MA). The open reading frame of the human SRRM4 was amplified from the cDNA clone BC152471 (TransOMIC Technologies, Huntsville, AL) using attB-flanked primers, listed in Supplementary Table S1. The amplicon was cloned into the entry vector pDON201 (BP reaction) and subsequently recombined with the destination vector pcDNA5/FRT/TO/N-GFP (LR reaction) to generate expression clones. The gene of interest was verified using Sanger sequencing in both plasmids. The pcDNA5/FRT/TO/GFP-cTAF1 (NM_004606) was available in the laboratory and contains two silent changes introducing an AatII site for cloning purposes. The pcDNA5/FRT/TO/GFP-TAF1-34ʹ was generated via site-directed mutagenesis. Mutagenesis primers are listed in Supplementary Table S1. The TAF1 mini-gene was generated using PCR amplification and classical cloning. The portion of the human TAF1 locus cloned into the pcDNA5/FRT/TO/N-GFP vector has the following GRch38 coordinates: chrX:71,454,175–71,458,366.
The HeLa Flp-In T-REx parental cell line [Citation43] was cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 4.5 g/litre of glucose (Lonza, Basel, Switzerland), supplemented with 10% (v/v) foetal bovine serum (Lonza), 200 µg/ml of Zeocin (Thermo Fisher Scientific) and 5 µg/ml of Blasticidin S (Invivogen, San Diego, CA), to select for the FRT site and the Tet repressor, respectively. To generate stable doxycycline (DOX)-inducible HeLa Flp-In T-REx cells expressing GFP-SRRM4, parental cells were seeded in 6-well plates and cultured for 24 h without antibiotics. Parental HeLa cells were then co-transfected with pOG44, an expression vector coding for the Flp recombinase, and the pcDNA5/FRT/TO/GFP-SRRM4, using polyethyleneimine (PEI). 48 h after transfection, the medium was supplemented with 250 µg/ml of hygromycin B (Roche Diagnostics, Basel, Switzerland) and 5 µg/ml of Blasticidin S (Invivogen), to select for the Flp-mediated recombination with the pcDNA5/FRT/TO/GFP-SRRM4 plasmid and for Tet repressor expression, respectively. The cell line was validated using immunoblot and immunofluorescence, after 18–24 h induction with 1 µg/ml DOX (MP Biomedical, Santa Ana, CA).
The procedure described above was also used to generate RPE1 and U-2 OS derivatives expressing pcDNA5/FRT/TO/GFP-SRRM4. Flp-In T-REx derivatives of human RPE1 and U-2 OS cells were kind gifts from Dr. Kops (Hubrecht Institute, Utrecht, The Netherlands) and Dr. Buhl (Harvard University, Cambridge, USA), respectively.
Neuroblastoma cell lines SH-SY5Y were a kind gift from Dr. Timmerman (University of Antwerp, Belgium). The cells were cultured in 1:1 DMEM containing 4.5 g/litre of glucose (Lonza)/F12 supplement (Lonza), supplemented with 10% (v/v) foetal bovine serum (Lonza).
Human embryonic kidney cells 293T were cultured in DMEM containing 4.5 g/litre of glucose (Lonza), supplemented with 10% (v/v) foetal bovine serum (Lonza). Transient transfection was performed using FuGENE HD Transfection Reagent (Promega) following manufacturer’s instructions.
Lund human mesencephalic (LUHMES) cells were cultured and differentiated as described in [Citation17]. Briefly, NunclonTM flasks were pre-coated with 50 µg/ml poly-L-ornithine (Sigma, St. Louis, MO) and 1 µg/ml fibronectin (Sigma) in H2O at 37°C overnight, washed once with H2O and air-dried. For culturing of cells proliferation medium with Advanced DMEM/F12 (Gibco, Thermo Fisher Scientific), 1x N-2 supplement (Gibco), 2 mM L-glutamine (Gibco) and 40 ng/ml recombinant basic fibroblast growth factor (R&D Systems, Minneapolis, MN) was used. Differentiation was induced by changing to differentiation medium containing DMEM/F12, 1x N-2 supplement, 2 mM L-glutamine, 1 mM dibutyryl cAMP (Sigma), 2 ng/ml recombinant human GDNF (R&D Systems) and 1 µg/ml tetracycline (Sigma).
Immunoblot
HeLa GFP-SRRM4 cells were seeded in 6-well plates at the density of 30,000 cells per well and were induced with 1 µg/ml DOX for 18 to 24 h. Cells were lysed in sample buffer (40 mM Tris-HCl pH 6.8, 1% SDS, 5% glycerol, 0.0125% bromophenol blue, 100 mM DTT), and equal amounts of protein were analysed by SDS-PAGE, followed by immunoblotting. After transfer to a nitrocellulose membrane, proteins were identified with the indicated antibodies and detected using Clarity ECL (Bio-Rad, Hercules, CA) and the ChemiDoc Touch system (Bio-Rad). The quantification of band intensity was performed with the ‘mean pixel gray’ values of the selected region of interest using ImageJ software after Gaussian blur and background subtraction.
Immunofluorescence and confocal microscopy
GFP-SRRM4 expressing cells were seeded in 12-well plates containing 16-mm glass coverslips (Marienfeld, Lauda-Königshofen, Germany), at the density of 10,000 cells per well. After DOX induction cells were fixed in 2% paraformaldehyde (Thermo Fisher Scientific) in PBS for 20 min at room temperature. Immunofluorescence was performed using a standard protocol. Primary and secondary antibodies were incubated for 1 h at room temperature. Nuclei were stained with 2 µg/ml DAPI (4,6-diamidino-2-phenylindole, Sigma); the cytoplasm was stained with Alexa Fluor 633 Phalloidin (Thermo Fisher Scientific). Coverslips were mounted on microscopy slides (Roth, Karlsruhe, Germany) using fluorescence mounting medium (Dako, Agilent Technologies, Santa Clara, CA) and kept overnight at 4°C.
Confocal images were taken with the Zeiss LSM710 confocal microscope, using the 63x/1.4 oil plan-apochromat objective, using 405-nm violet diode, 488-nm argon and 561-nm solid state lasers.
Antibodies
The production of cTAF1- and TAF1-34ʹ-specific antibodies was performed in collaboration with Cambridge Research Biochemical and Abcam. To obtain antisera specific for the cTAF1 and TAF1-34ʹ proteins, rabbits were immunized with peptides (cTAF1: [C]-TPGPYTPQPPDLY-amide or TAF1-34ʹ: [C]-TPGPYTPQAKPPDLY-amide) coupled via their N-terminal Cys residue to keyhole limpet haemocyanin (KLH) or bovine serum albumin (BSA). The selected epitopes have 100% identity between human and mouse proteins. Rabbits were immunized by injections with KLH conjugates followed by booster injections with the BSA conjugates at regular intervals. Antibody responses were monitored by ELISA using the two peptides. Based on these ELISA results, polyclonal sera were collected from the best and most specific responders and affinity-purified using magnetic beads (Dynabeads M-280, ThermoFisher) coated with biotinylated derivatives of the immunized peptides. For immunohistochemistry on mouse tissue and HeLa cells, affinity-purified cTAF1 and TAF1-34ʹ antibodies and SRRM4 antibody [Citation44] were used, followed by biotinylated secondary anti-rabbit antibody (Vector Laboratories, 1:500 dilution).
For immunoblot and immunofluorescence, the following primary antibodies were used: GFP (JL-8, Clontech, Mountain View, CA), SRRM4 [Citation44], α-tubulin (DM1A, CP06, Calbiochem, San Diego, CA), SRSF2 (SC-35, S4045, Sigma), histone H3 (ab1791, Abcam, Cambridge, UK) and vinculin (7F9, sc-73,614, Santa Cruz). Notably, the SRRM4 antibody used was raised against the first 82 amino acids of the mouse protein [Citation44] and the identity between the mouse and the human epitope is 84%. Secondary antibodies used were: α-mouse-HRP conjugate (Bio-Rad) and α-rabbit-HRP conjugate (Bio-Rad), α-rabbit Alexa Fluor 568 (Thermo Fisher Scientific) and the α-mouse Alexa Fluor 568 (Thermo Fisher Scientific).
Expression analysis with RT-PCR and RT-qPCR
Total RNA was isolated from the GFP-SRRM4 expressing cell lines using the RNeasy kit (Qiagen, Venlo, The Netherlands), including an over-column DNase treatment using RNase-free DNase (Qiagen), according to the manufacturer’s instructions. The total RNA isolation from human brain specimens was performed using the RNeasy Lipid Tissue kit (Qiagen), with subsequent DNAse treatment performed with the TURBO DNAse treatment kit (Thermo Fisher Scientific). RNA integrity was analysed using Bioanalyzer (Agilent Technologies) and RNA Integrity Numbers (RINs) were above 7 for all samples.
Total RNA (1 µg) was used for cDNA synthesis (Superscript II, Thermo Fisher Scientific) using random hexamer primers. TAF1 and Lysine Demethylase 1A (KDM1A) amplicons were analysed on a 6% acrylamide gel and Dishevelled Associated Activator of Morphogenesis 1 (DAAM1) amplicon was analysed on a 2.5% agarose gel. Gels were stained with ethidium bromide. RT-PCR was performed using the primer pairs listed in Supplementary Table S1. In the TAF1 RT-PCRs, plasmids containing cTAF1 and TAF1-34ʹ cDNAs were used as size controls. Percentage-Spliced-In (PSI) was calculated as the fraction of TAF1 cDNA containing microexon 34ʹ to the sum of the inclusion and exclusion TAF1 cDNA. The quantification of band intensity was performed using ImageJ as described in the immunoblot paragraph.
RT-qPCR was performed in a CFX384 Real-Time system (Bio-Rad) using iQ SYBR Green Supermix (Bio-Rad). Primers used are listed in Supplementary Table S1. The relative expression level of SRRM4 mRNA was evaluated with the ΔΔCt method, using ACTB as reference gene. The reactions were performed in technical triplicates.
Nuclear extract and GFP affinity purification
Cytoplasmic and nuclear extracts were prepared from GFP-SRRM4 expressing HeLa cells as previously described [Citation45]. After Bradford measurement, 1 mg of nuclear extract was used for GFP affinity purification [Citation45], in the absence of ethidium bromide. The experiment was performed in triplicate with GFP-trap beads (gta-20, Chromotek, Planegg-Martinsried, Germany) and control agarose beads (bab-20, Chromotek). After trypsin-digestion, the peptides were desalted using C18 matrix in StageTips [Citation46]. Peptides were eluted in 80% acetonitrile and concentrated using a SpeedVac. The lyophilized samples were diluted to the desired volume in 0.1% trifluoroacetic acid and analysed by mass spectrometry.
Quantitative mass spectrometry (qMS)
For LC-MS/MS analysis, peptide samples were analysed by an Orbitrap Q-Exactive Plus (Thermo Fisher Scientific) mass spectrometer coupled to an Easy nano-LC 1000 (Thermo Fisher Scientific) with a flow rate of 300 nl/min. Buffer A was 0.1% (v/v) formic acid and buffer B was 0.1% (v/v) formic acid in 80% acetonitrile. For reversed phase chromatography, a gradient of increasing acetonitrile proportion was applied in combination with a C18 separating column (2 µm particle size, 100 Å pore size, length 150 mm, inner diameter 50 µm, Thermo Fisher Scientific). The mass spectrometer operated in data dependent mode with a maximum of 10 MS/MS scans following each MS1 scan. Peptide identification was performed with the Andromeda search engine using a human proteome database containing reviewed Uniprot sequences without isoforms downloaded from Uniprot 15 April 2016 (human, 20,193 entries). Peptide spectrum matching and relative protein quantitation were performed using MaxQuant v.1.5.2.8 using a false discovery rate (FDR) of 1%. Perseus v.1.6.0.7 was used to filter contaminants and reverse hits. Label-free quantification values were log2 transformed and groups based on beads type were defined. Identified proteins were accepted when measured three times within at least one combination of beads, resulting in a total of 917 proteins. To determine the relative stoichiometry of the identified interactors, intensity based absolute quantification (iBAQ) values were used [Citation47], after normalizing to the bait protein (GFP-SRRM4). FDR was set at 0.001 and the minimal fold change (s0) was set at 2.
Data availability
The proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [Citation48] partner repository with the dataset identifier PXD009905.
Keypoints
cTAF1 and TAF1-34ʹ mRNA and protein distinguish neurogenic from post-mitotic neurons
Splicing factor SRRM4/nSR100 is required and sufficient to direct microexon 34ʹ splicing into TAF1 mRNA
SRRM4-dependent splicing of microexon 34ʹ relies on UGC-containing 3ʹ sequences
Supplemental Material
Download MS Word (1.7 MB)Acknowledgments
We thank the present and past members of the Timmers laboratory for their support, discussions and critical reading of this manuscript. This work also benefitted from discussions with Dr. Paul Coffer and his group members (University Medical Center Utrecht, The Netherlands). We also thank Samitha Venu, Tomoko Yoshida, Marie Follo and Courtney Anderson, Emily Park, Mindy Wang, and Wei Wei from ACDBio for technical assistance. We thank Marcel Leist (University of Konstanz, Germany) for providing the LUHMES cells. This work was financially supported by the Collaborative Center for X-linked Dystonia Parkinsonism (CCXDP). We are particularly grateful for the stimulating discussions with all members of the CC-XDP and its inspirational leaders.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Irimia M, Weatheritt RJ, Ellis JD, et al. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell. 2014;159(7):1511–1523.
- Raj B, Irimia M, Braunschweig U, et al. A global regulatory mechanism for activating an exon network required for neurogenesis. Mol Cell. 2014;56(1):90–103.
- Quesnel-Vallieres M, Irimia M, Cordes SP, et al. Essential roles for the splicing regulator nSR100/SRRM4 during nervous system development. Genes Dev. 2015;29(7):746–759.
- Quesnel-Vallieres M, Dargaei Z, Irimia M, et al. Misregulation of an Activity-Dependent Splicing Network as a Common Mechanism Underlying Autism Spectrum Disorders. Mol Cell. 2016;64(6):1023–1034.
- Burley SK, Roeder RG. Biochemistry and structural biology of transcription factor IID (TFIID). Annu Rev Biochem. 1996;65:769–799.
- Makino S, Kaji R, Ando S, et al. Reduced neuron-specific expression of the TAF1 gene is associated with X-linked dystonia-parkinsonism. Am J Hum Genet. 2007;80(3):393–406.
- O’Rawe JA, Wu Y, Dorfel MJ, et al. TAF1 Variants Are Associated with Dysmorphic Features, Intellectual Disability, and Neurological Manifestations. Am J Hum Genet. 2015;97(6):922–932.
- Aneichyk T, Hendriks WT, Yadav R, et al. Dissecting the Causal Mechanism of X-Linked Dystonia-Parkinsonism by Integrating Genome and Transcriptome Assembly. Cell. 2018;172(5):897–909 e21.
- Lee LV, Pascasio FM, Fuentes FD, et al. Torsion dystonia in Panay, Philippines. Adv Neurol. 1976;14:137–151.
- Lee LV, Rivera C, Teleg RA, et al. The unique phenomenology of sex-linked dystonia parkinsonism (XDP, DYT3, “Lubag”). Int J Neurosci. 2011;121(Suppl 1):3–11.
- Goto S, Kawarai T, Morigaki R, et al. Defects in the striatal neuropeptide Y system in X-linked dystonia-parkinsonism. Brain. 2013;136(Pt 5):1555–1567.
- Goto S, Lee LV, Munoz EL, et al. Functional anatomy of the basal ganglia in X-linked recessive dystonia-parkinsonism. Ann Neurol. 2005;58(1):7–17.
- Erben L, He MX, Laeremans A, et al. A Novel Ultrasensitive In Situ Hybridization Approach to Detect Short Sequences and Splice Variants with Cellular Resolution. Mol Neurobiol. 2017;55(7):6169–6181.
- Hubbard KS, Gut IM, Lyman ME, et al. Longitudinal RNA sequencing of the deep transcriptome during neurogenesis of cortical glutamatergic neurons from murine ESCs. F1000Res. 2013;2:35.
- Lotharius J, Falsig J, van Beek J, et al. Progressive degeneration of human mesencephalic neuron-derived cells triggered by dopamine-dependent oxidative stress is dependent on the mixed-lineage kinase pathway. J Neurosci. 2005;25(27):6329–6342.
- Scholz D, Poltl D, Genewsky A, et al. Rapid, complete and large-scale generation of post-mitotic neurons from the human LUHMES cell line. J Neurochem. 2011;119(5):957–971.
- Gonatopoulos-Pournatzis T, Wu M, Braunschweig U, et al. Genome-wide CRISPR-Cas9 Interrogation of Splicing Networks Reveals a Mechanism for Recognition of Autism-Misregulated Neuronal Microexons. Mol Cell. 2018;72(3):510–24 e12.
- Nolte D, Niemann S, Muller U. Specific sequence changes in multiple transcript system DYT3 are associated with X-linked dystonia parkinsonism. Proc Natl Acad Sci U S A. 2003;100(18):10347–10352.
- Herzfeld T, Nolte D, Muller U. Structural and functional analysis of the human TAF1/DYT3 multiple transcript system. Mamm Genome. 2007;18(11):787–795.
- Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136(4):701–718.
- Daguenet E, Dujardin G, Valcarcel J. The pathogenicity of splicing defects: mechanistic insights into pre-mRNA processing inform novel therapeutic approaches. EMBO Rep. 2015;16(12):1640–1655.
- Barbosa-Morais NL, Irimia M, Pan Q, et al. The evolutionary landscape of alternative splicing in vertebrate species. Science. 2012;338(6114):1587–1593.
- Raj B, Blencowe BJ. Alternative Splicing in the Mammalian Nervous System: recent Insights into Mechanisms and Functional Roles. Neuron. 2015;87(1):14–27.
- Vuong JK, Lin CH, Zhang M, et al. PTBP1 and PTBP2 Serve Both Specific and Redundant Functions in Neuronal Pre-mRNA Splicing. Cell Rep. 2016;17(10):2766–2775.
- Muller F, Zaucker A, Tora L. Developmental regulation of transcription initiation: more than just changing the actors. Curr Opin Genet Dev. 2010;20(5):533–540.
- Mal TK, Masutomi Y, Zheng L, et al. Structural and functional characterization on the interaction of yeast TFIID subunit TAF1 with TATA-binding protein. J Mol Biol. 2004;339(4):681–693.
- Bhattacharya S, Lou X, Hwang P, et al. Structural and functional insight into TAF1-TAF7, a subcomplex of transcription factor II D. Proc Natl Acad Sci U S A. 2014;111(25):9103–9108.
- Wang H, Curran EC, Hinds TR, et al. Crystal structure of a TAF1-TAF7 complex in human transcription factor IID reveals a promoter binding module. Cell Res. 2014;24(12):1433–1444.
- Chalkley GE, Verrijzer CP. DNA binding site selection by RNA polymerase II TAFs: a TAF(II)250-TAF(II)150 complex recognizes the initiator. Embo J. 1999;18(17):4835–4845.
- Louder RK, He Y, Lopez-Blanco JR, et al. Structure of promoter-bound TFIID and model of human pre-initiation complex assembly. Nature. 2016;531(7596):604–609.
- Curran EC, Wang H, Hinds TR, et al. Zinc knuckle of TAF1 is a DNA binding module critical for TFIID promoter occupancy. Sci Rep. 2018;8(1):4630.
- Chen X, Hiller M, Sancak Y, et al. Tissue-specific TAFs counteract Polycomb to turn on terminal differentiation. Science. 2005;310(5749):869–872.
- Pointud JC, Mengus G, Brancorsini S, et al. The intracellular localisation of TAF7L, a paralogue of transcription factor TFIID subunit TAF7, is developmentally regulated during male germ-cell differentiation. J Cell Sci. 2003;116(Pt 9):1847–1858.
- Wang PJ, Page DC. Functional substitution for TAF(II)250 by a retroposed homolog that is expressed in human spermatogenesis. Hum Mol Genet. 2002;11(19):2341–2346.
- Herrera FJ, Yamaguchi T, Roelink H, et al. Core promoter factor TAF9B regulates neuronal gene expression. Elife. 2014;3:e02559.
- Buljan M, Chalancon G, Eustermann S, et al. Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol Cell. 2012;46(6):871–883.
- Ellis JD, Barrios-Rodiles M, Colak R, et al. Tissue-specific alternative splicing remodels protein-protein interaction networks. Mol Cell. 2012;46(6):884–892.
- Yang X, Coulombe-Huntington J, Kang S, et al. Widespread Expansion of Protein Interaction Capabilities by Alternative Splicing. Cell. 2016;164(4):805–817.
- Sako W, Morigaki R, Kaji R, et al. Identification and localization of a neuron-specific isoform of TAF1 in rat brain: implications for neuropathology of DYT3 dystonia. Neuroscience. 2011;189:100–107.
- Ito N, Hendriks WT, Dhakal J, et al. Decreased N-TAF1 expression in X-linked dystonia-parkinsonism patient-specific neural stem cells. Dis Model Mech. 2016;9(4):451–462.
- Rusconi F, Paganini L, Braida D, et al. LSD1 Neurospecific Alternative Splicing Controls Neuronal Excitability in Mouse Models of Epilepsy. Cereb Cortex. 2015;25(9):2729–2740.
- Ohnishi T, Shirane M, Nakayama KI. SRRM4-dependent neuron-specific alternative splicing of protrudin transcripts regulates neurite outgrowth. Sci Rep. 2017;7:41130.
- van Nuland R, Smits AH, Pallaki P, et al. Quantitative dissection and stoichiometry determination of the human SET1/MLL histone methyltransferase complexes. Mol Cell Biol. 2013;33(10):2067–2077.
- Calarco JA, Superina S, O’Hanlon D, et al. Regulation of vertebrate nervous system alternative splicing and development by an SR-related protein. Cell. 2009;138(5):898–910.
- Baymaz HI, Spruijt CG, Vermeulen M. Identifying nuclear protein-protein interactions using GFP affinity purification and SILAC-based quantitative mass spectrometry. Methods Mol Biol. 2014;1188:207–226.
- Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2007;2(8):1896–1906.
- Smits AH, Jansen PW, Poser I, et al. Stoichiometry of chromatin-associated protein complexes revealed by label-free quantitative mass spectrometry-based proteomics. Nucleic Acids Res. 2013;41(1):e28.
- Vizcaino JA, Csordas A, Del-Toro N, et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44(22):11033.